

Abstract
Motivation
Chromatin accessibility plays a key role in epigenetic regulation of gene activation and silencing. Open chromatin regions allow regulatory elements such as transcription factors and polymerases to bind for gene expression while closed chromatin regions prevent the activity of transcriptional machinery. Recently, Methyltransferase Accessibility Protocol for individual templates-Bisulfite Genome Sequencing (MAPit-BGS) and nucleosome occupancy and methylome sequencing (NOMe-seq) have been developed for simultaneously profiling chromatin accessibility and DNA methylation on single molecules. Therefore, there is a great demand in developing computational methods to identify chromatin accessibility from MAPit-BGS and NOMe-seq.
Results
In this article, we present CAME (Chromatin Accessibility and Methylation), a seed-extension based approach that identifies chromatin accessibility from NOMe-seq. The efficiency and effectiveness of CAME were demonstrated through comparisons with other existing techniques on both simulated and real data, and the results show that our method not only can precisely identify chromatin accessibility but also outperforms other methods.
Availability and Implementation
CAME is implemented in java and the program is freely available online at http://sourceforge.net/projects/came/
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Chromatin is a fundamental structure for compactly packaging a genome and reducing its volume in eukaryotic cells, and consists of nucleosomes composed of ∼147 bp DNA wrapped around core histone proteins (Richmond and Davey, 2003; Struhl and Segal, 2013). Chromatin accessibility plays a key role in epigenetic regulation of gene activation and silencing. In other words, open chromatin regions (OCRs) allow regulatory molecules such as transcription factors and polymerases to bind for gene expression while closed chromatin regions (CCRs) prevent the activity of the transcriptional machinery. It is well known that chromatin accessibility is highly correlated with DNA methylation and histone modifications such as methylation, acetylation and phosphorylation (Thurman et al., 2012) and is the key ‘on-off’ switch of mRNA transcription. In addition to its role in modulating gene transcription, chromatin accessibility can also affect other cellular processes such as DNA replication and recombination, and RNA splicing (Liu G, et al., 2016). Numerous studies have shown that aberrant alterations in chromatin accessibility cause disease (Hon et al., 2012; Simon et al., 2014; Suvà et al., 2013). For instance, decreased nucleosome occupancy proximal to mis-spliced exons was observed in human kidney tumors carrying mutations in histone H3K36 methyltransferase SETD2 (Simon et al., 2014). The chromatin accessibility changes caused by lack of H3K36me3 correlated with widespread RNA processing defects in kidney tumors. Furthermore, chromatin accessibility mapping revealed subtype-specific epigenome signatures and transcription regulatory networks in chronic lymphocytic leukemia (Rendeiro et al., 2016). Therefore, the identification of chromatin accessibility and nucleosome occupancy and the understanding of the underlying epigenetic mechanism are essential for deciphering the chromatin function in various pathophysiological processes.
With advances in next generation sequencing technologies, chromatin accessibility and nucleosome occupancy (positioning) can be assessed using FAIRE-seq (Giresi et al., 2007), DNase-seq (Song and Crawford, 2010) and MNase-seq (Barski et al., 2007). Recently, Methyltransferase Accessibility Protocol for individual templates-Bisulfite Genome Sequencing (MAPit-BGS) and nucleosome occupancy and methylome sequencing (NOMe-seq) have been developed for simultaneously profiling of chromatin accessibility and DNA methylation on single molecules (Kelly et al., 2012; Pondugula and Kladde, 2008). MAPit-BGS and NOMe-seq use a GpC methyltransferase (M.CviPI; Xu et al., 1998) to methylate GpCs in OCRs followed by bisulfite sequencing that measures the de novo methylation of cytosines by M.CviPI. Since the methylation of GpCs and CpGs represent chromatin accessibility and DNA methylation, respectively, NOMe-seq can footprint active (unmethylated and nucleosome-depleted), repressed (unmethylated and nucleosome-occupied) and silent (methylated and nucleosome-occupied) promoters. Using deep bisulfite sequencing of amplicons or long paired-end sequencing of shotgun libraries, it is possible to detect the minority subpopulations of tumor cells that display different chromatin and DNA methylation profiles from the bulk tumor population using MAPit-BGS and NOMe-seq (Kelly et al., 2012; Nabilsi et al., 2014).
To the best of our knowledge, there is no standard method for de novo identification of chromatin accessibility from NOMe-seq data. Kelly et al. (2012) analyzed nucleosome occupancy only for given gene promoters in the −100 to +50 bps region of transcription start sites by averaging methylation scores of trimer GpCpHs (hereinafter GCHs) and assessing the significance of the average methylation using the binomial test. CpG_MPs (Su et al., 2013) has been developed for identifying unmethylated and methylated regions based on a hotspot extension algorithm from bisulfite sequencing data. Although it was designed for analyzing CpG methylation, it can be easily applied to GpC methylation. Briefly, CpG_MPs divided GpC dinucleotides into four groups based on their methylation scores: unmethylated, partially unmethylated, partially methylated and methylated. Hotspots can be defined as genomic regions that contain more than N unmethylated or methylated GpCs. Then, unmethylated genomic regions are identified by extending hotspots until reaching a methylated or partially methylated GpC. A max-gap-min-run segmentation algorithm was used to identify transcriptionally active regions from tiling microarray data (Cawley et al., 2004; Emanuelsson et al., 2007). We applied it (Emanuelsson et al., 2007) to detect open (closed) chromatin regions from NOMe-seq data by changing the concept of max-gap and min-run. During scanning GpCs from the left to right direction, unmethylated regions can be determined by merging GpCs with smaller (larger) methylation score than a cutoff. GpCs that have larger (smaller) score than a cutoff can be included if they are in a specific distance defined as maxgap. The length of detected regions is required to be greater than a threshold defined as minrun.
In this study, we applied two existing algorithms and present a novel method, CAME, for analyzing NOMe-seq data. CAME uses a seed-extension approach and non-parametric mixture model to identify open and closed chromatin regions. As shown in experimental results with both simulated and real datasets, CAME yielded good results and outperformed the existing approaches.
2 Methods
There are two main steps in CAME: (i) seed detection and (ii) seed extension. In this section, we will describe each step in detail. As mentioned in the previous section, M.CviPI treatment methylates GpC dinucleotides in open chromatin regions (OCRs) while those in closed chromatin regions (CCRs) are unchanged, i.e. unmethylated. Therefore, CCRs and OCRs can be identified by searching unmethylated and methylated regions of GCHs (GpCs not followed by G), respectively. GCGs cannot be used because it is impossible to distinguish whether the methylation of GCGs represents endogenous methylation or is changed by M.CviPI treatment. Note that CpGs in CpG islands are generally unmethylated while those in the other regions are methylated in mammals. In contrast, CpHs, i.e. CpAs, CpCs, or CpTs, are largely unmethylated except in stem cells.
2.1 Seed detection
We first detect seeds to predict CCRs from NOMe-seq. Raw sequence reads from NOMe-seq are mapped to a reference genome and then the methylation score of all GCHs are calculated by dividing the number of methylated reads into the number of methylated and unmethylated reads. Note that a read could not carry a methylation state due to sequencing errors or polymorphisms, i.e. neither C nor T in each strand. Let N denote the total number of GCHs and βi the methylation score of a GCH at position i. Seeds are defined as GCHs with methylation score < a user-defined threshold δ (Supplementary Algorithm 1 and Fig. 1). In other words, a GCH is a seed if the methylation score is smaller than a user defined threshold. Next, since the length of nucleosome is fixed in general, i.e. 147 bp, adjacent seeds are merged if their distance is smaller than a specific distance d. For each merged seed, the methylation score is recalculated by averaging their methylation scores.
Fig. 1.
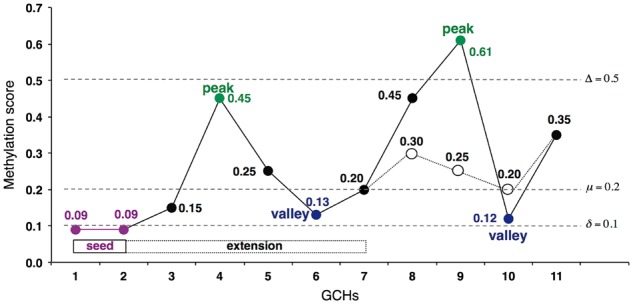
Illustration of seed-extension algorithm. The x-axis indicates 11 GCHs (from GCH1 to GCH11). GCH1 and GCH2 are seeds, merged and extended to GCH7 by the proposed algorithm (Color version of this figure is available at Bioinformatics online.)
2.2 Seed extension
The next step is to determine the boundary of CCRs by extending each seed in the order of ascending average methylation scores (Supplementary Algorithm 2). The main strategy of our extension algorithm is to iteratively search local peaks and valleys to decide whether a seed should be extended or not. A local peak GCHp is defined as the first GCH that βp > βp + 1, whereas a local valley GCHv is defined as the first GCH that satisfies βv < βv + 1. Initially, our extension algorithm searches the nearest local peaks on both left and right directions and compares the methylation score of the peaks to a threshold value. If the score is smaller than a specific threshold Δ, then the average methylation score is examined for extension decision. Otherwise the extension is stopped and the end point is decided. In the former case, the algorithm recalculates the average methylation score of the region from seed to the valley. If the score is smaller than a specific threshold μ, it repeats the procedure by considering the next nearest peak and valley until the extension stop condition is met. Otherwise the extension is stopped. When the extension is stopped, the extended seed includes valleys in the end and is further extended if the next GCHs have lower methylation score than one standard deviation of the methylation scores of GCHs in CCRs from the average methylation score of the seed. Since the standard deviation of methylation scores of GCHs within the CCR is unknown, we apply a non-parametric mixture model based on an expectation-maximization (EM) algorithm to estimate the mean and standard deviation of the methylation scores of GCHs in CCRs and OCRs. For extension decision, another threshold ε can be used to exclude seeds when the average methylation score of the extended region is calculated.
To better understand the extension algorithm, we illustrate the procedure using a simple example. If we have 11 GCHs, denoted as GCH1 to CGH11, which are sorted in ascending order of their genomic positions and all located closely enough, i.e. the distance between adjacent GCHs are smaller than d. The methylation scores of the GCHs are shown in Figure 1. Assuming that the input parameters are δ = 0.1, μ = 0.2, Δ = 0.5, only GCH1 and GCH2 are seeds and merged because their scores are smaller than δ and they are adjacent. The new average methylation score of the seed is 0.09. The nearest local peak is GCH4, and its methylation score is smaller than Δ, i.e. β4 = 0.45 < 0.5. Therefore, the algorithm searches the nearest valley GCH6 and then calculates the average methylation score of the GCH sites that precede it, including the seed sites, which is (0.09 + 0.09 + 0.15 + 0.45 + 0.25 +0.13)/6 = 0.19. Since the average score is smaller than μ = 0.2, the end point is updated from GCH2 to GCH6. In the same manner, GCH9 is selected as the next nearest peak in the second iteration. Since its methylation score β9 = 0.61 is larger than Δ, the extension is stopped and goes to the step to decide the end point. In the other example as drawn in the thin line for GCH8 to GCH11, the next peak GCH8 is smaller than Δ, but the average score to the next valley GCH10, (0.09 + 0.09 + 0.15 + 0.45 + 0.25 + 0.13 + 0.20 + 0.30 + 0.25 + 0.20)/10 = 0.211, is larger than μ. Therefore, the extension is stopped at GCH6. The final step is to decide the end point of the extension using the mean and standard deviation estimated by a non-parametric mixture model. Assuming that those of the methylation scores of GCHs in CCRs are 0.2 and 0.1, respectively, then, the end point is updated as long as the methylation score is smaller than 0.2 + 0.1 = 0.3. In this example, GCH7 is the end point. As a result, the genomic region from GCH1 to GCH7 is the final CCR detected by our algorithm.
2.3 Simulation model
It is challenging to benchmark CCRs and OCRs using real data since there is no gold standard data for the mammalian genome. In addition, robust evaluation measures such as sensitivity and specificity are needed to assess the performance of detection methods. To address these issues, we propose a simulation model. Since methylation scores range from 0 to 1, beta distribution is natural for modeling observed methylation scores. Normal distribution cannot be used to generate the scores because the scores may have the values under 0 or over 1. As shown below, the mean and standard deviation of normal distribution can be defined using the shape parameters of beta distribution to control alpha and beta values for a simulation. For the coordinate of CCRs given, our simulation model generates the methylation scores of GCHs in CCRs and OCRs using and , respectively. The shape parameters were computed from which are input parameters of our simulation model based on the following equations:
| (1) |
| (2) |
where and ( and ) are the mean and standard deviation of the methylation scores of GpCs within OCRs (CCRs), respectively.
2.4 Correlation to DNA methylation
Since chromatin accessibility is highly correlated to DNA methylation, CAME has a function to correlate chromatin accessibility to DNA methylation. To this end, the average methylation of trimer HpCpGs (hereinafter HCGs) in OCRs and CCRs are computed and then used to separate OCRs and CCRs based on thresholds for hyper- and hypo-methylation. Finally CAME outputs 4 groups of regions: hyper-methylated OCRs, hypo-methylated OCRs, hyper-methylated CCRs, and hypo-methylated CCRs, and visualizes them using smoothed scatter plots. Those groups can be further analyzed for functional analysis using DAVID (Huang et al., 2008) and Panther (Thomas et al., 2003).
3 Results
3.1 Simulation
To simulate realistic data, input CCRs were derived from micrococcal nuclease (MNase) sequencing data in the NCBI Sequence Read Archive (SRX021427). Raw sequence reads were mapped to the human reference genome (hg19) using Bowtie2 (Langmead and Salzberg, 2012) and then DANPOS (Chen et al., 2013) was used to identify nucleosome positions with parameter -jd 70 (Fig. 2). Finally we chose 550 089 CCRs and 550 088 OCRs in chromosome 1 from this data, resulting in 12 974 716 and 5 800 580 GCHs, respectively. From those CCRs and OCRs, we generated four datasets using the simulation model described in the previous section with different parameters: D1 (, , ,), D2 (, , ,), D3 (, , ,) and D4 (, , ,). Figure 3 shows the density curves of the methylation scores of GCHs for the generated datasets. As shown in the figure, the closer input parameters μO and μC were, the more two groups overlapped. These simulation datasets can extensively assess the performance of CAME from well separated case to highly overlapped case.
Fig. 2.
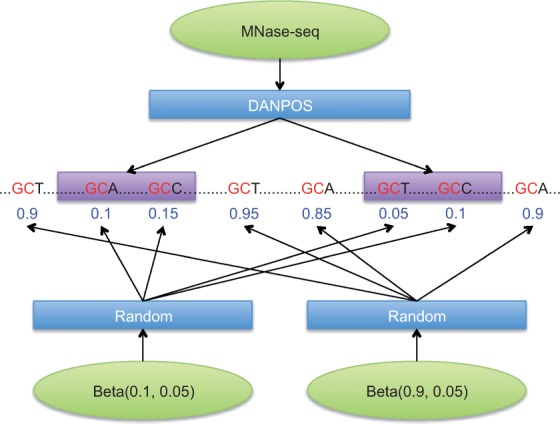
Simulation data was generated from CCRs identified by DANPOS from MNase-seq. The methylation score of GCHs in CCRs and OCRs were randomly chosen based on beta distribution with two means and standard deviations, e.g. (0.1, 0.05) and (0.9, 0.05), respectively (Color version of this figure is available at Bioinformatics online.)
Fig. 3.
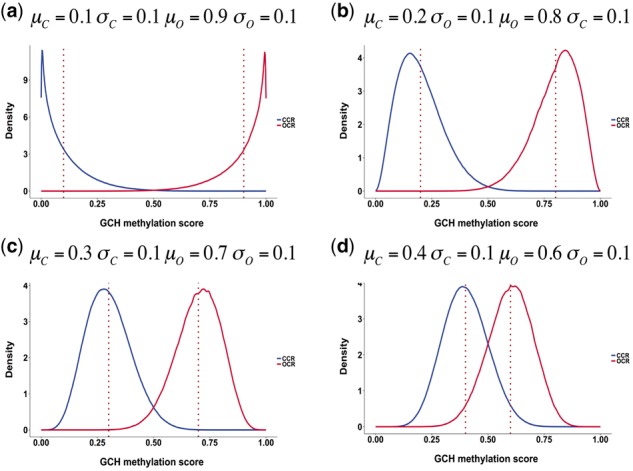
Density curves of the methylation scores of GCHs in CCRs (blue) and OCRs (red) for four simulated datasets with different means and standard deviations. The dotted red lines indicate the true average methylation scores in the two groups (Color version of this figure is available at Bioinformatics online.)
As mentioned in the previous section, CAME decides final extension end points based on sum of mean and standard deviation of the methylation scores of GCHs within CCRs. To estimate these values, we adopted npEM function in the mixtools R package (Benaglia et al., 2009). Table 1 shows the estimated mean and standard deviation of the methylation scores of GCHs in both CCRs and OCRs. The estimation results were accurate with error range ±0.02 compared with actual values, confirming that the EM-based non-parametric mixture model used in our algorithm accurately predicts the true statistics of GCH methylation. Similar results can also found in the estimation for the OCRs.
Table 1.
Estimated mean (Ave.) and standard deviation (Std.) of the methylation of scores of GCHs in CCRs and OCRs
| CCRs |
OCRs |
|||
|---|---|---|---|---|
| Ave. | Std. | Ave. | Std. | |
| D1 | 0.0980 | 0.0967 | 0.8990 | 0.1040 |
| D2 | 0.2000 | 0.0979 | 0.8010 | 0.1010 |
| D3 | 0.2920 | 0.0909 | 0.6920 | 0.1010 |
| D4 | 0.3880 | 0.0838 | 0.5960 | 0.0922 |
We first evaluated the performance of CAME by individual GCHs in terms of sensitivity and specificity on simulated datasets. True positives (TPs) and false negatives (FNs) represent correct prediction and wrong rejection of GCHs because they are in CCRs, respectively. True negatives (TNs) and false positives (FPs) represent correct rejection and wrong prediction of GCHs because they are in OCRs, respectively. Sensitivity, specificity and accuracy are defined as TP/(TP + FN), FP/(FP + TN) and (TP + TN)/(TP + FP + FN + TN), respectively. Table 2 shows a contingency table on different datasets with parameter d = 150, Δ = 0.6 and (δ, ε) = (0.1, 0.4), (0.2, 0.5), (0.3, 0.5) and (0.4, 0.5) for D1, D2, D3 and D4 respectively. While the sensitivities were 0.9954, 0.9964, 0.9941 and 0.9470, the specificities were 0.9999, 0.9997, 0.9928 and 0.9271 for D1, D2, D3 and D4, respectively (Supplementary Table S1 and Supplementary Fig. S1). The sensitivities were very high (>97%) for all the datasets and the specificities were also extremely high for all the datasets except D4 which is the most challenging dataset because the distribution of the methylation scores of GCHs in CCRs and OCRs overlap much (Fig. 3(d)). It makes our method difficult to accurately predict the regions. Then we performed ROC (Receiver Operating Characteristic) analysis and compared the results with that of CpG_MPs and max-gap-min-run. Figure 4 shows the ROC curves of three methods for different datasets. The curves were generated using ROCR R package (Sing et al., 2005). From the figure, we can easily see that CAME significantly outperforms the other two methods on all datasets.
Table 2.
Contingency table. The sensitivities were 0.9954, 0.9964, 0.9941 and 0.9470, and the specificities were 0.9999, 0.9997, 0.9928 and 0.9271 for D1, D2, D3 and D4, respectively
| D1 | Condition |
D2 | Condition |
||||
|---|---|---|---|---|---|---|---|
| CCR | OCR | CCR | OCR | ||||
| Prediction | CCR | 12 915 487 | 537 | Prediction | CCR | 12 928 007 | 1739 |
| OCR | 59 229 | 5 800 043 | OCR | 46 709 | 5 798 841 | ||
| D3 |
Condition |
D4 | Condition |
||||
|---|---|---|---|---|---|---|---|
| CCR | OCR | CCR | OCR | ||||
| Prediction | CCR | 12 897 794 | 41 893 | Prediction | CCR | 12 287 053 | 422 909 |
| OCR | 76 922 | 5 758 687 | OCR | 687 663 | 5 377 671 | ||
|
| |||||||
Fig. 4.
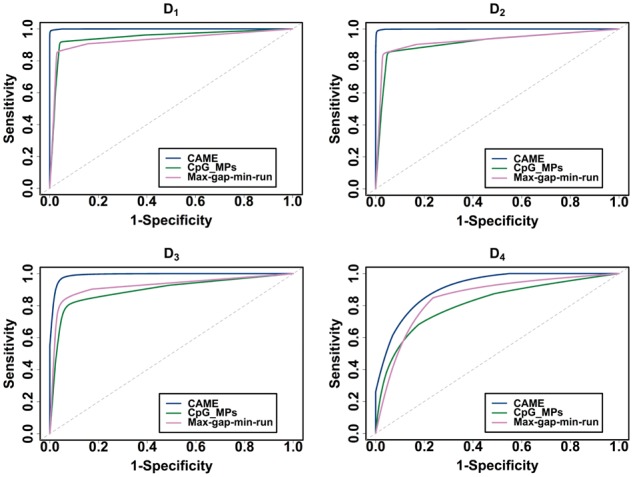
ROC curves of CAME, CpG_MPs and max-gap-min-run on different datasets (Color version of this figure is available at Bioinformatics online.)
Next, we examined how much predicted CCRs and OCRs overlap with true regions identified from MNase-seq. Figure 5 illustrates the histogram of overlapping fraction of CCRs detected by CAME (blue), CpG_MPs (green) and max-gap-min-run (pink) to true CCRs (top row) and true OCRs (bottom row) on four simulated datasets. The overlapping fraction was calculated as the length of overlapping region divided by the length of true region using BEDTools (Quinlan and Hall, 2010). Thus, a value of 1 indicates perfect overlap while a value of 0 means no common regions. As shown in the top plots, the number of complete overlapping (fraction 1) regions derived from CAME is higher than that of CpG_MPs and max-gap-min-run on all the simulated data, which verifies CAME more accurately identifies CCRs than the other two methods. However, the result of overlapping fraction between predicted CCRs to true CCRs was affected by the length of the predicted region. In particular, longer predicted regions result in higher counts for complete overlap, i.e. overlapping fraction will be 1 for all CCRs if all of GCHs are merged into one region. To avoid this issue, we also investigated the overlapping fraction between CCRs to true OCRs, which was shown in bottom plots. It is observed that most of CCRs predicted by CAME do not overlap with OCRs while that of the other two methods do overlap.
Fig. 5.

Histogram of overlapping fraction of CAME (blue), CpG_MPs (green) and max-gap-min-run (pink) on four simulated datasets. The top figures indicate results between predicted CCRs and true CCRs, the bottom figures show the results between predicted CCRs and true OCRs (Color version of this figure is available at Bioinformatics online.)
Finally, we tested and evaluted the algorithm with different detection strategies and thresholds. Supplementary Table S1 shows the results using CCR (left) and OCR (right) detection methods, which first detect CCRs and OCRs, respectively, using the algorithm explained in Section 2. There was trade-off between sensitivity and specificity, and overall performance in terms of accuracy was quite similar. The average methylation score of extended regions μ and jump methylation score Δ were tested. The sensitivity and specificity was not changed much.
3.2 hct116
We also tested CAME on our own NOMe-seq and BS-seq data from the human HCT116 cell line, a colon cancer cell line widely used by cancer researchers. Colon cancer cell line, HCT116, was treated GpC methyltransferase (M.CviPI) to methylate all GpC sites in the open chromain. The genomic DNA was then analyzed by targeted bisulfite sequencing. Oligo capture probes are designed to target 84 Mb sequences covering 3.7 million CpGs including CpG islands, cancer- or tissue-specific DMRs, Gencode promoters, DMRs or regulatory feature in CpG shores and shelves, DNase I hypersensitive sites, Refseq genes and Ensembl regulatory features. This dataset, which was deposited to NCBI GEO (GSE86823), is very useful because it includes M.CviPI-treated and -untreated data for the same cell line. Sequencing reads were mapped to hg19 using BWA (Li and Durbin, 2010) with in silico conversion. As a result, 21 578 352 GCHs were reported in the untreated data and 46 637 807 GCHs were reported in the treated data. Among them, 10 750 538 GCHs were found in both the untreated and treated sample (Table 3). Note that we only considered the common GCHs since we cannot determine the methylation changes for GCHs that are not found in both samples. Thus, the gold standard chromatin accessibility was determined as follows. If the methylation score difference of a GCH between enzyme treated and control was 0.2 or less, it was considered to be unmethylated, thus belonging to the CCRs, otherwise, it was considered to be methylated, and belonging to the OCRs. Subsequently, as shown in Table 4, 74% of GCHs belong to CCRs and 26% of GCHs belong to OCRs in chromosome 1. We chose the methylation score 0.2 as the methylated and unmethylated boundary for gold standard construction since the proportion of sequences within nucleosomes on chromosome 1 from the MNase-seq was approximately 70%, which was similar to our result of 74%.
Table 3.
Summary of HCT116 colon cancer data
| # of GCHs in chr1 | # of GCHs in all chr | |
|---|---|---|
| Control | 1 992 885 | 21 578 352 |
| Enzyme treated | 3 975 423 | 46 637 807 |
| Common | 1 044 073 | 10 750 538 |
Table 4.
Brief summary of gold standard for chr1
| # of GCHs | Percentage | |
|---|---|---|
| CCRs | 772 569 | 74% |
| OCRs | 271 504 | 26% |
Then, we ran CAME only on the enzyme treated data with parameters d = 150, Δ = 0.25, δ = 0.15, μ = 0.2 and ε = 0.2 and assessed the performance in terms of sensitivity and specificity based on the gold standard, and then compared the results with those of CpG_MPs and max-gap-min-run. Figure 6 shows the results of three methods evaluated by individual GCHs. The sensitivity and specificity of CAME were found to be 0.985 and 0.960, respectively, which was significantly better than max-gap-min-run. The sensitivity of CpG_MPs was slightly better than ours, however, the specificity was extremely low. This is because the length of regions reported in CpG_MPs was too long, which means it tends to merge too many GCHs into one CCR. Similar results can also be found in region-based evaluation, which is shown in Figure 7.
Fig. 6.
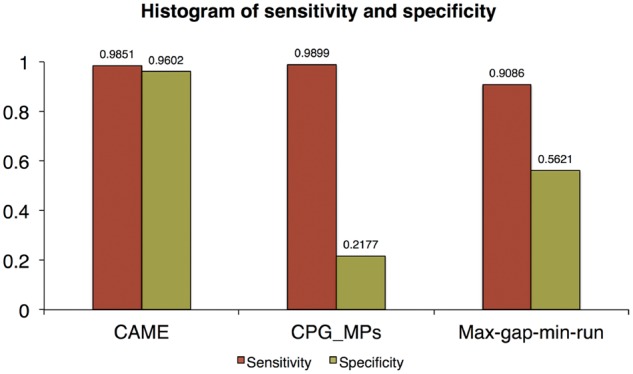
Sensitivity and specificity of three methods on HCT116 colon cancer data (Color version of this figure is available at Bioinformatics online.)
Fig. 7.
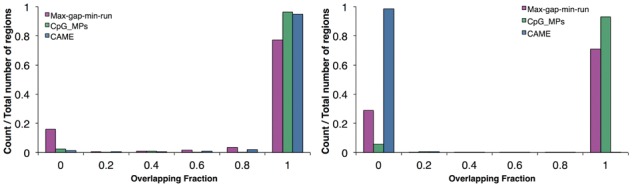
Histogram of overlapping fraction of CAME (blue), CpG_MPs (green) and max-gap-min-run (pink). The left plot indicates the fraction between predicted CCRs and true CCRs, the right one indicates the fraction between predicted CCRs and true OCRs (Color version of this figure is available at Bioinformatics online.)
3.3 imr90
To confirm the reliability of regions identified by CAME, we identified CCRs and OCRs using seed extension in IMR90 NOMe-seq data. The NOMe-seq raw reads on IMR90 cells were downloaded from the Sequence Read Archive (SRX186031), and were mapped to hg19 using BWA (Li and Durbin, 2010) with in silico conversion. We obtained in total 191 962 355 GCHs (15 722 569 for chr1) and identified CCRs with parameter d = 150, Δ = 0.3, δ = 0, μ = 0.1 and ε = 0.1. Then we compared CCR calls with nucleosome positions detected from MNase-seq and OCRs with that called from DNase-seq on the same cell type. The nucleosome positions from MNase-seq were achieved as mentioned in the previous section (see 3.1). The DNase-seq was downloaded from the UCSC genome browser (http://hgdownload.cse.ucsc.edu/goldenPath/hg19/enco deDCC/wgEncodeOpenChromDnase/wgEncodeOpenChromDnaseImr90Pk.narrowPeak.gz). We only examined the results on chromosome 1. Figure 8 shows the overlapping fraction of CCRs from CAME to MNase-seq (left) and OCRs from CAME to DNase-seq (right). From the figure, it is observed that 20% of nucleosome-occupied regions from MNase-seq were the same as OCRs detected by CAME, and 64% overlapped with at least 60% of lengths. Moreover, 30% of OCRs (peaks) from DNase-seq were exactly identified by CAME and 84% overlapped with at least 60% of lengths.
Fig. 8.
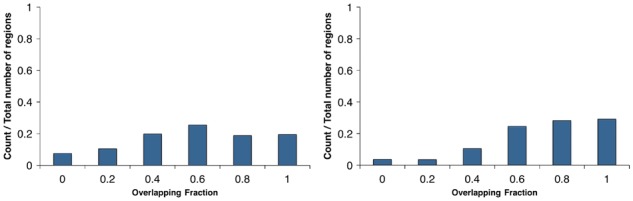
Histogram of overlapping fraction. The left figure shows the overlapping fraction between predicted CCRs by CAME and nucleosome positions from MNase-seq. The right one indicates the fraction between predicted OCRs and DNase-seq OCRs (Color version of this figure is available at Bioinformatics online.)
The predicted OCRs were further compared to nucleosome-depleted CTCF sites, TSS sites, and enhancers published in Kelly et al. As shown in Figure 9B, CAME completely identified all TSS sites and enhancers, and 81% of CTCF sites. Panel (A) shows a screenshot of IGV in chr1:182 359 738–182 373 646 with those tracks while panel (C) shows that most of nucleosome-depleted TSS sites were hypo-methylated.
Fig. 9.
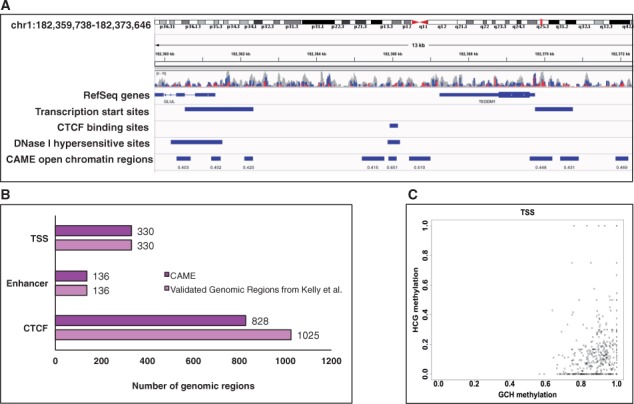
Comparison results of the predicted OCRs by CAME to public datasets. (A) A screenshot of IGV with tracks for nucleosome-depleted TSS and CTCF sites published in Kelly et al., DNase hypersensitive clusters in ENCODE, and OCRs predicted by CAME. (B) CAME can well identify the nucleosome-depleted regions published in Kelly et al. (C) Most of nucleosome-depleted TSS sites were hypo-methylated (Color version of this figure is available at Bioinformatics online.)
The OCRs and CCRs identified by CAME were further correlated to DNA methylation. As shown in Supplementary Figure S4, CCRs and OCRs were clearly separated and their methylation was mostly hyper- and hypo-methylated, respectively. DAVID analysis clearly showed that hyper-methylated CCRs and OCRs, and hypo-methylated CCRs and OCRs were enriched in different biological process based on GO terms (Supplementary Fig. S5).
4 Discussion
Recently, it was reported that nucleosome positioning affects DNA methylation patterns throughout the genome, which means that these two important epigenetic mechanisms are closely associated rather than independent (Chodavarapu et al., 2010; Portela et al., 2013; Taberlay et al., 2014). To this end, MAPit-BGS and NOMe-seq are innovative technologies since they measure DNA methylation and nucleosome occupancy simultaneously at the single molecule level using GpC methyltransferase and bisulfite sequencing.
In this study, we presented a novel algorithm, namely CAME, for identifying chromatin accessibility from MAPit-BGS and NOMe-seq. CAME first identifies seeds that are very likely GCHs in CCR, next extends seeds as long as the average of GCH methylation scores are smaller than a threshold, and finally decides the end point of the extended seeds using the predicted mean and standard deviation of methylation scores based on non-parametric mixture model. CAME also has function to correlate predicted chromatin accessibility to DNA methylation. Using different simulated datasets, we demonstrated that CAME was very effective for detecting open and closed chromatin regions, and significantly outperformed existing approaches. In the application to our experimental HCT116 colon cancer cell line dataset, CAME precisely identified most of the important regions verified by experimental data (enzyme treated – control) with >96% of sensitivity and specificity. This represents a crucial achievement since there is no need to make the additional effort to perform biological experiments to generate control data for verification of methylation status changes. Furthermore, comparative analysis of CAME’s results to nucleosome positions from MNase-seq and to accessible regions identified by DNase-seq on IMR90 cell reveals that many detected regions were completely or partially overlapping although these assays have different technical variations, i.e. DNase-seq has a preference for GC-rich regions (Taberlay et al., 2014).
The seed detection cutoff δ in CAME is an important parameter and may directly affect the final results, and thus should be carefully determined. Although we already set reasonable values as default, they may not be always appropriate for all datasets. Based on our experiences, we can make a general recommendation to obtain a reliable result. The seed detection threshold could be set as the predicted mean methylation scores M estimated from mixture model or could be selected from the ranges of M-0.1 to M + 0.1. The seed extension cutoff parameters are also important. We recommend M + S to M + 2S for the average methylation score of extended regions including and excluding seeds, i.e. μ and ε where S represents the standard deviation. CAME is implemented in Java and it takes approximately 6 minutes on genome-wide IMR90 NOMe-seq data with 1.18 billion reads resulted in 191 962 355 GCHs using 2.7 GHz Inter Core i5 CPU and 2 GB of memory running under Mac operating system.
As nucleosomes are dynamic structures (nucleosomes can displace or move along chromosomes), chromatin accessibility is also subject to a dynamic change, which could impact the predictability of the model that we developed (Flores et al., 2014). However, one of the advantages of NOMe-seq is that it reflects the chromatin accessibility at the single molecule level. The patterns of the GpC in a group of sequencing reads present the chromatin accessibility of a group of single cells. Therefore, if there are dynamic changes among populations of cells, NOMe-seq can detect these changes. Our model is well prepared to deal with these possible dynamic changes that may occur in a population of cells.
In summary, there has been increasing interest in determining genome-wide chromatin accessibility for deciphering important epigenetic changes in cell differentiation, environmental signaling and disease development. We believe that the excellent performance of CAME will greatly facilitate the understanding of the roles of chromatin and DNA methylation in various cellular functions and disease processes.
Supplementary Material
Funding
This work was partially supported by the National Institutes of Health [CA114229, AA019976, CA185833 and CA190429], Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (No. 2013R1A2A2A01068923), the MSIP (Ministry of Science, ICT and Future Planning), Korea, under the ITRC (Information Technology Research Center) support program (IITP-2016-H8501-16-1013) supervised by the IITP (Institute for Information & communication Technology Promotion) and Brain Korea 21.
Conflict of Interest: none declared.
References
- Barski A. et al. (2007) High-resolution profiling of histone methylations in the human genome. Cell, 129, 823–837. [DOI] [PubMed] [Google Scholar]
- Benaglia T. et al. (2009) mixtools: an R package for analyzing finite mixture models. J. Stat. Softw., 32, 6. [Google Scholar]
- Cawley S. et al. (2004) Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell, 116, 499–509. [DOI] [PubMed] [Google Scholar]
- Chen K. et al. (2013) Dynamic analysis of nucleosome position and occupancy by sequencing. Genome Res., 23, 341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chodavarapu R.K. et al. (2010) Relationship between nucleosome positioning and DNA methylation. Nature, 466, 388–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuelsson O. et al. (2007) Assessing the performance of different high-density tiling microarray strategies for mapping transcribed regions of the human genome. Genome Res., 17, 886–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores O. et al. (2014) Fuzziness and noise in nucleosomal architecture. Nucleic Acids Res., 42, 4934–4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giresi P.G. et al. (2007) FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res., 17, 877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D.W. et al. (2008) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc., 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Hon G.C. et al. (2012) Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome Res., 22, 246–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly T.K. et al. (2012) Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res., 22, 2497–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. (2012) Fast gapped-read alignment with Bowtie 2. Nat. Methods, 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin H. (2010) Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics, 26, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G. et al. (2016) A deformation energy-based model for predicting nucleosome dyads and occupancy. Sci. Rep., 6, 24133.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabilsi N.H. et al. (2014) Multiplex mapping of chromatin accessibility and DNA methylation within targeted single molecules identifies epigenetic heterogeneity in neural stem cells and glioblastoma. Genome Res., 24, 329–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pondugula S., Kladde M.P. (2008) Single-molecule analysis of chromatin: changing the view of genomes one molecule at a time. J. Cell. Biochem., 105, 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portela A. et al. (2013) DNA methylation determines nucleosome occupancy in the 5'-CpG islands of tumor suppressor genes. Oncogene, 32, 5421–5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan A.R., Hall I.M. (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics, 15, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rendeiro A.F. et al. (2016) Chromatin accessibility maps of chronic lymphocytic leukemia identify subtype-specific epigenome signatures and transcription regulatory networks. Nat. Commun., 7, 11938.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond T.J., Davey C.A. (2003) The structure of DNA in the nucleosome core. Nature, 423, 145–150. [DOI] [PubMed] [Google Scholar]
- Simon J.M. et al. (2014) Variation in chromatin accessibility in human kidney cancer links H3K36 methyltransferase loss with widespread RNA processing defects. Genome Res., 24, 241–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sing T. et al. (2005) ROCR: visualizing classifier performance in R. Bioinformatics, 21, 3940–3941. [DOI] [PubMed] [Google Scholar]
- Song L., Crawford G.E. (2010) DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harbor Protoc., 2, pdb–prot5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl K., Segal E. (2013) Determinants of nucleosome positioning. Nat. Struct. Mol. Biol., 20, 267–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su J. et al. (2013) CpG_MPs: identification of CpG methylation patterns of genomic regions from high-throughput bisulfite sequencing data. Nucleic Acids Res., 41, gks829.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suvà M.L. et al. (2013) Epigenetic reprogramming in cancer. Science, 29, 1567–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taberlay P.C. et al. (2014) Reconfiguration of nucleosome-depleted regions at distal regulatory elements accompanies DNA methylation of enhancers and insulators in cancer. Genome Res., 24, 1421–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman R.E. et al. (2012) The accessible chromatin landscape of the human genome. Nature, 489, 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas P.D. et al. (2003) PANTHER: a browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucleic Acids Res., 31, 334–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M. et al. (1998) Cloning, characterization and expression of the gene coding for a cytosine-5-DNA methyltransferase recognizing GpC. Nucleic Acids Res., 26, 3961–3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.