

Abstract
The mutational patterns of large tandem arrays of short sequence repeats remain largely unknown, despite observations of their high levels of variation in sequence and genomic abundance within and between species. Many factors can influence the dynamics of tandem repeat evolution; however, their evolution has only been examined over a limited phylogenetic sample of taxa. Here, we use publicly available whole-genome sequencing data of 85 haploid mutation accumulation lines derived from six geographically diverse Chlamydomonas reinhardtii isolates to investigate genome-wide mutation rates and patterns in tandem repeats in this species. We find that tandem repeat composition differs among ancestral strains, both in genome-wide abundance and presence/absence of individual repeats. Estimated mutation rates (repeat copy number expansion and contraction) were high, averaging 4.3×10−4 per generation per single unit copy. Although orders of magnitude higher than other types of mutation previously reported in C. reinhardtii, these tandem repeat mutation rates were one order of magnitude lower than what has recently been found in Daphnia pulex, even after correcting for lower overall genome-wide satellite abundance in C. reinhardtii. Most high-abundance repeats were related to others by a single mutational step. Correlations of repeat copy number changes within genomes revealed clusters of closely related repeats that were strongly correlated positively or negatively, and similar patterns of correlation arose independently in two different mutation accumulation experiments. Together, these results paint a dynamic picture of tandem repeat evolution in this unicellular alga.
Keywords: k-Seek, mutation accumulation, mutation rate, tandem repeat, Chlamydomonas, Illumina
Introduction
Tandemly arrayed repeats of short DNA sequences are a major, but understudied, component of eukaryotic genomes. These arrays, termed satellite DNA, can span many kilobases in the genome and are often associated with essential chromosomal structures such as centromeres (Iwata-Otsubo et al. 2017). Yet, satellites vary in both sequence and abundance across even closely related species (Jagannathan et al. 2017), suggesting rapid evolution. The extent to which this rapid evolution is due to high, but neutral, mutation rates as opposed to natural selection remains an open question. Tandem repetitive DNA might experience different mutation rates and patterns of evolution than single-copy euchromatic sequence because of its enrichment in heterochromatic, low recombining regions of the genome, its potential functions (or lack of), and its propensity to undergo unequal crossing-over and replication slippage (Charlesworth et al. 1994; Henikoff et al. 2001; Flynn, Caldas, et al. 2017). Determining the mutational and evolutionary processes generating satellite DNA variation is important, as variation in satellite arrays has been associated with disease, genome instability, differences in genome-wide gene expression, and reproductive isolation between species (reviewed in Garrido-Ramos 2017).
To understand both the mechanistic mutational processes and ultimate evolutionary forces generating satellite DNA diversity, it is essential to assess and compare genome-wide satellite DNA across individuals, populations, and species. However, because satellites are repetitive and large, they cannot be mapped or assembled in the genome using traditional computational methods (reviewed in Treangen and Salzberg 2011), and thus are typically ignored in genomic studies (Hoskins et al. 2007). Furthermore, most studies have focused on a single satellite sequence or a single family of sequences (Gall and Atherton 1974; Rojo et al. 2015; Samoluk et al. 2017). With tools to analyze these repetitive parts of the genome from short-read sequencing data becoming available (Garrido-Ramos 2017), a few recent studies have documented genome-wide tandem repeat composition and abundance across multiple species and populations, resulting in a dynamic picture of satellite evolution in nature. For example, Wei et al. (2014) investigated tandem repeat composition across lines of Drosophila melanogaster derived from five different geographic populations, and found great differences in the abundances of specific repeat sequences, including polymorphism for the presence and absence of certain repeats. These results suggest that tandem repeats are able to expand and contract in copy number at very high rates to produce the observed levels of diversity among populations within a single species, though the relative contributions of natural selection and neutral mutational processes to this diversity remains unknown.
A powerful method to disentangle mutation from selection is to estimate neutral mutation rate using a mutation accumulation (MA) experiment, where mutations accumulate almost neutrally because frequent bottlenecks reduce selection to a minimum (Simmons and Crow 1977; Halligan and Keightley 2009). Widely used for assessing genome-wide single nucleotide mutation rates, data from these experiments can also be used to assess tandem-repeat mutation rates, including change in copy number (expansion and contraction) and gain/loss of repeat sequences, by comparing whole genome sequencing data before and after MA. For example, Flynn, Caldas, et al. (2017) compared abundances of tandem repeats across MA lines of Daphnia pulex and found extremely high rates of expansions and contractions in tandem repeats as compared with other types of mutation (single nucleotide mutations, insertion/deletion: Flynn, Chain, et al. 2017; copy number variation: Seyfert et al. 2008; Keith et al. 2016; microsatellites: Seyfert et al. 2008), with rates varying across different repeats from 0.29–105 copies per generation. This suggests that high mutation rates could largely explain the commonly observed rapid divergence in genome-wide repetitive DNA across populations and species.
However, it seems the potential for divergence in tandem repeats is not realized in all taxa. In locust, only slight differences in satellite DNA content and abundance were found between geographically separated lineages (Ruiz-Ruano et al. 2016). The rates of tandem repeat evolution were found to be heterogeneous across different species groups of Drosophila, where some groups exhibited rapid rates of divergence in tandem repeat composition, while others were quite stagnant (Wei et al. 2018). Different levels of selection or different rates of mutation could explain these patterns. Likewise, evidence from comparing a population under selection to MA lines suggested that stabilizing selection reduced the variation in the copy number of tandem repeats in D. pulex population (Flynn, Caldas, et al. 2017). Additionally, different tandem repeat sequence arrays in the same genome may interact with each other to produce genomic trade-offs or constraints, perhaps dependent on their relative genomic location or sequence similarity (Wei et al. 2014; Flynn, Caldas, et al. 2017). Different environmental, physiological, or genomic conditions could also result in differing levels of selection on repetitive DNA across species (Charlesworth et al. 1994).
The effectiveness of selection can also influence the outcome of changes in copy numbers of tandem repeats. Taxa with higher effective population sizes (Ne) experience a weaker influence of drift and a stronger efficiency of selection (Lynch and Conery 2003). If repetitive DNA is mainly a burden to the host, then taxa with higher Ne might contain lower genome-wide amounts of repetitive DNA (Ohno 1972; Orgel and Crick 1980; Lynch and Conery 2003; Petit and Barbadilla 2009). Further, if expansions and contractions in tandem repeat copy number are deleterious on an average (Flynn, Caldas, et al. 2017), organisms with higher Ne would also be expected to have evolved lower rates of expansion and contraction. These predictions are concordant with the “drift-barrier hypothesis,” which posits that a trait, in this case the molecular mutation rate, can be limited in its ability to achieve its optimum by the efficiency of selection governed by Ne (Sung et al. 2012). The predicted inverse relationship between mutation rate and Ne has been demonstrated for single nucleotide mutation rates across various taxa (Sung et al. 2012), and this pattern might also apply to different types of mutation such as those affecting the copy number of tandem repeats.
Investigating genome-wide tandem repeats in a species with different characteristics such as life cycle, evolutionary history, effective population size, and genome organization could shed light on the reasons for different patterns of repetitive DNA evolution. Little is known about tandem repeats or their diversity in the unicellular green alga, Chlamydomonas reinhardtii. High amounts of tandem arrays of the dinucleotide repeat (AC)n were found in the nuclear genome using targeted molecular probes and cloning (Kang and Fawley 1997), but genome-wide satellite sequences have not previously been assayed in this species. Chlamydomonasreinhardtii has a large genome compared with other unicellular eukaryotes, at 121 Mb (Merchant et al. 2007). Chlamydomonasreinhardtii also possesses a 200-kb chloroplast genome that was found to be composed of at least 5% repeats, which were mostly interspersed throughout the genome (Maul et al. 2002). Chlamydomonasreinhardtii’s effective population size in nature was estimated to be on the order of 107 (Ness et al. 2016), thus we expect mutation rates might be lower than those of multicellular eukaryotes. MA lines have been used to estimate single nucleotide mutation (SNM) rates in this species (Ness et al. 2012; Sung et al. 2012). Ness et al. (2015) found rates to vary significantly across six different MA ancestors from different geographic populations. Concordant with the drift-barrier hypothesis, C. reinhardtii single nucleotide substitution rates were found to be lower than multicellular eukaryotes such as D. pulex (Sung et al. 2012; Ness et al. 2015; Flynn, Caldas, et al. 2017). Whether or not tandem repeat mutation rates also follow this pattern is unknown. Additionally, limited sample sizes of different types of mutations (SNMs, indels, copy number mutations) have prevented the evaluation of correlations between mutation types within a genome.
Here, we investigate population-level polymorphism in repeat composition and mutation rates of expansions/contractions using published whole-genome sequence data from 85 Chlamydomonas reinhardtii MA lines originating from six different ancestral strains (Morgan et al. 2014; Ness et al. 2015). 1) We assess the diversity of tandem repeat composition across lines and ancestral strains in this to-date uncharacterized taxon using k-Seek, a software designed to detect short simple sequence repeats in unassembled short read sequencing data. 2) We infer the evolutionary origins of repeat sequences using metrics of sequence similarity, GC content, and repeat length. 3) We investigate variation in mutation rate, estimated as the rate of change in copy number and unique repeat sequence gain and loss, across the six independent ancestral strains using the data from their derived MA lines. Finally, we 4) identify correlations in repeat abundances that suggest constraints on repeat evolution.
Materials and Methods
Mutation Accumulation Lines and Sequencing
We assessed short sequence repeats in publicly available Illumina whole-genome sequencing reads from 85 haploid MA lines derived from six genetically diverse strains (Morgan et al. 2014; Ness et al. 2015). Briefly, 15 replicate MA lines were generated from single colonies of each ancestral strain: CC-1373 (Massachusetts), CC-1952 (Minnesota), CC-2342 (Pennsylvania-1), CC-2344 (Pennsylvania-2), CC-2931 (North Carolina), CC-2937 (Quebec). A single colony from each line was transferred to a new plate every 3–5 days for 85 transfers (430–1,130 generations, depending on the line). This equated to a bottleneck of Ne= 6.5 each generation. The total number of generations was estimated separately for each line using growth curves at the end of the experiment. 85 of the 90 starting MA lines survived to the end of the experiment and were used for sequencing. DNA extraction was from whole cells; the chloroplast and nuclear genomes were not separated. Illumina libraries were constructed using a modified PCR protocol accounting for C.reinhardtii’s high GC content (∼63.9%) and then sequenced on the Illumina GAII platform with 100-bp paired-ends reads to an average of 30× depth. Full methods of MA line construction, sequencing, and generation estimation are available (Morgan et al. 2014; Ness et al. 2015).
Tandem Repeat Quantification
We downloaded the raw fastq files for each MA line from EBI ENA (accession: PRJEB9934) and assessed them for quality using fastqc (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/, last accessed October 10, 2016). Reads were quality filtered and trimmed to remove the first 9 bp which showed a strong bias in GC content using Trimmomatic v0.36 (Bolger et al. 2014; parameters: PE -phred 33 ILLUMINACLIP: 2: 30: 10 HEADCROP: 9 SLIDINGWINDOW: 4: 20 MINLEN: 50 AVGQUAL: 20). We identified and quantified tandem repeats using k-Seek (Wei et al. 2014) (https://github.com/weikevinhc/k-seek, last accessed August 16, 2017) following the procedure in (Flynn, Caldas, et al. 2017). k-Seek uses a hash table approach to count the number of all tandemly repeated sequences of 1–20 bp (kmers) that encompass at least 50 bp on a single read. These counts are tabulated across all reads in a sequencing library to obtain estimates of total genomic abundance (kmer copy number). As estimates of genomic abundance are expected to be influenced by both sequencing effort and biased PCR amplification of sequencing fragments based on GC content (Benjamini and Speed 2012), we normalized the kmer counts by both library depth and kmer sequence GC content using a previously published correction script (Flynn, Caldas, et al. 2017) (https://github.com/jmf422/Daphnia-MA-lines/tree/master/GC_correction, last accessed August 2, 2017). As inputs, this program requires 1) alignment files of uniquely mapping reads (bam file), 2) the read length (average 86 bp after trimming), and 3) the mean length of the fragments from which reads were derived. To generate these inputs, BAM files were generated using Bowtie2 v2.2.8 (Langmead and Salzberg 2012) to map reads to the C. reinhardtii v5.0 reference genome (Merchant et al. 2007) downloaded from Phytozome (Goodstein et al. 2012), with manual addition of the mitochondrial (NCBI: NC_00138) and chloroplast (NCBI: NC_005353) assemblies. Mean fragment length was estimated using a publicly available Python script (https://gist.github.com/davidliwei/2323462, last accessed August 2, 2017). The GC correction factor generated was then applied to kmer counts according to their GC content as in (Flynn, Caldas, et al. 2017). Only nuclear scaffolds were used for the GC correction as the GC contents of the mitochondrial and chloroplast genomes are drastically different from that of the nuclear genome, and reads from the organellar genomes could be overrepresented in the sequencing library due to organelle copy number.
Assessment of Repeat Abundance and Annotation
The final output from the above steps was a table containing corrected copy number counts of each kmer identified in each of the MA lines. We used these data to assess the variation in repeat abundance among the MA lines originating from different ancestral strains. We initially examined kmer abundance and diversity using only kmers with at least two copies across all MA lines derived from a single ancestor to control for potential spurious kmers in the data set. Thus, though these kmers were shared across all lines derived from an ancestor, they could be polymorphic in their presence/absence across different ancestral strains. Using this filtered data set, we also estimated the total kmer composition of each ancestor. As sequencing data from the ancestral strains was not available, we inferred the ancestral copy number as the mean copy number for each kmer across the MA lines derived from a single ancestor.
Subsequently, to concentrate our analysis on kmers involved in larger tandem arrays rather than smaller microsatellites, we focused our analysis on the kmers with an average of at least 100 copies in at least one inferred ancestor, termed high-abundance repeats. While these kmers had at least 100 copies in one inferred ancestor, there could be considerably fewer copies in the other ancestors. If a kmer had <15 copies in any of the other ancestors, it was considered absent in that ancestor.
To evaluate whether some of the tandem repeats k-Seek identified could have originated from the chloroplast genome, we used Tandem Repeats Finder (TRF) (Benson 1999) to find repeats of unit length no longer than 20 bp in the chloroplast assembly (parameters: maxperiod = 20), concordant with the kmer lengths identified by k-Seek (command: trf chlamy_chloroplast_assembly.fasta 2 7 7 80 10 100 20 -h). We then used a custom python script that identified shared kmer sequences between the data set of kmers identified by TRF from the chloroplast assembly and the data set of kmers detected with k-Seek from Illumina data, searching all rotations and reverse complements of the repeat sequences (supplementary file S4, Supplementary Material online). In order to assess the genome-wide tandem repeats that were not captured with k-Seek (including complex repeats > 20 bp) we used TRF (parameters 2 7 7 80 10 100 500 -h) and Phobos (http://www.ruhr-uni-bochum.de/ecoevo/cm/cm_phobos.htm, last accessed April 6, 2018 -U 200 parameter) on the genome assembly. We also used TAREAN (Novák et al. 2017) on a subset of raw reads (randomly subsampled to 0.37×) from a single sequencing library.
Mutation Rate Estimation
We examined repeat mutation rate in two ways: 1) change in copy number of each unique repeat sequence (expansion/contraction) and 2) change in the number of unique repeat sequences (gain/loss), estimated for each ancestral strain from its derived MA lines. All mutation rates are expressed on a per-generation basis.
Copy Number Change (Expansion/Contraction)
In the context of tandem repeats, the expansion/contraction rate is the number of changes in copy number of a given repeat on an average per genome per generation. This includes all changes in copy number affecting a certain tandem repeat sequence, which may encompass multiple loci genome-wide. Thus, for each repeat in each of the six independent MA experiments, we calculated expansion and contraction rates as the deviation from the ancestral copy number for each individual MA line, divided by the number of generations propagated, using the following equation:
where ui, m is the mutation rate of kmer i in MA line m, mi is the abundance of kmer i in MA line m, Ai is the abundance in the inferred ancestor of MA line m for kmer i, and Gm is the number of generations propagated for MA line m. u could be negative (for contractions) or positive (for expansions). We used 81 of the 85 MA lines for this mutation rate analysis so as to only include MA lines with a confident estimate of the number of generations diverged from the ancestral strain (Ness et al. 2015). Note that implicit in this approach is the assumption that multiple mutation events within a given repeat array are rare enough in the span of the experiment to be ignored.
Because expansion/contraction rates have been shown to be positively linearly correlated with copy number (Flynn, Caldas, et al. 2017) and because it is informative to calculate absolute change in copy number to quantify the overall magnitude of change, we also calculated an abundance-normalized absolute copy number change rate for each repeat. This allows direct comparison of expansion/contraction rates of repeats of different genomic abundances across taxa. Below is the equation we used to calculate copy-number normalized absolute mutation rates:
Change in Number of Unique Repeats (Gain/Loss)
Aside from changes in repeat abundance we were interested in processes contributing to repeat gain and loss within the ∼1,000 generations of the MA experiment. We inferred a gain if the kmer was absent from all lines from a given ancestor, except one line having at least 3 copies. We inferred a loss if one line had 0 copies of a kmer that was present in all other lines of the ancestor; only considering kmers that had at least 3 copies in at least one line.
Interspersion Analysis
To verify that k-Seek and our filtering criteria identified repeats derived from larger tandem arrays, we analyzed the kmer composition of paired-end reads. If repeats were present in both reads of a pair, we inferred that repeats encompassed the entire sequencing fragment (∼455 bp), and that the fragment was likely derived from a larger tandem array in the genome. This analysis also enabled investigation of kmers that co-occur on the same fragment, and thus represent repeats that are interspersed with one another in the genome.
Given the low measured mutation rates, it is unlikely that ∼1,000 generations of MA would result in considerable differences in genomic colocalization of tandem repeats among MA lines derived from the same ancestor. Instead we were interested in the differences between ancestors from different geographic populations that have diverged much further back than within each MA experiment. Therefore, we randomly chose one MA line from each ancestor in which to analyze the interspersion levels of kmers. We used the same interspersion metric in (Wei et al. 2014), which compares the number of reads that a kmer is interspersed with to the total number of reads containing that kmer. Details of the interspersion analysis are located in supplementary file S2, Supplementary Material online.
Analysis
To draw inferences on processes that could constrain repeat copy number change, we tested the homogeneity of correlations among expansion/contraction rates across MA lines derived from each ancestor using R. We used RStudio version 0.99.903 for all analyses, including investigating the tandem repeat composition of the six ancestral strains, and determining the rates and patterns of tandem repeat mutation over the course of the MA experiment. All scripts, including R script files, and additional bash and python scripts are deposited on Github (https://github.com/jmf422/Chlamy-repeats-mutation).
Results
Diversity in Tandem Repeat Composition
We scanned over 2 billion reads across 85 MA lines derived from six ancestral strains and identified almost five million reads (0.18%) composed of short simple tandem repeats. Normalizing for sequencing effort and GC bias, and filtering out low-abundance repeats (kmers < 2 copies in at least one MA line per ancestor) resulted in 480 unique kmer sequences identified across all MA lines (mean: 160, range: 134–198 kmers averaged across lines from a single ancestor, supplementary fig. S1, Supplementary Material online), accounting for an average of 180.48 kb of genomic content (range: 149.29–192.69 kb averaged across lines from a single ancestor). Thus, given a genome size of 121 Mb (http://plants.ensembl.org/Chlamydomonas_reinhardtii/Info/Annotation/, last accessed January 12, 2018), these repeats account for ∼0.15% of the genome. This is a lower proportion of simple repeats than what has been found for other organisms studied, and lower than the total repetitive DNA content estimated by Merchant et al. (2007). Tandem Repeats Finder and Phobos did not find high amounts of repeats 1–20 bp long in the reference genome not found in our analysis of Illumina reads with k-Seek. However, there were moderate amounts of complex repeats (units longer than 20 bp) present in the assembly and raw reads (with TAREAN) that we did not analyze here (supplementary tables S1 and S2, Supplementary Material online).
While most short kmers were present at modest (mean < 100 copies) to high (mean 100–999 copies) abundance, on an average, there were 3 to 7 kmers present at very high abundance (mean ≥1,000 copies) across lines derived from a single ancestor. Notably, the dimer AC had the highest copy number of all kmers by an order of magnitude (mean: 30,592 copies; range 26,422–35,291 copies per ancestral genome). The third most abundant repeat was AAAACCCT, which is the telomeric repeat in C. reinhardtii (characterized as TTTTAGGG in Sykorová et al. 2006). Using the mean across lines derived from an ancestor to infer ancestral abundance, there was a significant difference in the copy number of the telomere repeat across ancestral strains (supplementary fig. S2, Supplementary Material online; ANOVA, P < 2×10−16). In particular, the copy number of the telomere repeat in CC-2344 was significantly higher (approximately double: ∼4,000 vs. ∼2,000 copies) than that in all of the other inferred ancestors (post hoc Tukey test, P < 10−7). This suggests that strains differ widely in their telomere lengths. Almost all of the kmers (40/46) were interspersed to some extent with the telomere repeat from paired-end analysis (supplementary file S2, Supplementary Material online).
Principal components analysis using the abundances of kmers with at least 2 copies in each ancestral strain (including kmers absent in some ancestors) showed that lines derived from a single ancestor clustered together on the two largest components that together explained 37% of the variance (supplementay fig. S3, Supplementary Material online). PCA plots using different subsets of kmers yielded similar patterns (supplementary file S1, Supplementary Material online). Thus, while it is possible that the mean copy number of a given kmer across lines could have shifted over the ∼1,000 generations of MA and not accurately represent the actual copy number in the ancestor, the observed copy number changes that occurred during the MA were not enough to obscure the relatedness among lines. This suggests that any MA copy number changes were considerably less than the copy number differences that have evolved since the divergence of the geographically diverse ancestral strains.
Inferred Ancestral Abundant Repeats Show 6-Mer Propensity and High Presence/Absence Polymorphism
To focus on kmers likely involved in larger tandem arrays rather than smaller microsatellites, we restricted further analysis to the 46 unique kmers with an average copy number of at least 100 in at least one inferred ancestor, which we refer to henceforth as being “high-abundance.” If, in any of the other inferred ancestors, the kmer had fewer than 15 copies, it was considered absent from that ancestor. Each inferred ancestor contained 26–31 high-abundance kmers, with 18 being high-abundance in all six ancestors. Among the 46 kmers, 6-mers were by far the most common, with 19 distinct 6-mer repeat sequences.
Most high-abundance kmers had relatively uniform abundances among inferred ancestors, while others showed extreme presence/absence polymorphism (fig. 1A and supplementary file S1, Supplementary Material online). 17 of the 46 kmers were present at high abundance in some inferred ancestors but absent in others (fig. 1B). Of these 17 polymorphic kmers, 10 were AT-rich with unit length >10 bp, discussed in the following section. Inferred ancestors CC-2342 (Pennsylvania-1) and CC-2931 (North Carolina) had the most similar copy number profile of the 17 polymorphic kmers (Pearson correlation 0.9), whereas all other pairs of inferred ancestral lineages had at most a 0.32 correlation. Notably, some kmers had >1,000 copies in one to two inferred ancestors while being completely absent from others. For example, AAATAGCAGTATAT had 1,926 and 1,896 copies in CC-2342 and CC-2931, respectively, but was absent in the other lineages. AAATAACAAT had 1,203 copies in CC-1373 (Massachusetts) but was absent from other lineages. In total, five of the six inferred ancestral strains contained one to three unique high-abundance kmers that were absent from other strains.
Fig. 1.

—Polymorphism in tandem repeat abundance and presence/absence. (A) Mean copy number of five tandem repeats in the MA lines from six different ancestral strains. (B) Mean copy number of the 17 repeats that are polymorphic in presence/absence in the inferred six ancestral strains. Table cells are color-coded in a gradient by kmer abundance, and values were rounded down to 0 if the mean copy number was <15.
Highly Polymorphic AT-Rich Kmers May Originate from the Chloroplast Genome
To identify if high presence/absence polymorphic kmers were unusual due to their AT-richness and relatively long unit length (>10 bp), we examined their GC content and length distribution relative to all other kmers and the nuclear and organellar genomes. The GC content and unit length distribution of all kmers split into two distinct groups: short kmers with high average GC, and long kmers with low GC (fig. 2). Specifically, there were 36 kmers 1–8 bp long with an average GC of 67.5%, and 10 kmers 10–20 bp long with an average GC content of 17.4% and a maximum GC content of 35%. Since the chloroplast genome is low GC (35%) and the nuclear genome is high GC (64%), and interspersed simple repeats have been characterized in the chloroplast genome (Maul et al. 2002), we considered the possibility that these long AT-rich kmers originated from the chloroplast. We then searched the chloroplast assembly for tandem repeats with units 1–20 bp and found three repeats: AAAATAAAGTGT, AAAATATATAAATATAGCT, and AAATACCTTACGGGAATAT. All three were included in the set of high-abundance, high presence/absence polymorphism kmers.
Fig. 2.
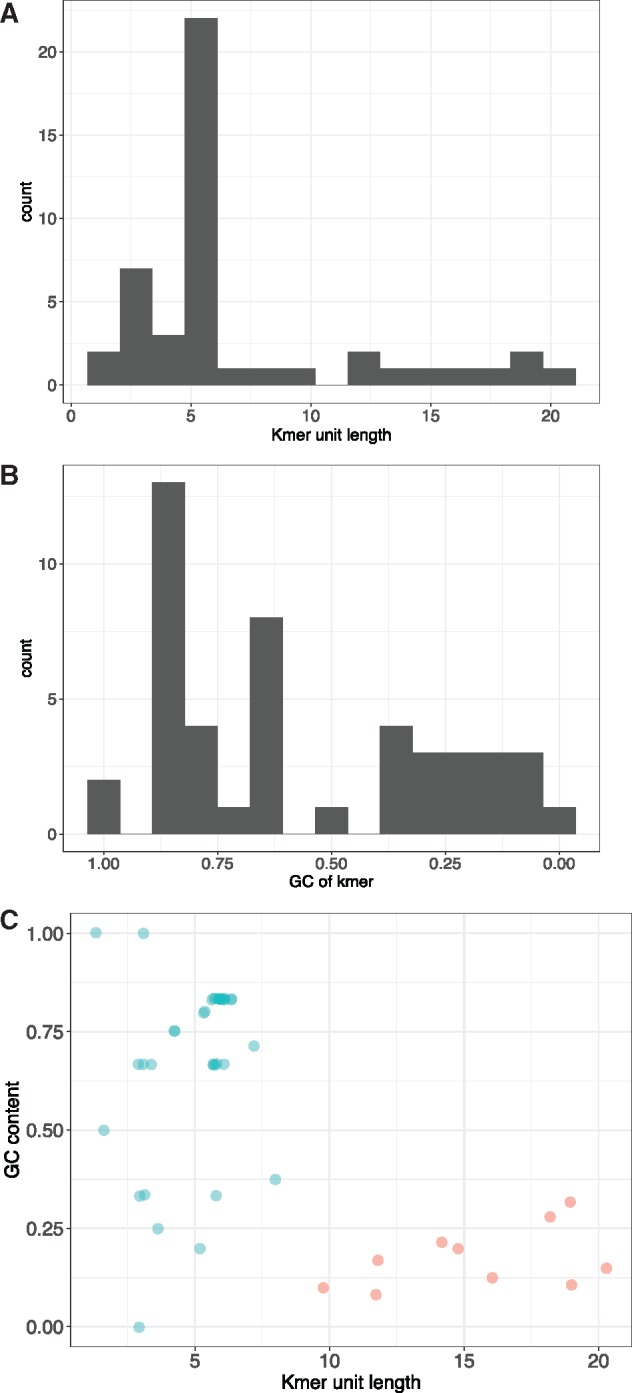
—Long kmers have a lower GC content. (A) Bimodal distribution of kmer unit length. (B) Bimodal distribution of kmer GC content. (C) Short kmers (<10 bp, blue) occupy the full range of GC contents, but long kmers (≥10 bp, pink) all have low GC content. Long kmers might have originated from the AT-rich chloroplast genome.
To determine if these repeats of putative chloroplast origin were, in fact, located in the nuclear genome, we analyzed both the forward and reverse of each paired read for presence of sequences diagnostic to the nuclear genome. Unexpectedly, all three of these putative chloroplast repeats had reads that were on the same DNA fragment as the telomere repeat. In fact, all 10 of the kmers that were abundant, polymorphic for presence/absence, AT-rich, and 10–20 bp long (fig. 2) were interspersed with the telomere repeat, supported by a total of 949 interspersed reads (1–12.5% of reads per kmer, supplementary file S2, Supplementary Material online), with 44% of those (416 reads) attributed to the three repeats found in the chloroplast assembly. This indicates that putative chloroplast genome repeats are sometimes found in genomic locations within 500 bp of telomere repeats.
Many Repeat Sequences Are Related by a Single Substitution or Indel
Since new repeat sequences are likely generated from existing ones (Wei et al. 2014; Flynn, Caldas, et al. 2017), we looked for relatedness between the 46 high-abundance kmers. Remarkably, we found 27 GC-rich kmers forming two distinct groups that are related mainly by a single mutational step (fig. 3). Eight of the kmers with presence/absence polymorphism (fig. 1B) clearly fit into the network. This suggests that the polymorphic kmer was likely present in the ancestor of all strains and was lost from some lineages; or alternatively that a kmer gained in a certain lineage is derived from a kmer already present in high abundance.
Fig. 3.

—Relationship network of 27/46 high-abundance kmers. Most kmers group into one of the two main distinct groups, related by a single nucleotide polymorphism (SNP, black dot) or indel (red dot). Kmers that are only present in one or a few strains are outlined in a colored line (see in-line legend).
We were able to infer the substitutions that potentially occurred to generate the diverse set of related repeat sequences. We assumed the consensus base at each polymorphic site was the ancestral state, and considered all 16 single nucleotide mutations (SNMs) in the network. All 16 SNMs were at C: G sites (where “:” represents complementary base pairing). Twelve of the 16 were C → G and G → C mutations, a result consistent with Ness et al. (2015), who found that genome-wide, based on 5,716 mutations accumulated under minimal selection, substitutions at C: G sites were 4.2× more frequent than expected based on the base composition than substitutions at A: T sites. The substitution pattern we found between kmer sequences was not significantly different from the neutral genome-wide substitution spectrum of Ness et al. (2015) (χ2 = 1.10, P = 0.294), showing that base substitution patterns in tandem repeats are consistent with substitutions in the rest of the genome.
Expansion and Contraction Rates of Repeats
Differences in the copy number of repeats among MA lines from a given ancestor represent mutational changes that have occurred during the MA experiment. These genome-wide copy number changes include changes potentially involving multiple loci and are inferred to be predominantly caused by replication slippage. We estimated copy number mutation rates per genome for each ancestor for all high-abundance kmers (26–31 kmers for each ancestor). The per kmer absolute mutation rates (including expansions and contractions) ranged from 0.016–7.63 copies per generation across kmers, not including the telomere repeat. We found that the kmer copy number was positively linearly related to its mutation rate in all of the ancestors (P < 1×10−15). Thus, to be able to compare and visualize mutation rates among kmers, and compare the mutation rates here to what was found in other species, we normalized the rates for each repeat by its ancestral copy number. After normalization, mutation rates ranged across ancestors from 2.64×10−4 to 4.75×10−4 copies per generation per copy. The poly-C repeat had the highest mutation rate in five of the six ancestors, with an average rate of 1.2×10−3 copies per generation per copy. The AGC repeat had the lowest mutation rate of all the kmers in all six ancestors, having an average rate of 4.28×10−5 (fig. 4).
Fig. 4.

—Variation in mutation rates. (A) Normalized expansion/contraction rates of 17 high-abundance kmers in all strains. ANOVA analysis did not suggest a significant difference in mutation rate between ancestors or kmer sequences. (B) No significant relationship between single nucleotide mutation (SNM) rate and repeat expansion/contraction rate. (C) No significant relationship between indel rate and repeat expansion/contraction rate. Each point is the mutation rate of a different MA line, color-coded by ancestral strain. SNM and indel rates are from Ness et al. (2015).
Some MA lines had much higher mutation rates across many kmers than the other lines within their ancestral group (table 1). Although we could not directly determine the directionality of change (expansion/contraction) from the MA ancestor since we did not have the sequence of the ancestor before MA, we inferred expansion if the copy number was above the mean of the MA lines derived from that ancestor and contraction if the copy number was below the mean. Interestingly, most outlier MA lines experienced high rates of both expansions and contractions by this metric, except for CC-1373-MA10 and CC-1373-MA11, which had a clear bias toward expansions. Remarkably, CC-1952-MA8 had the highest or second highest mutation rate for 23/30 kmers considered. This resulted in a mutation rate 3.6 times higher than the other MA lines from the same ancestor, caused by both expansions and contractions (table 1).
Table 1.
High Mutation Rate MA Lines for Each Inferred Ancestor
| Ancestor | Highest u | Number Kmers Highest | Kmers Considered | % Higher | Kmers Exp/Con |
|---|---|---|---|---|---|
| CC-1373 | MA-10 | 10 | 27 | 15.4 | 9/1 |
| MA-11 | 11 | 22.5 | 11/0 | ||
| CC-1952 | MA-8 | 23 | 30 | 205 | 11/12 |
| CC-2342 | MA-3 | 17 | 29 | 67.5 | 10/7 |
| MA-4 | 14 | 63.7 | 9/5 | ||
| CC-2344 | MA-11 | 10 | 25 | 81.5 | 8/2 |
| CC-2931 | MA-4 | 8 | 28 | 25.1 | 6/2 |
| MA-12 | 9 | 19.4 | 3/6 | ||
| CC-2937 | MA-8 | 11 | 28 | 82.3 | 4/7 |
Note.—Highest u lists the MA line(s) within an MA experiment that had the highest or second highest mutation rate (u) for each kmer considered. % higher refers to how much higher the focal MA line’s average mutation rate across all kmers was than the mean of that of all MA lines from that ancestor. The final column indicates how many of the high mutation rate kmers were expansions (exp) and how many were contractions (con).
Ness et al. (2015) found a significant difference in the single nucleotide mutation (SNM) rates of single-copy sequences between ancestral strains; specifically, CC-1373 had a SNM rate over three times higher than the mean of the other ancestors. We used a two-way ANOVA to test if there were significant differences in mutation rates between kmers, ancestors, or an interaction between kmer and ancestor. We found no significant difference in either of the terms. However, when grouping kmers into two categories (AT-rich kmers 10 bp or longer, vs. the rest, fig. 2), we found that the AT-rich repeats had higher mutation rates (supplementary fig. S4, Supplementary Material online, ANOVA P = 2.5×10−11, supplementary file S3, Supplementary Material online). Some MA lines were found to be outliers in SNM rates from Ness et al. (2015), potentially caused by an evolved hypermutator allele. None of these lines had particularly high or low rates of tandem repeat mutations (supplementary file S3, Supplementary Material online). In fact, when examining all 81 MA lines used for mutation rate analysis, there was no detectable relationship between SNM rate and repeat expansion/contraction rate (fig. 4B, linear model R2 = 0.02, P = 0.1, Pearson correlation = 0.18). There was also no relationship between indel rate and repeat mutation rate (fig. 4C, linear model R2 =−0.01, P = 0.81, Pearson correlation =−0.03). We conclude that copy number mutations in tandem repeats are influenced by different factors than SNM and indel mutations.
Repeats Gained and Lost during MA
In the approximately 1,000 generations of MA, it is possible that short sequences initially present in one or a few tandem copies in the genome could expand to reach the detection threshold of k-Seek in a single MA line. We searched for tandem repeats gained de novo within an MA experiment, where a tandem repeat with at least two normalized copies (and in an array at least 50 bp in a read) is unique to a single MA line (table 2). We found that the number of kmers gained was variable among ancestral groups, ranging from 3 to 40 new tandem repeats per MA experiment. For most of the ancestors, gain of new kmers was dominated by one or two MA lines (table 2). Only four of the total 79 gained kmers were gained in parallel in multiple MA experiments (supplementary file S3, Supplementary Material online).
Table 2.
Tandem Repeats Gained and Lost during MA
| Ancestor | Number Gained | Dominating MA Lines (Number Gained) | Number Lost | Dominating MA Lines (Number Lost) |
|---|---|---|---|---|
| CC-1373 | 7 | None | 16 | MA-4 (8) |
| CC-1952 | 17 | MA-13 (8), MA-8 (4) | 33 | MA-8 (23) |
| CC-2342 | 40 | MA-5 (29) | 17 | MA-3 (10) |
| CC-2344 | 4 | MA-4 (2) | 14 | MA-4 (4), MA-6 (3) |
| CC-2931 | 13 | MA-15 (6) | 12 | None |
| CC-2937 | 3 | None | 5 | None |
Note.—Some MA experiments had one or two MA lines that contributed highly (dominating) to the number of kmers gained or lost.
We also looked for repeats that were lost (or contracted to below k-Seek’s detection threshold) in a single MA line. The number of kmers lost in total across an ancestral group ranged from 5 to 33 (table 2). Interestingly, CC-2937 had the smallest number of kmers both gained and lost. Similarly to new kmers, the same MA line within an ancestral group tended to lose multiple kmers. In most cases it was not the same MA line that gained or lost multiple kmers. However, CC-1952-MA8 both gained and lost multiple kmers (with an especially high number of losses). Most kmers that were lost were rare to begin with, so drifting to absence in an MA line is not surprising. However, the poly-C repeat, which was highly abundant in all MA ancestors, was lost in one MA line in two independent MA experiments. Upon manual inspection of fastq files, it was clear that these MA lines had markedly fewer and shorter poly-C repeats than the other MA lines, and that the loss was not an artefact of the detection threshold of k-Seek or the GC correction used. Only three of the total 92 lost kmers were lost in parallel in multiple MA experiments (supplementary file S3, Supplementary Material online).
Correlation and Interspersion Structure among Repeats
Tandem repeats can be correlated in their mutation patterns (Wei et al. 2014; Flynn, Caldas, et al. 2017). If two (or more) repeats are located physically close to each other, this might cause their rates of copy number change to be correlated, and repeats similar in sequence are often located close to each other (Flynn, Caldas, et al. 2017). We used the normalized mutation rates to calculate correlation matrices between the mutation rates of the 29 kmers that are present in all six ancestors, ordering kmer sequences based on relatedness (fig. 5). Most strikingly, the ancestral strains CC-1373 (Massachusetts) and CC-2344 (Pennsylvania-2) showed similar patterns in their correlation matrices. Specifically, both possess a block of positively correlated kmers including five kmers from one of the two main families shown in figure 3 (AGGC, AGGCGG, ACGC, ACGCC, ACGCCC) and six of the kmers from the other main family in figure 3 (ACCGCC, ACCCGC, ACACGC, ACAGGC, ACAGGG, ACCAGC). AGGCGC had negative correlations with the above listed kmers in both CC-1373 and CC-2344 MA lines. Since the mutational correlations were calculated from the deviation from the mean, we checked if these two ancestors had the most similar means in the kmers listed about. However, this was not the case (supplementary file S3, Supplementary Material online). Furthermore, we found no clear relationship between the presence of interspersion between kmers and the presence of either positive or negative correlations (supplementary file S3, Supplementary Material online).
Fig. 5.

—Correlation of changes in repeat copy number. Kmers are ordered by relatedness based on figure 3, and ordering is the same in each diagram for each MA ancestor. Input data to these correlations plots were matrices of mutation rate for each kmer in each MA line of the given strain.
Another noteworthy set of correlations was seen in the descendants of CC-2342, which showed multiple smaller blocks of three to five closely related kmers having strong positive or negative correlations, with the most striking demonstration of negative correlations observed in all ancestors (fig. 5C). CC-1952 had some clear clustering of positively and negatively correlated kmers based on sequence similarity, but not nearly as pronounced as CC-2342. CC-2937 has even fewer strong correlations and less clustering with relatedness, and CC-2931 has very little correlation structure at all.
Paired-End Analysis of Repeats
Our filtering methods were effective at identifying repeats that likely exist in blocks of heterochromatic satellite arrays. Using analysis of paired-end reads, we found that all 46 high-abundance kmer sequences had tandem repeats on both reads of the pair in at least one ancestor. Per ancestor, 17–26% of paired-end reads contained repeats of either the same or different sequence on both mate pairs. It is likely that these repeats encompass at least the length of the sequenced fragments, which was ∼455 bp. The percent of reads with repeats on both pairs varied among kmers, with some kmers having <10% of their reads coming from inferred large repetitive blocks, and other kmers having 100% (supplementary fig. S5, Supplementary Material online). There was no difference in the rate of copy number change of repeats that are in long arrays (defined by having the same repeat on both paired-end reads) versus those that are in shorter arrays (ANOVA P = 0.87, supplementay fig. S6a, Supplementary Material online). There was also no difference in the inferred directionality of copy number change, that is, expansions versus contractions, between long array repeats and short array repeats (ANOVA P = 0.92, supplementary fig. S6b, Supplementary Material online).
Discussion
In this study, we characterized genome-wide tandem repeats and estimated mutation rates in them using MA experiments of six different C. reinhardtii strains. We focused on long arrays of short tandem repeats, with repeat units 1–20 bp that span at least 50 bp in tandem. A high percentage of the repeats we found spanned a length at least as long as the sequenced fragment. The genome-wide repertoire of tandem repeats had never been characterized in C. reinhardtii. We identified 46 kmers that were found to have at least 100 copies per 1× coverage in at least one of the six ancestral strains.
Patterns of Polymorphism among the Six Ancestral Strains
There were high levels of polymorphism in both presence/absence and copy number of kmers across the six ancestral strains. Surprisingly, there were 17 kmers present in high abundance (> 100 copies per genome) in some ancestral strains and absent from others. This was unexpected because recently gained repeats are likely to have a low copy number, and low copy repeats are more susceptible to being lost by drift in diverging populations. These tandem repeats must have undergone many generations of mutation in order to expand or contract to high or very low abundance, as opposed to low copy number repeats that have a more substantial probability of being lost in very few generations. Unfortunately, the divergence time of the ancestral lineages is not known, nor is the generation time of Chlamydomonas in nature (Ness et al. 2016). However, with the average mutation rate we estimated, a repeat has the potential to increase from 2 copies to 1,000 copies in ∼19,000 generations by a stepwise model and no constraint from selection.
The ancestors CC-2342 and CC-2931 were correlated in their presence/absence polymorphisms (fig. 1B), suggesting that they are more closely related than other pairs of ancestral strains. However, these strains are not the closest geographically, with one originating in Pennsylvania and the other in North Carolina. These strains may share a common ancestor sooner than expected based on a simple isolation by distance model.
Comparison to Estimates from MA Studies in Other Organisms
Flynn, Caldas, et al. (2017) used Daphnia pulex MA lines to characterize genome-wide tandem repeats and estimate mutation rates in this species. Firstly, the preferred unit length of repeats appears to vary across species. In D. pulex and Drosophila melanogaster, 5-mers and multiples of 5-mers (i.e., 10mers, 15mers, 20mers) were overrepresented (Wei et al. 2014; Flynn, Caldas, et al. 2017). In this study of Chlamydomonas reinhardtii, we instead found that 6-mers were the most common of the abundant kmers. Other species of Drosophila have a different preferred length of tandem repeats, for example, 7-mers in Drosophila virilis (Gall and Atherton 1974; Wei et al. 2018). The apparent bias favoring a particular unit size could be by chance; however it is unlikely that different tandem repeats of the same length would randomly prevail in the genome compared with repeats of other lengths. The periodicity of repeat lengths of a certain size might be selected for if it creates a preferred higher order structure or enables ideal periodicity for histone wrapping (Wu and Crothers 1984; Langley et al. 2014). Chlamydomonasreinhardtii and D. pulex also shared a pattern of sequence similarity between kmers, apparently related to each other by one to two single nucleotide mutations or an indel (fig. 3). This further supports our hypothesis that new repeat sequences are generated from mutation in existing ones followed by copy number expansion (Flynn, Caldas, et al. 2017).
The number of kmers with an average at least 100 copies, and at least 2 copies, was similar in C. reinhardtii as D. pulex (26–31 here vs. 39, and 134–198 vs. 162, respectively). However, in C. reinhardtii there were fewer kmers with an average of at least 1,000 copies. There were also fewer long kmers >15 bp units. In total, these differences resulted in almost an order of magnitude difference in the genome-wide tandem repeat content with C. reinhardtii having on an average between 148–198 kb and D. pulex having ∼1 Mb. It is a possibility that different library preparation methods could have confounded this comparison between D. pulex and C. reinhardtii. However, in support of C. reinhardtii having a true paucity of simple satellite repeats, our finding of a high copy number of AC repeats and the telomere repeat is concordant with findings in past studies (Kang and Fawley 1997; Sykorová et al. 2006).
One curious difference between these species was the difference in the expansion/contraction rates in their tandem repeats. Even after normalizing the mutation rates based on the copy number of the tandem repeats, the expansion/contraction rates were still an order of magnitude lower in C. reinhardtii than in D. pulex. Combined with overall lower abundance of tandem repetitive DNA in C. reinhardtii, these results are consistent with the hypothesis that tandem repeats are burdensome to C. reinhardtii, and copy number mutations are overall deleterious. Furthermore, since C. reinhardtii has a 10× higher effective population size than D. pulex (107, Ness et al. 2016; vs. 106, Paland et al. 2005), our findings are concordant with the drift-barrier hypothesis (Lynch and Conery 2003; Sung et al. 2012). In theory, a lower abundance of genome-wide repeats and lower mutation rates in these repeats would be preferred. Chlamydomonasreinhardtii, with a larger Ne, is able to get closer to the optimum lower mutation rate and genome-wide repetitive DNA content than D. pulex. Similarly, the single nucleotide mutation rate in C. reinhardtii is also lower than that in D. pulex, and on a similar scale (Ness et al. 2012, 2015; Keith et al. 2016; Flynn, Caldas, et al. 2017). Of course, there are other factors that could be driving the mutation rate differences between these two divergent taxa.
Relationship with Single Nucleotide Mutation Rates and Hypermutators
Genotypes that are lower in fitness have been observed in the laboratory to have higher rates and different patterns of mutation (Sharp and Agrawal 2012, 2016). This phenomenon might affect multiple classes of mutation. Ness et al. (2015) found CC-1373 to have a higher single nucleotide mutation rate than the other five strains, and it was also the strain that was observed to be in the worst culture condition of the experiment. However, we did not find variation in mutation rates between the different ancestral strains. This demonstrates that being in worse condition, although related to higher SNM rates, was not related to increased rates of expansion and contraction in tandem repeats.
Alleles that alter mutation rates have been observed in MA and experimental evolution studies (i.e., hyper or hypo mutators), presumably caused by mutations in cellular machinery that influence DNA damage or repair (Avila 2006; Tenaillon et al. 2016). Ness et al. (2015) found mutation rates to vary substantially among MA lines from the same ancestor, with some having very low or very high mutation rates. None of these outlier lines in SNMs deviated far from the average in tandem repeat mutations. In fact, repeat expansion/contraction rates were not correlated with single nucleotide or indel mutation rates, using all 81 MA lines with mutation rate estimates (supplementary file S3, Supplementary Material online). This is consistent with the notion that SNMs and satellite expansion/contractions are caused by different biochemical processes that work independently, at least in these C. reinhardtii strains. However, we did find putative tandem repeat hypermutator lines that acquired many more copy number changes than the other lines within their ancestral group (tables 1 and 2). Most striking was CC-1952-MA8, which also experienced high levels of de novo kmer gain and loss, and had a 3.6-fold higher mutation rate than the mean of the other lines from CC-1952. This line had an approximately equal balance of expansions and contractions, contrary to previous work with Daphnia (Flynn, Caldas, et al. 2017), where the MA line with the highest mutation rate was dominated by expansions only. Two MA lines, although with less extreme mutation rates, did follow this pattern of more expansions than contractions (CC-1373-MA10 and CC-1373-MA11, table 1).
Possible Repetitive DNA Exchange between the Nuclear and Chloroplast Genomes
We found three high-abundance AT-rich repeats linked to telomere repeats that are also present as tandem repeats in the chloroplast genome assembly. All other AT-rich kmers we discovered were also interspersed with the telomere repeat. The GC content of the nuclear genome is 64%, while the chloroplast genome is 35%. AT-rich repeats have been previously reported in the chloroplast genome (Maul et al. 2002). The clear separation between the kmers of different lengths and GC contents might correspond to repeats originating from both the nuclear and chloroplast genomes (fig. 2). Three possibilities could potentially explain the interspersion of AT-rich and telomeric repeats. 1) The AT-rich repeats are actually located on the nuclear genome, and the chloroplast genome is partially misassembled. Although we cannot exclude this possibility, matching patterns of GC content and kmer unit length make it less likely. 2) DNA segments, including the telomere tandem repeat, migrated from the nuclear to chloroplast genome. To our knowledge, this direction of DNA transfer has not been reported in plants. 3) Repetitive DNA from the chloroplast got incorporated into the nuclear genome. This is the most likely possibility, since genetic transfer from the chloroplast to nuclear genome has occurred repeatedly in plants, although this phenomenon was found to be rare or even undetectable in C. reinhardtii (Lister et al. 2003).
One caveat is if the repeats actually were sequenced from chloroplast rather than nuclear DNA (possibility 1 or 2), the copy numbers we estimated for these repeats may not be accurate. This is because we computed the depth and GC correction based on the nuclear genome; and the chloroplast genome is expected to be sequenced to a higher depth than the nuclear genome. Even if possibility (3) is correct, we may have some reads coming from the chloroplast and some coming from the nuclear genome, making it difficult to correct for.
Patterns of Correlation in Copy Number Changes of Tandem Repeats
We found interesting patterns of correlation in mutation rates between kmers, which differed among ancestors (fig. 5). There were clusters of strong correlations in kmer sequences that are closely related by single nucleotide mutations (fig. 3). Positive correlations might be caused by physical linkage between kmers. Since the correlations are clustered by sequence similarity, and similar kmers were probably derived from one another through mutation, similar kmers may be physically linked. Negative correlations are more puzzling, and we can only speculate on what causes them. In our previous work, we hypothesized that negative correlations occurred between repeats in conflict with each other, particularly when selection is at play (Wei et al. 2014; Flynn, Caldas, et al. 2017). Differences in the genomic distribution of kmers among ancestors or cryptic selection in different MA experiments might partially explain these patterns.
It was quite surprising to observe the similarity in the correlation plots of ancestors CC-1373 (Massachusetts) and CC-2344 (Pennsylvania-2). These correlations were calculated based on the deviation from the mean copy number, and the mean copy numbers varied between these ancestors. This finding suggests that these tandem repeats are evolving similarly and under similar constraints in these two lineages. These two lineages did not share any other similarities in polymorphic kmers, making it unlikely that this pattern arose because of a more recent common ancestor between these lineages.
Conclusion
In this study, we found high levels of variation in tandem repeat content between six strains of Chlamydomonas reinhardtii. Mutation rates of expansion and contraction were high, although an order of magnitude lower than the crustacean Daphnia pulex, which has a lower effective population size. Our data revealed evidence of potential exchange between chloroplast and nuclear genome repeats. We also found that rates of expansion and contraction were unrelated to rates of single nucleotide mutation and small insertions and deletions. Finally, we demonstrated a parallelism in the evolution and constraint patterns of expansion and contraction of tandem repeats. In total, using short-read sequencing of MA lines, we were able to answer questions about the dynamics of tandem repeat evolution in the C. reinhardtii genome. More extensive phylogenetic comparisons will further elucidate the role of neutral mutational processes in determining abundances, correlations and turnover of tandem repeats in eukaryotic genomes.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
The authors would like to thank Rob Ness for data sharing and discussions about the study. This work was supported by the National Institutes of Health (NIGMS R01GM119125 to D.A.B. and A.G.C.; Ruth L. Kirschstein National Research Service Award F32GM126736 to S.E.L.) and the Natural Sciences and Engineering Research Council (Postgraduate Scholarship-Doctoral to J.M.F.).
Literature Cited
- Avila V. 2006. Increase of the spontaneous mutation rate in a long-term experiment with Drosophila melanogaster. Genetics 173(1):267–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Speed TP.. 2012. Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Res. 40(10):e72.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson G. 1999. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27(2):573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B.. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Sniegowski P, Stephan W.. 1994. The evolutionary dynamics of repetitive DNA in eukaryotes. Nature 371(6494):215–220. [DOI] [PubMed] [Google Scholar]
- Flynn JM, Caldas I, Cristescu ME, Clark AG.. 2017. Selection constrains high rates of tandem repetitive DNA mutation in Daphnia pulex. Genetics 207(2):697–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn JM, Chain FJJ, Schoen DJ, Cristescu ME.. 2017. Spontaneous mutation accumulation in Daphnia pulex in selection-free vs. competitive environments. Mol Biol Evol. 34(1):160–173. [DOI] [PubMed] [Google Scholar]
- Gall JG, Atherton DD.. 1974. Satellite DNA sequences in Drosophila virilis. J Mol Biol. 85(4):633–664. [DOI] [PubMed] [Google Scholar]
- Garrido-Ramos MA. 2017. Satellite DNA: an evolving topic. Genes 8(9):230.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodstein DM, et al. 2012. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 40(Database issue):D1178–D1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halligan DL, Keightley PD.. 2009. Spontaneous mutation accumulation studies in evolutionary genetics. Annu Rev Ecol Evol Syst. 40(1):151–172. [Google Scholar]
- Henikoff S, Ahmad K, Malik HS.. 2001. The centromere paradox: stable inheritance with rapidly evolving DNA. Science 293(5532):1098–1102. [DOI] [PubMed] [Google Scholar]
- Hoskins RA, et al. 2007. Sequence finishing and mapping of Drosophila melanogaster heterochromatin. Science 316(5831):1625–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata-Otsubo A, et al. 2017. Expanded satellite repeats amplify a discrete CENP-A nucleosome assembly site on chromosomes that drive in female meiosis. Curr Biol. 27(15):2365–2373.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagannathan M, Warsinger-Pepe N, Watase GJ, Yamashita YM.. 2017. Comparative analysis of satellite DNA in the Drosophila melanogaster species complex. G3 (Bethesda) 7(2):693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang TJ, Fawley MW.. 1997. Variable (CA/GT)n simple sequence repeat DNA in the alga Chlamydomonas. Plant Mol Biol. 35(6):943–948. [DOI] [PubMed] [Google Scholar]
- Keith N, et al. 2016. High mutational rates of large-scale duplication and deletion in Daphnia pulex. Genome Res. 26(1):60–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley SA, Karpen GH, Langley CH.. 2014. Nucleosomes shape DNA polymorphism and divergence. PLoS Genet. 10(7):e1004457.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL.. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9(4):357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister DL, Bateman JM, Purton S, Howe CJ.. 2003. DNA transfer from chloroplast to nucleus is much rarer in Chlamydomonas than in tobacco. Gene 316:33–38. [DOI] [PubMed] [Google Scholar]
- Lynch M, Conery JS.. 2003. The origins of genome complexity. Science 302(5649):1401–1404. [DOI] [PubMed] [Google Scholar]
- Maul JE, et al. 2002. The Chlamydomonas reinhardtii plastid chromosome: islands of genes in. A sea of repeats. Plant Cell 14(11):2659–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merchant SS, et al. 2007. The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science 318(5848):245–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan AD, Ness RW, Keightley PD, Colegrave N.. 2014. Spontaneous mutation accumulation in multiple strains of the green alga, Chlamydomonas reinhardtii. Evolution 68(9):2589–2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ness RW, Kraemer SA, Colegrave N, Keightley PD.. 2016. Direct estimate of the spontaneous mutation rate uncovers the effects of drift and recombination in the Chlamydomonas reinhardtii plastid genome. Mol Biol Evol. 33(3):800–808. [DOI] [PubMed] [Google Scholar]
- Ness RW, Morgan AD, Colegrave N, Keightley PD.. 2012. Estimate of the spontaneous mutation rate in Chlamydomonas reinhardtii. Genetics 192(4):1447–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ness RW, Morgan AD, Vasanthakrishnan RB, Colegrave N, Keightley PD.. 2015. Extensive de novo mutation rate variation between individuals and across the genome of Chlamydomonas reinhardtii. Genome Res. 25(11):1739–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novák P, et al. 2017. TAREAN: a computational tool for identification and characterization of satellite DNA from unassembled short reads. Nucleic Acids Res. 45(12):e111.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno S. 1972. So much ‘junk’ DNA in our genome. Brookhaven Symp Biol. 23:366–370. [PubMed] [Google Scholar]
- Orgel LE, Crick FH.. 1980. Selfish DNA: the ultimate parasite. Nature 284(5757):604–607. [DOI] [PubMed] [Google Scholar]
- Paland S, Colbourne JK, Lynch M.. 2005. Evolutionary history of contagious asexuality in Daphnia pulex. Evolution 59(4):800–813. [PubMed] [Google Scholar]
- Petit N, Barbadilla A.. 2009. Selection efficiency and effective population size in Drosophila species. J Evol Biol. 22(3):515–526. [DOI] [PubMed] [Google Scholar]
- Rojo V, et al. 2015. Evolutionary dynamics of two satellite DNA families in rock lizards of the genus Iberolacerta (Squamata, Lacertidae): different histories but common traits. Chromosome Res. 23(3):441–461. [DOI] [PubMed] [Google Scholar]
- Ruiz-Ruano FJ, López-León MD, Cabrero J, Camacho JPM.. 2016. High-throughput analysis of the satellitome illuminates satellite DNA evolution. Sci Rep. 6:28333.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samoluk SS, Robledo G, Bertioli D, Seijo JG.. 2017. Evolutionary dynamics of an at-rich satellite DNA and its contribution to karyotype differentiation in wild diploid Arachis species. Mol Genet Genomics 292(2):283–296. [DOI] [PubMed] [Google Scholar]
- Seyfert AL, et al. 2008. The rate and spectrum of microsatellite mutation in Caenorhabditis elegans and Daphnia pulex. Genetics 178(4):2113–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp NP, Agrawal AF.. 2012. Evidence for elevated mutation rates in low-quality genotypes. Proc Natl Acad Sci U S A. 109(16):6142–6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp NP, Agrawal AF.. 2016. Low genetic quality alters key dimensions of the mutational spectrum. PLoS Biol. 14(3):e1002419.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons MJ, Crow JF.. 1977. Mutations affecting fitness in Drosophila populations. Annu Rev Genet. 11:49–78. [DOI] [PubMed] [Google Scholar]
- Sung W, Ackerman MS, Miller SF, Doak TG, Lynch M.. 2012. Drift-barrier hypothesis and mutation-rate evolution. Proc Natl Acad Sci U S A. 109(45):18488–18492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykorová E, et al. 2006. Minisatellite telomeres occur in the family Alliaceae but are lost in Allium. Am J Bot. 93(6):814–823. [DOI] [PubMed] [Google Scholar]
- Tenaillon O, et al. 2016. Tempo and mode of genome evolution in a 50,000-generation experiment. Nature 536(7615):165–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treangen TJ, Salzberg SL.. 2011. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet. 13(1):36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei KH-C, et al. 2018. Variable rates of simple satellite gains across the Drosophila phylogeny. Mol Biol Evol. 35(4):925–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei KH-C, Grenier JK, Barbash DA, Clark AG.. 2014. Correlated variation and population differentiation in satellite DNA abundance among lines of Drosophila melanogaster. Proc Natl Acad Sci U S A. 111(52):18793–18798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HM, Crothers DM.. 1984. The locus of sequence-directed and protein-induced DNA bending. Nature 308(5959):509–513. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.