

Abstract
Serum amyloid A (SAA) is an acute-phase protein whose action in innate immunity and lipid homeostasis is unclear. Most circulating SAA binds plasma high-density lipoproteins (HDL) and reroutes lipid transport. In vivo SAA binds existing lipoproteins or generates them de novo upon lipid uptake from cells. We explored the products of SAA-lipid interactions and lipoprotein remodeling in vitro. SAA complexes with palmitoyl-oleoyl phosphocholine (POPC) were analyzed for structure and stability using circular dichroism and fluorescence spectroscopy, electron microscopy, gel electrophoresis and gel filtration. The results revealed the formation of 8–11 nm lipoproteins that were ~50% a-helical and stable at near-physiological conditions but were irreversibly remodeled at Tm ~ 52 °C. Similar HDL-size nanoparticles formed spontaneously at ambient conditions or upon thermal remodeling of parent lipoproteins containing various amounts of proteins and lipids, including POPC and cholesterol. Therefore, such HDL-size particles formed stable kinetically accessible structures in a wide range of conditions. Based on their size and stoichiometry, each particle contained about 12 SAA and 72 POPC molecules, with a pro-tein:lipid weight ratio circa 2.5:1, suggesting a structure distinct from HDL. High stability of these nanoparticles and their HDL-like size suggest that similar lipoproteins may form in vivo during inflammation or injury when SAA concentration is high and membranes from dead cells require rapid removal. We speculate that solubilization of membranes by SAA to generate lipoproteins in a spontaneous energy-independent process constitutes the primordial function of this ancient protein, providing the first line of defense in clearing cell debris from the injured sites.
Keywords: Protein-lipid surface interactions; Lipoprotein structure, function and stability; Lipid homeostasis in acute-phase response; Clearance of dead cells and cell repair; Lipoprotein remodeling, fusion and fission; Thermodynamic and kinetic stability
1. Introduction
Serum amyloid A (SAA, ~12 kDa) is an ancient family of proteins highly conserved since the Cambrian explosion (Uhlar and Whitehead, 1999; Kisilevsky and Manley, 2012). SAA has been studied for over four decades as a protein precursor of amyloid A (AA) and an enigmatic biomarker of inflammation, yet its beneficial functions remain subject of debate (Urieli-Shoval et al., 2000; Kisilevsky and Manley, 2012; Eklund et al., 2012; Ye and Sun, 2015). In acute inflammation, infection, or injury, plasma levels of inducible SAA increase swiftly and reversibly up to 1000-fold reaching ~1 mg/ml; the benefit for the host survival of this dramatic but transient increase is unclear. Furthermore, plasma SAA is elevated in infections such as tuberculosis, in autoimmune disorders such as rheumatoid arthritis and Crohn’s disease, and in various cancers (Urieli-Shoval et al., 2000; Ye and Sun, 2015). Elevated SAA has emerged as a causal risk factor for atherosclerosis (Dong et al., 2011; Thompson et al., 2015). Moreover, elevated SAA can lead to reactive AA amyloidosis, a life-threatening complication of chronic inflammation and a major human systemic amyloidosis world-wide, wherein N-terminal fragments of SAA, termed AA, deposit in kidneys and other organs and damage them (Westermark et al., 2015). Our ultimate goal is to understand the delicate balance between the normal functions of SAA in innate immunity and lipid homeostasis and the pathologic pathway of SAA misfolding and deposition in AA amyloidosis. Both normal and pathologic effects of SAA are critically hinged upon its binding to numerous ligands, particularly lipids (Frame and Gursky, 2016; Kollmer et al., 2016; Tanaka et al., 2017) that are in the focus of the current study.
The best-understood function of SAA is its ability to mobilize phospholipids and cholesterol for cell repair (Kisilevsky and Manley, 2012), which is critically hinged on SAA binding to its major plasma carrier, high-density lipoproteins (HDL) (Benditt and Eriksen, 1977; Coetzee et al., 1986). HDL are heterogeneous particles, 8–12 nm in diameter, that remove excess cell cholesterol and protect against atherosclerosis (van der Westhuyzen et al., 2007; Getz and Reardon, 2008; Phillips, 2013). Each HDL particle from normal plasma contains several proteins, mainly apolipopro-tein A-I (apoA-I, 28 kDa), and several hundred lipids, mainly phosphatidylcholines (PCs) and cholesterol. Nascent HDL is comprised of a cholesterol-containing phospholipid bilayer with amphipathic a-helices of apoA-I wrapped around the perimeter, while mature HDL contains a core of apolar lipids and additional apolipoproteins. HDL solubility is conferred by its polar surface comprised of PC head groups along with the polar faces of the apolipoprotein a-helices, while the apolar moieties are sequestered in the lipoprotein interior (Phillips, 2013; Bibow et al., 2017). ApoA-I can rever-sibly bind to and dissociate from the HDL surface via the apolar faces of its amphipathic α-helices that form the major lipid surface-binding motif in the apolipoprotein family (Segrest et al., 1992).
In acute phase response, SAA is secreted mainly by the liver into plasma, binds HDL, possibly displaces some apoA-I, and thereby reroutes the transport of HDL lipids by interacting with cell receptors that normally do not bind HDL, such as CD36, LOX-1, and RAGE (van der Westhuyzen et al., 2007; Kisilevsky and Manley, 2012; Ye and Sun, 2015; Perrone et al., 2008). Relative to normal HDL that contain comparable mass of protein and lipid, acute-phase HDL are enriched in SAA and have a distinct lipid composition (Coetzee et al., 1986; Abe-Dohmae et al., 2006; Hu et al., 2008; de Beer et al., 2011). In mouse model studies, SAA-only HDL have been found in plasma (Cabana et al., 1999, 2004). The detailed structural and stability properties of SAA-containing HDL are unclear and are beginning to be elucidated by several teams including ours (Wang and Colón, 2004; Takase et al., 2014; Colón et al., 2015; Jayaraman et al., 2015).
Apart from binding to existing lipoproteins, SAA can also generate lipoproteins de novo by sequestering lipids from the plasma membrane. Cell studies showed that, similar to normal apoA-I-containing HDL, the biogenesis of SAA-only HDL is facilitated by ATP-binding cassette (ABC) lipid transporters such as ABCA1 (Stonik et al., 2004; van der Westhuyzen et al., 2005; Abe-Dohmae et al., 2006; Hu et al., 2008; de Beer et al., 2011; Qian et al., 2017). At least one study reported that, unlike apoA-I, SAA can also promote efflux of phospholipid and cholesterol from cells in the absence of transporters, suggesting that SAA may act as a unique acceptor for lipid removal from the inflammation sites (Stonik et al., 2004). However, other studies have questioned this finding (Abe-Dohmae et al., 2006; Hu et al., 2008). The current work explores the transporter-independent ability of SAA to solubilize lipid bilayers, and determines the structural properties and stability of the resulting particles.
Although the atomic structure of lipid-bound SAA is not available, important insights were obtained from the ~2.2 Å resolution crystal structures of two lipid-free SAA proteins, human isoform 1 (hSAA1) and murine isoform 3 (mSAA3) (Lu et al., 2014; Derebe et al., 2014). The structures of these proteins have been determined in three different oligomeric forms and in different space groups, yet they showed a very similar monomer fold, with a unique Y-shaped 4-helix bundle. Remarkably, the interior of the bundle is unusually polar (Derebe et al., 2014), while the apolar faces of amphipathic α-helices h1 (residues 1–27) and h3 (residues 50–69) are solvent-exposed, forming an extended concave hydrophobic surface. Notably, the radius of curvature of this surface, r ~4.2 nm, is commensurate with the HDL curvature. We proposed that this surface forms the lipid binding site in the SAA monomer, with a particular preference for HDL (Frame and Gursky, 2016).
The crystal structures of SAA have 80–85% a-helical content, which likely represents the maximally folded protein state. In stark contrast, lipid-free SAA in solution is intrinsically disordered, i.e. in the absence of ligands under near-physiologic buffer conditions and temperatures it can be fully or partially unfolded depending on the species and the isoform. We postulated that helices h1 and h3 are folded when bound to the lipid surface, while other regions of the SAA molecule fold upon binding other ligands in a manner typical of other intrinsically disordered proteins (Frame and Gursky, 2016).
The current study explores the structure, stability and thermal remodeling of SAA-only lipoproteins. Previously, we showed that structural stability of all major lipoprotein classes involves high kinetic barriers, and that thermal remodeling of various lipoproteins mimics important aspects of their metabolic remodeling by plasma factors (Gursky, 2015 and references therein). In fact, both thermal and metabolic remodeling of HDL involves protein release in a lipid-poor form and fusion of protein-depleted lipoproteins into larger particles; such fusion alleviates the imbalance between the polar surface of a lipoprotein and its apolar interior (Jayaraman et al., 2006, 2012). Therefore, thermal remodeling provides a useful tool to explore structural stability of lipoproteins and their morphologic transformations that mimic their functional remodeling in vivo (Gursky, 2015).
In the current study we used binary complexes of mSAA1, a major inducible amyloidogenic isoform that binds HDL, and a physiologically relevant zwitterionic phospholipid, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC; C16:0, C18:1). Similar to POPC, phospholipids in HDL and in the plasma membrane are mainly PCs enriched in 18-carbon acyl chains one of which is unsaturated. We also have explored ternary complexes of SAA, POPC and cholesterol, an essential constituent of HDL and cell membranes which is critical for cell viability and tissue repair. The results revealed spontaneous formation of stable SAA-lipid nanoparticles that are similar in size to HDL but have a different stoichiometry, suggesting a structure distinct from any other lipoproteins. Our findings suggest a new primordial function of SAA in clearing plasma membranes of dead cells via a spontaneous energy-independent process.
2. Materials and methods
2.1. Proteins, lipids and lipoproteins
Recombinant mSAA1 (103 a.a., 11.6 kDa, termed SAA for brevity) was expressed in E-coli and purified to 95% purity as previously described (Kollmer et al., 2016). Lipids, including POPC and unes-terified cholesterol, were 97% + pure from Avanti Polar Lipids. All other chemicals were of highest purity analytical grade. Protein concentration was determined using modified Lowry assay with bovine serum albumin as a standard (Markwell et al., 1978). Phospholipids were quantified using an enzymatic method (Wako Diagnostics phospholipids-C assay) wherein choline-phospholipid was monitored by absorbance (Bergmeyer, 1985).
Protein stock solutions were prepared by dissolving lyophilized SAA in water. All dilutions were made in 10 mM sodium phosphate at pH 7.5, which was the standard buffer used throughout this study. Homogenous small unilamellar vesicles (SUV, ~25 nm in size) of POPC in standard buffer were prepared by sonication following published protocols (McLean and Phillips, 1984). For spontaneous reconstitution of SAA-POPC complexes, SAA stock solution was added to POPC SUV at a protein:lipid molar ratio ranging from 1:0.5 to 1:80. The mixtures were incubated at 23 °C for up to 12 h and used for further studies. Lipoprotein reconstitution by cholate dialysis method (Jonas, 1986), which was used to obtain binary and ternary complexes with controlled protein and lipid composition, was carried out following published protocols (Cavigiolio et al., 2008). No protein degradation in lipoproteins was detected by SDS PAGE.
2.2. Circular dichroism (CD) and fluorescence spectroscopy
CD data were recorded by using an AVIV 400 spectropolarime-ter to monitor protein secondary structure and thermal stability. Far-UV CD spectra were recorded at 185–250 nm from samples containing 0.1 mg/ml SAA and varying amounts of lipid. Melting data, Θ222(T), were recorded at 222 nm to monitor a-helical unfolding during heating and cooling at a constant rate of either 60 or 10 °C/h. No significant scan rate effects on the heating curves were observed. The melting temperature, Tm, corresponding to the midpoint of the unfolding transition was determined by sigmoidal fitting of the Θ222(T) data. The accuracy in the determination of Tm, which is reported as a standard deviation of the mean obtained from 3 to 5 independent experiments, was ±1.5 °C or better. Following the buffer baseline subtraction, the CD data were normalized to protein concentration and were reported as molar residue ellipticity, [Θ]. The α-helical content was estimated from the value of [Θ222] (Mao and Wallace, 1984) with an accuracy of 5–7%. ORIGIN software was used for the data analysis and display.
Intrinsic Trp fluorescence was monitored at either 5 °C or 25 °C using a Varian Cary Eclipse spectrofluorimeter. The samples contained 0.3 mg/ml SAA and varying amounts of lipid. The excitation wavelength was λex = 295 nm and the emission was recorded at 310–450 nm with 5 nm excitation and emission slit widths. The wavelength of maximal fluorescence, kmax, was determined by Boltzmann fitting of the integrated peak intensity with an accuracy of ±2 nm or better.
2.3. Gel electrophoresis and size exclusion chromatography (SEC)
For non-denaturing polyacrylamide gel electrophoresis (PAGE), Novex™ 4–20% Tris-glycine gels (Invitrogen) were loaded with 5 μg protein per lane and run to termination at 1500 V h under non-denaturing conditions in Tris-glycine buffer. The gels were stained with EZ-Run protein gel staining solution (Fisher Bioreagents). Lipoprotein size was assessed by comparison with molecular standards from the HMW Native Marker Kit (GE Healthcare Life Sciences): thryoglobin (Mw = 669 kDa), ferritin (440 kDa), catalase (232 kDa), lactate dehydrogenase (140 kDa), and albumin (66 kDa). Stokes diameters of these folded globular proteins were indicated on the gels. SEC was performed with a Superose 6 10/300 GL column controlled by an AKTA UPC 10 FPLC system (GE Healthcare). Samples were eluted in phosphate buffer saline at pH 7.5 at a flow rate of 0.5 ml/min.
2.4. Negative stain electron microscopy (EM)
For transmission EM, a 4 μl drop containing ~0.1 mg/ml protein and varying amounts of lipid in standard buffer was deposited onto an EM grid. The grids were stained with 2% uranyl acetate and blotted as previously described (Gursky et al., 2002). Electron micrographs were collected at a 45,000 magnification using a CM12 transmission electron microscope (Philips Electron Optics, the Netherlands) equipped with a Teitz 2Kx2K CCD camera (TVIPS, Germany).
Unless otherwise stated, all experiments reported in this study were repeated three to six times using three different protein batches to ensure reproducibility.
3. Results and discussion
3.1. SAA and POPC spontaneously form HDL-size nanoparticles at ambient conditions
In the first series of experiments, SAA was incubated with POPC SUV at 1:0.5–1:80 protein:lipid molar ratios at 23 °C, and the incubation mixtures were analyzed for the lipoprotein structure and stability. The particle size distribution was assessed by native PAGE. Lipid-free protein migrated as a single band with a hydrodynamic size circa 7.3 nm indicating oligomeric SAA (Fig. 1A). Upon addition of lipid, two new bands were observed whose relative populations depended strongly on the protein:lipid ratio (Fig. 1A). First, an additional band corresponding to particle sizes of 7.6–8.3 nm was observed at any protein:lipid ratios. Initially, this band progressively increased in intensity, but not in the particle size, upon addition of lipid. At 1:5 protein:lipid, the amount of free protein abruptly decreased while the hydrodynamic size in the new band slightly increased to about 8.5–11 nm, which is comparable to the size of plasma HDL (Fig. 1B); this particle size remained invariant upon further addition of lipid (Fig. 1A). Further, at 1:20 ratio another band was observed with particle sizes over 20 nm corresponding to a protein bound to SUV. Importantly, both native PAGE and EM (described below and shown in Fig. 1C) clearly showed the absence of lipid vesicles at 1:0.5 to 1:15 SAA:POPC, indicating that all lipid in these samples was incorporated into HDL-size particles.
Fig. 1.
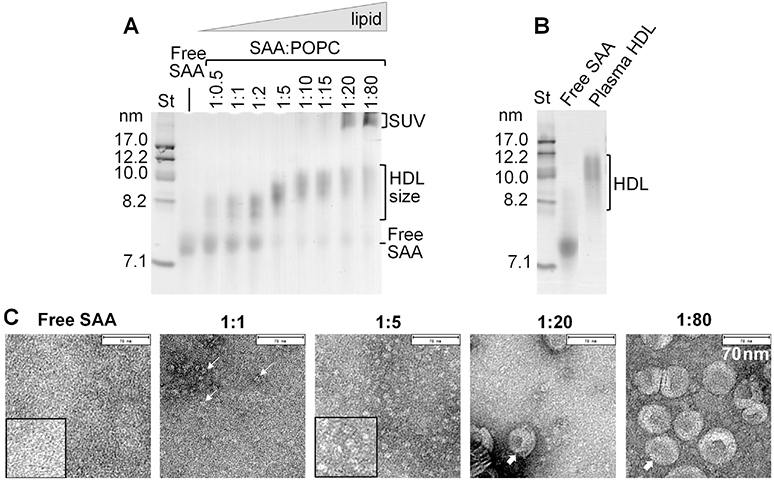
Particle size and morphology of SAA-POPC complexes reconstituted spontaneously at ambient conditions. SAA was incubated with POPC SUV at 23 °C and complete incubation mixtures were analyzed. SAA:POPC molar ratios ranged from 1:0.5 to 1:80 as indicated. Lipid-free SAA is shown for comparison. (A) Non-denaturing gel electrophoresis (E-Z Run protein stain) shows formation of SAA-POPC complexes in two distinct size ranges, 8–11 nm (HDL-size) and >22 nm (SUV-size), in addition to free SAA (~7.3 nm). (B) Non-denaturing gel electrophoresis shows free SAA and human plasma HDL. (C) Negatively stained electron micrographs of representative samples from panel A. Selected HDL-size particles and SUVs are shown by thin and thick arrows, respectively. Bar size is 70 nm. Inserts show zoomed-in portions of selected micrographs.
These results revealed that co-incubation of SAA with POPC led to SAA binding to SUVs and their spontaneous remodeling into HDL-size particles. Similar results were observed at incubation times varying from 30 min to 12 h, indicating that POPC remodeling by SAA was complete in 30 min. Unlike SAA, apoA-I (28 kDa) did not significantly remodel POPC even after 12 h incubation (Jayaraman et al., 2015), although smaller, less stable, hydrophobic apolipoproteins such as apoCs (6–9 kDa) are capable of such remodeling (data not shown).
To visualize the particles, SAA-POPC incubation mixtures were analyzed by negative stain EM; representative data are shown in Fig. 1C. EM results in Fig. 1C were in excellent agreement with native PAGE in Fig. 1A and showed no lipid vesicles at 1:0.5 to 1:15 SAA:POPC, a few collapsed vesicles at 1:20, and numerous vesicles at 1:80 SAA:POPC. Importantly, heterogeneous particles in the 8–11 nm size range were observed by both EM and the native PAGE at any protein:lipid ratios explored, but not in lipid-free SAA (Fig. 1A, C). Together, these results clearly showed a spontaneous formation of HDL-size SAA-POPC particles. To our knowledge, generation of such nanoparticles in an energy-independent process has not been previously reported, except for our own study (Jayaraman et al., 2015).
Next, the secondary structure in SAA-POPC incubation mixtures was explored by far-UV CD at two temperatures, 5 °C (where lipid-free mSAA1 was ~25% α-helical) and 25 °C (where lipid-free mSAA1 was essentially unfolded). The spectra at 25 °C clearly showed progressive lipid-induced protein folding, from the largely unstructured to a highly α-helical conformation observed at increasing lipid concentrations (Fig. 2A). Adding lipid up to 1:5 SAA:POPC caused the greatest increase in the helical structure (Fig. 2A, green to blue), with little changes observed upon further addition of lipid (purple to red). A similar lipid-induced trend was observed at 5 °C (spectra not shown). This trend was illustrated in Fig. 2B showing the α-helical content as a function of pro-tein:lipid ratio determined from the CD spectra at 5 °C (grey) and at 25 °C (black). At both temperatures the largest increase in the helical structure was observed upon addition of lipid up to 1:5 SAA:POPC (Fig. 2B). Native PAGE showed that under these conditions free SAA was converted into HDL-size particles (Fig. 1A, B). Consequently, SAA binding to lipid was accompanied by helical folding. Once most free protein has been bound to lipid, which occurred circa 1:5 SAA:POPC, further addition of lipid caused little additional helical folding (Figs. 1A and 2B compared). We conclude that lipid-bound SAA had comparable a-helical content on HDL-size particles and on SUV. In the presence of the lipid, the maximal helical content reached 50% at 5 °C and 47% at 25 °C. In comparison, lipid-free SAA was only ~10% α-helical at 25 °C (Fig. 2B). Hence, approximately one third of all SAA amino acids underwent a lipid-induced random coil to α-helix transition at 25 °C.
Fig. 2.
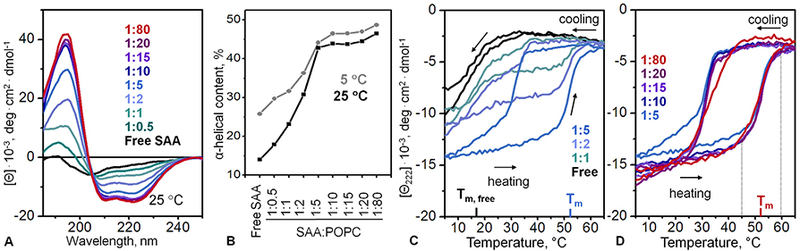
Secondary structure and thermal stability of SAA-POPC complexes obtained by spontaneous reconstitution. The data were recorded from SAA-POPC incubation mixtures prepared as described in Fig. 1. (A) Far-UV CD spectra at 25 °C. SAA:POPC molar ratios are indicated and color-coded; lipid-free SAA (black lines) is shown for comparison. The isodichroic point suggests significant population of two alternative protein conformations, α-helix and random coil. (B) Helical content at indicated SAA: POPC ratios at 25 °C (black) and 5 °C (grey), which was determined from far-UV CD spectra such as those shown in panel A. (C, D) Heating and cooling data recorded by CD spectroscopy at 222 nm to monitor α-helical unfolding and refolding. Arrows show directions of temperature changes. Melting temperatures for lipid-free SAA (Tm,free = 18 ± 1.5 °C) and POPC-bound SAA (Tm = 52.5 ± 1.4 °C) are indicated.
The structural stability of SAA-POPC complexes was explored by far-UV CD (Fig. 2C). Incubation mixtures containing various pro-tein:lipid ratios were heated and cooled at a constant rate of 60 °C/h, and helical unfolding and refolding was monitored as a function of temperature by CD at 222 nm, Θ222(T). Lipid-free SAA showed a reversible unfolding with a transition midpoint Tm=17 °C (Fig. 2C, black), in agreement with previous reports (Jayaraman et al., 2015). In contrast, at 1:5 SAA:POPC when most SAA was bound to HDL-size particles (Fig. 1A), the thermal unfolding shifted to much higher temperatures, with an apparent Tm = 52 °C, and became thermodynamically irreversible, as evident from the hysteresis in the heating and cooling data (Fig. 2C, blue).
Further, at intermediate SAA:POPC ratios of 1:0.5 to 1:2, when mixtures of free protein and HDL-size particles were observed by native PAGE (Fig. 1A), the heating data represented a weighted average of two transitions: a reversible transition with Tm = 17 °C involving free SAA and an irreversible transition with Tm = 52 °C involving SAA on HDL-size particles (Fig. 2C). At higher lipid concentrations corresponding to 1:10 to 1:80 SAA:POPC when little free protein was seen by native PAGE, only the high-temperature transition was observed by CD (Fig. 2D). These observations indicate comparable structural stability of SAA in HDL-size particles and in SUV-bound forms, which co-existed in different proportions at 1:20 and 1:80 ratios (Fig. 1A).
Taken together, the results in Figs. 1 and 2 consistently showed that, unlike lipid-free SAA that was natively unfolded at 25 °C, SAA in the presence of POPC acquired a stable helical structure that unfolded upon heating with Tm = 52 °C in a thermodynamically irreversible manner and refolded upon cooling with midpoint circa 37 °C (Fig. 2B). No significant scan rate effects on Tm were detected (data not shown) suggesting that the unfolding transition in SAA-POPC complexes did not involve high activation energy (Gursky et al., 2002). The secondary structure and thermal stability in lipid-bound SAA were similar on particles of different sizes.
3.2. Structure of lipoproteins containing 1:10 to 1:80 SAA:POPC
To further explore thermal remodeling of SAA-POPC complexes, we employed the cholate dialysis method that enabled us to obtain relatively large amounts of homogeneous lipoproteins with controlled stoichiometries. Various binary complexes of SAA and POPC as well as ternary complexes containing cholesterol were obtained by this method and used in the remainder of this study.
First, we reconstituted SAA-POPC complexes at protein:lipid molar ratios of 1:10, 1:30 or 1:80, and analyzed their size and structure (Fig. 3). Native PAGE showed that each sample ran as a broad band with the average particle size increasing with increased relative amount of lipid; a similar trend was observed in other lipoproteins (Lyssenko et al., 2013). Notably, at 1:10 SAA:POPC, the particles obtained by cholate dialysis and by spontaneous reconstitution ran very similarly on the gel, forming a broad band corresponding to 8–11 nm size (Figs. 3A and1A, 1:10 lanes). This result suggests that at 1:10 SAA:POPC ratio, HDL-size particles formed by different methods had similar stoichiometries.
Fig. 3.
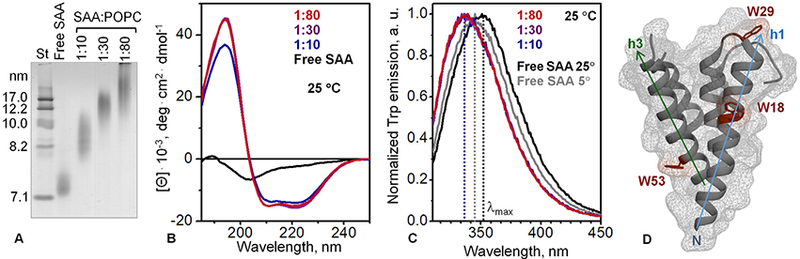
Structural properties of SAA-POPC complexes obtained by cholate dialysis method. SAA:POPC molar ratios are indicated and color-coded; lipid-free SAA is shown for comparison. (A) Non-denaturing gel electrophoresis (E-Z Run protein stain) shows that all protein was incorporated into SAA-POPC complexes whose size progressively increased with increasing lipid content. (B) Far-UV CD spectra of SAA-POPC complexes at 25 °C. (C) Intrinsic Trp fluorescence at 25 °C. Normalized emission spectra show a blue shift, with the wavelength of maximal fluorescence at 25 °C decreasing from λmax = 352 ±1.5 nm in free SAA to337±1.5 nm in SAA-POPC complexes (dotted lines). (D) X-ray crystal structure of mSAA3 (PDB ID 4Q5G) (Derebe et al., 2014); this isoform has 80% sequence similarity to mSAA1 isoform used in the current study, as assessed by Clustal W2 pairwise Needleman-Wunsch algorithm (Sievers et al., 2011). Three Trp in mSAA1, which are also present in mSAA3, are indicated. W18 and W53 are located in the hydrophobic faces of amphipathic α-helices h1 and h3 (residues 1–27 and 50–69), with W29 closely nearby. In SAA monomer these helical faces form an exposed hydrophobic surface that was proposed to bind lipids (Frame and Gursky, 2016; also shown in Fig. 8C). Face-up view of this hydrophobic surface is shown in panel D, with h1 (blue), h3 (green) and Trp (dark red) color-coded.
The structural similarity between the SAA-POPC complexes obtained by different methods was further supported by the similarity in their far-UV CD spectra. At 1:10 SAA:POPC, the a-helical content in both types of particles determined by CD was ~48%, with minor further changes at 1:80 SAA:POPC (blue and red lines in Figs. 2A and3B). The similarity in the size and secondary structure suggested that the complexes formed by cholate dialysis at 1:10 SAA:POPC ratio provided a useful structural model for spontaneously formed complexes.
Next, lipid-induced changes in the environment of the three tryptophans in mSAA1 (W18, W29 and W53) were monitored by intrinsic fluorescence at 5 °C and at 25 °C using λex = 295 nm to selectively excite Trp (Fig. 3C). At 25 °C in lipid-free SAA, which was largely unfolded, the wavelength of maximal fluorescence was λmax = 352 ± 1.5 nm, indicating solvent-exposed Trp. At 5 °C when free SAA acquired a partial a-helical conformation, a blue shift to λmax = 346 ± 1 nm was observed, indicating decreased Trp exposure to solvent (Fig. 1D). This result was consistent with the Trp locations in or near α-helices h1 and h3 seen in the X-ray crystal structure of mSAA3 (Fig. 3D), a protein that is highly homologous to mSAA1 used in our studies. Both mSAA1 and mSAA3 contain W18, W29 and W53. We conclude that partial folding of h1 and h3 helices led to partial burial of Trp at 5 °C. Compared to free SAA, SAA:POPC lipoproteins showed a 15 nm blue shift to λmax = 337 ±1.5 nm at 25 °C, indicating substantial burial of Trp upon lipid binding. The shift did not depend upon the SAA:POPC ratio in the range explored. This observation supported our idea that the apolar faces of the two amphipathic α-helices, h1 (residues 1–27) and h3 (50–69), formed an extended lipid binding site in SAA (Frame and Gursky, 2016). In fact, W18 and W53 are located in the apolar lipid-binding faces of h1 and h3, while W29 is in the nearby loop (Fig. 3D). Hence, lipid binding via these helical faces is expected to largely screen W18, W53 and perhaps W29 from the solvent, which is consistent with the observed value of λmax = 337 nm indicating largely buried Trp in lipid-bound SAA at 25 °C (Fig. 3C).
3.3. Thermal remodeling of lipoproteins containing 1:10 to 1:80 SAA: POPC
Next, we used CD to analyze thermal stability of SAA-POPC complexes containing 1:10, 1:30 or 1:80 SAA:POPC. The melting data of these complexes largely overlapped and showed a thermodynamically irreversible helical unfolding (Fig. 4A, colored lines), with the value of Tm (52.5 ±1 °C at 1:10, 53.4 ±1 °C at 1:30, and 51 ± 1.5 °C at 1:80) very similar to those observed in the incubation mixtures at these SAA:POPC ratios when all SAA spontaneously bound to lipid (Fig. 2D). Together, these results clearly showed that the secondary structural stability of lipid-bound SAA was very similar on the particles of different sizes, and was much higher than that of lipid-free SAA.
Fig. 4.
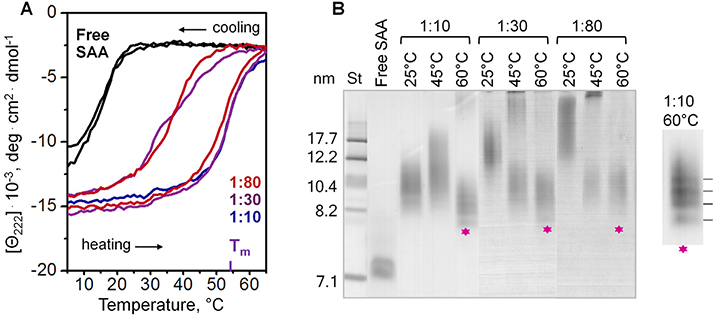
Thermal denaturation of SAA-POPC complexes obtained by cholate dialysis. SAA:POPC molar ratios are indicated; lipid-free SAA is shown for comparison. (A) Heat-induced changes in the a-helical structure of SAA monitored by CD at 222 nm. The melting data were recorded during sample heating and cooling from 5 °C to 65 °C at a rate of 60 °C/h. The melting temperature Tm of SAA-POPC complexes is indicated. (B) Non-denaturing gel electrophoresis (E-Z Run protein stain) of complexes containing various initial molar ratios of SAA:POPC (indicated), which have been incubated at 25 °C, 45 °C or 60 °C for 1 h. Asterisks indicate bands corresponding to HDL-size particles formed upon heating of larger SAA-POPC complexes to 60 °C followed by cooling. Zoomed-in view suggests that these HDL-size particles formed discrete populations with distinct stoichiometry.
To analyse thermal remodeling of these particles, the parent lipoproteins were incubated for 1 h at either 45 °C or 60 °C, i.e. close to either the onset or the completion of the helical unfolding transition, followed by native PAGE analysis (Fig. 4B). The results clearly showed that the helical unfolding was accompanied by remodeling of intact lipoproteins into larger and smaller particles. Larger particles, which were apparent products of the heat-induced lipoprotein fusion, showed a progressive increase in size upon increasing the amount of lipid, as well as upon heating to higher temperatures. Interestingly, smaller particles formed upon lipoprotein heating to 60 °C followed by cooling to ambient temperature showed very similar size of 8–11 nm (asterisks, Fig. 4B), even though the size of parent lipoproteins differed greatly for different SAA:POPC ratios. This observation suggested that, while the parent lipoproteins formed by cholate dialysis were kinetically trapped and were irreversibly remodeled upon heating and cooling into larger and smaller particles, the small HDL-size particles resulting from such remodeling represented thermodynamically stable species.
3.4. Detailed characterization of particles generated by thermal remodeling
The HDL-size particles obtained by thermal remodeling migrated on the native PAGE similarly to the parent lipoproteins prepared at 1:10 SAA:POPC (Fig. 4B). Therefore, to explore the properties of these HDL-size particles, we used 1:10 SAA:POPC ratio in the initial cholate dialysis preparation. The parent lipoproteins were subjected to thermal remodeling at 60 °C for 1 h followed by SEC analysis. The SEC profile of intact parent lipoproteins showed a single peak that eluted at 17 ml (Fig. 5A, black line). After heating, this major peak remained essentially invariant (peak I) and two additional minor peaks corresponding to larger and smaller particles were observed (peaks II and III in Fig. 5). Individual peak fractions were collected and analyzed by native PAGE and EM (Fig. 5B, C). The results clearly showed that peak I contained HDL-size particles of 8–11 nm (asterisk, Fig. 5B), which were slightly smaller and more homogeneous than the parent lipoproteins; peak II contained protein-containing vesicles ≥20 nm in size; and peak III contained few smaller particles.
Fig. 5.
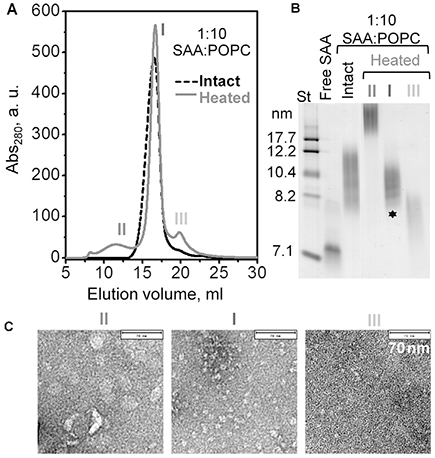
Characterization of lipoproteins obtained upon thermal remodeling of SAA-POPC complexes. Intact complexes were obtained by cholate dialysis using 1:10mol:mol SAA:POPC, and have been heated to 60 °C as described in Fig. 4. (A) SEC profile of intact (black) and heated samples (grey). Elution peaks in the heated sample are numbered. (B) Non-denaturing gel electrophoresis of intact complexes (total and isolated peak fraction) and of the peak fractions isolated from the heated sample (numbered as in panel A). Asterisk indicates HDL-size particles formed upon thermal remodeling of the parent lipoproteins. (C) Electron micrographs of negatively stained peak fractions isolated by SEC from the heated sample. Bar size is 70 nm.
The three isolated peak fractions were analyzed by CD spectroscopy for secondary structure and stability, and the results were compared with those of the parent lipoproteins (Fig. 6). Far-UV CD spectra and the heating and cooling data of peak I fraction and of the parent lipoproteins were very similar and showed Tm = 52 °C, indicating similar secondary structure and stability (black solid and dashed lines in Fig. 6A, B). Peak II fraction showed a lower helical content but a similar Tm (dark grey), while peak III fraction showed even less helical content that changed little upon heating (light grey). Notably, the results in Fig. 6 clearly showed that HDL-size particles formed upon thermal remodeling had similar helical content and thermal stability as their parent lipoproteins.
Fig. 6.
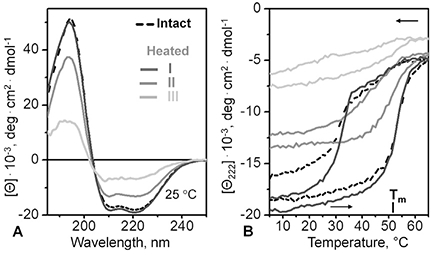
Secondary structure and thermal stability of SAA-POPC complexes from isolated peak fractions. Parent complexes, which were prepared by cholate dialysis using 1:10 mol:mol SAA:POPC, have been incubated at 60 °C for 1 h, and peak fractions I-III were isolated by SEC as described in Fig. 4. Data of intact parent SAA-POPC complexes are shown for comparison (dashed lines). (A) Far-UV CD spectra at 25 °C of isolated peak fractions I (dark grey), II (grey) and III (light grey). (B) CD melting data at 222 nm recorded during heating and cooling at a constant rate. The melting temperature Tm for peak fraction I is indicated; intact SAA-POPC complexes have a similar Tm.
Next, the biochemical composition of isolated peak fractions I, II and III was determined. Intact parent lipoproteins were used as a control; as expected, the SAA:POPC molar ratio measured in these parent lipoproteins was 1:10 (Table 1). Importantly, biochemical analysis of the major peak I revealed a 1:6 SAA:POPC molar ratio. This result was consistent with the native PAGE showing slightly smaller particle size (and hence, slightly smaller lipid content) in peak I fraction (Figs. 4B, 5B) as compared to parent lipoproteins. Furthermore, analysis of large particles in the peak II fraction showed a 1:25 SAA:POPC ratio, which was consistent with the protein-containing lipid vesicles observed by native PAGE and EM (Fig. 5B, C). In contrast, smaller particles in peak III showed 1:1 SAA:POPC molar ratio, which was insufficient to form a substantial population of stable SAA-POPC complexes, as suggested by both the CD (light grey lines, Fig. 6A, B) and the EM data of peak III (Fig. 5C).
Table 1.
Biochemical composition, size and secondary structure of SAA-POPC complexes obtained by thermal remodeling. Parent lipoproteins, which were prepared by cholate dialysis using 1:10 mol:mol SAA:POPC, were incubated at 60 °C for 1 h. Peak fractions I, II and III were isolated by SEC and analyzed as described in Figs. 5 and 6. Protein and lipid content in each fraction was determined as described in Methods. The biochemical analysis was repeated twice using two independent lipoprotein preparations, and the results agreed within better than 10%. Hydrodynamic size range of lipoproteins was determined by non-denaturing gel electrophoresis and confirmed by negative-stain EM (Fig. 5). Helical content was determined from far-UV CD spectra at 25 °C (Fig. 6A) based on the molar residue ellipticity at 222 nm (Mao and Wallace, 1984) with ±5% accuracy. These results were used to estimate the stoichiometry in the HDL-size particles (peak I). Since SAA in these particles is ~50% folded, the unfolded region is expected to increase the apparent Stokes diameter, leading to a potential overestimate in the particle size. This effect is probably not large since the particle size assessed from the native PAGE agrees with that observed by EM (Figs. 1, 5). Further, the biochemical analysis provides an average protein:lipid ratio that may not represent any particular sub-population of particles from the discrete bands seen on the native PAGE (Figs. 4B, 5B). With these caveats, the measured size and stoichiometry of the HDL-size particles suggest that, on average, each particle contains about 12 SAA and 72 POPC molecules.
| Lipoprotein sample | SAA:POPC, mol:mol | Size, nm | α-helical content |
|---|---|---|---|
| Intact | 1:10 | 8–12 | 53% |
| Heated | |||
| peak I | 1:6 | 8–11 | 55% |
| peak II | 1:25 | >20 | 25% |
| peak III | 1:1 | 7.5–8.5 | 41% |
Taken together, our results revealed that very similar 8–11 nm HDL-size complexes formed at various initial protein:lipid ratios either upon spontaneous reconstitution at ambient temperatures (Figs. 1, 2) or upon thermal remodeling of parent lipoproteins (Figs. 4 and 5, asterisks). Clearly, these HDL-size complexes represent thermodynamically stable species. The average stoichiometry of these complexes was 1:6 mol:mol SAA:POPC.
3.5. Effects of cholesterol on SAA-POPC complexes
Next, we tested whether similar SAA-containing complexes could form in the presence of cholesterol, an essential component of cell membranes and plasma HDL. To mimic cholesterol content in plasma HDL, we reconstituted ternary complexes containing either 1:30:2 or 1:80:4 SAA:POPC:cholesterol molar ratios as previously described (Cavigiolio et al., 2008), and compared them with binary complexes containing either 1:30 or 1:80 mol:mol SAA: POPC. Such complexes with POPC and cholesterol have been previously explored for other apolipoproteins but not for SAA. Native PAGE showed that incorporation of cholesterol led to a small but significant increase in the particle size (Fig. 7A). Far-UV CD spectra showed a small but significant decrease in amplitude suggesting a ~6% decrease in the a-helical content upon cholesterol incorporation (Fig. 7B). Intrinsic Trp fluorescence showed no significant spectral changes upon cholesterol incorporation (data not shown). Far-UV CD melting data also did not show any major changes upon cholesterol incorporation (Fig. 7C). These trends were consistent with the effects of cholesterol on the structure and stability of other apolipoprotein-PC complexes (Jayaraman et al., 2010). Furthermore, cholesterol did not affect the hysteresis in the melting data, suggesting similar thermal remodeling of cholesterol-containing and cholesterol-free particles (Fig. 7C).
Fig. 7.
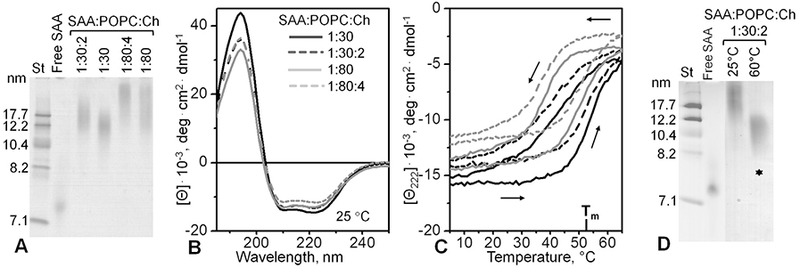
Effects of cholesterol on the overall structure and stability of SAA:POPC complexes. The complexes were prepared by cholate dialysis method using SAA:POPC: Cholesterol molar ratios of 1:30:2 or 1:80:4 as indicated. Cholesterol-free complexes containing 1:30 or 1:80mol:mol SAA:POPC are shown for comparison. These experiments were repeated twice using two independent sets of lipoprotein preparations. (A) Native PAGE of intact complexes (E-Z Run protein stain); lipid-free SAA is shown for comparison. (B) Far-UV CD spectra of intact complexes at 25 °C. (C) CD heating and cooling data of the complexes. Line coding is as in panel B. Arrows show directions of temperature changes. (D) Native PAGE of SAA:POPC and SAA:POPC:cholesterol complexes that have been either unheated (25 °C) or have been heated up to 60 °C. Asterisks indicate HDL-size particles formed upon thermal denaturation of intact lipoproteins.
This conclusion was confirmed by native PAGE of cholesterol-containing particles before and after incubation at 60 °C for 1 h (Fig. 7D). Intact lipoproteins containing 1:30:2 SAA:POPC:choles-terol were converted upon thermal remodeling into smaller particles of 8–11 nm (asterisks, Fig. 7D) and larger particles that did not enter the gel. Notably, the 8–11 nm particles migrated very similarly to their cholesterol-free counterparts (asterisks in Figs. 4B, 5B, and 7D). Consequently, such HDL-size particles could form not only in the binary SAA-POPC systems but also in the ternary systems that contained cholesterol. This result lends additional physiological relevance to our finding that SAA can form stable HDL-size nanoparticles in a wide range of conditions.
3.6. The structure of SAA-containing HDL-size particles is distinct from other lipoproteins
Despite their HDL-like size, the stoichiometry of the stable SAA-POPC complexes was different from HDL. The 1:6 SAA:POPC molar ratio determined from the biochemical analysis of these particles (peak I fraction in Fig. 5) corresponds to a 2.5:1 mg/mg protein: lipid ratio. In comparison, normal plasma HDL contain comparable amounts of protein and lipid by weight, while larger low-and very-low density lipoproteins contain much higher relative amounts of lipid. Consequently, the HDL-size particles in peak I are enriched in protein and depleted in lipid by more than twofold as compared to HDL or any other major class of plasma lipoproteins. Since SAA in these particles is approximately 50% folded, the unfolded region is expected to alter the electrophoretic mobility and lead to a potential overestimate in the particle size. Further, discrete bands seen on the native PAGE suggest different numbers of SAA copies per particle (Figs. 4B, 5B). With these caveats, the stoichiometry of the HDL-size particles suggests that each particle contains on average about 12 SAA and 72 POPC molecules, for the average molecular weight circa 200 kDa per particle.
In comparison, the smallest HDL-like particle reconstituted from apoA-I and POPC contains two apoA-I molecules that encircle 160 or more POPC molecules in a bilayer conformation to form a discoidal assembly (Fig. 8A). Such small bilayer-containing “nanodisks” are accepted models of nascent HDL (Phillips, 2013; Bibow et al., 2017), although other models have also been proposed (Sorci-Thomas et al., 2012).
Fig. 8.
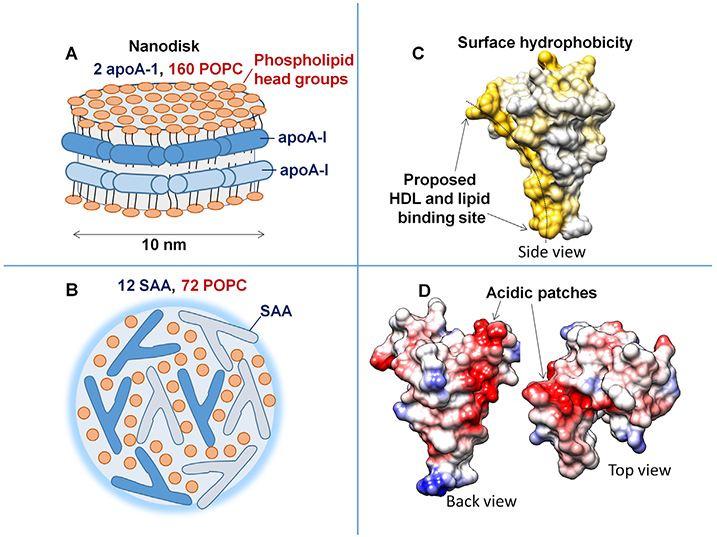
Key structural features of model lipoproteins and of SAA monomer. Cartoons illustrate the overall assembly of an apoA-I - POPC “nanodisk”, which represents nascent ‘‘discoidal” HDL (A), and of an HDL-size SAA - POPC complex uncovered in the current study (B). Phospholipid head groups are in dark red. Individual protein molecules are color-coded in dark and light blue. X-ray crystal structure of hSAAl (PDB ID: 4IP8) illustrates surface hydrophobicity (C) and Coulombic surface potential (D) in human SAA1 monomer. The figures in C, D were prepared using molecular graphics software Chimera. The concave hydrophobic surface (yellow, C), whose curvature is optimal for HDL binding, is proposed to form the lipid surface binding site (Frame and Gursky, 2016). Electronegative patches (red, D) on other faces are formed mainly by the largely conserved acidic residues from SAA helices h4 and h2. We propose that these acidic patches probably bind cell receptors such as CD36, SR-BI, LOX-1 or RAGE, because the basic apexes in these receptors form the ligand-binding sites (Ryeom et al., 1996; Chen et al., 2001; Jimenez-Dalmaroni et al., 2009; Koch et al., 2010; Neculai et al., 2013; Thakkar et al., 2015).
Compared to nascent HDL, SAA-POPC complexes revealed in this study contain less than one half of lipid molecules per particle, and at a 1:6 stoichiometry most of these lipids are expected to directly interact with the protein. Hence, these lipids are expected to be distorted from the bilayer conformation. This conclusion is consistent with the EM data showing irregularly shaped HDL-size particles face up (Figs. 1C, 5C).
Although the structure of the HDL-size SAA-POPC complexes uncovered in the current study remains to be determined, these complexes must contain a polar surface and an apolar interior like other lipoproteins. However, unlike nascent HDL, lipids in these complexes are unlikely to form a bilayer. We hypothesize that the surface of SAA-POPC complexes is comprised of phospholipid head groups, polar faces of helices h1 and h3 (which are probably folded on the lipid), and the rest of the SAA molecule (which is probably largely unfolded, accounting for the total α-helical content of ~50% observed by CD (Fig. 6, Table 1)). The apolar interior is expected to contain lipid acyl chains that interact with the apolar faces of the amphipathic α-helices h1 and h3; the latter is sup-ported by the Trp sequestration upon SAA binding to lipid observed by fluorescence (Fig. 3C, D). The cartoon in Fig. 8B illustrates a hypothetical SAA-POPC assembly that satisfies these requirements.
Such 8–11 nm SAA-POPC complexes can also be compared to lipid-poor apoA-I. Like SAA, most circulating apoA-I is bound to plasma HDL, yet it can be transiently released in a metabolically active lipid-poor state that contains one protein and several lipid molecules. Such lipid-poor apoA-I can be generated in vitro upon thermal remodeling of various HDL (Jayaraman et al., 2012). Compared with lipid-poor apoA-I, which is a ~35 kDa monomer, HDL-size SAA-POPC complexes of the current study are larger (~200 kDa) and contain multiple protein molecules. We conclude that such SAA-POPC complexes are distinctlydifferent from any other major classes of lipoproteins, including HDL and lipid-poor apoA-I.
3.7. Potential relevance of SAA structure to its function in lipid clearance
Previous cell-based studies reported de novo formation of SAA-containing HDL upon phospholipid and cholesterol efflux from plasma membranes of various cells to SAA (van der Westhuyzen et al., 2007; Abe-Dohmae et al., 2006; Hu et al., 2008; de Beer et al., 2011). This energy-dependent process was facilitated by ABC lipid transporters such as ABCA1 that perturbs the packing in the outer leaflet of the plasma membrane and thereby helps solubilize lipids ((Qian et al., 2017) and references therein). One team reported transporter-independent efflux of phospholipids and cholesterol from cells to SAA and suggested that this effect reflected the unique role of SAA in lipid removal from the inflammation sites (Stonik et al., 2004).
All these studies used healthy cells; however, upregulation of SAA in inflammation is triggered by cell injury and death. Since ABC transporters are not functional in dead cells, apoA-I cannot serve as their lipid acceptor; hence, the role of SAA as a lipid acceptor becomes particularly important. Therefore, we posit that the ability of SAA to spontaneously extract lipids from bilayers and repack them into nanoparticles is relevant to membrane clearance of dead cells, which is essential for tissue repair.
We hypothesize that SAA represents the first line of defense in clearing lipids from dead cells, which helps explain its dramatic upregulation in acute inflammation and injury. This idea is in accord with previous studies showing that SAA is expressed in a wide range of normal human tissues where it was proposed to play a house-keeping role as the first line of defense, possibly protecting from formation of reactive lipid species by sequestering membranes upon cell injury (Urieli-Shoval et al., 1998, 2000). Similarly, group IIA phospholipase A2, a small acute-phase reactant that is upregulated concomitantly with SAA, is implicated in rapid clearance of phospholipid debris (Birts et al., 2008), and may possibly act in synergy with SAA in rapid removal of membrane debris from injured sites.
We propose that the unique 3D structure of SAA monomer, with its exposed concave hydrophobic surface formed by h1 and h3 (Fig. 8C), explains how this small protein can provide the first line of defense against cell injury. We posit that several SAA molecules together spontaneously solubilize membranes by binding and encapsulating lipids via their curved hydrophobic surface to form nanoparticles such as those observed in the current study. Further, we hypothesize that the acidic patches observed in the crystal structure of hSAA1 (Fig. 8D), which are formed by largely conserved residues in h4 and h2, may undergo coupled folding and binding in solution and are likely candidates for binding to cell receptors. These acidic patches probably direct SAA-containing particles to receptors such as CD36, SR-BI, LOX-1 or RAGE, which are proposed to bind their ligands via the basic surface sites (Ryeom et al., 1996; Chen et al., 2001; Jimenez-Dalmaroni et al., 2009; Koch et al., 2010; Neculai et al., 2013; Thakkar et al., 2015). These receptors, which are expressed on the surfaces of various cells including macrophages, mediate internalization of the SAA-containing nanoparticles by these cells and thereby augment lipid clearance, sequestration and recycling for tissue repair. We speculate that this housekeeping function of SAA likely pre-dates its binding to HDL, and may represent a key primordial function of this ancient protein.
4. Conclusions
This study explores interactions between SAA, an acute-phase reactant that circulates mainly on plasma HDL in mice and men, and POPC, a model lipid that mimics the primary constituents of cell membranes and HDL. The key new finding is spontaneous formation of SAA-POPC complexes that are 8–11 nm in size, have up to 50% α-helical content, and are stable at near-physiological conditions but undergo a thermodynamically irreversible thermal remodeling at Tm = 52 °C, much higher than that in lipid-free SAA. Such SAA-POPC complexes can form either upon SAA incubation with POPC at ambient conditions (Fig. 1A) or upon thermal remodeling of parent lipoproteins containing various amounts of SAA and POPC(Fig. 4B). Similar complexes form upon thermal remodeling of parent lipoproteins containing cholesterol (Fig. 7D). Formation of SAA-containing HDL-size complexes observed in a wide range of temperatures, initial protein:lipid ratios, and lipid compositions suggests that these complexes form stable kinetically accessible structures. Such structures are expected to form in a wide range of conditions in vitro and, potentially, in vivo, perhaps contributing to the primordial function of SAA in rapid clearance of cell debris from the sites of injury. Moreover, relatively high stability of the SAA-lipid complexes helps protect this intrinsically disordered protein from rapid degradation and misfolding in AA amyloidosis.
Acknowledgements
We are indebted to Dr. Christian Haupt for providing us with ultrapure murine SAA1.1. In this inaugural issue, OG would like to thank her first teacher of structural biology, Dr. Donald L.D. Caspar, who trained her not only how to do structural biology but also how to think and write about it.
Funding
This work was supported by the National Institutes of Health grants RO1 GM067260 and T32 HL007969, and by the Stevens Family Amyloid Endowment Fund.
Abbreviations:
- SAA
serum amyloid A
- mSAA
murine serum amyloid A
- hSAA
human serum amyloid A
- AA
amyloid A
- ABC
ATP-binding cassette
- HDL
high-density lipoprotein
- apoA-I
apolipoprotein A-I
- PC
phosphatidylcholine
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- SUV
small unilamellar vesicles
- CD
circular dichroism
- EM
electron microscopy
- PAGE
polyacrylamide gel electrophoresis
- SEC
size-exclusion chromatography
Footnotes
This Special Issue was edited by Piotr Fajer, Alexei S. Soares and Kenneth Taylor and represents a Festschrift honoring Donald L. D. Caspar on the occasion of his 90th birthday, based on a meeting held at Florida State University in January 2017.
References
- Abe-Dohmae S, Kato KH, Kumon Y, Hu W, Ishigami H, Iwamoto N, Okazaki M, Wu CA, Tsujita M, Ueda K, Yokoyama S, 2006. Serum amyloid A generates high density lipoprotein with cellular lipid in an ABCA1- or ABCA7- dependent manner. J. Lipid Res 47,1542–1550. [DOI] [PubMed] [Google Scholar]
- Benditt EP, Eriksen N, 1977. Amyloid protein SAA is associated with high density lipoprotein from human serum. Proc. Natl. Acad. Sci. U.S.A 74, 4025–4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmeyer HU, 1985. Metabolites 3: lipids, amino acids and related compounds In: Bergmeyer J, Grassl M (Eds.), Methods of Enzymatic Analysis, vol. VIII VCH; Weinheim, Germany. [Google Scholar]
- Bibow S, Polyhach Y, Eichmann C, Chi CN, Kowal J, Albiez S, McLeod RA, Stahlberg H, Jeschke G, Güntert P, Riek R, 2017. Solution structure of discoidal high-density lipoprotein particles with a shortened apolipoprotein A-I. Nat. Struct. Mol. Biol 24 (2), 187–193. [DOI] [PubMed] [Google Scholar]
- Birts CN, Barton CH, Wilton DC, 2008. A catalytically independent physiological function for human acute phase protein group IIA phospholipase A2: cellular uptake facilitates cell debris removal. J. Biol. Chem 283 (8), 5034–5045. [DOI] [PubMed] [Google Scholar]
- Cabana VG, Reardon CA, Wei B, Lukens JR, Getz GS, 1999. SAA-only HDL formed during the acute phase response in apoA-I+/+ and apoA-I−/− mice. J. Lipid Res 40 (6), 1090–1103. [PubMed] [Google Scholar]
- Cabana VG, Feng N, Reardon CA, Lukens J, Webb NR, de Beer FC, Getz GS, 2004. Influence of apoA-I and apoE on the formation of serum amyloid A-containing lipoproteins in vivo and in vitro. J. Lipid Res 45 (2), 317–325. [DOI] [PubMed] [Google Scholar]
- Cavigiolio G, Shao B, Geier EG, Ren G, Heinecke JW, Oda MN, 2008. The interplay between size, morphology, stability, and functionality of high-density lipoprotein subclasses. Biochemistry 47 (16), 4770–4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Inoue K, Narumiya S, Masaki T, Sawamura T, 2001. Requirements of basic amino acid residues within the lectin-like domain of LOX-1 for the binding of oxidized low-density lipoprotein. FEBS Lett. 499 (3), 215–219. [DOI] [PubMed] [Google Scholar]
- Coetzee GA, Strachan AF, van der Westhuyzen DR, Hoppe HC, Jeenah MS, de Beer FC, 1986. Serum amyloid A-containing human high density lipoprotein 3. Density, size, and apolipoprotein composition. J. Biol. Chem 261, 9644–9651. [PubMed] [Google Scholar]
- Colón W, Aguilera JJ, Srinivasan S, 2015. Intrinsic stability, oligomerization, and amyloidogenicityofHDL-free serum amyloidA.Adv. Exp. Med. Biol 855,117–134. [DOI] [PubMed] [Google Scholar]
- de Beer MC, Ji A, Jahangiri A, Vaughan AM, de Beer FC, van der Westhuyzen DR, Webb NR, 2011. ATP binding cassette G1 dependent cholesterol efflux during inflammation. J. Lipid Res 52 (2), 345–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derebe MG, Zlatkov CM, Gattu S, Ruhn KA, Vaishnava S, Diehl GE, MacMillan JB, Williams NS, Hooper LV, 2014. Serum amyloid A is a retinol binding protein that transports retinol during bacterial infection. eLife 3, e03206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Z, Wu T, Qin W, An C, Wang Z, Zhang M, Zhang Y, Zhang C, An F, 2011. Serum amyloid A directly accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Mol. Med 17 (11–12), 1357–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eklund KK, Niemi K, Kovanen PT, 2012. Immune functions of serum amyloid A. Crit. Rev. Immunol 32 (4), 335–348. [DOI] [PubMed] [Google Scholar]
- Frame NM, Gursky O, 2016. Structure of serum amyloid A suggests a mechanism for selective lipoprotein binding and functions: SAA as a hub in macromolecular interaction networks. FEBS Lett. 590 (6), 866–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getz GS, Reardon CA, 2008. SAA, HDL biogenesis, and inflammation. J. Lipid Res 49, 269–270. [DOI] [PubMed] [Google Scholar]
- Gursky O, 2015. Structural stability and functional remodeling of high-density lipoproteins. FEBS Lett. 589 (19 Pt A), 2627–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gursky O, Ranjana, Gantz DL, 2002. Complex of human apolipoprotein C-1 with phospholipid: thermodynamic orkinetic stability? Biochemistry 41 (23), 7373–7384. [DOI] [PubMed] [Google Scholar]
- Hu W, Abe-Dohmae S, Tsujita M, Iwamoto N, Ogikubo O, Otsuka T, Kumon Y, Yokoyama S, 2008. Biogenesis of HDL by SAA is dependent on ABCA1 in the liver in vivo. J. Lipid Res 49, 386–393. [DOI] [PubMed] [Google Scholar]
- Jayaraman S, Gantz DL, Gursky O, 2006. Effects of salt on the thermal stability of human plasma high-density lipoprotein. Biochemistry 45, 4620–4628. [DOI] [PubMed] [Google Scholar]
- Jayaraman S, Benjwal S, Gantz DL, Gursky O, 2010. Effects of cholesterol on thermal stability of discoidal high-density lipoproteins.J. Lipid Res 51 (2), 324–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman S, Cavigiolio G, Gursky O, 2012. Folded functional lipid-poor apolipoprotein A-I obtained by heating of high-density lipoproteins: relevance to high-density lipoprotein biogenesis. Biochem. J 442 (3), 703–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman S, Haupt C, Gursky O, 2015. Thermal transitions in serum amyloid A in solution and on the lipid: implications for structure and stability of acute-phase HDL. J. Lipid Res 56 (8), 1531–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Dalmaroni MJ, Xiao N, Corper AL, Verdino P, Ainge GD, Larsen DS, Painter GF, Rudd PM, Dwek RA, Hoebe K, Beutler B, Wilson IA, 2009. Soluble CD36 ectodomain binds negatively charged diacylglycerol ligands and acts as a co-receptor for TLR2. PLoS One 4, e7411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas A, 1986. Reconstitution of high-density lipoproteins. Methods Enzymol. 128, 553–582. [DOI] [PubMed] [Google Scholar]
- Kisilevsky R, Manley PN, 2012. Acute-phase serum amyloid A: perspectives on its physiological and pathological roles. Amyloid 19, 5–14. [DOI] [PubMed] [Google Scholar]
- Koch M, Chitayat S, Dattilo BM, Schiefner A, Diez J, Chazin WJ, Fritz G, 2010. Structural basis for ligand recognition and activation ofRAGE. Structure 18(10), 1342–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollmer M, Meinhardt K, Haupt C, Liberta F, Wulff M, Linder J, Handl L, Heinrich L, Loos C, Schmidt M, Syrovets T, Simmet T, Westermark P, Westermark GT, Horn U, Schmidt V, Walther P, Fändrich M, 2016. Electron tomography reveals the fibril structure and lipid interactions in amyloid deposits. Proc. Natl. Acad. Sci. U.S.A 113 (20), 5604–5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Yu Y, Zhu I, Cheng Y, Sun PD, 2014. Structural mechanism of serum amyloid A-mediated inflammatory amyloidosis. Proc. Natl. Acad. Sci. U.S.A 111 (14), 5189–5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyssenko NN, Nickel M, Tang C, Phillips MC, 2013. Factors controlling nascent high-density lipoprotein particle heterogeneity:ATP-binding cassette transporter A1 activity and cell lipid and apolipoprotein AI availability. FASEB J. 27 (7), 2880–2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao D, Wallace BA, 1984. Differential light scattering and absorption flattening optical effects are minimal in the circular dichroism spectra of small unilamellar vesicles. Biochemistry 23 (12), 2667–2673. [DOI] [PubMed] [Google Scholar]
- Markwell MA, Haas SM, Bieber LL, Tolbert NE, 1978. A modification of the Lowry procedure to simplify protein determination in membrane and lipoprotein samples. Anal. Biochem 87 (1), 206–210. [DOI] [PubMed] [Google Scholar]
- McLean LR, Phillips MC, 1984. Cholesterol transfer from small and large unilamellar vesicles. Biochim. Biophys. Acta 776 (1), 21–26. [DOI] [PubMed] [Google Scholar]
- Neculai D, Schwake M, Ravichandran M, Zunke F, Collins RF, Peters J, Neculai M, Plumb J, Loppnau P, Pizarro JC, Seitova A, Trimble WS, Saftig P, Grinstein S, Dhe-Paganon S, 2013. Structure of LIMP-2 provides functional insights with implications for SR-BI and CD36. Nature 504 (7478), 172–176. [DOI] [PubMed] [Google Scholar]
- Perrone L, Peluso G, Melone MA, 2008. RAGE recycles at the plasma membrane in S100B secretory vesicles and promotes Schwann cells morphological changes. J. Cell. Physiol 217 (1), 60–71. [DOI] [PubMed] [Google Scholar]
- Phillips MC, 2013. New insights into the determination of HDL structure by apolipoproteins: thematic review series: high density lipoprotein structure, function, and metabolism. J. Lipid Res 54 (8), 2034–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian H, Zhao X, Cao P, Lei J, Yan N, Gong X, 2017. Structure of the human lipid exporter ABCA1. Cell 169, 1–12. [DOI] [PubMed] [Google Scholar]
- Ryeom SW, Silverstein RL, Scotto A, Sparrow JR, 1996. Binding of anionic phospholipids to retinal pigment epithelium may be mediated by the scavenger receptor CD36. J. Biol. Chem 271, 20536–20539. [DOI] [PubMed] [Google Scholar]
- Segrest JP, Jones MK, De Loof H, Brouillette CG, Venkatachalapathi YV, Anantharamaiah GM, 1992. The amphipathic helix in the exchangeable apolipoproteins: a review of secondary structure and function. J. Lipid Res 33 (2), 141–166. [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen DG, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins D, 2011. Fast, scalable generation ofhigh-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorci-Thomas MG, Owen JS, Fulp B, Bhat S, Zhu X, Parks JS, Shah D,Jerome WG, Gerelus M, Zabalawi M, Thomas MJ, 2012. Nascent high density lipoproteins formed by ABCA1 resemble lipid rafts and are structurally organized by three apoA-I monomers. J. Lipid Res 53 (9), 1890–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stonik JA, Remaley AT, Demosky SJ, Neufeld EB, Bocharov A, Brewer HB, 2004. Serum amyloid A promotes ABCA1-dependent and ABCA1-independent lipid efflux from cells. Biochem. Biophys. Res. Commun 321, 936–941. [DOI] [PubMed] [Google Scholar]
- Takase H, Furuchi H, Tanaka M, Yamada T, Matoba K, Iwasaki K, Kawakami T, Mukai T, 2014. Characterization of reconstituted high-density lipoprotein particles formed by lipid interactions with human serum amyloid A. Biochim. Biophys. Acta 1842, 1467–1474. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Nishimura A, Takeshita H, Takase H, Yamada T, Mukai T, 2017. Effect of lipid environment on amyloid fibril formation of human serum amyloid A. Chem. Phys. Lipids 202, 6–12. [DOI] [PubMed] [Google Scholar]
- Thakkar S, Wang X, Khaidakov M, Dai Y, Gokulan K, Mehta JL, Varughese KI, 2015. Structure-based design targeted at LOX-1, a receptor for oxidized low-density lipoprotein. Sci. Rep 5, 16740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JC, Jayne C, Thompson J, Wilson PG, Yoder MH, Webb N, Tannock LR, 2015. A brief elevation of serum amyloid A is sufficient to increase atherosclerosis. J. Lipid Res 56, 286–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlar CM, Whitehead AS, 1999. Serum amyloid A, the major vertebrate acute-phase reactant. Eur. J. Biochem 265, 501–523. [DOI] [PubMed] [Google Scholar]
- Urieli-Shoval S, Cohen P, Eisenberg S, Matzner Y, 1998. Widespread expression of serum amyloid A in histologically normal human tissues. Predominant localization to the epithelium. J. Histochem. Cytochem 46 (12), 1377–1384. [DOI] [PubMed] [Google Scholar]
- Urieli-Shoval S, Linke RP, Matzner Y, 2000. Expression and function of serum amyloid A, a major acute-phase protein, in normal and disease states. Curr. Opin. Hematol 7 (1), 64–79. [DOI] [PubMed] [Google Scholar]
- van der Westhuyzen DR, Cai L, de Beer MC, de Beer FC, 2005. Serum amyloid A promotes cholesterol efflux mediated by scavenger receptor B-I. J. Biol. Chem 280, 35890–35895. [DOI] [PubMed] [Google Scholar]
- van der Westhuyzen DR, de Beer FC, Webb NR, 2007. HDL cholesterol transport during inflammation. Curr. Opin. Lipidol 18 (2), 147–151. [DOI] [PubMed] [Google Scholar]
- Wang L, Colón W, 2004. The interaction between apolipoprotein serum amyloid A and high-density lipoprotein. Biochem. Biophys. Res. Commun 317 (1), 157–161. [DOI] [PubMed] [Google Scholar]
- Westermark GT, Fändrich M, Westermark P, 2015. AA amyloidosis: pathogenesis and targeted therapy. Annu. Rev. Pathol 10, 321–344. [DOI] [PubMed] [Google Scholar]
- Ye RD, Sun L, 2015. Emerging functions of serum amyloid A in inflammation. J. Leukoc. Biol 98 (6), 923–929. [DOI] [PMC free article] [PubMed] [Google Scholar]