

Abstract
In recent years the ubiquitin proteasome system (UPS) has garnered increasing interest as a target for chemotherapeutics. Due to the success of the proteasome inhibitors Bortezomib and Carfilzomib in the treatment of multiple myeloma, several new compounds have been developed to target E3 ubiquitin ligases and the proteasome in numerous human cancers. This has increased the need for new analytical methods to precisely measure intracellular enzyme activity in cells. A key component of a desired analytical method is a substrate that is capable of rapid intracellular ubiquitination yet easily incorporated into the next generation of more sophisticated UPS reporters. Portable degradation sequences, or degrons, have the ability to bind to E3 ligases and promote substrate ubiquitination when the sequence is presented in isolation or appended to other entities such as fluorescent peptide-based reporters. Previous work identified an E3 ligase (MDM2)-binding element at p53 amino acids 92-112, which was later demonstrated to be rapidly ubiquitinated in cytosolic lysates effectively functioning as a transportable degron. In this work, a shortened p53 sequence within amino acids 92-112 that displayed rapid ubiquitination kinetics was identified. A nine-member peptide library was synthesized using sequence elements of various sizes and lengths, all based on the initial 22 amino acid long sequence, containing a single ubiquitination site lysine. The ubiquitination kinetics were determined using a combination of gel electrophoresis and analytical high performance liquid chromatography (HPLC) to rank the members of the library and identify the optimal ubiquitination sequence. This analysis identified the five amino acid sequence, KGSYG, corresponding to residues 105-108 with an added N-terminal lysine, as a portable degron since this sequence demonstrated the most rapid ubiquitination kinetics.
Introduction
Protein ubiquitination is the primary cellular pathway responsible for the recognition and degradation of misfolded or damaged proteins1, 2. This is accomplished by a three-step enzymatic cascade consisting of E1 ubiquitin activating enzymes, E2 ubiquitin conjugating enzymes, and E3 ubiquitin ligases.3 The E3 ligases are the most abundant and diverse class of enzymes permitting precise identification and ubiquitination of target proteins through the recognition of specific degradation sequences or degrons.1, 4 After the E3 ligase binds to the degron, its ubiquitin cargo is transferred to a proximal lysine residue forming an isopeptide bond between the C-terminal carboxylate of ubiquitin and the ε-amino group of the lysine resulting in protein ubiquitin. Additional ubiquitin subunits are subsequently added to one of several possible lysines on this anchor ubiquitin to form a polyubiquitin chain in a process called polyubiquitination. The site on which the ubiquitin subunits bind in the chain often dictates the fate of the protein. For example, K48-linked polyubiquitin (polyUb) chains are normally targeted to the 26S proteasome for degradation while K63-linked chains play a role in further signal transduction cascades such as DNA damage repair.4 This process can be reversed by deubiquitinating enzymes (DUBs) which facilitate the removal of the polyUb chain to rescue the protein. Due to this precise control over intracellular protein levels, members of the ubiquitin proteasome system (UPS) have become attractive targets for molecularly-targeted therapeutics. Examples include the proteasome inhibitors Bortezomib and Carfilzomib, which have shown great success in the treatment of patients suffering from multiple myeloma.5 Based on this clinical success, new inhibitors are being developed to target E3 ligase, DUBs, and the proteasome in a number of human cancers.2
The onset of these molecularly-targeted therapeutics towards the UPS has created a significant need for new tools with the potential to act as subcomponents for reporting systems to precisely quantify intracellular enzyme activity in response to these inhibitors. Traditional methods of measuring enzyme activity include western blotting and ELISA, both of which rely on the analysis of bulk cell lysates. While effective for preliminary discovery, all heterogeneity amongst cells is lost since these methods report a population-averaged result. Thus valuable information about distinct subpopulations, such as clonal subgroups, is unavailable. To overcome this limitation, fluorescent peptide-based reporters have been coupled with single cell analytical methods such as capillary electrophoresis and microfluidic separation to provide a precise analytical readout of intracellular enzyme activity.6 Peptide reporters can be rapidly synthesized, easily modified, and do not require complex manipulations for incorporation into single, living cells. Previous work utilized peptide-based reporters to monitor peptidase, kinase7 and phosphatase8 activity in single cancer cells. One potential drawback with peptide-based reporters is the rapid degradation by intracellular peptidases and proteases. To overcome this limitation, investigators have coupled peptide-based substrates with β-hairpin ‘protectides’ to confer extended stability and lifetime to the peptide-based substrate.9, 10 However, in order for the protectide to be effective it needs to be conjugated to a substrate of minimal effective length to maximize the protective effect while still remaining an efficient substrate. For E3 ligase measurements, this has led to the search for portable degrons that can be transferred to reporters to produce substrates for E3 ligases and ultimately the proteasome. Recent work has demonstrated that incorporating these degradation signals into native proteins results in highly effective proteasomal degradation.11, 12 While several degrons have been identified by researchers, the ability of each sequence to serve as a portable degron varies greatly depending on the ubiquitination kinetics, the number of amino acids (e.g., size and length), and need for further post-translational modification (e.g., phosphorylation).13 A broader suite of portable degrons would significantly enhance efforts to generate sensors for both the ligases and proteasome.
The goal of this paper was to identify a shortened portable degron based on a previously identified sequence element derived from the tumor suppressor protein, p53. Gu et al. identified a sequence element from p53 that signals the E3 ligase, MDM2, to ubiquitinate p53 targeting this protein for degradation.14 Melvin et al. capitalized on this work to synthesize a peptide-based reporter capable of rapid ubiquitination in cytosolic lysates; however, the long amino-acid sequence (26 amino acids) of the reporter made its use challenging due to competition from the cytosolic peptidases.13 As such, this p53-derived degron provides a useful starting point to identify a shortened sequence from p53 that would remain a target for ubiquitination. This shorter degron sequence might then be more readily protected by entities such as protectides. To identify a shortened degron sequence from the p53 protein, a library of nine peptide substrates was synthesized consisting of peptides of various lengths all originating from the sequence element identified by Gu et al. Peptide ubiquitination was initiated using an in vitro reaction mixture and then assessed by gel electrophoresis. For precise quantification of peptide ubiquitination, reverse phase high performance liquid chromatography (RP-HPLC) was also implemented enabling quantification of the reaction kinetics. An identified five amino acid sequence was then assayed in cytosolic lysates with and without E1 enzyme and E3 ligase inhibitors to identify the potential range of enzyme for which the sequence acts as a substrate.
Materials and Methods
Substrate synthesis and purification
Peptides were synthesized either manually or by automated solid phase peptide synthesis on a Creosalus TetrasUI peptide synthesizer using Fmoc-protected amino acids on a CLEAR-amide resin purchased from Peptide Internationals. All natural Fmoc-[N]-protected amino acids and Fmoc-Lys(ivDde)-OH were acquired from Advanced Chem Tech. 6-Carboxyfluorescein was obtained from Chem Impex International. Activation of amino acids was performed with 4 eq HBTU (2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate) and 4 eq HOBt (hydroxybenzotriazole) in the presence of 5 eq DIPEA (N,N-Diisopropylethylamine) in DMF (dimethylformamide) and NMP (N-Methylpyrrolidone). Peptide deprotection was carried out in 2% DBU (1,8-diazobicyclie[5.4.0]undec-7-ene) and 2% piperidine in DMF for 2 cycles of 15 min each. Each natural amino acid coupling step was performed twice for 30 min or 1 h. For coupling of Fmoc-Lys(ivDde)-OH (4 eq) standard coupling agents were used for a single coupling of 4 h. All peptides were acetylated at the N-terminus with 5% acetic anhydride and 6% 2,6-lutidine in DMF for 35 min. Deprotection of the Fmoc-Lys(ivDde)-OH side chain was performed with 3% hydrazine monohydrate in DMF for 3 × 3 min. Removal of the ivDde protecting group was confirmed by the Kaiser test. Conjugation of 6-carboxyfluorescein (4 eq) was done using 4 eq PyBOP (benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate), HOBt (4 eq), and DIPEA (8 eq) in DMF and allowed to react overnight in the dark. Cleavage of the peptide from the resin was performed in 95:2.5:2.5 TFA (trifluoroacetic acid): TIPS (triisopropylsilane): dH2O for 3.5 h. TFA was evaporated and cleavage products were precipitated with cold ethyl ether. The peptide was extracted into water and lyophilized. It was then purified by reverse phase HPLC using an Atlantis C-18 semi-preparative column first with a gradient of 0 to 100% B over 60 min and second with a gradient of 0 to 100% B over 100 min, where solvent A was 95:5 water: acetonitrile with 0.1% TFA and solvent B was 95:5 acetonitrile: water with 0.1% TFA. After purification the peptide was lyophilized to a powder and identified using ESI or MALDI mass spectrometry. Purified peptides were reconstituted in 50 mM phosphate buffer (pH 8.0) and quantified using a Nanodrop 2000 spectrophotometer measuring the fluorescence of the 6-carboxyfluorescein tag at 492 nm. Peptide concentrations were determined using Beer’s Law.
Cell culture and lysate generation
HeLa S3 cells (ATCC) were maintained in Dulbecco’s modified eagle medium (DMEM) with 10% v/v bovine calf serum (HyClone) and maintained in a 37°C, 5% CO2 environment. All media components are from Cellgro unless otherwise noted. Unless otherwise noted all reagents used in following assays are from Sigma-Aldrich. HeLa S100 cytosolic lysates were generated from cells based on the Dignam protocol as previously described.13 Isolated HeLa S100 cytosolic lysates were quantified with a Nanodrop 2000 (Thermo Scientific), aliquoted, and stored at −80°C.
In vitro ubiquitination assay
p53-based peptide ubiquitination was evaluated using an in vitro ubiquitination assay. The reaction consisted of 10 mM Tris-HCl (pH 7.6), 2 mM MgCl2, 2 mM DTT, 300 μM ubiquitin (Boston Biochem), 1X ATP-Energy Regenerating Solution (ERS) (Boston Biochem), 1 μM Ube1 (E1, Boston Biochem), 10 μM UbcH5b (E2, Boston Biochem), 1 μM HDM2 (E3, Boston Biochem), and varying concentrations of peptide substrate (1 μM, 5 μM, 10 μM, 20 μM, and 30 μM) in a total reaction volume of 20 μL incubated at 30 °C for 2 hrs. The reaction was halted by the addition of 40 μL Tricine Sample Buffer (BioRad) followed by heating at 90°C for 5 min. For analysis by gel electrophoresis, samples were loaded onto SDS-PAGE gels (precast 16.5% Mini-PROTEAN Tris Tricine, Bio-Rad) using 1X tris-tricine running buffer and visualized with a Typhoon Imager (GE Healthcare Life Sciences).
Analysis of peptide ubiquitination by high performance liquid chromatography (HPLC)
Separation of parent peptide from ubiquitinated peptide (Ub~peptide) was accomplished using analytical HPLC set-up (an Agilent 100 HPLC system, a Phenomenex Jupiter 300 C-18 column, and fluorescence detector) based a previously established separation scheme.15 For this analysis, the in vitro ubiquitination reaction was performed as described above at concentrations ranging from 1-30 μM peptide and was halted by immediately placing the samples at −20°C and stored at this temperature until analysis by HPLC. Samples were thawed to room temperature, spiked with 10 μM fluorescein (internal standard), and injected into system for analysis. Parent and ubiquitinated peptide were separated using a gradient from polar/hydrophilic solvent A (99.9% dH2O, 0.1% TFA) to non-polar/hydrophilic solvent B (99.9% acetonitrile, 0.1% TFA) for 15 min. Example separations for peptides 2, 8, and 1 at increasing concentrations of initial parent peptide are shown in Figure 3. As a control, a sample of parent peptide was chromatographed prior to experiments to confirm the separation efficiency and provide the retention time for that experimental condition (Figure 3A, C, E, red). Chromatograms for all runs were analyzed using Origin (OriginLab) to calculate the peak areas for parent (AP) and Ub~peptide (AU) along with the fluorescein standard (to verify equal loading). The concentration of ubiquitinated peptide (CUb) was calculated as follows based on the fraction of ubiquitinated peptide (fUb).
| (Eq. 1) |
| (Eq. 2) |
where Co is the initial concentration of the peptide substrate. All experiments were performed in triplicate with corresponding error bars included for quantitative analysis with the exception of the negative control peptide 9. This peptide was only characterized in triplicate with a Co of 10 μM with no observable ubiquitination.
Figure 3. Quantification of p53-based substrate ubiquitination.
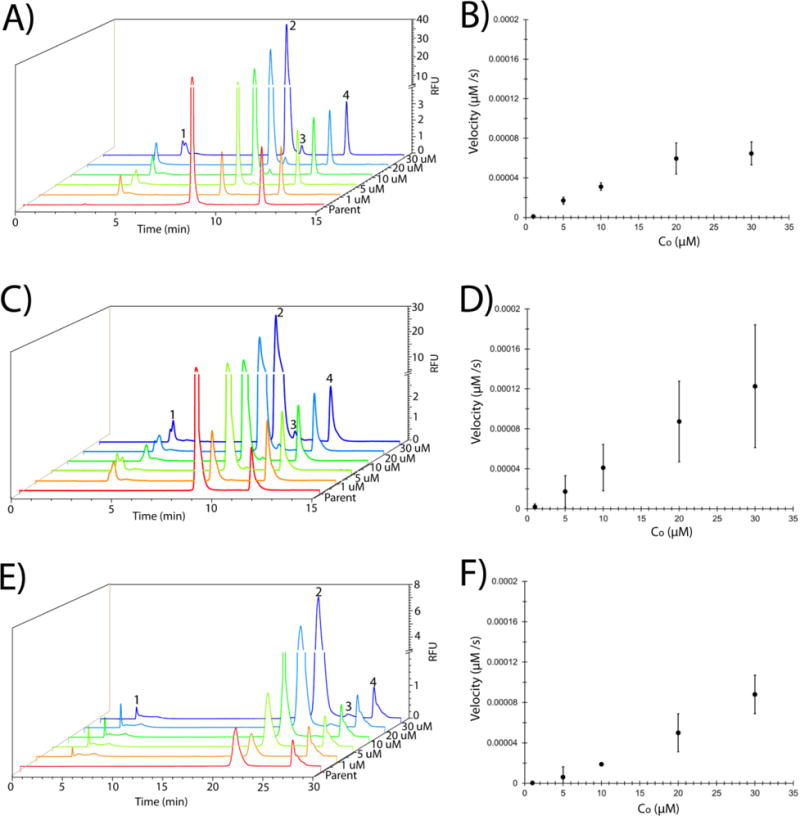
Chromatograms depicting the separation of parent peptide from Ub~peptide for peptides 2 (A), 7 (C), and 1 (E) using initial peptide concentrations of 1 μM (orange), 5 μM (yellow), 10 μM (green), 20 μM (blue), and 30 μM (indigo) as marked on the right side of each trace. Separation of parent peptide from the internal standard (FAM, carboxyfluorescein) was also performed (red) and is labeled “Parent”. Peaks are labeled as parent peptide (peak 2), Ub~peptide (peak 3), internal standard (FAM, peak 4), and the unidentified, non-reactive contaminant (peak 1). The initial concentration of peptides 2 (B), 7 (D), and 1 (F) against the velocity of peptide ubiquitination (μM/s). The data presented here is the average of three independent experiments with the standard deviation of the data points depicted by the error bars.
Additional analytical parameters were calculated to characterize the separation according to equations 3-6:
| (Eq. 3) |
| (Eq. 4) |
| (Eq. 5) |
| (Eq. 6) |
The average width (W1/2,Avg) was determined based on the width of the two peaks of interest: the peak for the parent peptide, W1/2,A and the ubiquitinated peptide, W1/2,B. This values was used to calculate the resolution (R) according to Eq. 5 using the difference in resolution between the two peaks (Δtr). These two terms were also used to calculate the number of theoretical plates (N) according to Eq. 5. Finally the theoretical plate height was calculated by Eq. 6 using the column length (L).
Ubiquitin pull down assay
Ubiquitination of the p53-based peptide substrates was performed using a pull down experiment as previously described.13 Briefly, a 100 μL reaction mixture that consisted of 10 mM Tris-HCl pH. 7.6, 5 mM MgCl2, 2 mM DTT, 20 μg/mL ubiquitin aldehyde (Boston Biochem), 400 μg/mL ubiquitin (Boston Biochem), 1X ATP ERS, 100 μM MG-132 (EMD Chemicals), 1 μL PhosSTOP (Roche), 1 μL ULTRA (Roche), 2 mg/mL HeLa S100 cytosolic lysates, and 10 μM peptide was incubated for 120 minutes at 37°C. Prior to initiating the reaction, the HeLa S100 lysates were pre-incubated for 60 min with each of the following inhibitor compounds (all obtained from LifeSensors) at three times the listed IC50 value for each compound: 30 μM serdementan, 0.27 μM nutlin-3, 150 μM SKPin C1, 300 μM SMER3, 90 μM thalidomide, 30 μM PYR-41, or a DMSO vehicle control. At the end of the reaction, samples were incubated with Control-Agarose beads (LifeSensors), diluted in TBS-T buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl, and 0.1% v/v Tween-20), for 60 min on a tube rotator at 4°C. Samples were subsequently centrifuged at 1800×g for 5 min to pellet and remove control beads. The supernatant was transferred to solution of Agarose-TUBES1 (LifeSensors) diluted in TBS-T and incubated overnight on a tube rotator at 4°C. Ubiquitin-bound beads were washed 5X with TBS-T and then the samples were eluted off of the bead with 2X tricine sample buffer, boiled for 5 min, and then isolated by centrifugation for 5 min at 13000×g. Samples were loaded onto an SDS PAGE gel, electrophoresed, visualized, and quantified as described above.
Results
Differential ubiquitination of peptide substrates based on the p53/MDM2 binding region
Previous work by Melvin et al. demonstrated that the 21 amino acid degron [KPLSSSVPSQKTYQGSYGFRLGK(FAM)KKK] derived from p53, residues 92-112, was rapidly ubiquitinated in a HeLa cytosolic lysate.13 The peptide library presented in this study was based on the sequence but with the three C-terminal lysines replaced with arginines to prevent ubiquitination on these residues and to eliminate multi-monoubiquitination of the peptides.13 The side chain of the lysine at the −4 position (from the C-terminus) of each peptide was used as a site for fluorescein conjugation leaving the N-terminal lysine as the only possible ubiquitination site. The N-terminal lysine was not a part of the sequence element identified by Gu et al., but was incorporated by Melvin et al. and identified to be the preferential site of ubiquitination.13 Nine peptides were designed based on these key sequence elements (Figure 1A). The shortened sequences were designed around the internal lysine residue (+11 in peptide 1) as this was the only lysine residue in the p53 sequence element. Peptides 2-8 varied in length from 8-14 amino acids containing a single ubiquitination site lysine either at the N-terminus (Figure 1A, Peptides 2, 4, 7-8) or towards the middle of the peptide sequence (Figure 1A, peptides 3, 5-6). The ninth control peptide (peptide 9) possessed a similar sequence to that of peptide 2, except that the N-terminal amino acid substitution of K→V was used so that peptide 9 served as a negative (non-ubiquitinatable) control.
Figure 1. In vitro ubiquitination of peptide substrates based on a p53/MDM2 binding element.
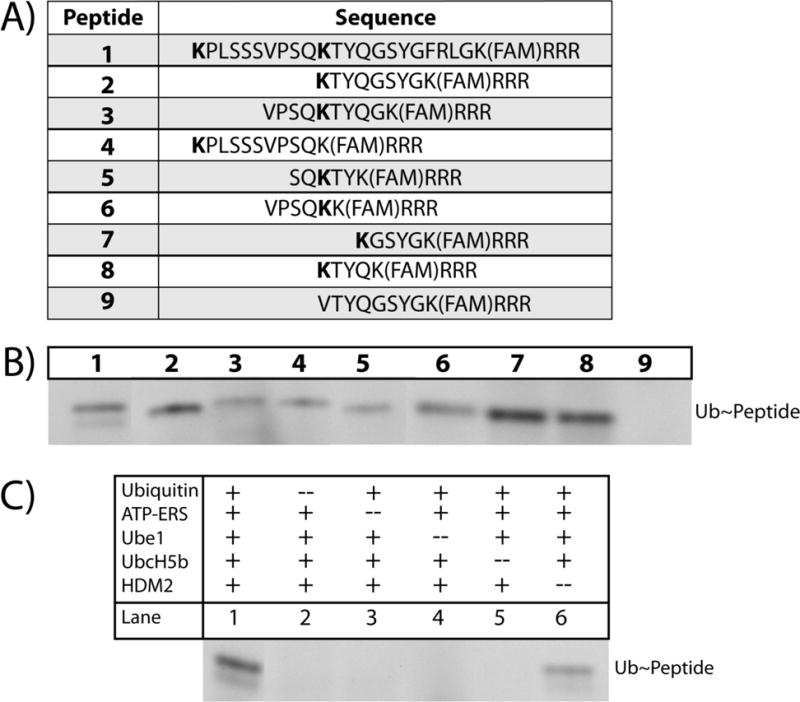
(A) Sequences of peptide substrates evaluated in this study. Bold amino acid residues correspond to ubiquitination-site lysines and FAM stands for 6-carboxyfluorescein. (B) In vitro ubiquitination of all members of the peptide library in the presence of E1 (Ube1), E2 (UbcH5b), and E3 (HDM2) enzymes followed by separation of unreacted (not shown) and ubiquitinated peptide by gel electrophoresis. Shown are fluorescent bands corresponding to 12 kDa. The upper row of numbers corresponds to the peptide number. (C) Control experiments were performed using peptide 1 with a single assay component removed (Lane 2-6) followed by gel electrophoresis. Shown are the fluorescent bands corresponding to ubiquitinated peptide.
In vitro ubiquitination of the peptides was assayed using purified proteins to simplify assay interpretation and avoid competing reactions such as degradation by intracellular peptidases. Each peptide was incubated with an E1 enzyme (Ube1), E2 enzyme (UbcH5b), and E3 ligase (HDM2, the human analog of MDM2) enzyme. The samples were analyzed by gel electrophoresis and peptides were detected by fluorescence measurement. As seen in Figure 1B, peptides 1-8 were all ubiquitinated, albeit to varying degrees, in the presence of the E1, E2, and E3 enzymes. Peptide 9 (the negative control) was not ubiquitinated, as was expected. Preliminary inspection of the differences in band intensity suggests that peptides 2, 7, and 8 are the most strongly ubiquitinated with the remaining peptides displaying decreased levels of ubiquitination (Figure 1B). Assays with peptide 1 as the substrate were performed to ensure that the observed results were due to peptide ubiquitination (Figure 1C, Lanes 2-6). Omission of ubiquitin, ATP-ERS, Ube1, or UbcH5b eliminated ubiquitination of peptide 1 (Figure 1C, Lanes 2-5). While the relative band intensities on the gels suggested that peptides 7 and 8 were the most efficiently ubiquitinated, precise quantification of peptide ubiquitination was not possible due to the poor signal-to-noise ratio of the gel images (data not shown).
Removal of HDM2 resulted in peptide 1 ubiquitination, although significantly decreased (Figure 1C Lane 1 vs. Lane 6). This observation was not entirely unexpected as previous studies have demonstrated examples of this phenomenon. For example, a deubiquitinating enzyme (Otub1) can be mono-ubiquitinated directly by the E2 enzyme UbcH5.16 Moreover, prior work has demonstrated that the rate-limiting step in p53 ubiquitination is the local accumulation of the UbcH5b~Ub complex, and that MDM2 serves as a ‘landing pad’ for this complex to facilitate substrate ubiquitination.17 It is possible due to the decrease in substrate length and the abundance of both E2 enzyme and ubiquitin supplied in the reaction that the peptide substrate interacted directly with the E2~Ub complex under these conditions, resulting in substrate ubiquitination. This is consistent with the fact that there was some ubiquitination, but to a substantially lesser degree as this reaction was not mediated by HDM2.
Separation of unmodified and ubiquitinated p53-degron based substrates by HPLC
Analytical, reverse phase high performance liquid chromatography (HPLC) has been used in numerous studies to assess protein degradation and posttranslational modifications such as phosphorylation18 and deubiquitination.19 Furthermore, reverse phase HPLC has been used to separate and quantify chemically synthesized ubiquitinated peptides.20 This analytical technique offers a high degree of precision and the ability to quantify protein concentrations through the use of internal standards or other established metrics. Here, analytical HPLC was utilized to quantify peptide ubiquitination in samples from the in vitro ubiquitination assay. Peptide 7 was used as a model substrate for optimization of the HPLC-based readout of the in vitro ubiquitination assay. Three distinct peaks were observed on the chromatogram (Figure 2A). The identity of the parent peptide peak (peak 2 at 8.81 min ± 0.05) was accomplished by co-mixing the reactants with an excess amount (10 μM) of parent peptide, followed by separation. Peak 3 (retention time of 9.75 min ± 0.11) was confirmed as Ub~peptide (MW 10054.24) by a combination of mass spectrometry (Figure 2B) and experimental controls (Figure 2C). The mass spectrometry data also demonstrated species with the molecular weights corresponding to unmodified parent peptide and free ubiquitin (data not shown). Assays omitting a required reactant (E1, E2, ubiquitin, or peptide) did not demonstrate peak 3, also supporting that peak 3 was indeed Ub~peptide. Moreover, the persistence of peak 1 (retention time of 3.64 min ± 0.41), even in the absence of parent peptide, suggested that this peak corresponded to a contaminant in the ubiquitination assay mixture. A small, unidentified peak was observed on the chromatogram in the absence of any peptide (Figure 2C, i); however, the area of this peak was significantly lower than that of the peptide peak (10 μM peptide). Thus this peak was likely a contaminant in the ubiquitin assay mixture. The quality and efficiency of the separation was given by a resolution of 3.37 ± 0.33 for the peptide and Ub~peptide, a theoretical plate number of 14157 ± 6360 for peak 2, and a theoretical plate height of 0.126 ± 0.13 for peak 2. These data demonstrated that reverse-phase HPLC was a suitable separation method for the p53-derived peptides and their ubiquitinated counterparts. Additionally, the data demonstrated that peptide 7 was ubiquitinated under these in vitro conditions.
Figure 2. Separation of parent peptide from Ub~peptide using reverse phase HPLC.
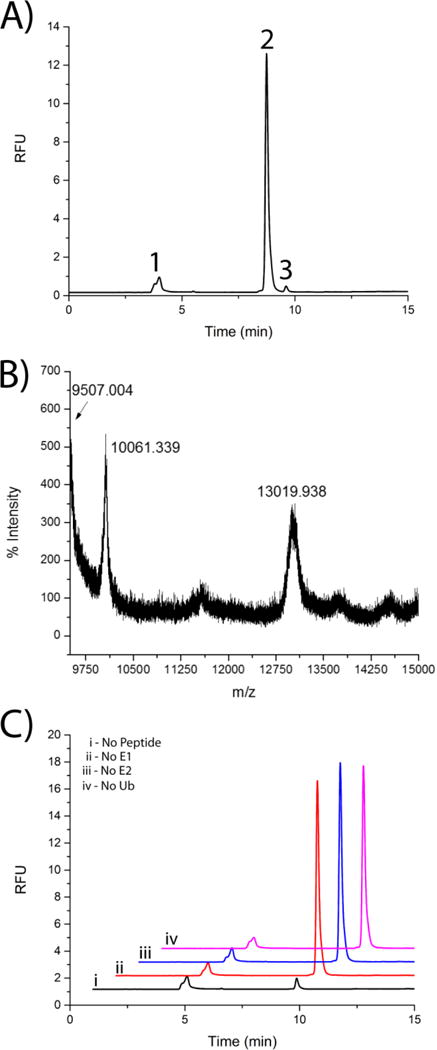
(A) Example chromatogram of the ubiquitination of peptide 7 (10 μM) demonstrating the separation of parent/unmodified peptide (Peak 2) from Ub~peptide (Peak 3) using a C18-bonded silica analytical column. Peak 1 was an impurity in the in the peptide stock. (B) Mass spectrometry analysis (MALDI-TOF) of ubiquitination assay mixture with peptide 7. (C) Chromatogram (10 μM peptide 7) under control conditions in the absence of peptide (i), E1 enzyme (ii), E2 enzyme (iii), or ubiquitin (iv).
Quantification of p53-derived peptide ubiquitination using HPLC
Concentration-dependent quantification of peptide ubiquitination was accomplished using analytical HPLC in order to determine the kinetic constants for each peptide and to identify the shortened p53-derived portable degron sequence. Separation of parent peptide (peak 2) and the ubiquitin-conjugated peptide (peak 3) was demonstrated for all peptides at all concentrations (Figure 3A, C, E and Supplemental Figure 1A, C, E, G, I). The concentration of the unmodified parent peptide (AP) and ubiquitinated peptide (AU) were calculated and then multiplied by the fraction of ubiquitinated peptide (equation 1) to estimate the concentration of Ub~peptide (equation 2). The rate of Ub~peptide formation was plotted against Co for each peptide (Figure 3B, D, F and Supplemental Figure 1B, D, F, H, J). As expected, the velocity of Ub~peptide formation increased with the initial concentration of parent peptide. The rate of reaction was approximated as linearly dependent on the parent peptide concentration. Calculation of the square of the Pearson product moment correlation coefficient (R2) demonstrated the high quality of the linear fit (Table 1). This linear approximation between the reaction velocity and substrate concentration at low substrate concentration for this multi-reaction system was previously demonstrated by Brazhnik and colleagues for MDM2-dependent ubiquitination of full length p53.21 Thus, the linear approximation observed in this study suggested that the combined reactions could be described using pseudo-Michaelis-Menten kinetics at a Km >>C. For this reason, an aggregate vmax/Km to describe the combined reactions was calculated to quantify the suitability of peptides 1-8 as substrates. A summary of the values for the aggregate vmax/Km are included in Table 1.
Table 1.
Summary of kinetic constants for p53-based substrate ubiquitination.
| Peptide | Vmax/Km {1/min)*1000 |
R2 |
|---|---|---|
| 1 | 0.184 ±0.028 | 0.987 |
| 2 | 0.134 ±0.022 | 0.932 |
| 3 | 0.120 ±0.064 | 0.994 |
| 4 | 0.114 ±0.029 | 0.981 |
| 5 | 0.058 ± 0.009 | 0.976 |
| 6 | 0.175 ±0.084 | 0.979 |
| 7 | 0.255 ±0.117 | 0.997 |
| 8 | 0.169 ±0.024 | 0.998 |
Peptide 7 offered the best kinetic values (vmax/Km = 2.55 × 10−4 min−1), which matched the qualitative observations from the gel electrophoresis. These results indicated that substrate length, make-up, and location of ubiquitination site lysine all impact ubiquitination efficiency. The difference between peptide 7 and the remaining peptides implied that the sequence GSYG (which is missing from peptides 3, 4, 5, 6, and 8) is important for peptide ubiquitination. Moreover, the fact that peptides 1 and 2 exhibited decreased kinetics even with the GSYG binding sequence would suggest that substrate length and the location of the ubiquitination site lysine both play a role in substrate ubiquitination. The fact that this sequence element was found in the peptide substrate previously characterized by Melvin et al13 highlights the importance of this specific sequence in the recognition by the ubiquitination machinery. Taken together, the kinetic analysis performed here demonstrates that peptide 7 behaves as a shortened portable degron that is ubiquitinated in an MDM2-dependent manner.
Ubiquitination of the p53-derived portable degron in cytosolic lysates
While using a cell-free, enzyme-only ubiquitination assay has several advantages (e.g., known enzyme concentrations and identities and no peptidases or proteases to degrade the peptide substrates), this system does not yield a complete picture of how a substrate will behave under in vivo conditions. Since peptide 7 offered the best kinetics, the degree of ubiquitination of this peptide was assayed in HeLa S100 cytosolic lysates. The assay was performed similar to that above except that the lysates served as the source of the E1, E2, and E3 enzymes. After peptide incubation with the lysates, ubiquitinated molecules were isolated from the reaction mixture using tandem ubiquitin binding entity (TUBE) agarose beads as previously described.13 The assay was performed in the presence of a number commercially available E1 enzyme and E3 ligase inhibitors to determine whether ubiquitination was dominated by a single enzyme group and to evaluate the utility of the shortened p53-based substrate as a portable degron. Six inhibitor compounds were selected including serdemetan,22 nutlin-3,23 SKPin C1,24 SMER3,25 and thalidomide26 in addition to the E1 enzyme inhibitor PYR-41 (Figure 4). Each inhibitor was added at a concentration three times that of its published IC50 value to insure adequate inhibition of the targeted enzyme. An advantage of the peptide-based assay is the direct incorporation of the fluorescein tag which eliminated the need for western blotting following immunoprecipitation of the ubiquitin-bound peptide. This enabled facile and direct visualization of the fluorescent peptides on the gels. The result from this experiment indicated that peptide 7 was readily ubiquitinated in cytosolic lysates, as observed by the bands corresponding to mono-, di-, tri-, and tetra-ubiquitinated species (Figure 4, Lane 1). The inhibitor compounds nutlin-3 (Figure 4, Lane 2), serdementan (Figure 4, Lane 3), and thalidomide (Figure 4, Lane 6) had no apparent effect on the ubiquitination of peptide 7. Interestingly, the SKPin C1 compound, which inhibits the cullin-RING ubiquitin E3 ligase SCF-Skp2, slightly reduced the polyubiquitination of peptide 7, but not mono-ubiquitination (Figure 5, Lane 4). Additionally, the SMER3 (small molecule enhancer of rapamycin 3) compound, which is an inhibitor of the E3 ligase SCF-MET30, significantly reduced both the poly- and mono-ubiquitination of peptide 7 (Figure 5, Lane 5). These results suggested that peptide 7, the shortened degron isolated from the MDM-p53 binding element, could potentially act as a substrate for the SCF family of E3 ubiquitin ligases. This group of multi-protein E3 ligases contain three core subunits including Skp1 (the bridging protein), cullin (the major structural scaffold of the complex), and RBX1 (the RING finger zinc-binding domain responsible for E2 ubiquitin conjugating enzyme binding). However, each member of the SCF family contains different F-box containing proteins (e.g., Skp2 or β-TrCP) which recognize and bind to specific target proteins enabling for the specificity of this class of E3 ligases. The results presented here suggest that peptide 7 can be recognized and ubiquitinated by an SCF complex. Interestingly, the GSYG motif identified here appears similar in sequence to other portable degrons described in the literature that are ubiquitinated by members of the SCF family of E3 ligases including β-Catenin (DSGIHSG), IFNAR1 (DSGNYS), and TAZ (HSREQSTDSG).13 One significant difference between the GSYG motif and the other portable degrons is that GSYG did not require the incorporation of a phosphorylated serine residue. The three phospho-degrons from β-Catenin, IFNAR1, and TAZ all require that one or both of the serine residues be phosphorylated for ubiquitination in cytosolic lysates. While these results suggested that the shortened substrate loses specificity towards MDM2, the broader substrate behavior of the isolated degron relative to that of the full length p53 protein is an attractive feature for reporters designed to assess overall ubiquitination activity within a cell and is consistent with the absence of a secondary structure that may act to limit substrate suitability to a single enzyme. These results also verify that the shortened portable degron based on the p53-MDM2 binding region can be ubiquitinated in cellular lysates, which lends credence to its potential use as a reporter for an analytical readout of members of the UPS.
Figure 4. Effect of inhibitors on the ubiquitination of a p53-based peptide substrate in cell lysates.
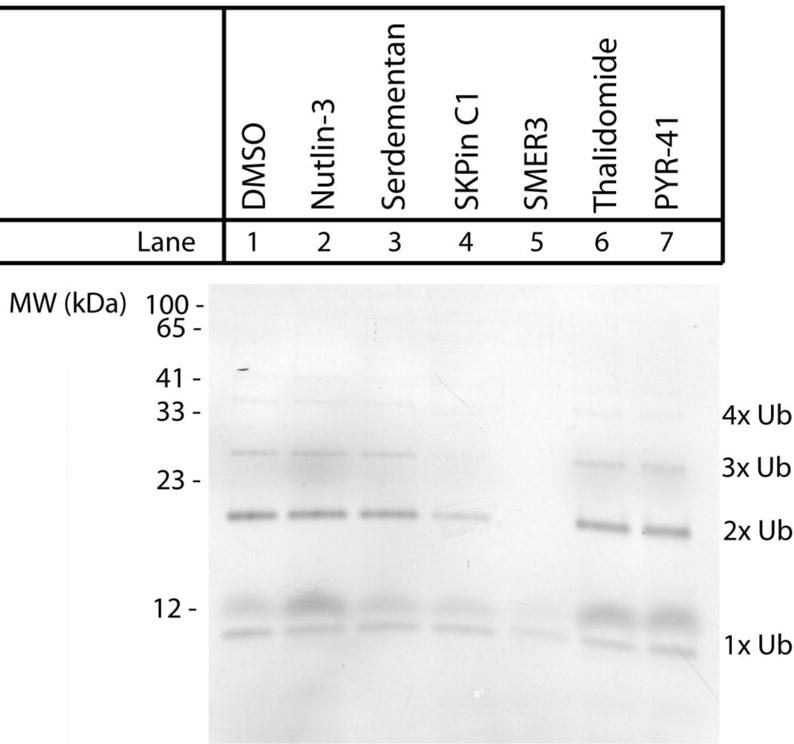
Peptide 7 (10 μM) was incubated in HeLa S100 lysates for 2 h at 37°C in the presence of commercially available E1 and E3 enzyme inhibitors. Each inhibitor was added with the following concentrations: nutlin-3 (0.27 μM), serdementan (30 μM), SKPin C1 (150 μM), SMER3 (300 μM), thalidomide (90 μM), PYR-41 (30 μM), or a DMSO vehicle control. Ubiquitin-conjugated peptides were purified using ubiquitin-binding beads, separated by SDS-PAGE, and visualized using the fluorescein tag. Relative protein sizes are compared to values obtained from a fluorescent marker (left side). Fluorescent species at the appropriate molecular weight for mono-, di-, tri-, and tetra-ubiquitinated peptides are labeled accordingly.
Discussion
The goal of this work was to identify a shortened portable degron based on a previously identified MDM2/p53 binding element that exhibited optimal ubiquitination kinetics.14 A peptide library of nine peptide substrates was synthesized based on different sequence elements of the p53/MDM2 binding element and the degree of ubiquitination was quantified. The top-performing library member, peptide 7, was readily ubiquitinated in a cytosolic lysate demonstrating promise as a portable degron substrate for E3 ligases or for incorporation into more complex proteasome reporters. Additionally, the ubiquitination of peptide 7 was found to be disrupted in the presence of inhibitors of the SCF family of E3 ligases suggesting that this sequence might serve as a reporter for this class of enzymes. The shortened size should enable the degron to be incorporated into longer peptide-based reporters by straight forward solid-phase peptide synthesis or post-synthetic conjugation to a β-hairpin protectide to limit off-target enzymatic modifications in the cytosolic environment.9, 10 With further study and modifications, it is envisioned that ligase substrates such as that developed may act as valuable sensors of E3 ligase or proteasome activity in single cancer cells providing new insight in to the actions of chemotherapeutic inhibitors. These shortened degrons may have additional utility when incorporated into other constructs since shortened proteasome substrates have been shown to be useful tools in the disruption of proteasomal degradation.11 Finally, the length of a degron has recently been linked to its ability to be mono- versus polyubiquitinated, suggesting that the shortened sequence might be suitable as a probe to further understand ubiquitin chain length in protein fate.27
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Biomedical Imaging and Bioengineering, R01EB011763 (NLA), and the National Cancer Institute, F32CA162574 (ATM) and R01CA177993 (NLA). LDD was supported in part by a Summer Undergraduate Research Fellowship from the Office of Undergraduate Research at the University of North Carolina at Chapel Hill.
Abbreviations
- MDM2
murine double minute 2
- Ub
ubiquitin
- HPLC
high performance liquid chromatography
- SPPS
solid phase peptide synthesis
- TFA
trifluoroacetic acid
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
References
- 1.Schrader E, Harstad K, Matouschek A. Nature Chemical Biology. 2009;5:815–822. doi: 10.1038/nchembio.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoeller D, Dikic I. Nature. 2009;458:438–444. doi: 10.1038/nature07960. [DOI] [PubMed] [Google Scholar]
- 3.Grabbe C, Husnjak K, Dikic I. Nature Reviews Molecular Cell Biology. 2011;12:295–307. doi: 10.1038/nrm3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ravid T, Hochstrasser M. Nature Reviews Molecular Cell Biology. 2008;9:679–U625. doi: 10.1038/nrm2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mattern M, Wu J, Nicholson B. Biochimica Et Biophysica Acta-Molecular Cell Research. 2012;1823:2014–2021. doi: 10.1016/j.bbamcr.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kovarik M, Allbritton N. Trends in Biotechnology. 2011;29:222–230. doi: 10.1016/j.tibtech.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Proctor A, Wang Q, Lawrence D, Allbritton N. Analytical Chemistry. 2012;84:7195–7202. doi: 10.1021/ac301489d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Phillips R, Bair E, Lawrence D, Sims C, Allbritton N. Analytical Chemistry. 2013;85:6136–6142. doi: 10.1021/ac401106e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cline L, Waters M. Biopolymers. 2009;92:502–507. doi: 10.1002/bip.21266. [DOI] [PubMed] [Google Scholar]
- 10.Yang S, Proctor A, Cline L, Houston K, Waters M, Allbritton N. Analyst. 2013;138:4305–4311. doi: 10.1039/c3an00874f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bonger K, Chen L, Liu C, Wandless T. Nature Chemical Biology. 2011;7:531–537. doi: 10.1038/nchembio.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Neklesa T, Tae H, Schneekloth A, Stulberg M, Corson T, Sundberg T, Raina K, Holley S, Crews C. Nature Chemical Biology. 2011;7:538–543. doi: 10.1038/nchembio.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Melvin A, Woss G, Park J, Dumberger L, Waters M, Allbritton N. PLOS One. 2013;8:e78082. doi: 10.1371/journal.pone.0078082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gu J, Chen D, Rosenblum J, Rubin R, Yuan Z. Molecular and Cellular Biology. 2000;20:1243–1253. doi: 10.1128/mcb.20.4.1243-1253.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mant C, Cepeniene D, Hodges R. Journal of Separation Science. 2010;33:3005–3021. doi: 10.1002/jssc.201000518. [DOI] [PubMed] [Google Scholar]
- 16.Li Y, Sun X, Elferich J, Shinde U, David L, Dai M. Journal of Biological Chemistry. 2014;289:5097–5108. doi: 10.1074/jbc.M113.533109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ranaweera R, Yang X. Journal of Biological Chemistry. 2013;288:18939–18946. doi: 10.1074/jbc.M113.454470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yan J, Packer N, Gooley A, Williams K. Journal of Chromatography a. 1998;808:23–41. doi: 10.1016/s0021-9673(98)00115-0. [DOI] [PubMed] [Google Scholar]
- 19.Layfield R, Franklin K, Landon M, Walker G, Wang P, Ramage R, Brown A, Love S, Urquhart K, Muir T, Baker R, Mayer R. Analytical Biochemistry. 1999;274:40–49. doi: 10.1006/abio.1999.4234. [DOI] [PubMed] [Google Scholar]
- 20.Kumar K, Spasser L, Ohayon S, Erlich L, Brik A. Bioconjugate Chemistry. 2011;22:137–143. doi: 10.1021/bc1004735. [DOI] [PubMed] [Google Scholar]
- 21.Brazhnik P, Kohn K. Mathematical Biosciences. 2007;210:60–77. doi: 10.1016/j.mbs.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 22.Kojima K, Burks J, Arts J, Andreeff M. Molecular Cancer Therapeutics. 2010;9:2545–2557. doi: 10.1158/1535-7163.MCT-10-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vassilev L, Vu B, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu E. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 24.Wu L, Grigoryan A, Li Y, Hao B, Pagano M, Cardozo T. Chemistry & Biology. 2012;19:1515–1524. doi: 10.1016/j.chembiol.2012.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aghajan M, Jonai N, Flick K, Fu F, Luo M, Cai X, Ouni I, Pierce N, Tang X, Lomenick B, Damoiseaux R, Hao R, del Moral P, Verma R, Li Y, Li C, Houk K, Jung M, Zheng N, Huang L, Deshaies R, Kaiser P, Huang J. Nature Biotechnology. 2010;28:738–U1750. doi: 10.1038/nbt.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ito T, Ando H, Suzuki T, Ogura T, Hotta K, Imamura Y, Yamaguchi Y, Handa H. Science. 2010;327:1345–1350. doi: 10.1126/science.1177319. [DOI] [PubMed] [Google Scholar]
- 27.Shabek N, Herman-Bachinsky Y, Buchsbaum S, Lewinson O, Haj-Yahya M, Hejjaoui M, Lashuel H, Sommer T, Brik A, Ciechanover A. Molecular Cell. 2012;48:87–97. doi: 10.1016/j.molcel.2012.07.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.