

Abstract
Losses of the p-arm of chromosome 8 are frequently observed in breast, prostate, and other types of cancers. Using the Systematic Multiplex RT-PCR (SM RT-PCR) method and the DNA microarray hybridization method, we examined the expression of 273 genes located on the p-arm of chromosome 8 in five breast and three prostate human cancer cell lines. We observed frequent decreases in expression of two dozen genes and increases in expression of several genes on this chromosomal arm. These changes in gene expression of the cell lines were later confirmed by real-time qRT-PCR. Additionally and more importantly, we found that a number of these variations were also observed in the majority of clinical cases of breast cancer we examined. These included downregulation of the MYOM2, NP_859074, NP_001034551, NRG1, PHYIP (PHYHIP), Q7Z2R7, SFRP1, and SOX7 genes, and upregulation of the ESCO2, NP_115712 (GINS4), Q6P464, and TOPK (PBK) genes.
Key words: Systematic Multiplex RT-PCR (SM RT-PCR), Real-time qRT-PCR, DNA microarray hybridization, Chromosome 8p, Chromosomal scanning, Gene expression, Breast cancer
INTRODUCTION
The activation of oncogenes and the inactivation of tumor suppressor genes both play important roles in carcinogenesis. Most changes in these activating/inactivating processes occur in copy number, gene expression, or nucleotide/amino acid sequences. Therefore, the determination of copy number and gene expression, together with nucleotide sequencing, assists in the identification of both oncogenes and tumor suppressor genes. For the activation of an oncogene, a monoallelic dominant change is often sufficient. Examples of monoallelic activation include transcriptional activation of the BCL2 (B-cell leukemia 2) gene by the t(14;18) translocation that places this gene next to an active promoter in follicular lymphoma (33), MYCN gene amplification that is concomitant with an increased gene expression in neuroblastoma (16), and activating mutations in the KRAS2 gene in cancers of the lung, colon, and pancreas, among others (2,21,29). However, for the inactivation of a tumor suppressor gene, haplo-insufficiency is rare and the disruption of both alleles (biallelic inactivation) is usually necessary.
Quantitative analysis of copy number progressed when the comparative genomic hybridization (CGH) method was invented based on two-color fluorescence in situ hybridization (FISH) (13). In CGH, genomic DNA from a test sample is labeled with one fluorescent color, a reference genomic DNA is labeled with another color, and they are mixed and hybridized with metaphase chromosomal spreads of normal cells. The ratio of the two fluorescence intensities, rather than the absolute intensity, is used to monitor the difference in copy number. Using this technique, many maps of chromosomal alterations in cancer were produced. It was shown that there was a significant degree of heterogeneity among a variety of tumors, as well as within the same type of tumor. Chromosomal gains and losses, which are indicative of the presence of oncogenes and tumor suppressor genes, respectively, were located on the chromosomes. For example, frequent gains in chromosomal arms 1q, 3q, 8q, 16p, 17q, and 20q and losses in 1p, 6q, 8p, 13q, 16q, 17p, 18q, 22q, and X were reported in breast cancer (5,8). Chromosomal losses were more frequent than gains in prostate cancer and observed with chromosomal arms 1p, 5q, 6q, 8p, 10q, 13q, 16q, and 18q (3,6). The use of BAC clone DNA microarrays (1,24) and cDNA fragment microarrays (25,26) for the CGH karyotyping analysis of copy number has produced a more powerful and high-resolution arrayCGH method.
Several tumor suppressor genes have been identified in the chromosomal regions of losses. These include CDH1 on 16q22 (22) and PTEN on 10q23 (18). The inactivation of those genes was considered to be the selective force that resulted in the loss of the corresponding chromosomal regions. This is because of the frequent abnormalities and functional failure of the proteins encoded by those genes. Aiming to identify the novel genes with tumor suppressor activity, we started gene expression analysis. We chose the p-arm of chromosome 8 because this arm is one of the chromosomal arms most frequently lost in breast and prostate cancers, which strongly suggests that the region may harbor tumor suppressor genes involved in the pathogenesis of those cancers (4,8). Although breast and prostate cancers both progress from an early, sex hormone-dependent, organ-confined disease to a highly invasive, hormone-independent, metastatic disease, they arise in two different organs. By pursuing tumor suppressor genes common to cancers of these two different organs, we speculated that the exclusion of inappropriate genes, whose expression is specific to either mammary or prostate normal epithelial cells, would be easier. Here, we report the results obtained by analysis of gene expression on the chromosomal arm by the combined use of the moderately high-throughput Systematic Multiplex RT-PCR (SM RT-PCR) method (37–40) and the high-throughput DNA microarray hybridization method (27,28).
MATERIALS AND METHODS
ArrayCGH Experiments to Measure Copy Number of the Genes on the p-Arm of Chromosome 8
The copy number of three breast cancer cell lines (MCF7, MDA-MB468, and BT-20) was analyzed at NimbleGen (Madison, WI) by DNA microarray hybridization using the arrayCGH method (1,24,25). Genomic DNA from normal females was used as a reference for those hybridization experiments. Intensity of fluorescence was measured that was hybridized to 385,000 isothermal long oligonucleotide probes tiled through genic and intergenic regions at a median probe spacing of 6 kilo base pairs. Copy number was determined over the entire human genome by calculating relative fluorescence intensity.
SM RT-PCR Experiments to Measure Expression of the Genes on the p-Arm of Chromosome 8
The following RNA samples were used for gene expression analysis: both normal and primary tumor tissue from the breast of a patient with invasive ductal carcinoma, and both normal and primary carcinoma tissue of the prostate from a patient with prostate cancer, normal prostate tissue, and hyperplastic prostate tissue, five mammary (BT-20, MCF7, MDA-MB-231, MDA-MB-468, and T-47D) and three prostate (DU145, LNCaP, and PC3) cancer cell lines, and primary cultures of normal mammary and prostate epithelial cells. cDNA was prepared by reverse transcription of total RNA using oligo dT as a primer and the Advantage RT-for-PCR Kit (BD Biosciences-Clontech).
We followed the SM RT-PCR experimental protocols described previously (37–40). Briefly, the genes on 8p were categorized into groups of ∼10 genes, and PCR primers were designed to amplify different sizes of DNA fragments from single exons of the genes in a group. After using the multiplex reactions of genomic DNA from normal human tissues as a control, the concentrations of the primers were adjusted to produce bands of similar intensities. Once the conditions were elaborated, cDNA samples from human cells and tissues were used as templates to examine gene expression. Small aliquots of the SM RT-PCR reaction products were loaded onto an 8% polyacrylamide gel and electrophoresed. The gels were stained with ethidium bromide, gel pictures were taken, and the images were saved in TIFF format. The band intensity was measured using Image-Quant software (Amersham Biosciences) and normalized by adjusting the average band intensities of individual gels.
DNA Microarray Hybridization Experiments to Determine Gene Expression
In order to compare and complement the SM RT-PCR experiments, we also performed DNA micro-array hybridization experiments. Illumina’s Sentrix Human-6 Expression BeadChips, which contain probes from the entire 23,000 RefSeq collection and an additional 23,000 other expressed sequences, were used. The following RNA samples were analyzed: normal breast tissue, normal prostate tissue, primary cultures of normal mammary and prostate epithelial cells, and five mammary (BT-20, MCF7, MDA-MB-231, MDA-MB-468, and T-47D) and three prostate (DU145, LNCaP, and PC3) cancer cell lines. The same preparations of RNA that were used in SM RT-PCR were used in the microarray hybridization experiments. Following Illumina’s protocol, biotinylated cRNA was prepared and hybridized with the BeadChips. After washing, the BeadChips were treated with Cy3-labeled streptavidin, washed, dried, and scanned for fluorescence intensity with Illumina’s BeadStation 500. Raw data were generated and normalized using the Beadscan 3.0 software. The gene expression data for the genes on the p-arm of chromosome 8 were extracted.
Real-Time qRT-PCR Experiments to Measure Gene Expression
Real-time qRT-PCR of the selected genes was performed using the same set of cDNA from the cells and tissues that were analyzed by the DNA micro-array hybridization experiments, together with genomic DNA control. The same preparations of cDNA that were used in the SM RT-PCR were used in the real-time qRT-PCR experiments. In addition to this subset of cDNA samples, additional cDNA samples prepared from 12 matched pairs of normal and tumor breast tissues were also analyzed by real-time qRT-PCR. The same primer pairs that were used in the SM RT-PCR experiments were also used in the realtime qRT-PCR experiments. The reagent from the Power SYBR Green PCR Master Mix (Applied Bio-systems) was used, and the yields of the PCR products were monitored using the Mx3000p system (Stratagene) under default conditions, with the exception that the annealing temperature was raised to 60°C instead of 55°C. Data were analyzed using the MxPro software, and the Ct values were obtained for the individual reactions. The Ct values of the ubiquitously expressed ASAH1 gene, which is located on 8p, were subtracted from those values and normalized.
RESULTS
ArrayCGH Analysis of the Genes on Chromosome 8
We analyzed the copy number of the MCF7, MDA-MB468, and BT-20 breast cancer cell lines that were previously shown to exhibit decreased copy number on the 8p chromosomal arm (8). Data on fluorescence intensity were extracted for the probes corresponding to the sequences on chromosome 8. The log2 values of relative intensity to normal female genomic DNA were then plotted on the y-axis along with the chromosomal location on the x-axis. Results are shown from left (pter) to right (qter) in Figure 1. The results confirmed that the copy number of the 8p region was reduced in those cell lines.
Figure 1.
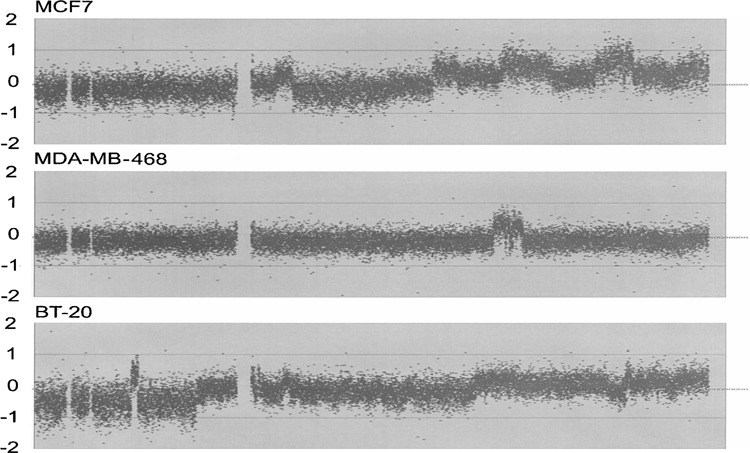
Copy number changes in chromosome 8. Relative copy number was analyzed for the sequences on chromosome 8. The results of the arrayCGH experiments are shown with three breast cancer cell lines: MCF7 (top), MDA-MB-468 (middle), and BT-20 (bottom). The entire length of chromosome 8 is 146,274,826 base pairs, and the centromeric region is located between 44 and 47 Mb from pter.
SM RT-PCR Analyses of the Genes on the 8p Chromosomal Arm
We established the SM RT-PCR system consisting of 254 genes. They were categorized into 26 groups. The list of the genes is shown in Supplementary Table 1 (available from the author’s website: http://fiyamamoto.googlepages.com/geneexpressionsupplementaryinformation), together with the nucleotide sequences and concentrations of the primers used in this study, and the sizes of the amplified DNA fragments. We examined the expression of those 254 genes in normal and cancerous breast and prostate cells and tissues. Results are shown in Figure 2. Because the PCR conditions were elaborated so that small amounts of genomic DNA would produce bands, the absence of at least one band was considered to confirm the absence of contaminating geno-mic DNA in the cDNA specimens. We found that approximately 42% of the genes were abundantly expressed in all of the cells and tissues that were examined. We also observed that approximately 30 genes were not expressed or rarely expressed in either normal or cancerous breast/prostate cells/tissues. The remaining genes were differentially expressed in some of the cDNA samples examined.
Figure 2.

SM RT-PCR results of breast and prostate cells and tissues. The results of the SM RT-PCR experiments are shown. There are a total of 26 sets. SM RT-PCR was performed to examine gene expression changes in breast and prostate cancer cells and tissues. The sources of cDNA are abbreviated: a normal sample (NB) and primary tumor (TB) of breast tissue from an individual; a normal sample (NP), and primary tumor tissues (TP) of prostate from an individual; a normal prostate tissue (NP) from a third individual; a hyperplastic prostate tissue (HyP) from a fourth individual; primary cultures of normal mammary (MP) and prostate (PP) epithelial cells; and MCF7 (MCF), MDA-MB-468 (468), MDA-MB-231 (231), BT-20 (BT), T-47D (T47), PC3 (PC), DU145 (DU), and LNCaP (LN) cancer cell line cells. The locations of the DNA fragments amplified from the individual genes are also shown at the left side of the gel pictures. The symbol M denotes DNA fragment size markers, and the symbol G shows the results of genomic DNA control.
Among them, we identified a dozen genes that exhibited unidirectional changes in gene expression in both breast and prostate cancer cell lines. Compared to the expression in normal epithelial cells, genes that were found downregulated in all five breast and three prostate cancer cell lines include: GON1 (GNRH1) (set 16), NRG1 (set 18), PIWL2 (PIWL2) (set 13), and Q7Z2R7 (sets 16 & 18). Genes that were found upregulated in all cell lines include: ESCO2 (set 20), GSHR (GSR) (set 26), NP_115712 (GINS4) (set 23), Q6P464 (CDCA2) (set 16), and TOPK (PBK) (set 17). We also identified additional genes that exhibited changes in a majority of either breast, prostate, or both cancer cell lines. These include the following genes: CH012 (C8orf12) (set 4), CHO13 (set 5), DEF1 (DEFA1) (set 1), EGR3 (set 20), ENST357748 (ENST000000357748) (set 11), FBX25 (FBX025) (set 1), MYOM2 (set 2), NP_065895 (set 7), NP_859074 (set 19), NP_001034551 (NP_ 1034551) (set 6), NPM2 (set 19), PHYIP (PHYHIP) (set 14), Q8NEP6 (set 5), Q96KT8 (set 6), SFRP1 (set 13), SOX7 (set 1), TPA (set 24), TR10D (TN FRSF10D) (set 15), and XR_017857 (C8orf48) (set 6). We measured the intensity of the SM RT-PCR bands for quantification. The results were then aligned by the chromosomal locations of the genes. Out of 254 genes, 238 genes were mapped on the p-arm of chromosome 8 in the newest version of Ensembl (version 43) and the results of those 238 genes are shown in the left column of Supplementary Figure 1 (available from the author’s website: http://fiyamamoto.googlepages.com/geneexpressionsupplementaryinformation).
DNA Microarray Hybridization Analysis of Gene Expression of the Genes on 8p
In addition to the SM RT-PCR experiments, we also performed genome-wide gene expression analysis by the DNA microarray hybridization method using Illumina’s BeadChips. Data for the genes on 8p were extracted and aligned based on the chromosomal locations of the genes. Because the PCR primers for SM RT-PCR were designed based on sequences that were not alternatively spliced, only the data using the “singular” or “all” probes (those that detect all the messages from the corresponding genes) were extracted from the Illumina data. The average fluorescence signal intensity of >30 beads was extracted and the results are shown, side-by-side, with the data from the SM RT-PCR experiments, in the right column of Supplementary Figure 1 (available from the author’s website: http://fiyamamoto.googlepages.com/geneexpressionsupplementaryinformation). The expression data were obtained for the 230 genes on the p-arm of chromosome 8, 195 of which overlapped with the genes whose expression was determined by SM RT-PCR. Compared to SM RT-PCR, the results from the DNA microarray hybridization experiments exhibited a wider range of intensity, as anticipated. There were 25 genes whose messages were not detected (fluorescence intensity below 10 in all the specimens). The number increased to 34 when the cut-off for fluorescence intensity was set at 15. Together with the results from the SM RT-PCR experiments, a total of 273 genes were covered in our study.
Gene Expression Analysis of the Selected Genes by Real-Time qRT-PCR
We performed real-time qRT-PCR to reexamine the expression of genes that exhibited consistent changes in expression by the SM RT-PCR method and/or the DNA microarray hybridization method. The same set of cells and tissues that was analyzed by DNA microarray hybridization was examined for the expression of the CH012, CH013, DEF1, EGR3, ESCO2, FBX25, GON1, GSHR, MYOM2, NP_065895, NP_115712, NP_859074, NP_001034551, NPM2, NRG1, PHYIP, PIWL2, Q6P464, Q7Z2R7, Q8NEP6, Q96KT8, SFRP1, SOX7, TOPK, TPA, TR10D, and XR_017857 genes. Because of high expression of the messages in all the samples in both the SM RT-PCR and the DNA microarray hybridization experiments, we selected, as a control gene, the ASAH1 gene (15). This gene encodes N-acylsphingosine amidohydrolase, also called acid ceramidase (AC; EC 3.5.1.23), which catalyzes the synthesis and degradation of ceramide. The Ct values obtained by real-time qRT-PCR were plotted on the x-axis. The log2 values calculated of the measured band intensity and fluorescence intensity of the genes from the corresponding cDNA samples were plotted on the y-axis. Results from all the selected genes of importance are shown in Figure 3. The results are shown on the same scale, and the result of the ASAH1 gene was also enlarged and shown next to the original figure on the top row. A higher degree of linearity was observed between the results from the SM RT-PCR experiments and the results from the real-time qRT-PCR experiments than between the DNA microarray hybridization experiments and the real-time qRT-PCR experiments. This is reasonable considering that both the SM RT-PCR and real-time qRT-PCR are PCR based and the same primers were used in those experiments. The differences observed by SM RT-PCR were confirmed to be real by real-time qRT-PCR, although some of them were not observed by DNA microarray hybridization.
Figure 3.
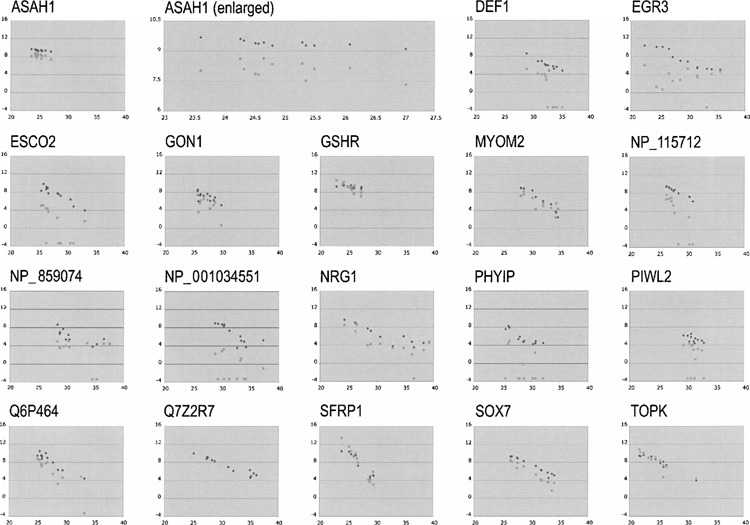
Correlation between the band intensity obtained from the SM RT-PCR or fluorescence intensity obtained from DNA microarray hybridization and the Ct values obtained from the real-time qRT-PCR experiments. The log2 values of the band intensity and fluorescence intensity were calculated and plotted along the y-axis with black diamonds and gray squares, respectively, against the Ct values on the x-axis. The ASAH1 gene was used as a control, because this gene was ubiquitously expressed in large quantity in all the cells and tissues that were examined in both the SM RT-PCR and the DNA microarray hybridization experiments. Negative and zero values obtained by micro-array hybridization experiments were assigned the value of 0.1 for these graphs. The portion of the ASAH1 results was enlarged and is also shown on the top row.
We next examined whether the same differences in the expression level that were observed in breast and prostate cancer cell lines were also present in the clinical specimens of cancer. Our primary research focus was breast cancer, so we analyzed the expression of the selected genes in matched normal and tumor pairs of breast tissues from a dozen breast cancer patients by real-time qRT-PCR. To normalize the Ct values of the individual specimens, we subtracted the Ct values of the ubiquitously expressed ASAH1 gene from those values. Downregulation was observed with the following genes in a majority of 12 breast cancer cases: MYOM2, PHYIP, SOX7 (10 cases), DEF1, FBX25, NP_001034551 (9 cases), CH012, GON1, NP_859074, NRG1, PIWL2, Q7Z2R7, SFRP1 (8 cases), Q8NEP6, Q96KT8, and XR_017857 (7 cases). Similarly, upregulation was observed in a majority of breast cancer cases with the following genes: TOPK (10 cases), Q6P464 (9 cases), ESCO2, NP_115712 (8 cases), GSHR (7 cases). For the remaining genes, downregulation was observed in 4 (NP_065895), 5 (TPA), or 6 cases (CH013, EGR3, NPM2, TR10D). Figure 4 illustrates several significant results with the plotting of normalized Ct values of tumor tissues less the corresponding values of the normal adjacent tissues.
Figure 4.
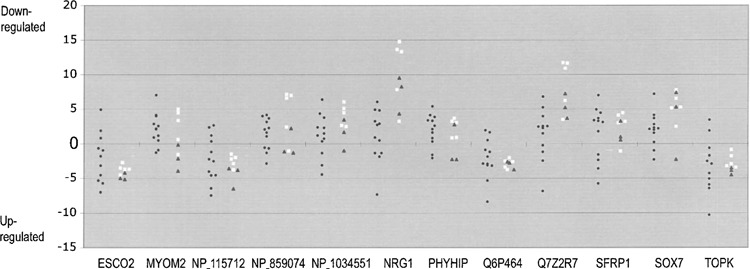
The expression of the selected genes in matched normal and cancer breast tissues and cell lines. The gene expression was determined for the selected genes with real-time qRT-PCR using cDNA prepared from 12 matched normal and cancer breast tissues. The Ct values of individual genes were normalized by subtracting the Ct values of ASAH1 from the same specimens. The difference in gene expression among normal and tumor tissues was calculated by subtracting the normalized normal Ct values from the normalized tumor Ct values from the same individuals. The results are shown in filled circles. The difference was also calculated for cancer cell lines using the Ct values shown in Figure 3. The normalized Ct values of primary cultures of normal cells were used for the subtraction segment, and the results are shown in open squares and filled triangles for breast and prostate, respectively. The dots above the zero line indicate downregulation in tumor, whereas dots below denote upregulation.
DISCUSSION
Previously, we established the SM RT-PCR systems of families of glycosyltransferases (40), HOX homeoproteins (38), and integrins (39). We also gradually increased the size of coverage from a few dozen genes (37) to more than a hundred genes in a few cytobands (36). Here we attempted to establish the SM RT-PCR system of more than 200 genes on the entire arm of a chromosome. Excluding the DNA microarray hybridization approach, this SM RT-PCR analysis is one of the largest attempts to understand the expression of the genes on a chromosomal arm-wide scale. We aimed to incorporate as many genes as possible into the system.
Because the defensin genes were highly homologous to one another and possessed short coding sequences, we were unable to design primers that selectively amplified a single species of the gene members for several members of the defensin gene family. Nonetheless, we included 254 genes in 26 multiplex reactions as shown in the primer list in Supplementary Table 1 (available from the author’s website: http://fiyamamoto.googlepages.com/geneexpressionsupplementaryinformation) and the genomic DNA lanes (G) in Figure 2. However, the Ensembl database was not finished at the time that we retrieved the gene and sequence information. When we aligned our results in the most recent version, 43, we found that 11 genes that were previously mapped in the region did not exist any longer. Furthermore, a few dozen additional genes that were not previously mapped have been added. These include novel protein-coding genes, pseudogenes, miRNA genes, snRNA genes, and sno RNA genes; 195 of the 238 genes whose expression was determined by SM RT-PCR overlapped with the genes whose expression was determined by DNA microarray hybridization experiments. A generally good correlation was observed in the results between the SM RT-PCR and DNA microarray hybridization experiments, except that the expression of approximately 40 genes were detected only by SM RT-PCR. We think that the probes of these genes used in the DNA microarray hybridization were either inappropriate or not functioning as expected.
The results shown in Figure 3 also illustrate this problem. To calculate the log2 values of fluorescence intensity from the DNA microarray hybridization results, we used 0.1 for the values below this number. Still, when the fluorescence signal was weak, as in the cases of NP_001034551 and PHYIP, no correlation was observed between real-time qRT-PCR and DNA microarray hybridization. However, when the fluorescence signal was strong, both SM RT-PCR and DNA microarray hybridization exhibited linear correlation with real-time qRT-PCR as shown with the GSHR, SFRP1, and TOPK genes. We concluded that the use of two different methods was beneficial in increasing the coverage of genes and decreasing the chance of missing potentially important candidate genes.
In addition to the cell lines, we also examined the expression of the selected genes in the clinical specimens of breast cancer. As opposed to the in vitro cultured cancer cell lines, which consist of a relatively uniform population of cells, tissues are made of several different types of cells and their ratios vary among different specimens. Therefore, measurement of the Ct values without standardization was not informative. We used the expression of the highly and ubiquitously expressed ASAH1 gene as a standard. By subtracting the Ct values of the ASAH1 gene, we compared the relative ratios of the gene messages among different specimens. Rather than comparing normal and cancer tissue specimens in two groups, instead we plotted the results from the individual pairs of cancer tissue specimens and their corresponding normal adjacent tissue specimens by subtracting the normalized normal value from the normalized tumor value. The tendencies of up- and downregulation in gene expression were easily confirmed with most of the genes examined. For several genes, the tendencies were not clear with the breast clinical specimens. Several potential reasons can be speculated. One possibility is that the cancer cell lines may have acquired downregulation in expression of those genes after they were brought into in vitro culture. Another possibility is that cells, other than cancer cells, that were present in the tumor tissues express these genes. Therefore, losses/decreases in cancer cells may have been masked.
Among the genes that exhibited a matched tendency of upregulation in the breast tumor tissues and breast cancer cell line cells, the tendency was striking with the ESCO2, TOPK, NP_115712, and Q6P464 genes. Because the ESCO2 gene is required for the establishment of sister chromatid cohesion during the S phase of the cell cycle (12), the TOPK gene encodes serine/threonine kinase that binds to the PDZ2 domain of Drosophila Discs-large (Dlg) tumor suppressor protein that regulates the cell cycle and/or cellular proliferation (9), the NP_115712 protein is a component of the GINS complex that is essential for the initiation of DNA replication (32), and the Q6P464 gene is associated with cell division cycle, upregulation of these genes in tumors and cancer cell lines may simply be a reflection of a higher number of dividing cells in those specimens. These four genes were elevated in gene expression in all five breast and three prostate cancer cell lines that were examined, suggesting that this is a likely possibility.
Among the genes that were downregulated in a majority of breast cancer cases, four genes, GON1, NRG1, PIWL2, and Q7Z2R7, were also downregulated in all five breast and three prostate cancer cell lines. Additionally, four genes (NP_859074, NP_001034551, PHYIP, and SOX7) exhibited downregulation in all five breast cancer cell lines examined and one gene (SFRP1) exhibited downregulation in a majority of breast and prostate cancer cell lines. Two genes, CH012 and XR_017857, exhibited downregulation in a minority of cell lines, and five genes (DEF1, FBX25, MYOM2, Q8NEP6, and Q96KT8) showed decreased expression only in the breast cancer cell lines. Among these downregulated genes, statistically significant tendencies were observed with the MYOM2, NP_859074, NP_001034551, NRG1, PHYIP, Q7Z2R7, SFRP1, and SOX7 genes, shown in Figure 4.
The tumor-suppressing role has been well established for the NRG1 gene. The gene encodes neuregulin 1 (heregulin) that interacts with the NEU/ERBB2 receptor tyrosine kinase to increase its phosphorylation on tyrosine residues (11). The purified protein induces phenotypic differentiation of breast and prostate cancer cells and inhibits cell growth (10,23). The SFRP1 and SOX7 genes play a similar role in carcinogenesis by repressing the Wnt signaling inside the cell. The SFRP1 gene encodes a secreted apoptosis-related protein that interferes with the Wnt-frizzled signaling pathway (7,20). The potential role of the SFRP1 gene in tumor suppression of breasts and prostates was previously suggested (19,34). The SOX7 gene encodes a transcription factor that possesses a functional transactivation domain in the C-terminus and significantly reduces Wnt/beta-catenin-stimulated transcription (31). The identification of three genes, NRG1, SFRP1, and SOX7, which are known to be involved in carcinogenesis among the candidates, indicates that the approach is working as expected.
In addition to those cancer-related genes, we also identified MYOM2, PHYIP, and three poorly characterized candidate genes. The MYOM2 and PHYIP genes encode myomesin 2, an M-band protein of sarcomeres (35), and phytanoyl-CoA hydroxylase-inter-acting protein (17), respectively. The NP_859074 gene predicts to encode a protein with the EF-hand domain (14), and the NP_001034551 and Q7Z2R7 genes predict proteins of 121 and 83 amino acid residues, respectively. Little else is known of those genes; however, the Q7Z2R7 gene is separated from the NRG1 gene by only 1577 bp and the expression profiles of those genes were similar in the cells and tissues that were examined. Because the orientations of these genes are the same, there is a possibility that the Q7Z2R7 sequence may be transcribed run-off in the 3′ untranslated region of the NRG1 gene messages rather than transcribed independently from its own promoter. Preliminary attempts to identify somatic mutations in the downregulated genes in several clinical cases of breast cancer were unsuccessful (data not shown). Even if we increase the number of specimens to be analyzed, we may not find any mutations, considering that none of those genes are included on the lists of cancer genes with mutations that were identified from a massive sequencing effort of 13,023 genes in 11 breast cancer cases (30) or that have been deposited in the Catalogue Of Somatic Mutations In Cancer (COSMIC) database (http://www.sanger.ac.uk/genetics/CGP/cosmic/). Nonetheless, the candidacy of those genes remains intact without devaluation because downregulation alone or downregulation in combination with decreased copy number is sufficient for inactivation of tumor suppressor activity. Final determination will require further studies based on functional characterization of the individual candidate genes.
In summary, we have shown that the SM RT-PCR approach is successful in the identification of genes with altered expression through scanning of the genes at the subchromosomal level. It should be emphasized that by performing multiplex reactions of 10 genes, on average, the number of reactions was reduced by 10 times in the SM RT-PCR experiments, compared with real-time qRT-PCR of individual genes. Together with more flexibility in designing the SM RT-PCR system than DNA microarrays and using preconfirmed primers, this advantage may allow the SM RT-PCR system to find its niche between the discovery method of higher-throughput DNA micro-array hybridization and the quantification method of more quantitative real-time qRT-PCR.
ACKNOWLEDGMENTS
We are thankful to Dr. Kang Liu for technical assistance in DNA microarray hybridization experiments using Illumina’s Sentrix Human-6 Expression BeadChips and Mr. Lloyd Slivka and Ms. Lauren Dahlquist for editorial assistance. We thank the Cooperative Human Tissue Network (CHTN) for providing matched normal and cancer breast tissues. This work was supported by NIH grant 1R01 CA087069 and the DOD BCRP grant W81XWH-05-1-0317 to F.Y.
REFERENCES
- 1. Albertson D. G.; Ylstra B.; Segraves R.; Collins C.; Dairkee S. H.; Kowbel D.; Kuo W. L.; Gray J. W.; Pinkel D. Quantitative mapping of amplicon structure by array CGH identifies CYP24 as a candidate oncogene. Nat. Genet. 25:144–146; 2000. [DOI] [PubMed] [Google Scholar]
- 2. Capon D. J.; Seeburg P. H.; McGrath J. P.; Hayflick J. S.; Edman U.; Levinson A. D.; Goeddel D. V. Activation of Ki-ras2 gene in human colon and lung carcinomas by two different point mutations. Nature 304:507–513; 1983. [DOI] [PubMed] [Google Scholar]
- 3. Cher M. L.; MacGrogan D.; Bookstein R.; Brown J. A.; Jenkins R. B.; Jensen R. H. Comparative genomic hybridization, allelic imbalance, and fluorescence in situ hybridization on chromosome 8 in prostate cancer. Genes Chromosomes Cancer 11:153–162; 1994. [DOI] [PubMed] [Google Scholar]
- 4. Clark J.; Edwards S.; Feber A.; Flohr P.; John M.; Giddings I.; Crossland S.; Stratton M. R.; Wooster R.; Campbell C.; Cooper C. S. Genome-wide screening for complete genetic loss in prostate cancer by comparative hybridization onto cDNA microarrays. Oncogene 22:1247–1252; 2003. [DOI] [PubMed] [Google Scholar]
- 5. Courjal F.; Theillet C. Comparative genomic hybridization analysis of breast tumors with predetermined profiles of DNA amplification. Cancer Res. 57:4368–4377; 1997. [PubMed] [Google Scholar]
- 6. Dong J. T. Chromosomal deletions and tumor suppressor genes in prostate cancer. Cancer Metastasis Rev. 20:173–193; 2001. [DOI] [PubMed] [Google Scholar]
- 7. Finch P. W.; He X.; Kelley M. J.; Uren A.; Schaudies R. P.; Popescu N. C.; Rudikoff S.; Aaronson S. A.; Varmus H. E.; Rubin J. S. Purification and molecular cloning of a secreted, Frizzled-related antagonist of Wnt action. Proc. Natl. Acad. Sci. USA 94:6770–6775; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Forozan F.; Mahlamaki E. H.; Monni O.; Chen Y.; Veldman R.; Jiang Y.; Gooden G. C.; Ethier S. P.; Kallioniemi A.; Kallioniemi O. P. Comparative genomic hybridization analysis of 38 breast cancer cell lines: A basis for interpreting complementary DNA microarray data. Cancer Res. 60:4519–4525; 2000. [PubMed] [Google Scholar]
- 9. Gaudet S.; Branton D.; Lue R. A. Characterization of PDZ-binding kinase, a mitotic kinase. Proc. Natl. Acad. Sci. USA 97:5167–5172; 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Grasso A. W.; Wen D.; Miller C. M.; Rhim J. S.; Pretlow T. G.; Kung H. J. ErbB kinases and NDF signaling in human prostate cancer cells. Oncogene 15:2705–2716; 1997. [DOI] [PubMed] [Google Scholar]
- 11. Holmes W. E.; Sliwkowski M. X.; Akita R. W.; Henzel W. J.; Lee J.; Park J. W.; Yansura D.; Abadi N.; Raab H.; Lewis G. D.; Shepard H. M.; Kuang W-J.; Wood W.; Goeddel D. V.; Vandlen R. L. Identification of heregulin, a specific activator of p185erbB2. Science 256:1205–1210; 1992. [DOI] [PubMed] [Google Scholar]
- 12. Hou F.; Zou H. Two human orthologues of Eco1/Ctf7 acetyltransferases are both required for proper sister-chromatid cohesion. Mol. Biol. Cell 16:3908–3918; 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kallioniemi A.; Kallioniemi O. P.; Sudar D.; Rutovitz D.; Gray J. W.; Waldman F.; Pinkel D. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science 258:818–821; 1992. [DOI] [PubMed] [Google Scholar]
- 14. Katoh M.; Katoh M. Comparative genomics on FGF20 orthologs. Oncol. Rep. 14:287–290; 2005. [PubMed] [Google Scholar]
- 15. Koch J.; Gartner S.; Li C. M.; Quintern L. E.; Bernardo K.; Levran O.; Schnabel D.; Desnick R. J.; Schuchman E. H.; Sandhoff K. Molecular cloning and characterization of a full-length complementary DNA encoding human acid ceramidase. Identification of the first molecular lesion causing Farber disease. J. Biol. Chem. 271:33110–33115; 1996. [DOI] [PubMed] [Google Scholar]
- 16. Kohl N. E.; Kanda N.; Schreck R. R.; Bruns G.; Latt S. A.; Gilbert F.; Alt F. W. Transposition and amplification of oncogene-related sequences in human neuroblastomas. Cell 35:359–367; 1983. [DOI] [PubMed] [Google Scholar]
- 17. Lee Z. H.; Kim H.; Ahn K. Y.; Seo K. H.; Kim J. K.; Bae C. S.; Kim K. K. Identification of a brain specific protein that associates with a refsum disease gene product, phytanoyl-CoA alpha-hydroxylase. Brain Res. Mol. Brain Res. 75:237–247; 2000. [DOI] [PubMed] [Google Scholar]
- 18. Li J.; Yen C.; Liaw D.; Podsypanina K.; Bose S.; Wang S. I.; Puc J.; Miliaresis C.; Rodgers L.; McCombie R.; Bigner S. H.; Giovanella B. C.; Ittmann M.; Tycko B.; Hibshoosh H.; Wigler M. H.; Parsons R. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275:1943–1947; 1997. [DOI] [PubMed] [Google Scholar]
- 19. Lodygin D.; Epanchintsev A.; Menssen A.; Diebold J.; Hermeking H. Functional epigenomics identifies genes frequently silenced in prostate cancer. Cancer Res. 65:4218–4227; 2005. [DOI] [PubMed] [Google Scholar]
- 20. Melkonyan H. S.; Chang W. C.; Shapiro J. P.; Mahadevappa M.; Fitzpatrick P. A.; Kiefer M. C.; Tomei L. D.; Umansky S. R. SARPs: A family of secreted apoptosis-related proteins. Proc. Natl. Acad. Sci. USA 94:13636–13641; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nakano H.; Yamamoto F.; Neville C.; Evans D.; Mizuno T.; Perucho M. Isolation of transforming sequences of two human lung carcinomas: structural and functional analysis of the activated c-K-ras oncogenes. Proc. Natl. Acad. Sci. USA 81:71–75; 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Oka H.; Shiozaki H.; Kobayashi K.; Inoue M.; Tahara H.; Kobayashi T.; Takatsuka Y.; Matsuyoshi N.; Hirano S.; Takeichi M. Expression of E-cadherin cell adhesion molecules in human breast cancer tissues and its relationship to metastasis. Cancer Res. 53:1696–1701; 1993. [PubMed] [Google Scholar]
- 23. Peles E.; Bacus S. S.; Koski R. A.; Lu H. S.; Wen D.; Ogden S. G.; Levy R. B.; Yarden Y. Isolation of the neu/HER-2 stimulatory ligand: A 44 kd glycoprotein that induces differentiation of mammary tumor cells. Cell 69:205–216; 1992. [DOI] [PubMed] [Google Scholar]
- 24. Pinkel D.; Segraves R.; Sudar D.; Clark S.; Poole I.; Kowbel D.; Collins C.; Kuo W. L.; Chen C.; Zhai Y.; Dairkee S. H.; Ljung B. M.; Gray J. W.; Albertson D. G. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat. Genet. 20:207–211; 1998. [DOI] [PubMed] [Google Scholar]
- 25. Pollack J. R.; Perou C. M.; Alizadeh A. A.; Eisen M. B.; Pergamenschikov A.; Williams C. F.; Jeffrey S. S.; Botstein D.; Brown P. O. Genome-wide analysis of DNA copy-number changes using cDNA microarrays. Nat. Genet. 23:41–46; 1999. [DOI] [PubMed] [Google Scholar]
- 26. Pollack J. R.; Sorlie T.; Perou C. M.; Rees C. A.; Jeffrey S. S.; Lonning P. E.; Tibshirani R.; Botstein D.; Borresen-Dale A. L.; Brown P. O. Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional program of human breast tumors. Proc. Natl. Acad. Sci. USA 99:12963–12968; 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schena M. Genome analysis with gene expression microarrays. Bioessays 18:427–431; 1996. [DOI] [PubMed] [Google Scholar]
- 28. Schena M.; Shalon D.; Davis R. W.; Brown P. O. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 270:467–470; 1995. [DOI] [PubMed] [Google Scholar]
- 29. Shimizu K.; Birnbaum D.; Ruley M. A.; Fasano O.; Suard Y.; Edlund L.; Taparowsky E.; Goldfarb M.; Wigler M. Structure of the Ki-ras gene of the human lung carcinoma cell line Calu-1. Nature 304:497–500; 1983. [DOI] [PubMed] [Google Scholar]
- 30. Sjoblom T.; Jones S.; Wood L. D.; Parsons D. W.; Lin J.; Barber T. D.; Mandelker D.; Leary R. J.; Ptak J.; Silliman N.; Szabo S.; Buckhaults P.; Farrell C.; Meeh P.; Markowitz S. D.; Willis J.; Dawson D.; Willson J. K.; Gazdar A. F.; Hartigan J.; Wu L.; Liu C.; Parmigiani G.; Park B. H.; Bachman K. E.; Papadopoulos N.; Vogelstein B.; Kinzler K. W.; Velculescu V. E. The consensus coding sequences of human breast and colorectal cancers. Science 314:268–274; 2006. [DOI] [PubMed] [Google Scholar]
- 31. Takash W.; Canizares J.; Bonneaud N.; Poulat F.; Mattei M. G.; Jay P.; Berta P. SOX7 transcription factor: sequence, chromosomal localisation, expression, transactivation and interference with Wnt signalling. Nucleic Acids Res. 29:4274–4283; 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Takayama Y.; Kamimura Y.; Okawa M.; Muramatsu S.; Sugino A.; Araki H. GINS, a novel multi-protein complex required for chromosomal DNA replication in budding yeast. Genes Dev. 17:1153–1165; 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tsujimoto Y.; Finger L. R.; Yunis J.; Nowell P. C.; Croce C. M. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science 226:1097–1099; 1984. [DOI] [PubMed] [Google Scholar]
- 34. Ugolini F.; Charafe-Jauffret E.; Bardou V. J.; Geneix J.; Adelaide J.; Labat-Moleur F.; Penault-Llorca F.; Longy M.; Jacquemier J.; Birnbaum D.; Pebusque M. J. WNT pathway and mammary carcinogenesis: loss of expression of candidate tumor suppressor gene SFRP1 in most invasive carcinomas except of the medullary type. Oncogene 20:5810–5817; 2001. [DOI] [PubMed] [Google Scholar]
- 35. van der Ven P. F.; Speel E. J.; Albrechts J. C.; Ramaekers F. C.; Hopman A. H.; Furst D. O. Assignment of the human gene for endosarcomeric cytoskeletal M-protein (MYOM2) to 8p23.3. Genomics 55:253–255; 1999. [DOI] [PubMed] [Google Scholar]
- 36. Yamamoto F.; Yamamoto M. Scanning copy number and gene expression on the 18q21-qter chromosomal region by the systematic multiplex PCR and reverse transcription-PCR methods. Electrophoresis 28:1882–1895; 2007. [DOI] [PubMed] [Google Scholar]
- 37. Yamamoto M.; Ahn R. H.; Yamamoto F. Scanning copy number and gene expression on the 16p13.3-13.2 chromosomal region by the systematic multiplex polymerase chain reaction and reverse transcription-polymerase chain reaction methods. Electrophoresis 27:2529–2540; 2006. [DOI] [PubMed] [Google Scholar]
- 38. Yamamoto M.; Takai D.; Yamamoto F.; Yamamoto F. Comprehensive expression profiling of highly homologous 39 Hox genes in 26 different human adult tissues by the Modified Systematic Multiplex RT-PCR method reveals tissue-specific expression pattern that suggests an important role of chromosomal structure in the regulation of Hox gene expression in adult tissues. Gene Expr. 11:199–210; 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yamamoto M.; Yamamoto A.; Leung P. C.; Yamamoto F. Gene expression analysis of an integrin family of genes by Systematic Multiplex RT-PCR. Electrophoresis 25:2201–2211; 2004. [DOI] [PubMed] [Google Scholar]
- 40. Yamamoto M.; Yamamoto F.; Luong T. T.; Williams T.; Kominato Y.; Yamamoto F. Expression profiling of 68 glycosyltransferase genes in 27 different human tissues by the systematic multiplex reverse transcription-polymerase chain reaction method revealed clustering of sexually related tissues in hierarchical clustering algorithm analysis. Electrophoresis 24:2295–2307; 2003. [DOI] [PubMed] [Google Scholar]