

Abstract
Endometrial cancer is the most common malignancy of the female reproductive tract. In many cases the prognosis is favorable, but 22% of affected women die from the disease. We aimed to study potential differences in gene expression between endometrioid adenocarcinomas from survivors (5-year survival) and nonsurvivors. Forty-five patients were included in the investigation, of which 21 were survivors and 24 were nonsurvivors. The tumors were analyzed with genome-wide expression array analysis, represented by 13,526 genes. Distinct differences in gene expression were found between the groups. A t-test established that 218 genes were significantly differentially expressed (p < 0.001) between the two survival groups, and in a cross-validation test 40 of the 45 (89%) tumors were classified correctly. The 218 differentially expressed genes were subjected to hierachical clustering analysis, which yielded two clusters both exhibiting over 80% homogeneity with respect to survival. When the additional constraint of fold change (FC > 2) was added the hierachical clustering yielded similar results. Stage I tumors are expected to have a favorable prognosis. However, in our tumor material there were six nonsurvivors with stage I tumors. Five out of six stage I nonsurvivors clustered in the nonsurvival fraction. Our findings suggest that a subgroup of early stage endometroid adenocarcinomas can be correctly classified as potentially aggressive by using molecular biology in combination with conventional markers, thereby providing a tool for a more accurate classification and risk evaluation of the individual patient.
Key words: Expression array, Survival, Endometrial cancer
INTRODUCTION
Carcinoma of the endometrium, also known as endometrial or corpus cancer, is the malignancy of the female reproductive tract that is most frequently diagnosed in Western countries. Although the prognosis is favorable for the majority of cases, almost one fourth of the affected women will die from the disease (22). The underlying biology of endometrial tumors still needs to be clarified and it is important to increase our knowledge about possible biological differences among these tumors.
Today the prognosis is primarily based on the surgical stage, but grade, type, and ploidy are also important factors used to predict the course of the disease. Need for adjuvant treatment is decided based on the estimated risk (10). Treatment failures are thought to be due, at least in part, to biological heterogeneity of the carcinomas, emphasizing the need for further studies that correlate survival of the patient with biological properties of the tumors. Several authors have suggested that there are two, perhaps more, distinct types of endometrial carcinoma with respect to underlying molecular pathways. The majority of endometrial cancer tumors (70–80%) follow an estrogen-related pathway and are designated as type I (endometrioid and mucinous differentiation), whereas tumors following an estrogen-unrelated pathway (10–20% of the tumors) are designated as type II tumors (nonendometrioid differentiation, serous, and clear cell) (11,14). In a previous study we analyzed 98 endometrioid tumors with comparative genomic hybridization (CGH), and the pattern of chromosomal aberrations in tumors from survivors was compared to that in tumors from nonsurvivors. Significant differences were detected between the two groups (13).
There are only few reports presenting expression array data of endometrial carcinomas, and the ones published deal predominantly with the question of differences between histological groups or between endometrioid tumors and normal endometrium (2,15,18,25,26). To our knowledge there are no prior studies of gene expression at a genome-wide scale focusing on differences in relation to patient survival. We selected a homogenous group of endometrioid adenocarcinomas to be included in the study. Gene expression profiles from 45 tumors (21 from survivors and 24 from nonsurvivors) were studied using a genome-wide expression array. The data obtained were analyzed statistically to reveal possible differences between the groups.
MATERIALS AND METHODS
Tumor Material
Endometrioid adenocarcinomas were collected from 45 patients diagnosed between 1991 and 2000 at Sahlgrenska University Hospital, Gothenburg, Sweden. The tumors were removed at primary surgery and secured for pathological examination and RNA extraction. All but two tumors were obtained from postmenopausal patients. Representative imprints from each of the frozen tumors were stained with May-Grünwald-Giemsa (Chemicon, Temecula, CA, USA), and the proportion of cancer cells was determined in each sample. The presence of >70% cancer cells was required for the specimen to be included in the study.
The tumors were of various stage and grade, as shown in Table 1. The median age of the patient at the time of diagnosis was 67 years (ranging from 41 to 87 years). Depending on surgical stage, histopathological subgroup and DNA ploidy status, the patients were divided into three different treatment groups. I) Stage I patients with G1 or G2 tumors, infiltration of less than 50% of the uterine wall, and diploid DNA content were considered low-risk and were not given any further treatment after removal of the tumor (27 patients). II) If one of the following criteria was present: G3 (+undifferentiated), infiltration of more than 50% of the uterine wall, or aneuploidy, patients were considered moderate risk and were randomized to vaginal brachytherapy +/− external radiation (4 patients) (8). III) Patients presenting more than one of the above-mentioned risk factors were randomized to external radiation + brachytherapy or external radiation + brachytherapy + chemotherapy (10 high-risk patients) (23). Four patients had not been risk-group classified. Patients that survived at least 5 years after diagnosis were regarded as survivors, whereas patients that died of the disease within 5 years (median survival time of 2 years) were included in the ssnonsurvivor group.
TABLE 1.
THE DISTRIBUTION OF THE TUMOUR MATERIAL WITH RESPECT TO STAGE AND GRADE, AND IN RELATION TO 5-YEAR SURVIVAL
| Total No. (Survivors/Nonsurvivors) | |
|---|---|
| Total Material | 45 (21/24) |
| Stage | |
| I | 21 (15/6) |
| II | 6 (2/4) |
| III | 7 (1/6) |
| IV | 5 (–/5) |
| n.a. | 6 (3/3) |
| Differentiation | |
| High (G1) | 8 (5/3) |
| Moderate (G2) | 20 (11/9) |
| Poor (G3) | 12 (2/10) |
| n.a. | 5 (3/2) |
n.a., data not available.
RNA Extraction
Frozen tumor material was homogenized together with TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) in a Micro-Dismembrator S (B. Braun Biotech International, Melsungen Germany). Total RNA was extracted from the suspension with RNeasy mini kit (Qiagen, Crawley, West Sussex, UK) according to the manufacturer’s protocol. The quality of the RNA was ev aluated with the BioAnalyzer (Agilent Tecnologies, Palo Alto, CA, USA). RIN values were between 5.8 and 9.1 (median at 6.7).
Expression Array Analysis
The microarray analysis was carried out as previously described by Partheen et al. (17). Since SCIBLU Genomics DNA Microarray Resource Center, who produced our expression arrays, made changes in their protocols over time, our study was performed using two separate array platforms. In the first set of 19 tumors (11 samples from survivors and 8 samples from nonsurvivors) expression was determined on arrays consisting of 35,000 reporters, whereas for the second set of tumors (26 in total; 9 from survivors and 17 from nonsurvivors) analysis was performed on a second platform of arrays composed of 27,000 reporters. Labeled tumor cDNA and reference cDNA were coprecipitated and hybridized to a glass slide containing 70 mer oligonucleotide reporter spots (SCIBLU). The microarray slides were scanned with an Agilent DNA microarray scanner G2505B (Agilent Tecnologies) and image analysis was performed using the Genepix 6.0.0.45 software (Axon Instruments, Union City, CA, USA). The expression data are available online at the Gene Expression Omnibus repository (GSE21882).
Statistical Analysis
The expression raw data extracted from the microarray analysis were initially processed in BASE (BioArray Software Environmental) (19). Included in the analysis were the 17,189 reporters that were present on both platforms. The background intensities were removed by substracting median background from median foreground and flagged spots were excluded. Pin-based lowess normalization (27), eight blocks in each group, was applied to each array in order to remove intensity-dependent effects in the values. Prior to further analysis the tumors were subjected to a de-correlation step to minimize the differences between the two sets of arrays. When the flagged spots and reporters with presence below 80% had been removed 13,526 reporters remained for further statistical analysis.
A two-tailed Student’s t-test was applied to identify genes that were differentially expressed between the two groups. To reduce the false-positive rate, the selection threshold was set to p < 0.001, which means that approximately 13 false positives were expected by chance. Kaplan-Meier survival curves were produced in the SPSS version 16 software and p-values for the difference between the curves were calculated using the Breslow-Wilcoxon test (5). For some of the clustering analysis a more focused gene set was selected by also applying a fold-change (FC) threshold. If the average expression value in the nonsurvivor group was ENS and in the survivor group was ES, the FC was calculated as ENS/ES if ENS > ES, and as ES/ENS if ENS < ES. The FC threshold was set to FC = 2.
The expression data for the differentially expressed genes was imported into the PermutMatrix software version X.Y and normalized for clustering using the interaction terms (rows versus columns) (3). A two-way clustering of genes and samples was derived using agglomerative hierarchical clustering with the default settings (i.e., McQuitty’s criteria as linkage rule and Euclidean distance for calculation of expression profile dissimilarities) (1).
Two different formations of the tumor material were set up to perform classification using the Weka implementation (6) of the Voting Features Interval classification algorithm (VFI) (4). VFI was chosen because we have found in our previous work that it consistently appeared among the best performing algorithms in the Weka package. Briefly, VFI sets an interval for each feature/gene and the feature votes for a given class if the expression level falls within this interval, and otherwise for the other class. Each feature is also assigned a weight, and the overall classification of the sample is determined by summing up all weights for each class and choosing the highest total. A classifier was derived using the pin-based lowess normalized expression data of all significant genes. The classifier was trained and tested using the standard leave-one-out cross-validation procedure, in this case using 45 iterations where each cycle consisted of training the voting features thresholds using 44 samples and testing the derived classifier on the 45th sample. We also evaluated the classification results on an independent test set, the classifiers were instead trained on data from the larger tumor set (26 samples) and tested on data from the smaller tumor set (19 samples).
Quantitative Polymerase Chain Reaction (QPCR)
All tumors except one (tumor 684 excluded due to lack of material) were also subjected to QPCR analysis for seven different genes (APOD, HOXA11, ITM2B, KIAA0738, RAB7L1, RAG1AP1, RAMP1) using validated Taqman® Gene Expression Assays, including gene-specific primers and probes (Applied Biosystems, Foster City, CA). The QPCR was performed in 384-well plates using the ABI PRISM® 7900HT Sequence Detection System (Applied Biosystems), and a pipetting robot (Beckman Coulter, Bromma, Sweden) to set up the PCR. The amplification reactions (10 μl) were performed in triplicate using 2 μl cDNA template, 1× TaqMan Universal PCR Master Mix (Applied Biosystems), 1× FAM-labeled TaqMan® Gene Expression Assays Mix (Applied Biosystems) in the 384-well format. Thermal cycling was performed using the 7900HT Sequence Detection System (Applied Biosystems) with an initiation step at 95°C for 10 min, followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. In each assay, a twofold dilution series of five samples was recorded, and one no-template control was included.
RESULTS
Among the total of 45 patients, 21 were survivors and 24 were nonsurvivors. The material was statistically considered in two different ways. First, the total material was screened as one entity for genes differentially expressed between survivors and nonsurvivors, and then, in order to perform a verifying analysis, the material was divided in two groups that were analyzed separately. Since SCIBLU, who produced our expression arrays, made some changes in their protocols over time, our study was performed using two separate platforms. Thus, the material was naturally divided into two groups depending on the platform, and based on these two sets of 19 and 26 tumors, respectively, we were able to perform an external validation as a complement to the analysis of the material as one unit.
Identification of Genes Differentially Expressed Between Survivors and Nonsurvivors
The total material was explored using a t-test, which revealed that 218 genes were differentially expressed at p < 0.001 between the two groups (survivors vs. nonsurvivors). A hierarchical clustering analysis was performed based on the 218 genes and their expression levels (Supplemental Fig. 1, available from: http://www.oncology.gu.se/Forskning/Publicerade_data/). The 45 tumors fell into two clusters. One cluster of 22 tumors consisted mostly of survivors (S cluster: 18 survivors vs. 4 nonsurvivors), whereas the other cluster of 23 tumors consisted mostly of non-survivors (NS cluster: 3 survivors vs. 20 nonsurvivors). The survival rates in the two clusters is shown in the Kaplan-Meier curves of Figure 1. In addition, a classification test was performed using the VFI algorithm and as many as 40 of the 45 (89%) tumors were correctly classified with respect to survival in the cross-validation. The five misclassifications were evenly distributed, with three survivors being misclassified as nonsurvivors and two vice versa.
Figure 1.
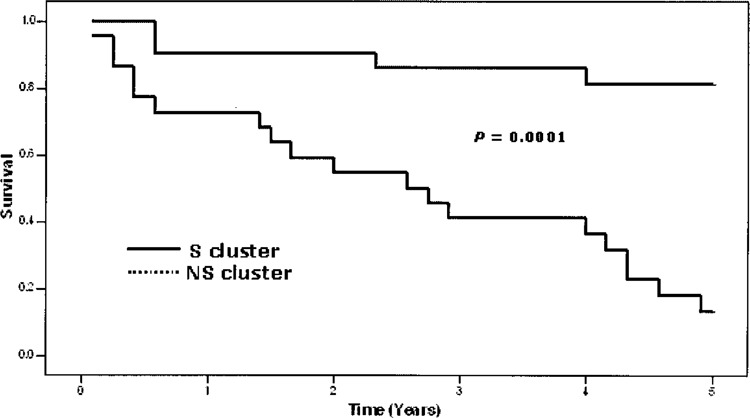
Kaplan-Meier curves showing the patient survival rates for the two clusters generated by the hierarchical clustering analysis based on 218 differentially expressed genes. The upper unbroken line represents the 22 patients in the S cluster, whereas the lower dotted line represents 21 of the 23 patients in the NS cluster. Two patients belonging to the NS cluster were known to be nonsurvivors, but the exact survival time had not been recorded. They were excluded from the diagram. The difference in survival rate between the two clusters is statistically highly significant (p = 0.0001) (7).
Stage I tumors are expected to have a favorable prognosis. However, in our tumor material there were six nonsurvivors with stage I tumors. In the clustering analysis five of them clustered in the NS cluster, suggesting that this type of analysis may have the potential to identify high-risk patients among those presenting with stage I tumors.
Among the 218 genes (p < 0.001) there were 64 genes with at least twofold difference in expression levels (FC > 2). For 45 of these genes expression was higher in the survivor group (Table 2) and the remaining 19 displayed higher expression in the non-survivor group (Table 3). Clustering based on this more focused set of genes again gave two main clusters, one containing 18 tumors, 16 from survivors (89%) and 2 from nonsurvivors (11%), and another containing 27 tumors, 5 from survivors (19%) and 22 from nonsurvivors (81%) (Fig. 2). Within this set of 64 genes, showing both significant differential expression and FC > 2, a number of known cancer-related genes (TACC1, APOD, and SALL4) were present. The set also included several genes that have been previously implicated in expression array studies of endometrial carcinoma (RAMP1, ACTA2, MYLK, REV3L, and UBE2C) (5,18,26).
TABLE 2.
LIST OF THE 45 GENES EXHIBITING HIGHER EXPRESSION LEVELS AMONG THE SURVIVORS COMPARED TO THE NONSURVIVORS (p < 0.001; FC > 2)
| Gene Symbol | Gene Name (Entrez GeneID) | FC |
|---|---|---|
| RBM24 | RNA binding motif protein 24 (221662) | 14.12 |
| RAMP1 | receptor (G protein-coupled) activity modifying protein 1 (10267) | 7.08 |
| GPR133 | G protein-coupled receptor 133 (283383) | 5.93 |
| MRGPRF | MAS-related GPR, member F (219928) | 5.85 |
| EDNRA | endothelin receptor type A (1909) | 5.51 |
| ACTA2 | actin, alpha 2, smooth muscle, aorta (59) | 5.15 |
| MYLK | myosin light chain kinase (4638) | 4.78 |
| SMOC2 | SPARC-related modular calcium binding 2 (64094) | 4.4 |
| HOXA11 | homeobox A11 (3207) | 4.39 |
| FREM1 | FRAS1-related extracellular matrix 1 (158326) | 4.15 |
| CGNL1 | cingulin-like 1 (84952) | 3.74 |
| TMOD1 | tropomodulin 1 (7111) | 3.66 |
| HOXD11 | homeobox D11 (3237) | 3.61 |
| FOXP2 | forkhead box P2 (93986) | 3.44 |
| APOD | apolipoprotein D (347) | 3.41 |
| PDE8B | phosphodiesterase 8B (8622) | 3.28 |
| GRAMD3 | GRAM domain containing 3 (65983) | 3.21 |
| HOXD9 | homeobox D9 (3235) | 3.18 |
| MITF | microphthalmia-associated transcription factor (4286) | 3.09 |
| HOXA10 | homeobox A10 (3206) | 3.07 |
| CDC42EP3 | CDC42 effector protein (Rho GTPase binding) 3 (10602) | 3.03 |
| CCND2 | cyclin D2 (894) | 2.88 |
| MAOB | monoamine oxidase B (4129) | 2.81 |
| PALLD | palladin, cytoskeletal associated protein (23022) | 2.79 |
| CACNA1H | calcium channel, voltage-dependent, T type, alpha 1H subunit (8912) | 2.78 |
| TACC1 | transforming, acidic coiled-coil containing protein 1 (6867) | 2.76 |
| CACNA1D | calcium channel, voltage-dependent, L type, alpha 1D subunit (776) | 2.72 |
| LOC91461 | hypothetical protein BC007901 (91461) | 2.71 |
| ISOC1 | isochorismatase domain containing 1 (51015) | 2.66 |
| Nbla00301 | unknown (79804) | 2.63 |
| PDE5A | phosphodiesterase 5A, cGMP-specific (8654) | 2.54 |
| PPM1K | protein phosphatase 1K (PP2C domain containing) (152926) | 2.54 |
| ADAMTSL1 | ADAMTS-like 1 (92949) | 2.52 |
| ZBTB16 | zinc finger and BTB domain containing 16 (7704) | 2.47 |
| PTPRB | protein tyrosine phosphatase, receptor type, B (5787) | 2.44 |
| GIMAP7 | GTPase, IMAP family member 7 (168537) | 2.24 |
| HNMT | histamine N-methyltransferase (3176) | 2.2 |
| TSPAN5 | tetraspanin 5 (10098) | 2.19 |
| COL15A1 | collagen, type XV, alpha 1 (1306) | 2.15 |
| UBL3 | ubiquitin-like 3 (5412) | 2.14 |
| OLFML3 | olfactomedin-like 3 (56944) | 2.11 |
| ATP1B2 | ATPase, Na+/K+ transporting, beta 2 polypeptide (482) | 2.11 |
| GNG11 | guanine nucleotide binding protein (G protein), gamma 11 (2791) | 2.11 |
| REV3L | REV3-like, catalytic subunit of DNA polymerase zeta (yeast) (5980) | 2.1 |
| ARRDC4 | arrestin domain containing 4 (91947) | 2.02 |
The genes are listed with their gene symbol, gene name, and the Entrez GeneID.
TABLE 3.
LIST OF THE 19 GENES EXHIBITING HIGHER EXPRESSION LEVELS AMONG THE SURVIVORS COMPARED TO THE NONSURVIVORS (p < 0.001; FC > 2)
| Gene Symbol | Gene Bame (Entrez GeneID) | FC |
|---|---|---|
| CLDN6 | claudin 6 (9074) | 4.23 |
| PITX1 | paired-like homeodomain 1 (5307) | 4.20 |
| CA9 | carbonic anhydrase IX (768) | 3.55 |
| IRX2 | iroquois homeobox 2 (153572) | 3.10 |
| L1CAM | L1 cell adhesion molecule (3897) | 2.84 |
| SALL4 | sal-like 4 (Drosophila) (57167) | 2.69 |
| PADI2 | peptidyl arginine deiminase, type II (11240) | 2.69 |
| PLEKHG4 | pleckstrin homology domain containing, family G member 4 (25894) | 2.43 |
| GABRE | gamma-aminobutyric acid (GABA) A receptor, epsilon (2564) | 2.25 |
| C16orf75 | chromosome 16 open reading frame 75 (116028) | 2.23 |
| VDR | vitamin D (1,25-dihydroxyvitamin D3) receptor (7421) | 2.17 |
| TAF4B | TAF4b RNA polymerase II, TATA box binding protein (TBP)-associated factor (6875) | 2.15 |
| TPX2 | TPX2, microtubule-associated, homolog (Xenopus laevis) (22974) | 2.14 |
| ECE2 | endothelin converting enzyme 2 (9718) | 2.11 |
| UBE2C | ubiquitin-conjugating enzyme E2C (11065) | 2.10 |
| EXOSC5 | exosome component 5 (56915) | 2.09 |
| RAG1AP1 | recombination activating gene 1 activating protein 1 (55974) | 2.08 |
| ATF5 | activating transcription factor 5 (22809) | 2.07 |
| GINS1 | GINS complex subunit 1 (Psf1 homolog) (9837) | 2.05 |
The genes are listed with their gene symbol, gene name, and the Entrez GeneID.
Figure 2.
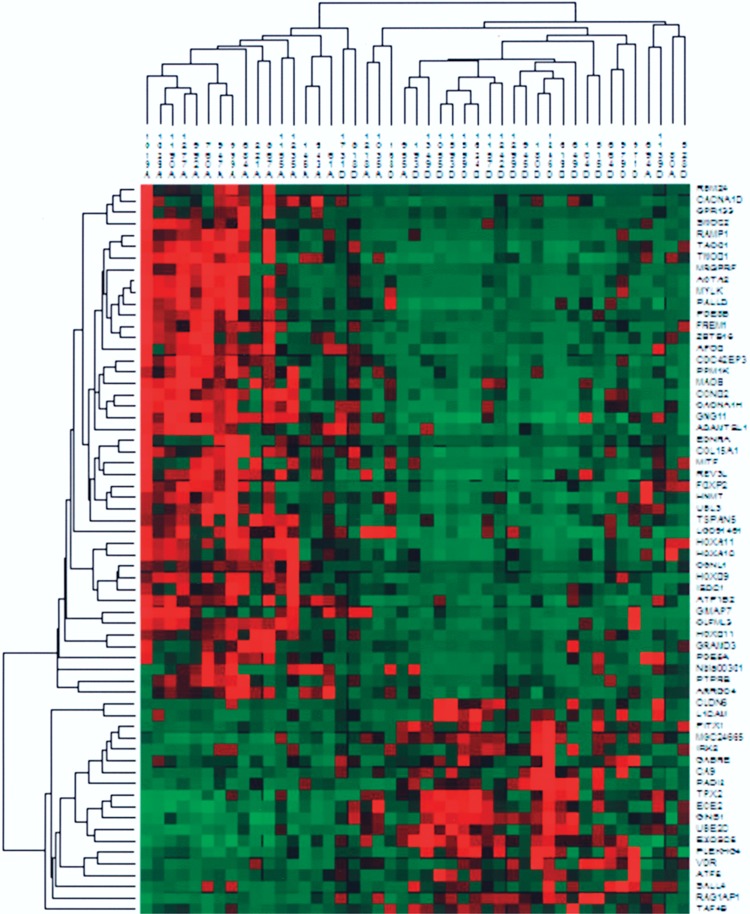
Hierarchical clustering of all 45 tumors based on 64 differentially expressed genes (p < 0.001; FC > 2; see also Tables 2 and 3). Green represents negative values compared to the reference; red represents positive values; and black represents 0. Two clusters are formed; one comprising 18 tumors, 16 from survivors (A) and 2 from nonsurvivors (D), whereas the other cluster comprises 27 tumors, 5 from survivors and 22 from nonsurvivors.
Tumors From Nonsurvivors Exhibited Overexpression of Genes Involved in Cell Cycle Control
To establish whether there were any known interactions among the 218 differentially expressed genes we examined potential connections in cellular pathways. Five of the genes (E2F1, E2F2, EXO1, MSH2, TP53BP2) that were expressed at higher levels among nonsurvivors (FC values between 1.53 and 1.72) are all connected to the RB/E2F pathway and are involved in cell cycle control (16). In addition, both REV1L (FC = 1.35) and REV3L (FC = 2.10) have been described to interact in the vicinity of this pathway. This pathway also involves other known cancer-related proteins such as TP53, RB1, MYC, MYCN, and BRCA1, but none of these were differentially expressed between the two groups in the present investigation.
Dividing the Material Into One Test Group and One External Validation Group: Classification of Tumors With up to 79% Accuracy
To be able to evaluate the classification results in an independent test set, the selection of significant genes and training of classifiers was repeated, this time using only one of the tumor sets. This set up gave the opportunity to use the other set of tumors for testing the accuracy of the classifiers. Applying the t-test to the larger training set (26 tumor samples), 92 genes were found to show significant differential expression between the 9 survivors and the 17 nonsurvivors (p < 0.001). Among these, there were 63 genes that were differentially expressed in at least 80% of the samples in each group (Supplemental Table 1, available from: http://www.oncology.gu.se/Forskning/Publicerade_data/). The overlap between the two gene sets (63 vs. 218 genes) was 43%. The normalized expression values for these genes in the larger training tumor set (26 samples) were used to train a VFI classifier, which was then tested on the smaller test set (19 tumor samples). The obtained accuracy of 74% (14 of 19 samples correctly classified) was significantly better than the baseline performance of 58% (11 of 19), which would be obtained by simply voting for the majority class for every sample. When testing a range of classification algorithms (including two types of decision trees, a multilayer perceptron and two types of Bayesian classifiers) accuracies in the range of 68% (13 of 19) to 79% (15 of 19) were obtained. We also tried using the larger tumor set for deriving genes only, and then performing both training and testing on the smaller tumor set, which gave similar results (data not shown).
Results From QPCR Analysis Supported the Microarray Findings
To evaluate the accuracy of the results from the microarray analysis we validated several transcripts with an alternative method for gene expression analysis. Transcripts for the QPCR analysis were selected from the gene list with the 218 genes expressed differentially at p < 0.001. Five of the genes displayed higher expression in tumors from survivors (APOD, HOXA11, ITM2B, KIAA0738, RAMP1), whereas two had higher expression in tumors from nonsurvivors (RAB7L1, RAG1AP1). The control genes used were selected from the microarray analysis and consisted of genes that had stable expression levels between the samples (BECN1, CXXC1, MTPN, WDR39), and, in addition, three genes were included in the analysis from the Taqman® endogenous controls (B2M, GUSB, TBP). Differential expression in all seven test genes showed concordance in the expression pattern between microarray and QPCR. The expression results from the tested genes were normalized to the geometric average of the seven control genes and the normalized values were subjected to a t-test. Six of seven genes showed significant difference in expression levels between the two groups (APOD, p < 0.01; HOXA11, p < 0.001; ITM2B, p < 0.01; RAMP1, p < 0.01; RAB7L1, p < 0.01; RAG1AP1, p < 0.05).
DISCUSSION
We performed microarray analysis in a selection of endometrial adenocarcinomas and were able to identify evident differences in the gene expression pattern between tumors from survivors and tumors from nonsurvivors.
We showed that there were 218 genes that were differentially expressed at the level of p < 0.001 with respect to survival in the tumor material. Among them were several genes known to be cancer related. For instance, we detected lower levels of expression of APOD among tumors from nonsurvivors. It is known that estrogen plays an important role in the development and progression of endometrial carcinoma. Estrogen also has a negative effect on the expression of APOD with cell proliferation as a consequence (9,21). In addition, REV3L was found to have a lower expression level in tumors from the nonsurvivor group. The lower expression of REV3L detected in the more aggressive tumors agrees with the findings of Risinger et al., who reported downregulation of REV3L in endometrioid adenocarcinomas compared to normal tissue (18). Furthermore, TACC1, which is suspected to be involved in oncogenesis, was found to display higher expression among survivors compared to nonsurvivors (24). This finding and the fact that TACC1 was found to be expressed at a higher level among tumors with more differentiated cells compared to tumors with lower differentiation grade suggests that high expression of TACC1 could be a potential marker for better prognosis (5).
In the pathway analysis we identified five genes (E2F1, E2F2, EXO1, MSH2, TP53BP2) that are involved in the cell cycle regulation, all of which exhibited higher expression levels among the tumors from nonsurvivors. These five genes are connected in the important RB/E2F pathway, which is involved in cell cycle control and has a central role in relation to cancer development (16). It has been reported that the regulation of the G1/S transition and the activation of DNA replication is mainly under the control of the RB/E2F pathway (12). To our knowledge this pathway has not been implicated in relation to endometrial carcinoma previously. Clarification of its role could be beneficial for a more accurate classification of these tumors. Furthermore, in the vicinity of the RB/E2F pathway other recognized cancer-related genes interact (e.g., TP53, MYCN, and MYC) (16). In a previous study we detected increased copy numbers of MYCN in a group of tumors tested for gene amplification by FISH (20). We found no difference in MYCN expression between survivors and nonsurvivors, but this does not rule out the possibility of a general amplification/overexpression of MYCN in endometrial carcinomas. Because increased gene expression may not immediately lead to increased protein production, it would be of interest to determine possible differences in protein expression between the survival groups for the proteins involved in the RB/E2F pathway. Any differences in expression that may be found potentially could be valuable as prognostic markers.
We performed a hierarchal clustering analysis based on the 218 differentially expressed genes and two clusters were formed with survival homogeneity of more than 80% in both clusters. Furthermore, in the classification test as many as 89% of the tumors were correctly classified. This definitely points towards the possible use of this gene panel as a prognostic tool. In particular, five of the six stage I tumors from patients who died clustered together with the nonsurvivors, suggesting that gene expression analysis may be useful to identify difficult-to-detect, high-risk patients who have small but aggressive tumors. Normally, stage I patients are only surgically treated, but if aggressive tumors could be identified at an early stage, adjuvant treatment could be introduced immediately for the benefit of the patient.
In the classification test performed for the external validation in the independent test set five out of 19 tumors were misclassified (one survivor and four nonsurvivors). The one survivor that clustered together with the nonsurvivors came from a patient that only survived for slightly over 5 years. The four non-survivors that clustered in the survival cluster had a survival time from 5 months up to 4 years and the median age was high among these women (83.5 years; ranging from 82 to 87). The marginally lower accuracies (78%) that can reasonably be considered as expected, given the smaller number of samples available for training (26 samples instead of 45), the smaller number of significantly expressed and frequently present genes (65 instead of 218), and the inevitable accidental biases introduced by splitting a patient group into two smaller subgroups. It is also possible that differences between the two versions of the microarray platform, or between the hybridization experiments, may have contributed to the digression. However, the average accuracies from the independent test set evaluation are still clearly better than the baseline. This indicates that the identified genes do have a predictive value.
CONCLUSION
In this article we analyzed endometrioid adenocarcinomas in relation to survival and found distinct gene expression differences between tumors from survivors compared to tumors from nonsurvivors. Using the micorarray technique we identified multiple genes with differences in expression between tumors from survivors and tumors from nonsurvivors with endometrial carcinoma. An intriguing finding was the upregulation of genes related to the RB/E2F pathway in the group of nonsurvivors, suggesting an interesting target for further investigation. Our findings contribute to the molecular characterization of this complex disease, and, in combination with current prognostic markers, they could help in predicting the outcome for individual patients and lead to improvements in treatment.
ACKNOWLEDGMENTS
We thank Ghita Fallenius for the cytological evaluation and the personnel at SCIBLU Genomics Centre at Lund University for technical advice concerning the microarray. The King Gustav V Jubilee Clinic Cancer Research Foundation supported this work. This study was approved (S154-02) by the local ethics committee Medical Faculty Research Ethics Committee, Göteborg, Sweden. The microarray dataset has been deposited GEO (Series GSE21882).
REFERENCES
- 1. Bair E.; Tibshirani R. Semi-supervised methods to predict patient survival from gene expression data. PLoS Biol. 2:E108; 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cao Q. J.; Belbin T.; Socci N.; Balan R.; Prystowsky M. B.; Childs G.; Jones J. G. Distinctive gene expression profiles by cDNA microarrays in endometrioid and serous carcinomas of the endometrium. Int. J. Gynecol. Pathol. 23:321–329; 2004. [DOI] [PubMed] [Google Scholar]
- 3. Caraux G.; Pinloche S. PermutMatrix: A graphical environment to arrange gene expression profiles in optimal linear order. Bioinformatics 21:1280–1281; 2005. [DOI] [PubMed] [Google Scholar]
- 4. Demiroz G.; Guvenir H. A. Classification by voting feature intervals. In: Lecture notes in computer science, vol. 1224. Berlin: Springer; 1997. [Google Scholar]
- 5. Ferguson S. E.; Olshen A. B.; Viale A.; Awtrey C. S.; Barakat R. R.; Boyd J. Gene expression profiling of tamoxifen-associated uterine cancers: Evidence for two molecular classes of endometrial carcinoma. Gynecol. Oncol. 92:719–725; 2004. [DOI] [PubMed] [Google Scholar]
- 6. Frank E.; Hall M.; Trigg L.; Holmes G.; Witten I. H. Data mining in bioinformatics using Weka. Bioinformatics 20:2479–2481; 2004. [DOI] [PubMed] [Google Scholar]
- 7. Gehan E. A. A generalized Wilcoxon test for comparing arbitrarily singly-censored samples. Biometrika 52:203–223; 1965. [PubMed] [Google Scholar]
- 8. Hogberg T. Adjuvant chemotherapy in endometrial carcinoma: Overview of randomised trials. Clin. Oncol. (R Coll. Radiol.) 20:463–469; 2008. [DOI] [PubMed] [Google Scholar]
- 9. Ito K. Hormone replacement therapy and cancers: The biological roles of estrogen and progestin in tumorigenesis are different between the endometrium and breast. Tohoku J. Exp. Med. 212:1–12; 2007. [DOI] [PubMed] [Google Scholar]
- 10. Jadoul P.; Donnez J. Conservative treatment may be beneficial for young women with atypical endometrial hyperplasia or endometrial adenocarcinoma. Fertil. Steril. 80:1315–1324; 2003. [DOI] [PubMed] [Google Scholar]
- 11. Lax S. F. Molecular genetic pathways in various types of endometrial carcinoma: From a phenotypical to a molecular-based classification. Virchows Arch. 444:213–223; 2004. [DOI] [PubMed] [Google Scholar]
- 12. Leone G.; DeGregori J.; Yan Z.; Jakoi L.; Ishida S.; Williams R. S.; Nevins J. R. E2F3 activity is regulated during the cell cycle and is required for the induction of S phase. Genes Dev. 12:2120–2130; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Levan K.; Partheen K.; Osterberg L.; Helou K.; Horvath G. Chromosomal alterations in 98 endometrioid adenocarcinomas analyzed with comparative genomic hybridization. Cytogenet. Genome Res. 115:16–22; 2006. [DOI] [PubMed] [Google Scholar]
- 14. Liu F. S. Molecular carcinogenesis of endometrial cancer. Taiwan J. Obstet. Gynecol. 46:26–32; 2007. [DOI] [PubMed] [Google Scholar]
- 15. Moreno-Bueno G.; Sanchez-Estevez C.; Cassia R.; Rodriguez-Perales S.; Diaz-Uriarte R.; Dominguez O.; Hardisson D.; Andujar M.; Prat J.; Matias-Guiu X.; Cigudosa J. C.; Palacios J. Differential gene expression profile in endometrioid and nonendometrioid endometrial carcinoma: STK15 is frequently overexpressed and amplified in nonendometrioid carcinomas. Cancer Res. 63:5697–5702; 2003. [PubMed] [Google Scholar]
- 16. Nevins J. R. The Rb/E2F pathway and cancer. Hum. Mol. Genet. 10:699–703; 2001. [DOI] [PubMed] [Google Scholar]
- 17. Partheen K.; Levan K.; Osterberg L.; Horvath G. Expression analysis of stage III serous ovarian adenocarcinoma distinguishes a sub-group of survivors. Eur. J. Cancer 42:2846–2854; 2006. [DOI] [PubMed] [Google Scholar]
- 18. Risinger J. I.; Maxwell G. L.; Chandramouli G. V.; Jazaeri A.; Aprelikova O.; Patterson T.; Berchuck A.; Barrett J. C. Microarray analysis reveals distinct gene expression profiles among different histologic types of endometrial cancer. Cancer Res. 63:6–11; 2003. [PubMed] [Google Scholar]
- 19. Saal L. H.; Troein C.; Vallon-Christersson J.; Gruvberger S.; Borg A.; Peterson C. BioArray Software Environment (BASE): A platform for comprehensive management and analysis of microarray data. Genome Biol. 3:SOFTWARE0003; 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Samuelson E.; Levan K.; Adamovic T.; Levan G.; Horvath G. Recurrent gene amplifications in human type I endometrial adenocarcinoma detected by fluorescence in situ hybridization. Cancer Genet. Cytogenet. 181:25–30; 2008. [DOI] [PubMed] [Google Scholar]
- 21. Simard J.; de Launoit Y.; Haagensen D. E.; Labrie F. Additive stimulatory action of glucocorticoids and androgens on basal and estrogen-repressed apolipoprotein-D messenger ribonucleic acid levels and secretion in human breast cancer cells. Endocrinology 130:1115–1121; 1992. [DOI] [PubMed] [Google Scholar]
- 22. Socialstyrelsen. Cancer incidence in Sweden 2006. Stockholm: Statistics, Sweden; 2007. [Google Scholar]
- 23. Sorbe B.; Horvath G.; Andersson H.; Boman K.; Lundgren K.; Pettersson B. External pelvic and vaginal irradiation versus vaginal irradiation alone as postoperative therapy in medium risk endometrial carcinoma—a prospective randomized study. Abstract, IGCS Meeting, Bangkok; 2008. [DOI] [PubMed] [Google Scholar]
- 24. Still I. H.; Hamilton M.; Vince P.; Wolfman A.; Cowell J. K. Cloning of TACC1, an embryonically expressed, potentially transforming coiled coil containing gene, from the 8p11 breast cancer amplicon. Oncogene 18:4032–4038; 1999. [DOI] [PubMed] [Google Scholar]
- 25. Sugiyama Y.; Dan S.; Yoshida Y.; Akiyama F.; Sugiyama K.; Hirai Y.; Matsuura M.; Miyata S.; Ushijima M.; Hasumi K.; Yamori T. A large-scale gene expression comparison of microdissected, small-sized endometrial cancers with or without hyperplasia matched to same-patient normal tissue. Clin. Cancer Res. 9:5589–5600; 2003. [PubMed] [Google Scholar]
- 26. Wong Y. F.; Cheung T. H.; Lo K. W.; Yim S. F.; Siu N. S.; Chan S. C.; Ho T. W.; Wong K. W.; Yu M. Y.; Wang V. W.; Li C.; Gardner G. J.; Bonome T.; Johnson W. B.; Smith D. I.; Chung T. K.; Birrer M. J. Identification of molecular markers and signaling pathway in endometrial cancer in Hong Kong Chinese women by genome-wide gene expression profiling. Oncogene 26:1971–1982; 2007. [DOI] [PubMed] [Google Scholar]
- 27. Yang Y. H.; Dudoit S.; Luu P.; Lin D. M.; Peng V.; Ngai J.; Speed T. P. Normalization for cDNA microarray data: A robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res. 30:e15; 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]