

Abstract
A number of hereditary syndromes have been found to be associated with pheochromocytoma development, but there is a paucity of data regarding secondary molecular events, such as downregulation of the p16 INK4A /Cdkn2a gene (hereafter p16), contributing to pheochromocytoma tumorigenesis. Using tissue microarray and immunohistochemistry, we evaluated the expression of p16 in 31 pheochromocytoma tumor specimens. Our results showed that the p16 gene was expressed at low level or even not expressed in all but one specimens [30/31 (96.8%)], indicative of the prevalence of p16 downregulation in pheochromocytomas. In contrast, high expression of p16 was observed in the majority of control “normal” specimens [5/7 (71.6%)]. To further investigate the molecular mechanisms underlying p16 downregulation in pheochromocytomas, we used quantitative real-time PCR, methylation-specific PCR, and direct DNA sequencing to analyze these specimens for potential genetic alterations of the p16 gene. Deletions and aberrant CpG methylation of p16 were identified in 9 (29.0%) and 11 (35.5%) specimens, respectively, while one specimen harbored a point mutation, Ala → Pro at residue 20 of P16, and this mutation led to an eightfold decrease in the CDK4-inhibitory activity of P16. The overall frequency of p16 genetic alterations is 67.7%. Taken together, our results demonstrate that reduced expression of p16 is a common event in human pheochromocytomas, and the primary cause for such downregulation is inactivating genetic abnormalities in the p16 gene.
Key words: p16, Pheochromocytomas, Expression downregulation, Genetic alterations
INTRODUCTION
Pheochromocytomas are rare neuroendocrine tumors histologically characterized by chromaffin tissue and comprised of catecholamine-containing neurosecretory granules (11,13). These tumors are primarily located in the adrenal medulla, but may also arise in the dorsal root ganglia of the sympathetic nervous system. Pheochromocytomas may present asymptomatically or as part of an endocrine dyscrasia precipitated by oversecretion of catecholamines. The most common sequellae of catecholamine abundance is hypertension, which can be sustained or paroxysmal and may lead to death from cardiovascular or cerebrovascular disease (11). When asymptomatic, they are often discovered during radiographic investigation of an unrelated abdominal complaint or during screening evaluation of a patient with multiple endocrine neoplasia (MEN) syndrome type 2 (13). Although pheochromocytomas are known to be associated with several hereditary syndromes, including MEN 2, von Hippel-Lindau (VHL) disease, neurofibromatosis type 1 (NF 1), hereditary paraganglioma, and SDHD gene-related tumors, there is little information about other genetic and epigenetic abnormalities.
The P16/CDK4/Rb/E2F pathway is a key component of the machinery controlling cell cycle progression (23). In the G1-to-S transition, cyclin-dependent kinase 4 (CDK4)-mediated phosphorylation of the retinoblastoma susceptible gene product (Rb) disrupts the incompetent Rb/E2F complexes, which leads to the release of active transcription factors E2Fs, thus triggering the transcription of genes required for entry into the S phase. The p16 INK4A /Cdkn2a/MTS1 gene (hereafter, abbreviated as p16) encodes a 16-kDa protein that binds to CDK4 and specifically inhibits CDK4-mediated phosphorylation of Rb as well as the subsequent E2F-mediated transcription (21). Perturbations in this intricate pathway, such as downregulation of p16, lead to uncontrolled cell proliferation and may subsequently promote the process of tumorigenesis.
In the past decade, it has been well demonstrated that p16 is a tumor suppressor frequently mutated or deleted in most of human cancers (7,8,16); thus, p16 could be a potential target for the development of novel cancer therapy and prevention strategies (14). While inactivation of p16 has been extensively studied in cancers such as oral, lung, liver, and pancreatic tumors (14,17), data about the role of p16 in pheochromocytomas are limited partly due to the paucity of such type of tumors (13). To date, only two studies have been reported about the genetic abnormalities of p16 in human pheochriomomas. In the first study, Aguiar et al. (1) used a semiquantitative multiplex PCR to detect p16 deletion in 26 pheochromocytomas specimens. The failure to find p16 deletions in any of these specimens led to a conclusion that the p16 gene is not deleted in either hereditary or sporadic pheochromocytomas, and therefore probably does not play a role in the pathogenesis of pheochromocytomas. In the second study, Dammann et al. (3) investigated the methylation status of the p16 gene in both hereditary and sporadic pheochromocytomas by methylation-specific PCR, and hypermethylation of the p16 gene was found in 24% of tested specimens (6/25), implying that inactivation of the p16 gene could contribute to the development of pheochromocytomas.
On one hand, there are “inconsistencies” between these two studies in regard to the role of p16 inactivation in the development of pheochromocytomas. On the other hand, multiple mechanisms, including homozygous/hemizygous deletions, aberrant methylation of 5′ CpG islands, and point mutations are known to be involved in genetic inactivation of the p16 gene in human tumors (14,19). Previous studies are not comprehensive, and deletions, mutations, and methylation of the p16 gene should be assessed concomitantly. Furthermore, overexpression of p16 has been found in a number of human tumors including colon, prostate, brain, lung, and oral cancers (12) and is related to poor prognosis of these tumors, indicating that in addition to genetic alterations, epigenetic changes of p16 could contribute to the development of human tumors.
In our current study, we investigated the expression profile of p16 as well as the underlying molecular mechanisms in a series of pheochromocytoma specimens obtained at the time of surgical resection at The Ohio State University Medical Center. Our results show that epigenetic silencing of p16 is prevalent in human pheochromocytomas and such silencing is mainly attributed to genetic inactivation of the p16 gene.
MATERIALS AND METHODS
Tumor Specimens
Thirty-one pheochromocytoma tumors from 27 patients were obtained at the time of surgical resection at The Ohio State University Medical Center. Following resection, the tissues were fixed in formalin and paraffin embedded according to routine protocol. The diagnosis of pheochromocytoma was made pre-operatively based on clinical presentation, urine biochemical analyses, and radiological imaging for tumor localization. Histologic confirmation of the tumor type was made by a single pathologist (W.L.F.) after hematoxylin and eosin staining. Seven specimens from histologically normal appearing adjacent adrenal tissues were collected as control. This study was approved by the Institutional Review Board of The Ohio State University.
Tissue Microarray (TMA) and Immunohistochemistry (IHC) for P16
The methods for TMA creation have been described previously (4). Briefly, paraffin-embedded tissues were obtained from the archival files at The Ohio State University Department of Pathology. Two tissue cores (1.5 mm diameter each) were punched out of each donor paraffin block and transferred to each of the recipient TMA blocks using a precision instrument (Beecher Instruments, Silver Spring, MD). Paraffin-embedded tissue was cut at 4 μm and placed on positively charged slides and then heated to 40°C for 30 min. After leveling paraffin and cores, the array was cooled to 4°C for 15 min.
TMA slides were placed in a 60°C oven for 1 h, cooled, deparaffinized, and rehydrated through xylene and graded ethanol solutions to water. All slides were quenched for 5 min in a 3% hydrogen peroxide solution in water to block for endogenous peroxidase. Antigen retrieval was performed by a heat method in which the specimens were placed in a citric acid solution (Target Retrieval Solution, pH 6.1, DakoCytomation, Carpinteria, CA) for 25 min at 94°C using a vegetable steamer and cooled for 15 min in solution. Slides were placed on a DakoCytomation Autostainer immunostaining system for use with IHC. The primary monoclonal anti-P16 antibody (Cell Marque, Hot Springs, AK) was added at a dilution of 1:20 and incubated for 60 min. A labeled streptavidin-biotin complex was used for detection, and visualization occurred after the application of 3,3′-diamino-benzidine chromogen. Slides were counterstained in Richard Allen hematoxylin, dehydrated through graded ethanol solutions, and cover-slipped. Positive and negative controls were stained appropriately. All TMA slides were read by a senior pathologist (W.L.F.). The numbers of cells with convincing nuclear P16 staining were determined for each sample and reported as a percentage. Duplicate slides were evaluated for all tumor samples, and the mean values of samples were used to categorize the samples as previously described (6): complete absence of nuclear immunostaining, P16-negative; 1–25% of positive nuclei, P16-low; >25% of positive nuclei, P16-high.
Genomic DNA Extraction From Tumor and Control Tissues
Genomic DNA was extracted from specimens using a ChargeSwitch gDNA mini tissue extraction kit (Invitrogen, Carlsbad, CA) as the manufacturer recommended. Two cores of tumor or control (10 mg) were separated from archived paraffin blocks and confirmed by the pathologist (W.L.F.). After digestion with proteinase K in the lysis buffer at 65°C overnight, the tumor samples were treated with RNaseA at room temperature for 1 h. Subsequently, the supernatants were separated through centrifugation and incubated with magnetic beads at room temperature for 20 min. The beads were separated on the Charge Switch MagnaRack and washed with the purification buffer twice before they were suspended in the elution buffer and incubated at 55°C overnight. Consequently, the beads were removed out of the solution using the MagnaRack, and the solutions containing genomic DNA were transferred into new tubes and stored at −20°C for further analyses. DNA yields were determined by spectrophotometry.
Real-Time PCR to Detect Homozygous Deletions in Human p16 Exon 2
Real-time PCR amplification reactions were carried out using the Smart Cycler (Cepheid, Sunnyvale, CA). Forward (5′-GGC TCT ACA CAA GCT TCC TTT CC-3′) and reverse (5′-TCA TGA CCT GCC AGA GAG AAC A-3′) primers (Life Technologies) were used at a final concentration of 0.2 μM. The dual-labeled fluorogenic probe (5′-FAM-CCC CCA CCC TGG CTC TGA CCA-TAMRA-3′) was used at a final concentration of 0.1 μM (2). Each reaction was performed in the presence of 200 μM dNTPs, 3 mM MgSO4, 1.25 units of Taq polymerase (Stratagene, LaJolla, CA), 1× additive (1 mg/ml BSA, 750 mM Trehalose, and 1% Tween 20), and 2.0 μl of genomic DNA (containing about 5–30 ng DNA). Cycling conditions were: 96°C for 1 min, followed by 55 cycles of 30 s at 95°C, 30 s at 62°C, 45 s at 68°C. A final extension step was performed at 68 °C for 5 min. A housekeeping gene, β-actin, was coamplified as an internal control. The primer and fluorogenic probe sequences used for the internal control were: forward primer, 5′-AGC GCG GCT ACA GCT TCA-3′; reverse primer, 5′-CGT AGC ACA GCT TCT CCT TAA TGC-3′; and the probe, 5′-TET-ATT TCC CGC TCG GCC GTG GT-TAMRA-3′. All experiments were performed in triplicate.
p16 Gene Deletion Analysis
The accuracy of the above real-time PCR assay was verified using a series of mixtures of genomic DNA from human Hela cells (+/+) (9) and Mia PaCa-2 cells (−/−) (20) at various ratios (Hela/Mia PaCa-2 = 100:0, 75:25, 50:50, 25: 75, 0:100). Multiplex real-time PCR was performed and the resulting cycle threshold (Ct) values were normalized using the following equation: ΔΔCt = ΔCtp16 – ΔCtβ-actin, where ΔCtp16 is equal to Ctp16 of a sample minus Ctp16 of Hela, and ΔCtβ-actin is the Ctβ-actin of a sample minus the Ctβ-actin of Hela (15). While ΔCtβ-actin correlates with the difference in total genomic DNA concentrations between the sample and Hela, ΔCtp16 is ascribed to the “total” difference in p16 gene concentrations between the sample and Hela (wild-type), including the contaminating p16 DNA and tumor p16 DNA. Therefore, ΔΔCt reflects the difference in the p16 gene dosage (15). When the ΔΔCt values were plotted against known ratios of normal p16 DNA (in exponential form), a linear graph with a correlation coefficient of 0.992 was obtained (Fig. 2), indicating that the relative concentration p16 can be accurately measured using this technique (2). On the basis of these observations, p16 gene dosage from pheochromocytoma tumor specimens was determined using the above real-time PCR assay and the results were interpreted as follows (12): relative concentration <25%, p16 homozygous deletion (−/−); relative concentration >25% and <75%, p16 hemizygous deletion (+/−); and relative concentration >75%, p16 wild-type (+/+).
Figure 2.
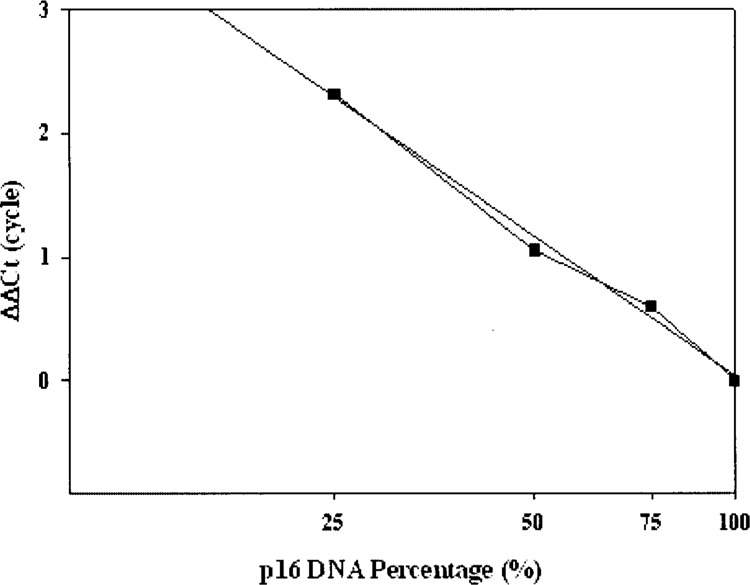
Standard curve for real-time PCR based human p16 gene deletion analysis. Multiplex real-time PCR for both human p16 and β-actin genes was performed with a series of mixtures of geneomic DNA from Hela (+/+ p16) and Mia PaCa-2 (−/− p16) cells at various rations. The resulting Ct values were normalized as described in Materials and Methods. The ΔCt values were plotted against the relative ration of normal p16 DNA in the mixtures (in exponential form) to get a standard curve with a linear correlation coefficient of 0.992.
Methylation-Specific PCR of a CpG Island in the Human p16 Promoter
Genomic DNA from tumor tissues were bisulfite modified using the CpGenome DNA modification kit (Chemicon International, Temecula, CA), and methylation at the promoter region of p16 was analyzed using the CpG WIZ® p16 Amplication Kit (Chemicon International). The PCR mixture contained 1× PCR buffer, 1× enhancer, 1.5 mM MgCl2, dNTPs (each at 1.25 mM), 1 unit of platinum Taq DNA polymerase (Invitrogen), primers (0.2 μM each), and 10 μl of bisulfite-modified DNA in a final volume of 50 μl. Amplification was performed in a GeneAmp 9700 Thermal Cycler (Applied Biosystems, Foster City, CA) with PCR conditions of 95°C for 2 min followed by 40 cycles of 95°C for 45 s, 60°C for 45 s, 72°C for 60 s, and a final elongation step of 72°C for 5 min. The primer sequences in this kit are not disclosed for proprietary reasons. The PCR products were analyzed by electrophoresis on a 10% PAGE gel. Fragments amplified from methylated and unmethylated p16 genomic DNA templates were 145 and 154 bp, respectively. Positive and negative controls were provided by the manufacturer.
Automatic Sequencing of p16 Exons 1 and 2
Human p16 exon 1 was amplified by PCR using the following intron-based primers: 5′-GCT GCG GAG AGG GGG AGA GCA GGC A-3′ (forward) and 5′-GCG CTA CCT GAT TCC AAT TC-3′ (reverse) (19). Exon 2 was amplified in two fragments (2a and 2b) with at least one of the primers in each set located within an intron in order to exclude coamplification of a similar gene family member or potential pseudo-genes. The primers for 2a were 5′-ACA AGC TTC CTT TCC GTC ATG CCG-3′ (forward) and 5′-CCA GGC ATC GCG CAC GTC CA-3′ (reverse), while the 2b primers were 5′-TTC CTG GAC ACG CTG GTG GT-3′ (forward) and 5′-TCT GAG CTT TGG AAG CTC TCA G-3′ (reverse). Potential mutations at exon 3 were not analyzed because exon 3 represents only 3% (12 bp) of the coding sequence of p16 and mutations in this region have not been shown to be pathogenetically significant (4). The PCR mixture contained 1× PCR buffer, 1.5 mM MgCl2, 1.25 mM dNTPs, 1.0 μM of each primer, 1 unit of Taq DNA polymerase (Invitrogen), 1.5× enhancer, and 4 μl of genomic DNA in a final volume of 50 μl. Cycling conditions were: 96°C for 2 min (1 cycle); 30 s at 95°C, 30 s at 56°C, and 30 s at 72°C (50 cycles); 5 min at 72°C (1 cycle). The PCR products were purified using a PCR Product Purification kit (Qiagen, Valencia, CA), and subsequently sequenced using an ABI 377A automated DNA sequencer (Applied Biosystems). Both strands of PCR-amplified fragments were sequenced in order to confirm potential mutations.
Mutant Protein Expression and Characterization
Human p16 Ala20Pro mutant was generated through PCR-mediated site-directed mutagenesis (Stratagene, La Jolla, CA), and the mutant protein was expressed and purified in bacteria as previously described (15,19). Briefly, P16 Ala20Pro mutant protein was expressed as glutathione S-transferase (GST)-fusion proteins in Esherichia coli BL21 (DE3) (Stratagene), and after affinity purification using reduced glutathione-agarose (Sigma, St Louis, MO), the GST tag was removed by digestion with PreScission protease (Amersham Biosciences, Piscataway, NJ). Free P16 Ala20 Pro protein was further purified with a S100 gel filtration column, and its function and structure were analyzed using an in vitro CDK4 kinase inhibition assay and NMR assays, respectively (15,19).
Statistical Analyses
The correlation between the frequency of promoter methylation, mutation, and deletion of p16 and age, and sporadic/familial tumors were examined with the chi-square test and the Student t-test. A two-sided value of p < 0.05 was considered statistically significant.
RESULTS
Patient Demographics
Thirty-one tumor specimens obtained from 27 patients were analyzed for p16 genetic alterations and altered expression by the techniques described above. Patient demographics are listed in Table 1. Four patients were found to have bilateral tumors and one patient was noted to have a functioning right retroperitoneal paraganglioma. The mean age was 43 years, and 40.7% of patients were males. Nineteen patients had sporadic tumors (70.4%) and eight had MEN 2-associated tumors (29.6%). Although patients with familial pheochromocytomas tended to be younger (mean age 38.9 vs. 44.7 years), this difference was not found to be statistically significant (p = 0.18).
TABLE 1.
PATIENT DEMOGRAPHICS
| Patient | Familial/Sporadic | Sex | Age (Years) | Location* |
|---|---|---|---|---|
| 1 | sporadic | M | 46 | L |
| 2 | sporadic | M | 51 | R |
| 3 | sporadic | F | 38 | L |
| 4 | MEN2 | M | 20 | L |
| 5 | sporadic | F | 37 | R |
| 6 | MEN2 | F | 49 | R |
| 7 | sporadic | F | 49 | L |
| 8 | sporadic | F | 71 | R |
| 9 | MEN2 | F | 47 | B |
| 10 | sporadic | F | 66 | L |
| 11 | sporadic | F | 49 | R |
| 12 | sporadic | F | 31 | R |
| 13 | sporadic | F | 50 | R |
| 14 | sporadic | F | 33 | R |
| 15 | sporadic | M | 23 | R |
| 16 | MEN2 | M | 37 | B |
| 17 | sporadic | M | 45 | L |
| 18 | MEN2 | F | 32 | B |
| 19 | sporadic | M | 35 | L |
| 20 | sporadic | F | 40 | P |
| 21 | sporadic | M | 43 | R |
| 22 | sporadic | F | 27 | R |
| 23 | sporadic | F | 83 | R |
| 24 | MEN2 | M | 49 | B |
| 25 | sporadic | M | 33 | L |
| 26 | MEN2 | F | 44 | L |
| 27 | MEN2 | M | 33 | R |
R: right-sided tumor; L: left-sided tumor; B: bilateral tumor; P: paraganglioma.
Tissue Microarray Analysis
IHC was used to evaluate the expression of p16 in these pheochromocytomas specimens. As summarized in Table 2, low nuclear expression of p16 was found in all tumor specimens but specimen T16L (Fig. 1). Among the control “normal” specimens, five of them exhibited high expression of p16, while expression of p16 at low level was found in the other two specimens. In comparison, reduced expression of p16 was observed in all seven tumor specimens matching these control “normal” specimens. While the relatively small population size of control specimens limits the power of statistical tests of the incidence of p16 downregulation between normal and tumor specimens (28.6% vs 96.8%), it is reasonable to conclude that downregulation of p16 is prevalent in human pheochromocytomas. Moreover, the two control “normal” specimens harboring low expression of p16 may not represent the “true” normal tissues in histology and molecular biology (this issue will be further addressed in the next section).
TABLE 2.
IMMUNOHISTOCHEMICAL AND GENETIC ANALYSES OF p16 TUMOR SUPPRESSOR GENE INACTIVATION IN PHEOCHROMOCYTOMA TUMOR SPECIMENS
| Specimen* | Expression of p16 Evaluated by IHC† | Genetic Analysis of the p16 Gene | |||
|---|---|---|---|---|---|
| Deletion‡ | Methylation | Point Mutation | Genetic Inactivation | ||
| T1 | P16-low | +/+ | no | no | no |
| T2 | P16-negative | +/+ | no | no | no |
| T3 | P16-low | +/+ | no | no | no |
| T4 | P16-negative | +/− | no | no | yes |
| T5 | P16-low | +/− | no | no | yes |
| T6 | P16-negative | +/+ | no | no | no |
| T7 | P16-low | +/− | no | no | yes |
| T8 | P16-low | +/+ | no | no | no |
| T9R§ | P16-low | +/+ | no | no | no |
| T9L§ | P16-negative | +/+ | yes | no | yes |
| T10 | P16-low | +/+ | yes | no | yes |
| T11 | P16-low | −/− | no | no | yes |
| T12 | P16-negative | +/− | no | no | yes |
| T13 | P16-negative | −/− | no | no | yes |
| T14 | P16-low | +/+ | yes | no | yes |
| T15 | P16-negative | +/+ | yes | no | yes |
| T16R§ | P16-low | +/+ | no | no | no |
| T16L§ | P16-high | +/+ | no | Ala 20 (GCT)/Pro (CCT) | yes |
| T17 | P16-negative | +/+ | yes | no | yes |
| T18R§ | P16-negative | −/− | no | no | yes |
| T18L§ | P16-low | +/+ | yes | no | yes |
| T19 | P16-negative | +/+ | yes | no | yes |
| T20 | P16-negative | +/− | no | no | yes |
| T21 | P16-low | −/− | no | no | yes |
| T22 | P16-negative | +/+ | no | no | no |
| T23 | P16-negative | +/+ | no | no | no |
| T24R§ | P16-low | +/+ | no | no | no |
| T24L§ | P16-low | +/+ | yes | no | yes |
| T25 | P16-low | +/+ | yes | no | yes |
| T26 | P16-low | +/+ | yes | no | yes |
| T27 | P16-low | +/+ | yes | no | yes |
| C3 | P16-high | +/+ | no | no | no |
| C4 | P16-low | +/+ | no | no | no |
| C9R | P16-high | +/+ | no | no | no |
| C9L | P16-high | +/+ | no | no | no |
| C22 | P16-high | +/+ | no | no | no |
| C25 | P16-high | +/+ | no | no | no |
| C27 | P16-low | +/+ | no | no | no |
T and C represent tumor and control “normal” specimens, respectively.
IHC immunohistochemistry. Based on the percentage of positive nuclear P16 staining by immunohistochemis-try, the samples were categorized as P16-negative (complete absence of nuclear immunostaining), P16-low (1–25% of positive nuclei) or P16-high (>25% of positive nuclei), as previously described.
+/+ wild-type, +/− hemizygous deletion, −/− homozygous deletion.
R: right-sided tumor, L: left-sided tumor.
Figure 1.
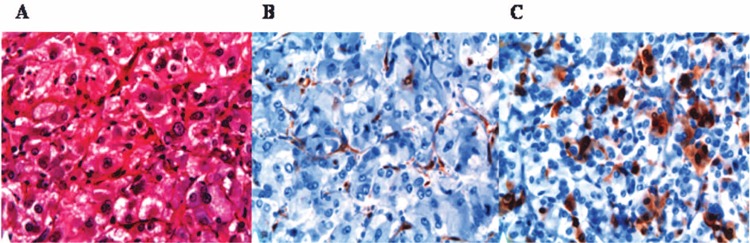
Pheochromocytoma immunohistochemical data. (A) H&E stain; (B) P16 negative (note that there is some staining in the endothelial cells); (C) P16 focally positive. All images are 40× magnification.
Genetic Alterations of p16
To explore the molecular mechanisms underlying the prevalent downregulation of p16 expression in tested pheochromocytomas specimens, we extracted genetic DNA from these specimens and investigated potential genetic abnormalities of the p16 gene including deletions, aberrant CpG methylation, and point mutations (Table 2). Real-time PCR-based deletion analysis (Fig. 2) indicated that five specimens harbored heterozygous deletions (+/−) and four specimens harbored homozygous deletions (−/−) for a total frequency of 29.0%. Methylation-specific PCR analysis indicated the presence 5′ CpG island methylation in 11 specimens (35.5%) (Fig. 3). Only one specimen, T16L, harbored aG → C transition, which resulted in an Ala → Pro missense mutation at amino acid residue 20 of P16 protein. Interestingly, this missense mutation has been reported in human gall bladder tumors, melanomas, and lung cancers (14). The potential influence of this Ala → Pro mutation was quantitatively evaluated using an in vitro kinase assay (19). As shown in Figure 4, the IC50 values of the above Ala → Pro P16 mutant protein and wild-type P16 were 620 ± 70 and 70 ± 15 nM, respectively, indicating that this mutation results in an eightfold decrease in the CDK4-inhibitory activity of P16. Further structural analyses using NMR spectroscopy demonstrated that this Ala → Pro mutation significantly perturbed the global structure of P16, thus impairing its function (data not shown). Overall, 21 of 31 tumor specimens (67.7%) demonstrated evidence of potentially inactivating p16 alterations. Interestingly, each of these 20 tumor specimens harbored either deletion or methylation or point mutation, suggesting that one mutagenic mechanism is sufficient to inactivate the p16 gene. In comparison, genomic DNAs from seven control “normal” specimens were analyzed, and none of these control specimens harbored potentially inactivating p16 abnormalities (Table 2). In addition, the frequencies of potentially inactivating p16 alterations in familial (50.0%) and sporadic (78.9%) tumors did not differ significantly (p > 0.9), suggesting that with regard to inactivating p16 alterations, there is no preference between familial and sporadic pheochromocytoma tumors.
Figure 3.
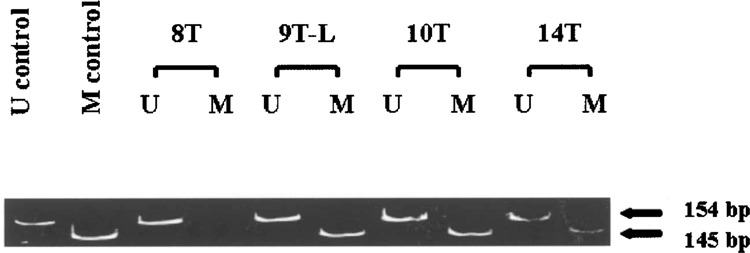
Representative methylation-specific PCR. The 145-bp band corresponds to the methylation-specific PCR product (M) and the 154-bp band corresponds to the unmethylated PCR product (U). Tumor #8 (T8) demonstrates amplification with only the unmethylated-specific primers indicating no 5′ CpG methylation. Tumors #9L, 10, and 14 (T9-L, T10, and T14) demonstrate amplification using both unmethylated and methylation-specific primers and represent specimens harboring p16 methylation.
Figure 4.
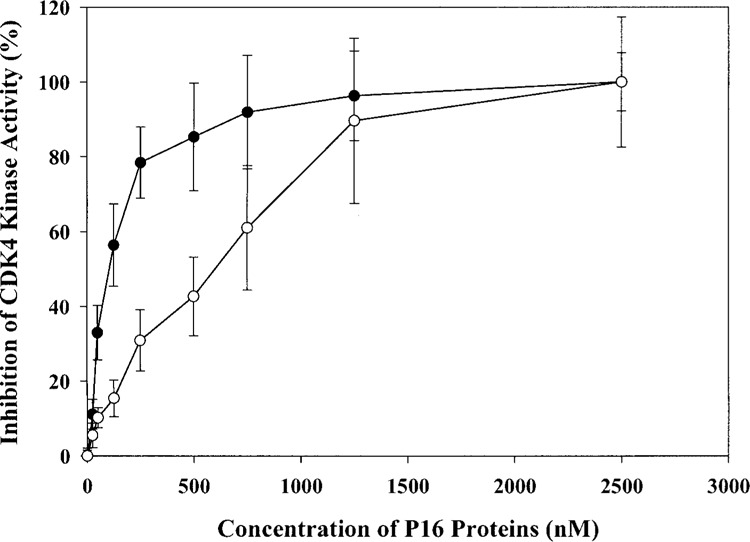
In vitro kinase assay to determine the CDK4-inhibitory activities of human P16 wild-type and mutant proteins. Each reaction mixture included 3 units of CDK4-cyclin D2 (∼0.15 μg), 50 ng of GST-Rb790-928, 5 μCi [γ-32P]ATP, and various amounts of P16 proteins. After reaction at 30°C for 15 min, the reaction mixtures were separated by 10% SDS-PAGE and the incorporation of γ-32P into GST-Rb protein was quantified using a PhoshoImager (Molecular Dynamics, Inc., Sunnyvale, CA). Filled circles represent hamster P16 wild-type protein, and open circels represent hamster P16 A20P mutant protein. All measurements were performed in triplicate.
DISCUSSION
Here we present the evaluation of the expression of p16 in pheochromocytoma specimens. Our results show that expression of p16 is downregulated in almost all tested tumor specimens (30 out of 31), suggesting that downregulation of p16 could play an important role in the development of human pheochromocytomas. Of the 30 tumor specimens with downregulated expression of p16, 20 of them harbored inactivating genetic alterations of p16 gene, including 9 specimens with deletions and 11 specimens with methylation. Therefore, inactivating genetic alterations of p16 gene, specifically, deletion and methylation are the primary cause of downregulated expression of p16 in pheochromocytomas. However, 10 tumor specimens, T1, T2, T3, T6, T8, T9R, T16R, T22, T23, and T24R, harbored no detectable p16 genetic alterations but the nuclear expression of p16 in these specimens was low or negative, suggesting that p16 expression may have been downregulated by mechanisms other than genetic alterations of p16 gene.
While the above premise remains to be verified, it has been reported recently that there exists a regulatory sequence in the upstream of the INK4A/ARF locus (6), alterations in which downregulated the transcription of three tumor suppressor genes encoded by the INK4A/ARF locus, namely, p15, p16, and p14ARF. Moreover, binding of Cdc6, an oncoprotein frequently overexpressed in non-small-cell lung carcinomas (NSCLCs), to this regulatory sequence transcriptionally repressed these three tumor suppressors (6). Presumably, in addition to direct inactivating p16 gene through deletion, methylation, and point mutation, potential mutations or polymorphorisms in the above regulatory sequence, and overexpression of cdc6 may constitute alternative pathways for the functional inactivation of p16 in human cancers. This could be also true with the two control “normal” specimens with reduced expression of p16, C4, and C27. These two specimens might not be true normals, and there could be some tumor-related molecular events happening in these tissues even though histologically they don’t look like tumor tissues and the p16 gene remains intact. Furthermore, the only tumor specimen with expression of p16 at high level was T16L. Interestingly, the p16 gene in this specimen was mutated, and the resultant P16 protein was partially inactivated (Fig. 4). However, it remains to be elucidated whether a compensation mechanism exists in cells, in which the decrease in the CDK4-inhibitory activity of P16 due to mutation can be “counteracted” by overexpression of the mutated p16 gene.
Genetic alterations of p16 gene have been found with a range of frequencies from 25% to 70% in leukemia, lymphomas, glioblastomas, and cancers of the head and neck, esophagus, biliary tract, lung, bladder, colon, liver, and breast, and most of these alterations are subject to deletion and methylation (14,17). In our current study, the p16 gene was frequently altered in pheochromocytomas through deletion (9 out of 31), methylation (11 out of 31), and point mutation (1 out of 31) with an overall frequency of 67.7%. Hence, our results support the notion that, like in other human tumors, the p16 gene plays an important role in the development of pheochromocytomas. Specifically, the frequency of p16 methylation identified in our study, 35.5%, is close to the previous finding showing that in 25 tested pheochromocytoma tumor specimens, six specimens harbored hypermethylated p16 gene (3). However, while neither homozygous nor hemizygous deletions were identified in pheochromocytomas in previous studies (1,3), the frequency of p16 inactivation by homozygous and hemizygous deletions is 29.0% in our study, suggesting that, like in other types of tumors, deletion is a major mechanism to “genetically” inactivate the p16 gene.
Our results in regard to the role of p16 deletion in the development of pheochromocytomas were further supported by a recent study using p16 +/− <PTEN +/−, p16 −/− PTEN +/−, and p16 +/+ PTEN +/− mice. That study demonstrated that there was functional synergy between p16 and PTEN, and mice harboring hemizygous, and particularly homozygous, p16 deletions showed increased predisposition towards pheochromocytoma development (24). Moreover, Dammann et al. (4) previously reported that hypermethylation of the p16 gene was noted to be particularly associated with hereditary pheochromocytomas, as 42% of MEN 2 tumors exhibited p16 hypermethylation compared to only 8% of sporadic tumors. In contrast, there is no significant difference between MEN 2 and sporadic tumors in frequencies of p16 alterations by methylation and deletion (p = 0.37 and 0.23, respectively) in our current study, implying that p16 inactivation is a common, not preferential, event in both forms of pheochromocytomas.
As for the discrepancy in familial/sporadic preference of p16 methylation between our current study and the previous study performed by Dammann et al. (4), it could be partially attributed to the likely differences in the development stages of those pheochromocytomas specimens. Methylation-associated silencing of p16 is regarded as a gradual process, during which a subset of cytosine residues within 5′ CpG islands become progressively methylated and tumors at later stages may have more extensive methylation of the 5′ CpG islands of p16 (18,20).
It is also worthwhile to note that human p16/Arf locus encodes two tumor suppressors, P16 and P14ARF, using distinct exon 1s (exon 1α vs. exon 1β) and alternate reading frames of exons 2 and 3. Alterations in the p16/Arf locus could consequently influence two prominent cancer-related pathways, the P16/CDK4/Rb/E2F pathway and the P14ARF/P53/ MDM2 pathway (5,22). In our current study, even though deletions in exon 2 of p16 gene also disrupted p14ARF gene (10), the epigenetic and genetic status of p14ARF gene in these pheochromocytomas specimens remains to be determined.
In summary, our study demonstrates that the expression of p16 gene is predominantly downregulated in human pheochromcytomas, and the primary cause underlying such transcriptional silencing is inactivating genetic alterations of p16 gene. Familial (MEN 2) versus sporadic tumor status does not appear to be associated with the frequency of p16 transcriptional silencing nor with the incidence of genetic abnormalities of p16 in pheochromocytomas. These findings support further evaluation of genetic and epigenetic alterations of p16 in pheochromocytomas and contribute to our current understanding of pheochromocytoma pathogenesis and intervention.
ACKNOWLEDGMENTS
The authors thank Dr. Thomas J. Knobloch in Division of Environmental Health Sciences for critical reading of the manuscript. This work was partly supported by a research grant from NIH (RO1 CA69472) to Dr. Ming-Daw Tsai at Departments of Chemistry and Biochemistry, The Ohio State University.
REFERENCES
- 1. Aguiar R. C.; Dahia P. L.; Sill H.; Toledo S. P.; Goldman J. M.; Cross N. C. Deletion analysis of the p16 tumour suppressor gene in phaeochromocytomas. Clin. Endocrinol. (Oxf) 45:93–96; 1996. [PubMed] [Google Scholar]
- 2. Carter T. L.; Watt P. M.; Kumar R.; Burton P. R.; Reaman G. H.; Sather H. N.; Baker D. L.; Kees U. R. Hemizygous p16(INK4A) deletion in pediatric acute lymphoblastic leukemia predicts independent risk of relapse. Blood 97:572–574; 2001. [DOI] [PubMed] [Google Scholar]
- 3. Dammann R.; Schagdarsurengin U.; Seidel C.; Trumpler C.; Hoang-Vu C.; Gimm O.; Dralle H.; Pfeifer G. P.; Brauckhoff M. Frequent promoter methylation of tumor-related genes in sporadic and men2-associated pheochromocytomas. Exp. Clin. Endocrinol. Diabetes 113:1–7; 2005. [DOI] [PubMed] [Google Scholar]
- 4. De Lott L. B.; Morrison C.; Suster S.; Cohn D. E.; Frankel W. L. CDX2 is a useful marker of intestinal-type differentiation: A tissue microarray-based study of 629 tumors from various sites. Arch. Pathol. Lab. Med. 129:1100–1105; 2005. [DOI] [PubMed] [Google Scholar]
- 5. Gil J.; Peters G. Regulation of the INK4b-ARF-INK4a tumor suppressor locus: All for one or one for all. Nat. Rev. Mol. Cell. Biol. 7:667–677; 2006. [DOI] [PubMed] [Google Scholar]
- 6. Gonzalez S.; Klatt P.; Delgado S.; Conde E.; Lopez-Rios F.; Sanchez-Cespedes M.; Mendez J.; Antequera F.; Serrano M. Oncogenic activity of Cdc6 through repression of the INK4/ARF locus. Nature 440:702–706; 2006. [DOI] [PubMed] [Google Scholar]
- 7. Gonzalez-Zulueta M.; Bender C. M.; Yang A. S.; Nguyen T.; Beart R. W.; Van Tornout J. M.; Jones P. A. Methylation of the 5′ CpG island of the p16/ CDKN2 tumor suppressor gene in normal and transformed human tissues correlates with gene silencing. Cancer Res. 55:4531–4535; 1995. [PubMed] [Google Scholar]
- 8. Herman J. G.; Merlo A.; Mao L.; Lapidus R. G.; Issa J. P.; Davidson N. E.; Sidransky D.; Baylin S. B. Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res. 55:4525–4530; 1995. [PubMed] [Google Scholar]
- 9. Jagasia A. A.; Sher D. A.; Le Moine P. J.; Kim D. H.; Moldwin R. L.; Smith S. D.; Diaz M. O. Deletion or lack of expression of CDKN2 (CDK4I/MTS1/ INK4A) and MTS2 (INK4B) in acute lymphoblastic leukemia cell lines reflects the phenotype of the uncultured primary leukemia cells. Leukemia 10:624–628; 1996. [PubMed] [Google Scholar]
- 10. Kees U.; Terry P.; Ford J.; Everett J.; Murvh A.; Klerk N.; Baker D. Detection of hemizygous deletions in genomic DNA from leukemia specimens for the diagnosis of patients. Leukemia Res. 29:165–171; 2005. [DOI] [PubMed] [Google Scholar]
- 11. Koch C. A.; Vortmeyer A. O.; Huang S. C.; Alesci S.; Zhuang Z.; Pacak K. Genetic aspects of pheochromocytoma. Endocr. Regul. 35:43–52; 2001. [PubMed] [Google Scholar]
- 12. Lang J. C.; Borchers J.; Danahey D.; Smith S.; Stover D. G.; Agrawal A.; Malone J. P.; Schuller D. E.; Weghorst C. M.; Holinga A. J.; Lingam K.; Patel C. R.; Esham B. Mutational status of overexpressed p16 in head and neck cancer: Evidence for germline mutation of p16/p14ARF. Int. J. Oncol. 21:401–408; 2002. [DOI] [PubMed] [Google Scholar]
- 13. Lenders J. W.; Eisenhofer G.; Mannelli M.; Pacak K. Phaeochromocytoma. Lancet 366:665–675; 2005. [DOI] [PubMed] [Google Scholar]
- 14. Li J.; Byeon I.-J. L.; Poi M. J.; Ericson K.; Selby T.; O’Maille P.; Qin D.; Tsai M.-D. The structure–function relationship of p16 tumor suppressor. In: Ehrlich M., ed. DNA alterations in cancer: Genetic and epigenetic changes. Westborough, MA: BioTechniques Books, Eaton Publishing; 2000:71–83. [Google Scholar]
- 15. Li J.; Weghorst C. M.; Tsutsumi M.; Poi M. J.; Knobloch T. J.; Casto B. S.; Melvin W. S.; Tsai M.-D.; Muscarella P. Frequent p16INK4A/CDKN2A alterations in chemically induced Syrian golden hamster pancreatic tumors. Carcinogenesis 25:263–268; 2004. [DOI] [PubMed] [Google Scholar]
- 16. Merlo A.; Herman J. G.; Mao L.; Lee D. J.; Gabrielson E.; Burger P. C.; Baylin S. B.; Sidransky D. 5′ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/ MTS1 in human cancers. Nat. Med. 1:686–692; 1995. [DOI] [PubMed] [Google Scholar]
- 17. Ortega S.; Malumbres M.; Barbacid M. Cyclin-dependent kinases, INK4 inhibitors and cancer. Biochim. Biophys. Acta 1602:73–87; 2002. [DOI] [PubMed] [Google Scholar]
- 18. Park T. J.; Kim H. S.; Byun K. H.; Jang J. J.; Lee Y. S.; Lim I. K. Sequential changes in hepatocarcinogenesis induced by diethylnitrosamine plus thioacetamide in Fisher 344 rats: Induction of gankyrin expression in liver fibrosis, pRB degradation in cirrhosis, and methylation of p16(INK4A) exon 1 in hepatocellular carcinoma. Mol. Carcinog. 30:138–150; 2001. [DOI] [PubMed] [Google Scholar]
- 19. Poi M. J.; Yen T.; Li J.; Song H.; Lang J. C.; Schuller D. E.; Pearl D. K.; Casto B. C.; Tsai M.-D.; Weghorst C. M. Somatic INK4a-ARF locus mutations: a significant mechanism of gene inactivation in squamous cell carcinomas of the head and neck. Mol. Carcinog. 30:26–36; 2001. [DOI] [PubMed] [Google Scholar]
- 20. Schutte M.; Hruban R. H.; Geradts J.; Maynard R.; Higers W.; Rabindran S. K.; Moskaluk C. A.; Hahn S. A.; Schwarte-Waldhoff I.; Schmiegel W.; Baylin S. B.; Kern S. E.; Herman J. G. Abrogation of the Rb/p16 tumor-suppressive pathway in birtually all pancreatic carcinomas. Cancer Res. 57:3126–3130; 1997. [PubMed] [Google Scholar]
- 21. Serrano M.; Hannon G. J.; Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 366:704–707; 1993. [DOI] [PubMed] [Google Scholar]
- 22. Sharpless N. E.; Depinho R. A. The INK4A/ARF locus and its two gene products. Curr. Opin. Genet. Dev. 9:22–30; 1999. [DOI] [PubMed] [Google Scholar]
- 23. Sherr C. J.; Roberts J. M. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 18:2699–2711; 2004. [DOI] [PubMed] [Google Scholar]
- 24. You M. J.; Castrillon D. H.; Bastian B. C.; O’Hagan R. C.; Bosenberg M. W.; Parsons R.; Chin L.; DePinho R. A. Genetic analysis of Pten and Ink4a/Arf interactions in the suppression of tumorigenesis in mice. Proc. Natl. Acad. Sci. USA 99:1455–1460; 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]