

Abstract
The inward rectifier potassium (Kir) channel Kir4.1 (KCNJ10) carries out important physiologic roles in epithelial cells of the kidney, astrocytes in the central nervous system, and stria vascularis of the inner ear. Loss-of-function mutations in KCNJ10 lead to EAST/SeSAME syndrome, which is characterized by epilepsy, ataxia, renal salt wasting, and sensorineural deafness. Although genetic approaches have been indispensable for establishing the importance of Kir4.1 in the normal function of these tissues, the availability of pharmacological tools for acutely manipulating the activity of Kir4.1 in genetically normal animals has been lacking. We therefore carried out a high-throughput screen of 76,575 compounds from the Vanderbilt Institute of Chemical Biology library for small-molecule modulators of Kir4.1. The most potent inhibitor identified was 2-(2-bromo-4-isopropylphenoxy)-N-(2,2,6,6-tetramethylpiperidin-4-yl)acetamide (VU0134992). In whole-cell patch-clamp electrophysiology experiments, VU0134992 inhibits Kir4.1 with an IC50 value of 0.97 µM and is 9-fold selective for homomeric Kir4.1 over Kir4.1/5.1 concatemeric channels (IC50 = 9 µM) at −120 mV. In thallium (Tl+) flux assays, VU0134992 is greater than 30-fold selective for Kir4.1 over Kir1.1, Kir2.1, and Kir2.2; is weakly active toward Kir2.3, Kir6.2/SUR1, and Kir7.1; and is equally active toward Kir3.1/3.2, Kir3.1/3.4, and Kir4.2. This potency and selectivity profile is superior to Kir4.1 inhibitors amitriptyline, nortriptyline, and fluoxetine. Medicinal chemistry identified components of VU0134992 that are critical for inhibiting Kir4.1. Patch-clamp electrophysiology, molecular modeling, and site-directed mutagenesis identified pore-lining glutamate 158 and isoleucine 159 as critical residues for block of the channel. VU0134992 displayed a large free unbound fraction (fu) in rat plasma (fu = 0.213). Consistent with the known role of Kir4.1 in renal function, oral dosing of VU0134992 led to a dose-dependent diuresis, natriuresis, and kaliuresis in rats. Thus, VU0134992 represents the first in vivo active tool compound for probing the therapeutic potential of Kir4.1 as a novel diuretic target for the treatment of hypertension.
Introduction
Inward rectifier potassium (Kir) channels Kir4.1 (encoded by KCNJ10) are critical for regulating potassium (K+) transport and electrolyte homeostasis in the central nervous system (CNS), retina, inner ear, and nephrons of the kidney (Hibino et al., 2010; Welling, 2016). In the brain and spinal cord, homotetrameric Kir4.1 channels are expressed almost exclusively in glia, oligodendrocytes, and astrocytes, although some evidence for neuronal expression has been reported (Zhang et al., 2011) (see below). Kir4.1 expression levels are variable between different brain and spinal cord regions, indicating that the contributions of the channel to CNS physiology are complex and anatomically heterogeneous (Nwaobi et al., 2016). Astroglial Kir4.1 channels are best known for their role in clearance, or “spatial buffering,” of extracellular K+ and glutamate in the CNS. By virtue of their prominent membrane expression and high open probability, Kir4.1 channels drive the membrane potential (Vm) of glial cells to negative voltages. When K+ is released from active neurons, thereby locally depolarizing the Nernst equilibrium potential for K+ with respect to the astrocyte Vm, an inwardly directed electrochemical driving force that promotes K+ and glutamate uptake into astrocytes is generated (Kofuji et al., 2002; Djukic et al., 2007; Kucheryavykh et al., 2007; Olsen and Sontheimer, 2008).
Alterations in Kir4.1 expression and function are associated with a number of human diseases (Nwaobi et al., 2016). Autosomal recessive mutations in KCNJ10 cause epilepsy ataxia sensorineural deafness tubulopathy (EAST) or seizures sensorineural deafness ataxia mental retardation electrolyte imbalances (SeSAME) syndrome (Bockenhauer et al., 2009; Scholl et al., 2009; Reichold et al., 2010). The clinical presentation of EAST/SeSAME syndrome is readily explained by the loss of Kir4.1 function in the CNS, inner ear, and kidney. In two separate mouse models of Huntington’s disease (i.e., R6/2 and Q175 mice), the functional expression of Kir4.1 in astrocytes is reduced, leading to elevated extracellular K+ concentration and an increase in neuronal excitability. Viral delivery of Kir4.1 channels to striatal astrocytes prolongs the lifespan and attenuates some of the motor deficits in R6/2 mice (Tong et al., 2014). Epigenetic changes in Kir4.1 expression have been linked to Rett syndrome, a neurologic disorder that affects cognitive, sensory, motor, and autonomic functions (e.g., cardiac function, digestion, and breathing). In most cases, Rett syndrome is caused by mutations in the methyl CpG binding protein 2 (MECP2) gene located on the X chromosome, which is responsible for the transcriptional regulation of dozens of genes, including KCNJ10 (Nwaobi et al., 2016). Zhang et al. (2011) proposed that the overexpression of Kir4.1 homotetrameric channels in respiratory-related neurons from MecP2 mice leads to a reduction in CO2/pH chemosensitivity and disruption of normal breathing. This overexpression could be restricted to respiratory neurons since recent data from Kahanovitch et al. (2018) suggest that there is a reduction in glial cell Kir4.1 from MeCP2 mice. An emerging body of literature has implicated Kir4.1 in autism spectrum disorder, sudden infant death syndrome, epilepsy, pain, and multiple sclerosis (Sicca et al., 2011, 2016), although in most cases a clear mechanistic link between the channel and these diseases has not yet been established.
In polarized epithelial cells of the distal convoluted tubule (DCT) and cortical collecting duct (CCD), Kir4.1 is expressed on the basolateral (i.e., blood-facing) membrane, predominantly in a heteromeric complex with Kir5.1 (encoded by KCNJ16) (Lourdel et al., 2002; Zhang et al., 2014; Welling, 2016). Kir5.1 does not produce functional K+ channels on its own, but serves to modify the functional (e.g., unitary conductance, activation kinetics, rectification) and regulatory (e.g., pH sensitivity) properties of Kir4.1-containing channels (Tucker et al., 2000; Pessia et al., 2001; Paulais et al., 2011). In the DCT, Kir4.1/5.1 channels: 1) recycle K+ ions across the basolateral membrane to sustain the activity of the basolateral Na+/K+/ATPase, which maintains a favorable chemical driving force for Na+ reabsorption across the luminal (urine-facing) membrane by the Na+/Cl− cotransporter NCC; and 2) create a negative Vm that promotes Cl− exit across the basolateral membrane. In the CCD, basolateral Kir4.1/5.1 and Kir4.1 channels hyperpolarize the basolateral Vm and electrically coupled luminal membrane to promote Na+ reabsorption via the epithelial Na+ channel (ENaC). Thus, Kir4.1-containing channels play key roles in promoting NaCl reabsorption in the DCT and CCD of the nephron (Welling, 2016).
The lack of specific pharmacological tool compounds has slowed progress in exploring the function, integrative physiology, and, in some cases, therapeutic potential of Kir4.1, leading us to carry out a high-throughput screen of 76,575 compounds for pharmacological modulators of Kir4.1. Here we report the discovery and characterization of VU0134992, the first in vivo active inhibitor of renal Kir4.1 channels.
Materials and Methods
Stable Cell Lines and Transient Transfection
Stably transfected monoclonal T-Rex-human embryonic kidney 293 (HEK-293) cell lines expressing Kir1.1, Kir2.2, Kir2.3, Kir4.1, Kir4.2, Kir6.2/sulfonylurea receptor 1 (SUR1), or Kir7.1-M125R from a tetracycline-inducible promoter were generated as described previously (Lewis et al., 2009; Raphemot et al., 2011, 2013; Swale et al., 2016). To promote cell surface expression and the function of Kir4.2, lysine 110 was mutated to asparagine and the most distal 22 amino acids were deleted from the C terminus (Kir4.2-K110N-Δ22), as described by Pearson et al. (2006). The day before Tl+ flux assays, cells were plated at a density of 20,000 cells/well in black-walled, clear-bottomed, amine-coated, 384-well plates (Purecoat; Corning, Corning, NY) in the presence of 1 µg/ml tetracycline to induce Kir channel expression. Constitutively expressing Kir3.1/3.2, Kir3.1/3.4 cells were used without any induction. Kir6.2/SUR1 was activated with 30 µM VU063 (Raphemot et al., 2014) for testing inhibitors. Transient transfections were performed using Lipofectamine-LTX (Thermo Fisher Scientific, Waltham, MA) according the manufacturer instructions. The pcDNA3.1-Kir4.1-5.1 concatemer plasmid was obtained from Chung Jiang (Georgia State University, Atlanta, GA) with permission from John Adelman (Oregon Health & Science University, Portland, OR).
Kir4.1 Primary Screen
Tl+ flux assays were performed as described previously (Lewis et al., 2009; Raphemot et al., 2011, 2013; Swale et al., 2016) with modifications to reagents optimized per cell line as noted below. Briefly, cells cultured overnight 37°C and 5% CO2 in 384-well plates were loaded with the thallium-sensitive dye Thallos-AM (TEFLabs, Austin, TX) in ambient conditions, washing before and after dye using Hanks’ balanced salt solution/20 mM HEPES (assay buffer). Media and buffer exchange was performed on the ELx405 plate washer (BioTek, Winooski, VT). Dye-loaded cells were then transferred to a Functional Drug Screening System 6000 (model FDSS6000; Hamamatsu, Tokyo, Japan) or Panoptic Kinetic Imaging Plate Reader (Wavefront Bioscience, Franklin, TN). Both instruments collect live measurements at 1 Hz (480/40 nm excitation and 540/40 nm emission) during simultaneous 384-well pipetting of 10 µM small molecules (0.1% dimethylsulfoxide assay buffer) or control (100 µM fluoxetine). Cells are treated 20 minutes, continuing ambient conditions, before adding 5× Tl+ stimulus buffer (125 mM NaHCO3, 1 mM MgSO4, 1.8 mM CaSO4, 5 mM glucose, 20 mM HEPES, and 1.8–6 mM Tl2SO4) to each well to initiate Tl+ flux. Screening data were collected into a custom database, and data reduction software was applied to merge individual compounds with the associated values on a well-to-well, then plate-by-plate basis. Fluorescence values for individual wells were normalized by dividing each data point for that well by the initial data point (F/F0). After normalization of all wells, the fluorescence data (i.e., fluorescence vs. time) from each of the designated vehicle-control wells were averaged, and this averaged control wave was subtracted from all wells on the plate to yield normalized, vehicle control–subtracted data. Fluorescence amplitudes (maximum-minimum) were extracted from the control-subtracted wells over a range from 0 to 200 seconds. Alternatively, the slope 5–20 seconds after Tl+ addition was calculated. Hit selections were made by analysis on a per plate basis evaluating 3 S.D. values from the mean of the compound population. Compounds that were tested in dose response were calculated as above, and reduced values were then plotted with GraphPad Prism version 5.01 (GraphPad Software, San Diego, CA) to generate estimated potencies of concentration-response curves (CRCs) fit using a four-parameter logistic model of nonlinear regression analysis.
Site-Directed Mutagenesis
Mutations in Kir4.1 were introduced using a QuickChange II Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA) according to the manufacturer instructions. The complete cDNA was sequenced to verify the introduction of the intended codon changes.
Patch-Clamp Electrophysiology
HEK-293T cells were transfected with wild-type (WT) or mutant pcDNA5-Kir4.1 (1 µg) and pcDNA3.1-EGFP (0.5 µg; transfection marker) using Lipofectamine LTX reagent according to the manufacturer’s instructions. The cells were dissociated the following day and plated on poly-l-lysine–coated coverslips and allowed to recover for at least 1 hour in a 37°C 5% CO2/95% air incubator before starting experiments. Patch-clamp experiments were performed essentially as described previously (Raphemot et al., 2013). Briefly, patch electrodes (2–3 MΩ) were filled with an intracellular solution containing 135 mM KCl, 2 mM MgCl2, 1 mM EGTA, 10 mM HEPES-free acid, and 2 mM Na2ATP (Roche Diagnostics, Risch-Rotkreuz, Switzerland), pH 7.3, 275 mOsmol/kg water. The standard bath solution contained 135 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 5 mM glucose, and 10 mM HEPES free acid, pH 7.4. Macroscopic currents were recorded under whole-cell voltage-clamp conditions using an Axopatch 200B Amplifier (Molecular Devices, Sunnyvale, CA). Cells were voltage clamped at a holding potential of −75 mV and stepped every 5 seconds to −120 mV for 200 milliseconds before ramping to 120 mV at a rate of 1.2 mV/ms. Data were collected at 5 kHz and filtered at 1 kHz. Data acquisition and analysis were performed using the pClamp 9.2 software suite (Molecular Devices). Pharmacology experiments were terminated by applying 2 mM barium (Ba2+) chloride to measure leak current. Cells exhibiting <90% block by Ba2+ were excluded from analysis. The mean current amplitude recorded over five successive steps to −120 mV in WT controls or mutants at a single concentration were expressed as the mean ± S.D. Statistical analysis was performed using one-way analysis of variance with Bonferroni multiple-comparisons test with statistical significance defined at P < 0.05. IC50 values were determined by fitting the Hill equation to CRCs using variable-slope nonlinear regression analyses. All the analyses were performed with GraphPad Prism version 5.01 (GraphPad Software).
Homology Modeling of Kir4.1
The Kir4.1 sequence (residues 28–360) was threaded onto the Kir2.2 crystal structure (PDB 3JYC) based on a sequence alignment generated by ClustalW. The sequence identity between Kir4.1 and Kir2.2 is 43.3%. Transmembrane segments for Kir4.1 were predicted using the OCTOPUS topology prediction web server (http://octopus.cbr.su.se/). Missing coordinates in the threaded Kir4.1 model were reconstructed using Rosetta with fragment insertion from the fragment libraries generated by the Rosetta server (Leaver-Fay et al., 2011). The modeling pipeline used RosettaMembrane (Barth et al., 2007) and RosettaSymmetry (King et al., 2012) in the Rosetta revision 58019. Loops were closed using the cyclic coordinate descent algorithm and refined using kinematic loop closure from the Rosetta Loop Modeling application (Mandell et al., 2009). One thousand models were generated, and the top eight models by score and root mean square deviation to Kir2.2 (models 1–8) were further relaxed using FastRelax in the Rosetta relax application producing 100 models each. The top three models from parent models 1, 2, 3, 5, and 7 were chosen for ligand-docking studies.
Docking VU0134992 in the Kir4.1 Channel Pore
VU0134992 was manually placed in a coordinate frame that corresponds to the pore cavity below the selectivity filter of Kir4.1. VU0134992 conformers were generated using BCL::Conf (Kothiwale et al., 2015). The top 15 homology models described above were used for ligand-binding studies with RosettaLigand (Meiler and Baker, 2006), producing 7500 VU0134992-Kir4.1 complexes. The top 10% of 7500 models as determined by Rosetta interface score were analyzed for favorable residue interactions (better than −1 Rosetta Energy Unit) and highest frequency interaction between residues of Kir4.1 and VU0134992.
Chemical Synthesis
Synthesis and Characterization of VU0134992.
The synthetic scheme of VU0134992 is shown in Supplemental Fig. 1 as an example of general synthetic scheme. The experimental procedure for VU0134992 is described below. Specific synthetic schemes for selected compounds are also shown in Supplemental Material (Supplemental Figs 2 and 3).
2-Bromo-4-isopropylphenol (2).
To a solution of 4-isopropylphenol (2.00 g, 14.7 mmol) in CH3CN (30 ml) was added N-bromosuccinimide (2.88 g, 16.2 mmol) at 0°C. After a resulting greenish solution was stirred at 0°C for 1.5 hours, ice/NaHCO3-aqueous (aq) was added to the reaction mixture, which was extracted with ethyl acetate twice. Combined organic extracts were washed with brine and dried over MgSO4. The filtrate was evaporated under reduced pressure to give crude residue, which was purified on silica gel chromatography (hexane/ethyl acetate) to yield 2-bromo-4-isopropylphenol 2 [723 mg (content 618 mg), 20% yield] as a pale yellow oil. Crude fraction collected was purified by a high-performance liquid chromatography separation system (Gilson, Middleton, WI) using (0.1% trifluoroacetic acid in water)/CH3CN as an eluent. Extraction from the collected fraction with dichloromethane (DCM) gave another batch of 2 [1.06 g (content 1.02 g), 32% yield] as a pale reddish oil. The total yield was 52%.
2-Chloro-N-(2,2,6,6-tetramethylpiperidin-4-yl)acetamide hydrochloride (4).
To a solution of 4-amino-2,2,6,6-tetramethylpiperidine 3 (2.345 g, 15.0 mmol) in DCM (30 ml) was added chloroacetyl chloride (1.19 ml, 15.0 mmol) at 0°C. After a resulting white suspension was stirred at 0°C for 40 minutes, precipitates were collected by filtration to yield 2-chloro-N-(2,2,6,6-tetramethylpiperidin-4-yl)acetamide hydrochloride 4 [4.052 g (content 3.908 g), 97% yield].
VU0134992.
To a solution of 2 (935 mg, 4.35 mmol) in dimethylformamide (30 ml) was added 4 (975 mg, 3.62 mmol), Cs2CO3 (2.95 g, 9.05 mmol), and NaI (179 mg, 1.19 mmol) at ambient temperature. After a resulting white suspension was stirred at 50°C for 18 hours, the reaction mixture was poured into cold 1 mol/l NaOH-aq (120 ml), and it was extracted with DCM three times (first, 50 ml; second, 50 ml; third, 30 ml). Combined organic extracts were washed with brine (40 ml) and dried over MgSO4. The filtrate was evaporated under reduced pressure. The residue was purified by the Gilson high-performance liquid chromatography separation system using (0.1% trifluoroacetic acid in water)/CH3CN as an eluent. Desired fractions were collected and concentrated up to about half the amount. CH3CN was added to the aqueous solution until precipitates were formed (water/CH3CN ratio, about 1:2). After the precipitate was collected by filtration, it was extracted with DCM from 1 mol/l NaOH-aq three times. Combined organic extracts were dried over MgSO4. The filtrate was evaporated under reduced pressure to yield VU0134992 (1.16 g, 78% yield) as a white powder. 1H NMR (400.1 MHz, CDCl3): 7.42 (d, J = 1.7 Hz, 1H), 7.13 (dd, J = 8.4, 1.7 Hz, 1H), 6.80 (d, J = 8.4 Hz, 1H), 6.74 (d, J = 8.0 Hz, 1H), 4.48 (s, 2H), 4.39-4.30 (m, 1H), 2.85 (sept, J = 6.9 Hz, 1H), 1.96-1.92 (m, 2H), 1.27 (s, 6H), 1.22 (d, J = 6.9 Hz, 6H), 1.14 (s, 6H), 1.03-0.97 (m, 2H). 13C NMR (100.6 MHz, CDCl3): 166.87, 151.81, 144.44, 131.41, 126.90, 113.87, 112.10, 68.51, 51.17, 45.27, 42.47, 35.10, 33.38, 28.73, 24.12. liquid chromatography-mass spectrometry: retention time (RT) = 0.912 minute, mass/charge ratio = 411 [M + H]+. High-resolution-mass spectrometry was calculated for C20H31BrN2O2 [M+], 410.1569 (found, 410.1571).
Drug Metabolism and Pharmacokinetics
Detailed methods for in vitro and in vivo drug metabolism and pharmacokinetics analyses are described in Supplemental Material.
Metabolic Cage Studies
All studies involving animals were approved by Institutional Animal Care and Use Committee. Metabolic cage studies were performed as described previously (Kharade et al., 2016). Briefly, male Sprague-Dawley rats (250–300 g) were allowed to acclimatize to single housing in metabolic cages for 2 hours before an experiment. Access to food and water was restricted during the entire experiment. Rats were given drug VU0134992 or vehicle (10% ethanol + 40% PEG400 + 50% saline) by oral gavage (by mouth). After 30 minutes, voiding was induced by giving each animal a saline load (18 ml/kg by oral gavage), and urine was collected at 2-, 4-, and 6-hour time points. Urine volumes were measured, centrifuged, aliquoted, and frozen at −80°C until analyzed. Urine Na+ and K+ concentrations were measured by flame photometry (model 943; Instrumentation Laboratories, Lexington, MA). The total number of moles of Na+ or K+ excreted over the 4 hours was normalized to animal body weight (i.e., µmol/100 g per 4 hours). The data are expressed as the mean ± S.D. and were analyzed using one-way analysis of variance with Bonferroni multiple-comparisons test with statistical significance defined at P < 0.05.
Results
Discovery of VU0134992.
Seventy-six thousand five hundred seventy-five structurally diverse small molecules from the Vanderbilt Institute of Chemical Biology library were screened against Kir4.1 using a Tl+ flux assay that we have described previously (Raphemot et al., 2013). Briefly, the assay reports the inwardly directed movement of Tl+ through the ion conduction pore of Kir4.1 using the intracellular, Tl+-sensitive dye Thallos. The mean Z' plate statistic for the primary screen was 0.78, indicating that the performance of the assay was robust and reproducible, and there was a hit rate of 0.8%. We selected 640 hit compounds resupplied from the vendor for confirmation and counter screening against induced and uninduced cells. Fifty-eight hits inhibited Tl+ flux in tetracycline-induced T-Rex-HEK-293-Kir4.1 cells in duplicate testing at 10 µM. Screening against uninduced cells identified 42 compounds that inhibited endogenous Tl+ flux pathways expressed in T-Rex-HEK-293 cells. This left 16 authentic inhibitors of Kir4.1 (0.02% success rate). We focused on VU0134992 because of its superior potency, selectivity, and chemical tractability. In gold standard patch-clamp electrophysiology experiments, VU0134992 markedly inhibited Kir4.1 at 3 µM with an IC50 value of 0.97 μM [95% confidence interval (CI), 0.5–1.7 µM] (Fig. 1). Because Kir4.1 forms heteromeric channels with Kir5.1 in the kidney (Tucker et al., 2000; Lachheb et al., 2008; Zhang et al., 2014, 2015), we evaluated the sensitivity of Kir4.1–Kir5.1 concatemeric channels to VU0134992. As shown in Fig. 1, VU0134992 inhibits Kir4.1–Kir5.1 concatemeric channels with an IC50 value of 9.05 µM (95% CI, 7.4–11.1 µM) at −120 mV, providing approximately 9-fold selectivity toward Kir4.1 over Kir4.1-5.1 channels at this test potential. The IC50 values for the inhibition of outward currents through Kir4.1 and Kir4.1–Kir5.1 at +120 mV were 1.2 (95% CI, −0.97 to 1.6 µM) and 26.8 µM (95% CI, 18.2–39.8 µM) (22-fold selectivity).
Fig. 1.

VU0134992 exhibits preference for Kir4.1 over Kir4.1-5.1 channels. (A) Chemical structure of VU0134992. (B) Representative traces showing inhibition of Kir4.1 homomeric and Kir4.1-5.1 heteromeric currents recorded in the absence (blue) or presence (red) of 3 µM VU0134992 and 2 mM BaCl2 (black). (C) Mean ± S.D. CRCs of VU0134992 for homotetrameric Kir4.1 and heterotetrameric concatemer of Kir4.1/5.1 currents measured at −120 mV (n ≥ 5). (D) Mean ± S.D. CRCs of VU0134992 for homotetrameric Kir4.1 and heterotetrameric concatemer of Kir4.1/5.1 currents measured at +120 mV (n ≥ 5).
VU0134992 Selectivity.
The selectivity of VU0134992 for Kir4.1 versus nine other members of the Kir channel family was evaluated at concentrations ranging from 0.3 nM to 30 µM in 11-point CRC experiments, using established Tl+ flux assays (Swale et al., 2016). The results are summarized in Table 1. VU0134992 exhibited no apparent activity toward Kir1.1, Kir2.1, or Kir2.2, and caused only partial inhibition of Kir2.3 (73% inhibition at 30 µM), Kir6.2/SUR1 (12% inhibition at 30 µM), and Kir7.1 (15% inhibition at 30 µM). However, upon further evaluation, we found that VU0134992 inhibits Kir3.1/Kir3.2 (92% inhibition at 30 µM, IC50 = 2.5 µM), Kir3.1/Kir3.4 (92% inhibition at 30 µM, IC50 = 3.1 µM), and Kir4.2 (100% inhibition at 30 µM, IC50 = 8.1 µM) with approximately the same efficacy and potency that VU0134992 inhibits Kir4.1 (100% at 30 µM, IC50 = 5.2 µM). The rightward shift in Kir4.1 IC50 values derived from Tl+ flux experiments compared with electrophysiology experiments is commonly observed with other Kir channels (Bhave et al., 2011; Raphemot et al., 2011, 2013; Swale et al., 2016).
TABLE 1.
Comparison of VU0134992 and antidepressant selectivity for Kir4.1 over other Kir channels
Data shown are mean IC50 micromolar values and maximal percentage inhibition at 30 and 90 µM in parentheses derived from Tl+ flux assays. Experiments were performed in triplicate on 2 separate days.
| Channel | VU0134992 | Amitriptyline | Nortriptyline | Fluoxetine |
|---|---|---|---|---|
| Kir1.1 | Inactive | 33.0 (16%, 58%) | 31.0 (6%, 50%) | 27.0 (6%, 30%) |
| Kir2.1 | Inactive | 35.0 (1%, 36%) | Inactive | 10.1 (10%, 86%) |
| Kir2.2 | Inactive | 41.7 (2%, 41%) | Inactive | Inactive |
| Kir2.3 | 13.2 (73%) | 21.3 (53%, 89%) | 19.4 (60%, 91%) | 7.1 (98%, 100%) |
| Kir3.1/3.2 | 2.5 (92%) | 13.6 (73%, 91%) | 16.1 (65%, 87%) | 3.4 (97%, 100%) |
| Kir3.1/3.4 | 3.1 (92%) | 12.0 (67%, 88%) | 15.0 (56%, 80%) | 2.4 (94%, 100%) |
| Kir4.1 | 5.2 (100%) | 89.0 (0%, 66%) | 41.7 (10%, 93%) | 31.2 (35%, 95%) |
| Kir4.2 | 8.1 (100%) | 42.7 (0%, 45%) | 46.7 (0%, 65%) | 20.2 (98%, 99%) |
| Kir6.2/SUR1 | 11.4 (12%) | 32.2 (4%, 64%) | 42.0 (0%, 52%) | 64 (4%, 65%) |
| Kir7.1 | 34.2 (15%) | 22.9 (65%, 89%) | 26.0 (50%, 82%) | 5.8 (75%, 92%) |
The antidepressants amitriptyline, nortriptyline, and fluoxetine have been used to inhibit Kir4.1 activity in heterologous expression and native cell systems (Ohno et al., 2007; Su et al., 2007; Furutani et al., 2009). However, these drugs have never been evaluated for selectivity toward Kir4.1 over several closely related Kir channels in a single, systematic study. Toward this end, and to provide a comparison for VU0134992, we evaluated the potency and efficacy of amitriptyline, nortriptyline, and fluoxetine toward Kir1.1, Kir2.1, Kir2.3, Kir3.1/3.2, Kir3.1/3.4, Kir4.1, Kir4.2, Kir6.1/SUR1, and Kir7.1-M125R, in 11-point CRCs at concentrations up to 90 µM. As summarized in Table 1, all three drugs exhibited weak and nonspecific activity toward most members of the Kir channel family at concentrations that also inhibit Kir4.1. Thus, VU0134992 is more potent and selective than amitriptyline, nortriptyline, and fluoxetine toward Kir4.1 channels.
VU0134992 Interacts with E158 and I159 to Block the Kir4.1 Channel Pore.
A common mechanism by which small molecules inhibit Kir channels is through blockade of the ion-conduction pore (Furutani et al., 2009; Swale et al., 2014, 2016; Kharade et al., 2017). Some pore blockers exhibit voltage-dependent “knockoff” behavior at test potentials more negative than the Nernst K+ equilibrium potential difference. We therefore evaluated the voltage and K+ dependence of VU0134992 inhibition of Kir4.1 to begin identifying the VU0134992 binding site. As shown in Fig. 2A, 30 µM VU0134992 fully inhibited Kir4.1-mediated outward and inward currents across all test potentials when cells were bathed in a solution containing 5 mM K+ and were patch clamped using an internal solution containing 135 mM K+ (Nernst equilibrium potential = −83 mV). However, when the extracellular K+ concentration was raised to 50 mM K+ (Nernst equilibrium potential = −25 mV), we observed a time-dependent knockoff of VU0134992 at strong hyperpolarizing potentials (Fig. 2) and statistically significant reduction of block between −120 and −100 mV (Fig. 2). These data suggested that the VU0134992 binding site is indeed located within the Kir4.1 channel pore.
Fig. 2.

VU0134992 exhibits voltage-dependent block of Kir4.1 (A) Representative whole-cell current responses to voltage steps (1 second) from a holding potential (−80 mV for 5 mM K+ or −25 mV for 50 mM K+) in the presence or absence of 30 µM VU0134992. (B) Percentage of VU0134992 inhibition was calculated at each voltage with 5 and 50 mM extracellular K+. The VU0134992-induced block of Kir4.1 was statistically significantly lower at −120 and −100 mV in 50 mM K+ compared with 5 mM K+ containing bath solution. *P < 0.05, statistically significantly different from the 5 mM K+ extracellular solution control (n ≥ 5).
To identify pore-lining residues that participate in VU0134992 binding in the pore, we generated a comparative homology model of Kir4.1 based on the human Kir2.2 crystal structure and employed in silico docking to identify energetically favorable VU0134992 docking sites within the channel pore. Analysis of the top scoring VU0134992-Kir4.1 complexes by Rosetta interface score identified four pore-lining residues in Kir4.1 that 1) showed high frequency (>10% of 750 models) interactions with VU0134992 and 2) were different between Kir4.1 and Kir1.1. These residues are in the rank order of glutamate (E)158 (39.5% of interactions) > threonine (T)162 (37.3% of interactions) >T155 (10.1% of interactions) > isoleucine (I)159 (10.0% of interactions). The interactions are a combination of hydrogen bonds and van der Waals interactions (Fig. 3A). The homology model in Fig. 3A shows the predicted location of the VU0134992 in the membrane-spanning pore (right), whereas the close-up view (left) identifies close interactions of VU0134992 with E158, the top-scoring residue. Interactions with E158 are dominated by hydrogen bonds. However, several van der Waals contacts exist in addition.
Fig. 3.

E158 and I159 are required for effective block of Kir4.1 by VU0134992. (A) (Left) Kir4.1 homology model showing the location of E158 (green), where it is predicted to interact with VU0134992. (Right) Magnified section of the Kir4.1 homology model (red square on left) showing the interaction of VU013492 with the pore-lining E158 residue in Kir4.1. (B) Mutagenesis studies were carried out to test the prediction of the homology model. Bar graph showing the percentage of inhibition of Kir4.1-WT (solid gray bar), and various mutant channels (colored or open bars) at 3 µM VU0134992. Kir4.1-carrying mutations at the E158 and I159 residue showed statistically significantly reduced sensitivity to VU0134992. (C) Representative traces showing the inhibition of WT and E158N/Q/D and I159S mutants in the absence (blue) or presence (red) of 3 µM VU0134992 and 2 mM BaCl2 (black). (D) VU0134992 CRCs comparing Kir4.1-WT, Kir1.1, and Kir4.1-E158N, E158D, E158Q and I159S mutants at −120 mV. (E) CRCs for the indicated channels measured at +120 mV. *P < 0.05, statistically significantly different from the WT control (n ≥ 5). NF, not functional.
We mutated these four amino acids to the corresponding residues in Kir1.1, which is virtually insensitive to VU0134992 (Fig. 3, B–E), and measured the inhibition of the mutant channels to 3 µM VU0134992 by whole-cell patch-clamp electrophysiology. As shown in Fig. 3B, 3 µM VU0134992 inhibited inward current at −120 mV carried by WT Kir4.1 by 85.9% ± 11% (n = 12). In striking contrast, the mutation of E158 to asparagine (E158N) dramatically reduced the sensitivity of the channel to VU0134992 (0.4% ± 12% inhibition, n = 8; IC50, ∼235 µM with only 38.2% ± 14.7% inhibition at 30 µM) at −120 mV. To determine whether this loss of sensitivity is caused by an alteration of the side chain charge or the structure, we evaluated channels in which E158 was mutated to either glutamine (E158Q) or aspartate (E158D). Both mutant channels exhibited a complete loss of sensitivity in response to 3 µM VU0134992, and a large rightward shift in IC50 (Kir4.1-E158Q = ∼28 mM; Kir4.1-E158D = no fit), suggesting that changes in the side-chain structure at position 158 are sufficient to affect VU0134992 block. Mutation of I159 to serine (I159S) caused a complete loss of block by 3 µM VU0134992 and large rightward shift in the CRC (9.5% ± 7.7%, n = 6; IC50 = ∼199 µM; 77% ± 20% inhibition at 30 µM) (Fig. 3, B and D). Kir4.1-T155V channels were not functional and therefore could not be evaluated. We also mutated nearby residues T162 to cysteine (T162C), T164 to alanine (T164A) and F165 to isoleucine (F165I); however, neither mutation affected VU0134992 sensitivity.
VU0134992 Structure-Activity Relationships.
We employed medicinal chemistry in an effort to improve the potency of VU0134992 and establish its structure-activity relationships (SARs) for the inhibition of Kir4.1. No modifications made to the scaffold of VU0134992 improved its activity toward Kir4.1. It was revealed that VU0134992 bears fundamental components for the inhibition of Kir4.1 channels (Supplemental Tables 1–3). An overview of the SAR is shown in Fig. 4. Although a flat SAR was observed at the 4 position in the left-hand part for inhibitory activity of Kir4.1 with an IC50 of 3.0–9.6 µM in the thallium flux assay [methyl-, ethyl-, isopropyl (VU0134992), tert-butyl, chloro, and phenyl substituents], the monobromine atom at the 2 position was critical for the potency [not only were F, Cl, and H substituents for Br all inactive (no fit), but also the addition of Br and Cl at the 6 position resulted in a notable decrease in potency] (Supplemental Table 1). Although the linker modification, such as the incorporation of dimethyl substituents at α-position of the amide, was tolerated (Supplemental Table 3), all of modifications in the right-hand part made the compound noticeably less active (Supplemental Table 2).
Fig. 4.

Overview of VU0134992 SAR. Inhibition of Tl+ flux toward Kir4.1 was evaluated for each compound synthesized according to Supplemental Figs. 1–3. Specific figures for IC50 values are described in Supplemental Tables 1–3. Veh, vehicle.
VU0134992 Serum Binding, Clearance, and Blood-Brain Barrier Permeability.
VU0134992 displayed a large free unbound fraction (fu) in rat plasma (fu = 0.213), a modest fu in rat brain (fu = 0.013), a moderate predicted hepatic clearance in humans (13.4 ml/min per kilogram), and a high predicted hepatic clearance (60.5 ml/min per kilogram) in rat liver microsomes. A good in vitro/in vivo correlation was noted, with clearance above cardiac output (>1000 m/min per kilogram) and a short half-life (10.6 minutes) driven by a large volume of distribution at steady state (13.5 l/kg). In a standard rat plasma/brain level cassette, which is routinely used to rapidly screen the brain distribution potential of compounds, VU0134992 levels were below the limit of quantification in the brain. We also measured plasma and brain levels after an oral dose of 50 mg/kg, a dosing regimen that was used in subsequent metabolic cage studies (see below). Total VU0134992 brain and plasma concentrations were 0.94 and 0.76 µM [partition coefficient (Kp) = 1.2], respectively, whereas unbound VU0134992 concentrations in brain and plasma were 0.012 and 0.16 µM [unbound Kp (Kp,uu) = 0.08].
Effects of VU0134992 on Renal Fluid and Electrolyte Excretion.
Given the important role of Kir4.1 in renal Na+ reabsorption and K+ secretion, we have postulated that small-molecule inhibitors of Kir4.1 will induce diuresis, natriuresis, and kaliuresis (Denton et al., 2013). To test this hypothesis, we evaluated the effects of VU0134992 on renal function in volume-loaded Sprague-Dawley rats. The rats were administered either vehicle or different doses of VU0134992 followed by volume loading. The effects of VU0134992 on urine output are summarized in Fig. 5. Consistent with the hypothesis, oral administration of VU0134992 led to a statistically significant increase in urine output compared with vehicle at doses of 50 and 100 mg/kg. Urine Na+ and K+ concentrations were measured to determine how VU0134992 alters electrolyte transport along the nephron. As shown in Fig. 5, B and C, VU0134992 statistically significantly (P < 0.05) increased urinary Na+ as well as K+ excretion at doses of 50 and 100 mg/kg compared with vehicle-treated control animals.
Fig. 5.

Dose-dependent effects of VU0134992 on renal excretion. Rats were given oral gavage of either vehicle alone or the indicated doses of VU0134992 followed by saline volume load and were placed in metabolic cages for collection of urine over a 4-hour period. VU0134992 statistically significantly (P < 0.05) increased urine volume (A), urine Na+ (B), and urine K+ (C) at doses of 50 and 100 mg/kg as compared with the vehicle control. *P < 0.05, statistically significantly different from the vehicle control (n ≥ 4).
Discussion
Here we report the discovery and characterization of VU0134992, the first Kir4.1 K+ channel inhibitor to emerge from a high-throughput screening campaign. VU0134992 affords approximately 9-fold to 22-fold selectivity between homotetrameric Kir4.1 and heterotetrameric Kir4.1/5.1 channels when evaluated at −120 and +120 mV, respectively, a property that could be useful for sorting out the relative contribution of these two channel populations in native cells. To our knowledge, VU0134992 is the first subtype-preferring Kir4.1 inhibitor to be reported. Efforts to understand the molecular basis of this selectivity and to identify VU0134992 analogs with improved potency and selectivity toward Kir4.1 and Kir4.1/5.1 channels are underway.
The pharmacology of Kir4.1 has been primarily limited to inorganic cations (e.g., Ba2+, cesium) and antidepressants that have relatively weak off-target activity toward Kir4.1. A summary of published small-molecule Kir4.1 inhibitors, their intended drug targets, and IC50 values for Kir4.1 are shown in Table 2. The tricyclic antidepressant drugs nortriptyline, amitriptyline, desipramine, and imipramine were the first small molecules shown to inhibit Kir4.1 (Su et al., 2007). The IC50 values for inhibition by nortriptyline are 16 and 38 µM at test potentials of +30 and −110 mV, respectively, and the block is relieved upon elevation of the extracellular K+ concentration. Kir4.1 is also inhibited by the selective serotonin reuptake inhibitor drugs fluoxetine, sertraline, and fluvoxamine (Ohno et al., 2007). Similar to nortriptyline, block by fluvoxamine is voltage dependent and reduced by membrane hyperpolarization. Mutational analysis revealed that Kir4.1 inhibition by fluoxetine and nortriptyline requires T128 and E158 (Furutani et al., 2009). More recently, the antimalarial drugs quinacrine and chloroquine were also shown to inhibit Kir4.1 in a voltage-dependent manner involving interactions with T128 and E158 (Marmolejo-Murillo et al., 2017a,b). The most potent intracellular pore blocker of Kir4.1 is currently pentamidine (Aréchiga-Figueroa et al., 2017). When applied to the intracellular face of excised membrane patches, pentamidine inhibits Kir4.1 at +80 mV with an IC50 of 97 nM. Similar to the other inhibitors, Kir4.1 block by pentamidine requires interactions with T127, T128, and E158. Unfortunately, pentamidine is a poor inhibitor of Kir4.1 in the whole-cell configuration (i.e., ∼25% inhibition by 1 µM pentamidine after 20 minutes) and presumably intact cells. Thus, pentamidine is a useful tool for probing the structure of the intracellular pore, but will be limited as an in vitro and in vivo tool compound for exploring the physiology and druggability of Kir4.1. In the present study, we found that the inhibition of Kir4.1 by VU0134992 exhibited voltage-dependent knock-off behavior at hyperpolarized test potentials that is consistent with block of the inner pore cavity. We employed in silico docking calculations to guide mutagenic analysis of potential pore-lining binding sites in Kir4.1. Mutation of two of the top-scoring residues led to a severe (E158N/Q/D, I159S) loss of VU0134992 sensitivity, lending experimental validation for this computational approach to identifying small-molecule binding sites in the Kir4.1 channel pore. Interestingly, VU0134992 inhibited Kir3.1/Kir3.2 and Kir3.1/Kir3.4 channels with similar potencies. Because Kir3.2 and Kir3.4 have asparagine (N) residues at positions equivalent to E158 in Kir4.1, we postulate that this cross-activity is due to D173 in Kir3.1 (also equivalent to Kir4.1-E158) in heteromeric Kir3.1/Kir3.2 and Kir3.1/Kir3.4 channels.
TABLE 2.
Published Kir4.1 inhibitors
| Compound | Structure | Drug Class | Kir4.1 IC50 |
Reference |
|---|---|---|---|---|
| µM | ||||
| Fluoxetine | 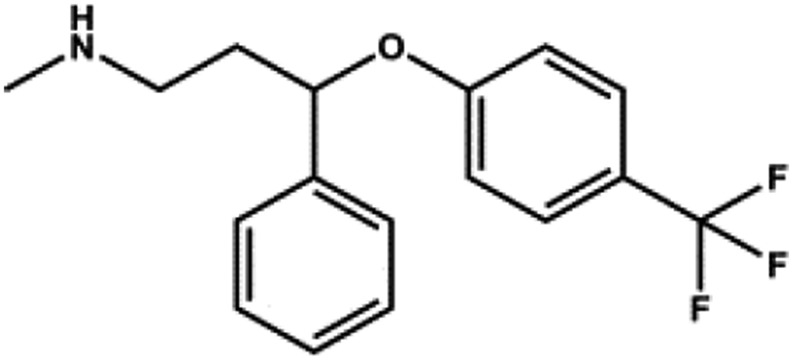 |
SSRI | 15 | Ohno et al. (2007) |
| Amitriptyline | 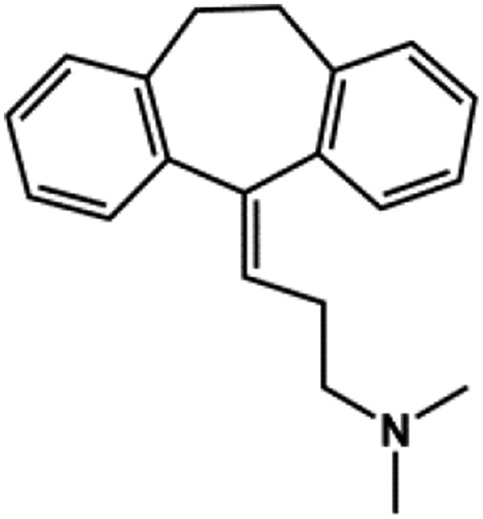 |
TCA | 89 | This study |
| Chloroquine | 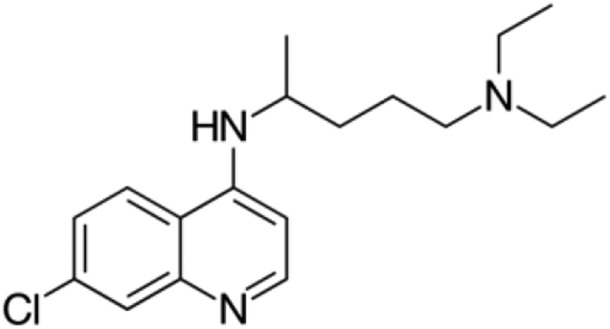 |
Antimalarial | 7 | Marmolejo-Murillo et al. (2017b) |
| Nortriptyline | 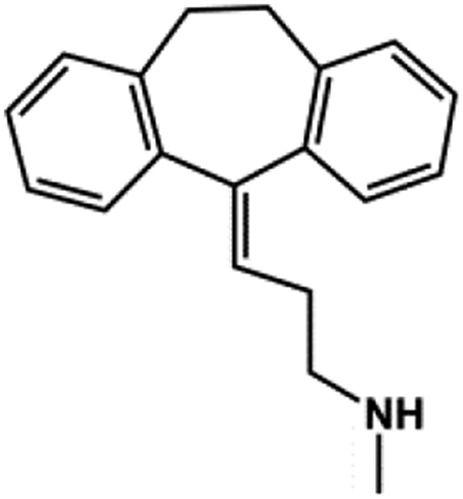 |
TCA | 16–38a | Su et al. (2007) |
| Pentamidine | 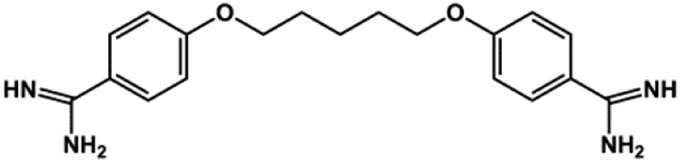 |
Antimicrobial | 0.097b | Takanari et al. (2013), Aréchiga-Figueroa et al. (2017) |
| Quinacrine | 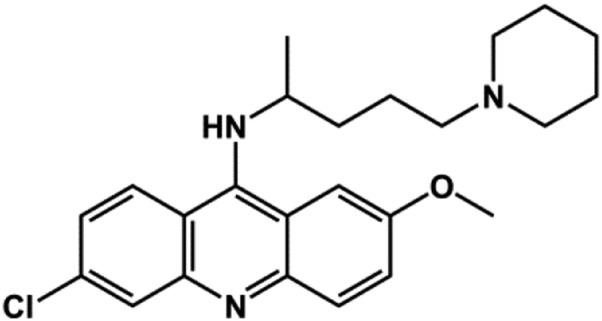 |
Antimalarial | ∼3 | Marmolejo-Murillo et al. (2017a) |
| VU717 | 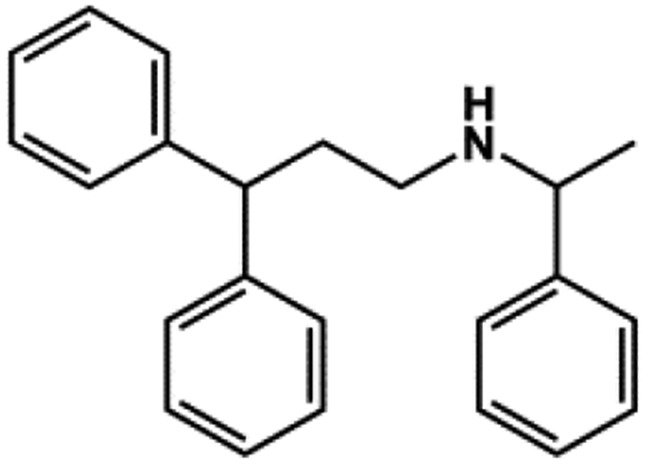 |
None | 6 | Raphemot et al. (2013) |
| VU0134992 | 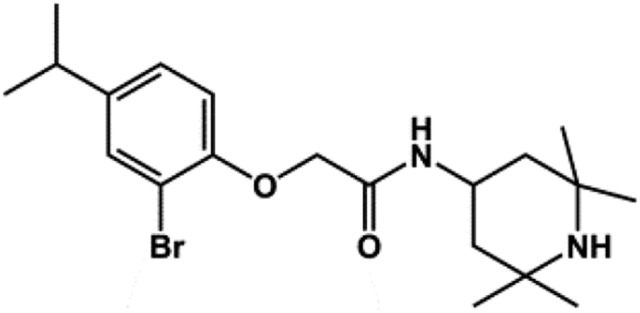 |
None | 0.97 | This study |
SSRI, selective serotonin reuptake inhibitor; TCA, tricyclic antidepressant.
Voltage-dependent block; calculated at +30 and −110 mV, respectively.
Voltage-dependent block; calculated at +80 mV in inside-out patch-clamp experiments.
Kir4.1 was the first K+ channel to be immunolocalized to the basolateral membrane of renal epithelial cells (Ito et al., 1996). The “silent” subunit, Kir5.1, was subsequently colocalized with Kir4.1 in the nephron, which, taken together with heterologous expression experiments showing that Kir4.1 and Kir5.1 form heteromeric channel complexes, suggested that the native channel subtype in the nephron is a Kir4.1-5.1 heteromer. Patch-clamp experiments on isolated nephron segments have identified a predominant ∼40-picosiemens (pS) basolateral K+ current that exhibits biophysical and regulatory properties that resemble heterologously expressed Kir4.1-5.1 (Lachheb et al., 2008) and is absent in KCNJ10 knockout mice (Zhang et al., 2014, 2015), and a less common ∼20 pS current that resembles homomeric Kir4.1 (Zaika et al., 2013; Zhang et al., 2013). Both of these channels appear to play important physiologic roles in the nephron (see below).
The basolateral K+ conductance of the cortical TAL is carried by a 40-pS channel that is likely composed of Kir4.1-5.1 heteromeric channels, and an 80-pS channel possibly carried by Na+-activated K+ channels (Fan et al., 2015; Zhang et al., 2015) (Fig. 6). Genetic ablation of KCNJ10 has no effect on the basolateral Vm in the TAL due to compensatory upregulation of the 80-pS channel (Zhang et al., 2015), suggesting that depolarization of the basolateral Vm in the TAL is unlikely to contribute to salt wasting in KCNJ10 knockout mice and patients with EAST/SeSAME syndrome (Bockenhauer et al., 2009; Scholl et al., 2009). Interestingly, however, the inhibition of the 40-pS channel with Ba2+ reduces the activity of basolateral ClC-Kb Cl− channels through mechanisms that are currently unclear but do not appear to involve membrane depolarization (Fan et al., 2015). These data suggest that the natriuretic and diuretic effects of VU0134992 could result, at least in part, in the inhibition of NaCl reabsorption in the TAL, as follows. NaCl reabsorption from the tubule fluid in the TAL is mediated by the electroneutral, loop diuretic–inhibitable, Na+-K+-2Cl− cotransporter NKCC2 (Fig. 6). Na+ is then pumped across the basolateral membrane through the Na+-K+-ATPase, and Cl− is conducted out of the cell through ClC-Kb Cl− channels. The indirect inhibition of ClC-Kb channels with VU0134992 should increase intracellular Cl− concentrations and thereby reduce the inwardly directed driving force for Cl− and coupled Na+ and K+ transport by NKCC2. In addition, basolateral K+ secretion provides substrate K+ ions that are needed to maintain the activity of the Na+-K+-ATPase. Provided that the 80-pS channel cannot compensate for the pharmacological inhibition of 40-pS Kir4.1-5.1 channels, VU0134992 should also slow the activity of the Na+-K+-ATPase, increase intracellular Na+, and further reduce the chemical driving force for NaCl reabsorption via NKCC2 (Fig. 6).
Fig. 6.

Regulation of renal ion transport by Kir4.1. In the TAL (left panel), heteromeric Kir4.1/5.1 channels recycle K+ across the basolateral membrane to maintain the activity of the Na+/K+-ATPase, which creates a favorable gradient for Na+ and K+ uptake via NKCC2. Kir4.1/5.1 is functionally coupled to basolateral ClC-Kb channels through unknown mechanisms. The inhibition of Kir4.1 should reduce Na+, Cl−, and K+ reabsorption in the TAL. In the DCT (middle panel), basolateral K+ secretion via Kir4.1/5.1 energizes the Na+/K+-ATPase to maintain a favorable chemical driving force for Na+ uptake by NCC. Kir4.1/5.1 also regulates the basolateral Vm and is functionally coupled to ClC-Kb. The inhibition of Kir4.1/5.1 should increase intracellular Cl− concentration and inhibit NCC function via the WNK-SPAK kinase pathway (data not shown; see Discussion). In the CCD (right panel), homomeric Kir4.1 and heteromeric Kir4.1/5.1 channels hyperpolarize the basolateral Vm and the electrically coupled apical Vm, and promote luminal Na+ reabsorption by ENaC. The inhibition of Kir4.1 or Kir4.1/5.1 channels in the CCD should depolarize the Vm and reduce the electrochemical driving force for Na+ reabsorption.
NaCl reabsorption in the DCT is mediated by the luminal membrane, electroneutral, thiazide-sensitive Na+-Cl− cotransporter NCC (Fig. 6). A rapidly developing body of evidence indicates that heterotetrameric Kir4.1-5.1 channels, which dominate the basolateral K+ conductance in the DCT, are critical regulators of NCC expression and activity (Zhang et al., 2014; Penton et al., 2016). Deletion of KCNJ10 in mice depolarizes the basolateral Vm, inhibits the basolateral Cl− conductance, and reduces the expression of NCC (Zhang et al., 2014). Furthermore, Penton et al. (2016) demonstrated that application of Ba2+ to kidney slices induced dephosphorylation and hence the inhibition of NCC. Taken together, these data are consistent with a model in which the inhibition of Kir4.1 activity increases intracellular Cl− concentration, which, in turn, inhibits NCC function through the Cl−-sensitive WNK/SPAK (with no lysine (K) kinase/Ste20-related proline/alanine-rich kinase) kinase pathway (reviewed in Su and Wang, 2016). Ongoing studies are testing whether VU0134992 administration induces the dephosphorylation of NCC in mice.
In the mouse CCD, Kir4.1 is expressed as a common 40-pS Kir4.1/5.1 current and a scarce 20-pS current carried by homomeric Kir4.1 channels (Zaika et al., 2013) (Fig. 6). Although the 40-pS channel is more common, the 20-pS current has a higher open probability and thus likely plays an important role in setting the resting Vm. In principle cells of the CCD, activation of insulin receptors stimulates the activity of Kir4.1-containing channels, which, in turn, increases the electrochemical driving force for Na+ reabsorption by the ENaC. In contrast, the inhibition of Kir4.1 and Kir4.1/5.1 channels by dopamine acting on D2-like receptors leads to reduced Na+ reabsorption via ENaC (Zaika et al., 2013). These data suggest that VU0134992 induces natriuresis at least in part through the inhibition of Kir4.1-containing channels expressed in the CCD (Fig. 6).
The diuretic and natriuretic responses induced by VU0134992 were accompanied by increases in urinary K+ excretion. In this regard, the renal actions of VU0134992 are reminiscent of those caused by loop or thiazide diuretics. The simplest interpretation is that the increased volume and Na+ load delivered to the distal nephron, secondary to the inhibition of Na+ reabsorption in the TAL and/or DCT, stimulates K+ secretion via flow-stimulated large-conductance “BK” K+ channels expressed in the CCD (Liu et al., 2007, 2009). These responses are different from those induced by inhibitors of the renal outer medullary K+ channel [ROMK (or Kir1.1)], another emerging diuretic target in the Kir channel family (Denton et al., 2013). We and others have shown that ROMK inhibition evokes diuresis and natriuresis without causing K+ wasting (Garcia et al., 2014; Kharade et al., 2016; Zhou et al., 2017). The K+-sparing effect appears to result predominantly from the inhibition of K+ secretion in the TAL with a relatively minor effect on distal K+ secretion (Kharade et al., 2016).
In conclusion, we have identified and characterized a moderately potent and selective inhibitor of Kir4.1 that exhibits a preference for the homotetrameric channel over heteromeric Kir4.1-5.1 channels. Oral administration of VU0134992 induces dose-dependent increases in urine volume, Na+ excretion, and K+ excretion in rats. We propose that the observed changes in renal excretion of water and electrolytes results from the inhibition of basolateral Kir4.1-containing channels somewhere along the nephron. The low unbound VU0134992 brain-to-plasma ratio (Kp,uu = 0.08) also supports the idea that the effects of VU0134992 on renal excretion is due to the inhibition of Kir4.1 channels in the kidney and not in the brain. Future studies will be aimed at characterizing the activity of VU0134992 in electrolyte transport in the TAL, DCT, and CCD. The data presented herein raise the intriguing possibility that the inhibition of basolateral Kir4.1-containing channels could be an effective way to lower blood pressure in hypertensive and heart failure patients. Targeting a basolateral membrane protein could provide therapeutic advantages over conventional diuretics. For example, loop (furosemide and bumetanide) and thiazide diuretics must be secreted into the tubule fluid before reaching their respective luminal target sites in the TAL and DCT (Uwai et al., 2000; Hasannejad et al., 2004; Hasegawa et al., 2007; Vallon et al., 2008). In clinical settings where diuretic secretion is compromised, such as chronic kidney disease or when other drugs compete for renal tubule secretion, the availability of a diuretic that acts basolaterally and independently of renal secretion would be more beneficial compared with conventional diuretics. Furthermore, a recent study by Rao et al. (2017) demonstrated that a majority of loop diuretic resistance cases in their cohort of patients with heart failure resulted from enhanced distal tubule Na+ reabsorption. The authors suggested that the administration of thiazide diuretics or a combination of thiazide and K+-sparing diuretics should help overcome loop diuretic resistance in some patients (Rao et al., 2017). An inhibitor of Kir4.1-containing channels would be potentially superior to thiazide or K+-sparing diuretics because it would simultaneously inhibit Na+ reabsorption all along the nephron without sparing K+. The development of VU0134992 provides the first in vivo active tool compound to enable these and other hypotheses to be tested.
Abbreviations
- aq
aqueous
- Ba2+
barium
- CCD
cortical collecting duct
- CI
confidence interval
- CNS
central nervous system
- CRC
concentration-response curve
- DCM
dichloromethane
- DCT
distal convoluted tubule
- EAST
epilepsy ataxia sensorineural deafness tubulopathy
- ENaC
epithelial Na+ channel
- fu
unbound free fraction
- HEK-293
human embryonic kidney 293 K+, potassium
- Kir
inward rectifier potassium
- Kp
partition coefficient
- Kp,uu
unbound partition coefficient
- MECP2
methyl CpG binding protein 2
- NCC
Na+-Cl− cotransporter
- NKCC2
Na+-K+-2Cl− cotransporter
- pS
picosiemens
- ROMK
renal outer medullary potassium channel
- SAR
structure-activity relationship
- SeSAME
seizures sensorineural deafness mental retardation electrolyte imbalances
- SPAK
Ste20-related protein proline/alanine-rich kinase
- SUR1
sulfonylurea receptor 1
- TAL
thick ascending loop
- Tl+
thallium
- Vm
membrane potential
- VU0134992
2-(2-bromo-4-isopropylphenoxy)-N-(2,2,6,6-tetramethylpiperidin-4-yl)acetamide
- WNK
lysine-deficient protein kinase
- WT
wild type
Authorship Contributions
Participated in research design: Kharade, Kurata, Blobaum, Figueroa, Duran, Satlin, Meiler, Weaver, Lindsley, Hopkins, and Denton.
Conducted experiments: Kharade, Kurata, Blobaum, Figueroa, Duran, Kramer, Days, Vinson, and Flores.
Contributed new reagents or analytical tools: Kurata, Bender, Meiler, and Hopkins.
Performed data analysis: Kharade, Kurata, Blobaum, Figueroa, Duran, Kramer, Days, and Flores.
Wrote or contributed to the writing of the manuscript: Kharade, Kurata, Blobaum, Figueroa, Duran, Vinson, Weaver, Lindsley, Hopkins, and Denton.
Footnotes
Work in the Denton laboratory was funded by National Institutes of Health (NIH) Grants [5R21-NS-073097-01S1 and R01-DK-082884]. E.F. was supported by NIH Training Grant [5T32- GM-007628]. Work in the Meiler laboratory is supported by NIH Grants [R01- GM-080403, R01-HL-122010] and National Science Foundation Grant [CHE 1305874]. Work in the Satlin laboratory is supported through NIH Grants [DK-038470 and P30-DK-079307, The Pittsburgh Center for Kidney Research]. The WaveFront Biosciences Panoptic kinetic imaging plate reader is housed and managed within the Vanderbilt High-Throughput Screening Core Facility, an institutionally supported core, and was funded by NIH Shared Instrumentation Grant [1S10-OD-021734]. C.D.W. is an owner of WaveFront Biosciences, manufacturer of the Panoptic plate reader, receives royalties from the sales of Thallos via a licensing agreement with Vanderbilt University. No other potential conflicts of interest relevant to this article are reported.
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Aréchiga-Figueroa IA, Marmolejo-Murillo LG, Cui M, Delgado-Ramírez M, van der Heyden MAG, Sánchez-Chapula JA, Rodríguez-Menchaca AA. (2017) High-potency block of Kir4.1 channels by pentamidine: molecular basis. Eur J Pharmacol 815:56–63. [DOI] [PubMed] [Google Scholar]
- Barth P, Schonbrun J, Baker D. (2007) Toward high-resolution prediction and design of transmembrane helical protein structures. Proc Natl Acad Sci USA 104:15682–15687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhave G, Chauder BA, Liu W, Dawson ES, Kadakia R, Nguyen TT, Lewis LM, Meiler J, Weaver CD, Satlin LM, et al. (2011) Development of a selective small-molecule inhibitor of Kir1.1, the renal outer medullary potassium channel. Mol Pharmacol 79:42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockenhauer D, Feather S, Stanescu HC, Bandulik S, Zdebik AA, Reichold M, Tobin J, Lieberer E, Sterner C, Landoure G, et al. (2009) Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med 360:1960–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton JS, Pao AC, Maduke M. (2013) Novel diuretic targets. Am J Physiol Renal Physiol 305:F931–F942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djukic B, Casper KB, Philpot BD, Chin LS, McCarthy KD. (2007) Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J Neurosci 27:11354–11365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan L, Wang X, Zhang D, Duan X, Zhao C, Zu M, Meng X, Zhang C, Su XT, Wang MX, et al. (2015) Vasopressin-induced stimulation of the Na(+)-activated K(+) channels is responsible for maintaining the basolateral K(+) conductance of the thick ascending limb (TAL) in EAST/SeSAME syndrome. Biochim Biophys Acta 1852:2554–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furutani K, Ohno Y, Inanobe A, Hibino H, Kurachi Y. (2009) Mutational and in silico analyses for antidepressant block of astroglial inward-rectifier Kir4.1 channel. Mol Pharmacol 75:1287–1295. [DOI] [PubMed] [Google Scholar]
- Garcia ML, Priest BT, Alonso-Galicia M, Zhou X, Felix JP, Brochu RM, Bailey T, Thomas-Fowlkes B, Liu J, Swensen A, et al. (2014) Pharmacologic inhibition of the renal outer medullary potassium channel causes diuresis and natriuresis in the absence of kaliuresis. J Pharmacol Exp Ther 348:153–164. [DOI] [PubMed] [Google Scholar]
- Hasannejad H, Takeda M, Taki K, Shin HJ, Babu E, Jutabha P, Khamdang S, Aleboyeh M, Onozato ML, Tojo A, et al. (2004) Interactions of human organic anion transporters with diuretics. J Pharmacol Exp Ther 308:1021–1029. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Kusuhara H, Adachi M, Schuetz JD, Takeuchi K, Sugiyama Y. (2007) Multidrug resistance-associated protein 4 is involved in the urinary excretion of hydrochlorothiazide and furosemide. J Am Soc Nephrol 18:37–45. [DOI] [PubMed] [Google Scholar]
- Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. (2010) Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev 90:291–366. [DOI] [PubMed] [Google Scholar]
- Ito M, Inanobe A, Horio Y, Hibino H, Isomoto S, Ito H, Mori K, Tonosaki A, Tomoike H, Kurachi Y. (1996) Immunolocalization of an inwardly rectifying K+ channel, KAB-2 (Kir4.1), in the basolateral membrane of renal distal tubular epithelia. FEBS Lett 388:11–15. [DOI] [PubMed] [Google Scholar]
- Kahanovitch U, Cuddapah VA, Pacheco NL, Holt LM, Mulkey DK, Percy AK, Olsen ML. (2018) MeCP2 deficiency leads to loss of glial Kir4.1. eNeuro 5:ENEURO.0194-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharade S, Sheehan J, Figueroa E, Meiler J, Denton J. (2017) Pore polarity and charge determine differential block of Kir1.1 and Kir7.1 potassium channels by small-molecule inhibitor VU590. Mol Pharmacol 92:338–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharade SV, Flores D, Lindsley CW, Satlin LM, Denton JS. (2016) ROMK inhibitor actions in the nephron probed with diuretics. Am J Physiol Renal Physiol 310:F732–F737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King NP, Sheffler W, Sawaya MR, Vollmar BS, Sumida JP, André I, Gonen T, Yeates TO, Baker D. (2012) Computational design of self-assembling protein nanomaterials with atomic level accuracy. Science 336:1171–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofuji P, Biedermann B, Siddharthan V, Raap M, Iandiev I, Milenkovic I, Thomzig A, Veh RW, Bringmann A, Reichenbach A. (2002) Kir potassium channel subunit expression in retinal glial cells: implications for spatial potassium buffering. Glia 39:292–303. [DOI] [PubMed] [Google Scholar]
- Kothiwale S, Mendenhall JL, Meiler J. (2015) BCL:Conf: small molecule conformational sampling using a knowledge based rotamer library. J Cheminform 7:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucheryavykh YV, Kucheryavykh LY, Nichols CG, Maldonado HM, Baksi K, Reichenbach A, Skatchkov SN, Eaton MJ. (2007) Downregulation of Kir4.1 inward rectifying potassium channel subunits by RNAi impairs potassium transfer and glutamate uptake by cultured cortical astrocytes. Glia 55:274–281. [DOI] [PubMed] [Google Scholar]
- Lachheb S, Cluzeaud F, Bens M, Genete M, Hibino H, Lourdel S, Kurachi Y, Vandewalle A, Teulon J, Paulais M. (2008) Kir4.1/Kir5.1 channel forms the major K+ channel in the basolateral membrane of mouse renal collecting duct principal cells. Am J Physiol Renal Physiol 294:F1398–F1407. [DOI] [PubMed] [Google Scholar]
- Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, Kaufman K, Renfrew PD, Smith CA, Sheffler W, et al. (2011) ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol 487:545–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis LM, Bhave G, Chauder BA, Banerjee S, Lornsen KA, Redha R, Fallen K, Lindsley CW, Weaver CD, Denton JS. (2009) High-throughput screening reveals a small-molecule inhibitor of the renal outer medullary potassium channel and Kir7.1. Mol Pharmacol 76:1094–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Morimoto T, Woda C, Kleyman TR, Satlin LM. (2007) Ca2+ dependence of flow-stimulated K secretion in the mammalian cortical collecting duct. Am J Physiol Renal Physiol 293:F227–F235. [DOI] [PubMed] [Google Scholar]
- Liu W, Wei Y, Sun P, Wang WH, Kleyman TR, Satlin LM. (2009) Mechanoregulation of BK channel activity in the mammalian cortical collecting duct: role of protein kinases A and C. Am J Physiol Renal Physiol 297:F904–F915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourdel S, Paulais M, Cluzeaud F, Bens M, Tanemoto M, Kurachi Y, Vandewalle A, Teulon J. (2002) An inward rectifier K(+) channel at the basolateral membrane of the mouse distal convoluted tubule: similarities with Kir4-Kir5.1 heteromeric channels. J Physiol 538:391–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell DJ, Coutsias EA, Kortemme T. (2009) Sub-angstrom accuracy in protein loop reconstruction by robotics-inspired conformational sampling. Nat Methods 6:551–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmolejo-Murillo LG, Aréchiga-Figueroa IA, Cui M, Moreno-Galindo EG, Navarro-Polanco RA, Sánchez-Chapula JA, Ferrer T, Rodríguez-Menchaca AA. (2017a) Inhibition of Kir4.1 potassium channels by quinacrine. Brain Res 1663:87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmolejo-Murillo LG, Aréchiga-Figueroa IA, Moreno-Galindo EG, Navarro-Polanco RA, Rodríguez-Menchaca AA, Cui M, Sánchez-Chapula JA, Ferrer T. (2017b) Chloroquine blocks the Kir4.1 channels by an open-pore blocking mechanism. Eur J Pharmacol 800:40–47. [DOI] [PubMed] [Google Scholar]
- Meiler J, Baker D. (2006) ROSETTALIGAND: protein-small molecule docking with full side-chain flexibility. Proteins 65:538–548. [DOI] [PubMed] [Google Scholar]
- Nwaobi SE, Cuddapah VA, Patterson KC, Randolph AC, Olsen ML. (2016) The role of glial-specific Kir4.1 in normal and pathological states of the CNS. Acta Neuropathol 132:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno Y, Hibino H, Lossin C, Inanobe A, Kurachi Y. (2007) Inhibition of astroglial Kir4.1 channels by selective serotonin reuptake inhibitors. Brain Res 1178:44–51. [DOI] [PubMed] [Google Scholar]
- Olsen ML, Sontheimer H. (2008) Functional implications for Kir4.1 channels in glial biology: from K+ buffering to cell differentiation. J Neurochem 107:589–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulais M, Bloch-Faure M, Picard N, Jacques T, Ramakrishnan SK, Keck M, Sohet F, Eladari D, Houillier P, Lourdel S, et al. (2011) Renal phenotype in mice lacking the Kir5.1 (Kcnj16) K+ channel subunit contrasts with that observed in SeSAME/EAST syndrome. Proc Natl Acad Sci USA 108:10361–10366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson WL, Skatchkov SN, Eaton MJ, Nichols CG. (2006) C-terminal determinants of Kir4.2 channel expression. J Membr Biol 213:187–193. [DOI] [PubMed] [Google Scholar]
- Penton D, Czogalla J, Wengi A, Himmerkus N, Loffing-Cueni D, Carrel M, Rajaram RD, Staub O, Bleich M, Schweda F, et al. (2016) Extracellular K+ rapidly controls NaCl cotransporter phosphorylation in the native distal convoluted tubule by Cl- -dependent and independent mechanisms. J Physiol 594:6319–6331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessia M, Imbrici P, D’Adamo MC, Salvatore L, Tucker SJ. (2001) Differential pH sensitivity of Kir4.1 and Kir4.2 potassium channels and their modulation by heteropolymerisation with Kir5.1. J Physiol 532:359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao VS, Planavsky N, Hanberg JS, Ahmad T, Brisco-Bacik MA, Wilson FP, Jacoby D, Chen M, Tang WHW, Cherney DZI, et al. (2017) Compensatory distal reabsorption drives diuretic resistance in human heart failure. J Am Soc Nephrol 28:3414–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raphemot R, Kadakia RJ, Olsen ML, Banerjee S, Days E, Smith SS, Weaver CD, Denton JS. (2013) Development and validation of fluorescence-based and automated patch clamp-based functional assays for the inward rectifier potassium channel Kir4.1. Assay Drug Dev Technol 11:532–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raphemot R, Lonergan DF, Nguyen TT, Utley T, Lewis LM, Kadakia R, Weaver CD, Gogliotti R, Hopkins C, Lindsley CW, et al. (2011) Discovery, characterization, and structure-activity relationships of an inhibitor of inward rectifier potassium (Kir) channels with preference for Kir2.3, Kir3.x, and Kir7.1. Front Pharmacol 2:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raphemot R, Swale DR, Dadi PK, Jacobson DA, Cooper P, Wojtovich AP, Banerjee S, Nichols CG, Denton JS. (2014) Direct activation of β-cell KATP channels with a novel xanthine derivative. Mol Pharmacol 85:858–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichold M, Zdebik AA, Lieberer E, Rapedius M, Schmidt K, Bandulik S, Sterner C, Tegtmeier I, Penton D, Baukrowitz T, et al. (2010) KCNJ10 gene mutations causing EAST syndrome (epilepsy, ataxia, sensorineural deafness, and tubulopathy) disrupt channel function. Proc Natl Acad Sci USA 107:14490–14495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl UI, Choi M, Liu T, Ramaekers VT, Häusler MG, Grimmer J, Tobe SW, Farhi A, Nelson-Williams C, Lifton RP. (2009) Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci USA 106:5842–5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicca F, Ambrosini E, Marchese M, Sforna L, Servettini I, Valvo G, Brignone MS, Lanciotti A, Moro F, Grottesi A, et al. (2016) Gain-of-function defects of astrocytic Kir4.1 channels in children with autism spectrum disorders and epilepsy. Sci Rep 6:34325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicca F, Imbrici P, D’Adamo MC, Moro F, Bonatti F, Brovedani P, Grottesi A, Guerrini R, Masi G, Santorelli FM, et al. (2011) Autism with seizures and intellectual disability: possible causative role of gain-of-function of the inwardly-rectifying K+ channel Kir4.1. Neurobiol Dis 43:239–247. [DOI] [PubMed] [Google Scholar]
- Su S, Ohno Y, Lossin C, Hibino H, Inanobe A, Kurachi Y. (2007) Inhibition of astroglial inwardly rectifying Kir4.1 channels by a tricyclic antidepressant, nortriptyline. J Pharmacol Exp Ther 320:573–580. [DOI] [PubMed] [Google Scholar]
- Su XT, Wang WH. (2016) The expression, regulation, and function of Kir4.1 (Kcnj10) in the mammalian kidney. Am J Physiol Renal Physiol 311:F12–F15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swale DR, Kharade SV, Denton JS. (2014) Cardiac and renal inward rectifier potassium channel pharmacology: emerging tools for integrative physiology and therapeutics. Curr Opin Pharmacol 15:7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swale DR, Kurata H, Kharade SV, Sheehan J, Raphemot R, Voigtritter KR, Figueroa EE, Meiler J, Blobaum AL, Lindsley CW, et al. (2016) ML418: the first selective, sub-micromolar pore blocker of Kir7.1 potassium channels. ACS Chem Neurosci 7:1013–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takanari H, Nalos L, Stary-Weinzinger A, de Git KC, Varkevisser R, Linder T, Houtman MJ, Peschar M, de Boer TP, Tidwell RR, et al. (2013) Efficient and specific cardiac IK1 inhibition by a new pentamidine analogue. Cardiovasc Res 99:203–214. [DOI] [PubMed] [Google Scholar]
- Tong X, Ao Y, Faas GC, Nwaobi SE, Xu J, Haustein MD, Anderson MA, Mody I, Olsen ML, Sofroniew MV, et al. (2014) Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat Neurosci 17:694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker SJ, Imbrici P, Salvatore L, D’Adamo MC, Pessia M. (2000) pH dependence of the inwardly rectifying potassium channel, Kir5.1, and localization in renal tubular epithelia. J Biol Chem 275:16404–16407. [DOI] [PubMed] [Google Scholar]
- Uwai Y, Saito H, Hashimoto Y, Inui KI. (2000) Interaction and transport of thiazide diuretics, loop diuretics, and acetazolamide via rat renal organic anion transporter rOAT1. J Pharmacol Exp Ther 295:261–265. [PubMed] [Google Scholar]
- Vallon V, Rieg T, Ahn SY, Wu W, Eraly SA, Nigam SK. (2008) Overlapping in vitro and in vivo specificities of the organic anion transporters OAT1 and OAT3 for loop and thiazide diuretics. Am J Physiol Renal Physiol 294:F867–F873. [DOI] [PubMed] [Google Scholar]
- Welling PA. (2016) Roles and regulation of renal K channels. Annu Rev Physiol 78:415–435. [DOI] [PubMed] [Google Scholar]
- Zaika OL, Mamenko M, Palygin O, Boukelmoune N, Staruschenko A, Pochynyuk O. (2013) Direct inhibition of basolateral Kir4.1/5.1 and Kir4.1 channels in the cortical collecting duct by dopamine. Am J Physiol Renal Physiol 305:F1277–F1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Wang L, Su XT, Lin DH, Wang WH. (2015) KCNJ10 (Kir4.1) is expressed in the basolateral membrane of the cortical thick ascending limb. Am J Physiol Renal Physiol 308:F1288–F1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Wang L, Thomas S, Wang K, Lin DH, Rinehart J, Wang WH. (2013) Src family protein tyrosine kinase regulates the basolateral K channel in the distal convoluted tubule (DCT) by phosphorylation of KCNJ10 protein. J Biol Chem 288:26135–26146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Wang L, Zhang J, Su XT, Lin DH, Scholl UI, Giebisch G, Lifton RP, Wang WH. (2014) KCNJ10 determines the expression of the apical Na-Cl cotransporter (NCC) in the early distal convoluted tubule (DCT1). Proc Natl Acad Sci USA 111:11864–11869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Su J, Cui N, Gai H, Wu Z, Jiang C. (2011) The disruption of central CO2 chemosensitivity in a mouse model of Rett syndrome. Am J Physiol Cell Physiol 301:C729–C738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Forrest MJ, Sharif-Rodriguez W, Forrest G, Szeto D, Urosevic-Price O, Zhu Y, Stevenson AS, Zhou Y, Stribling S, et al. (2017) Chronic inhibition of renal outer medullary potassium channel not only prevented but also reversed development of hypertension and end-organ damage in Dahl salt-sensitive rats. Hypertension 69:332–338. [DOI] [PubMed] [Google Scholar]