

Abstract
Acinar-to-ductal metaplasia (ADM) is a reversible epithelial trans-differentiation process that occurs in response to acute inflammation. ADM can rapidly progress toward pre-malignant pancreatic intraepithelial neoplasia (PanIN) lesions in the presence of mutant KRas and ultimately pancreatic adenocarcinoma (PDAC). In the present work we elucidate the role and related mechanism of Glycogen synthase kinase-3beta (GSK-3β) in ADM development using in vitro 3D cultures and genetically engineered mouse models. We show that GSK-3β promotes TGFα-induced ADM in 3D cultured primary acinar cells, whereas deletion of GSK-3β attenuates caerulein-induced ADM formation and PanIN progression in KRasG12D transgenic mice. Furthermore, we demonstrate that GSK-3β ablation influences ADM formation and PanIN progression by suppressing oncogenic KRas-driven cell proliferation. Mechanistically, we show that GSK-3β regulates proliferation by increasing the activation of S6 kinase. Taken together, these results indicate that GSK-3β participates in early pancreatitis-induced ADM and thus could be a target for the treatment of chronic pancreatitis and prevention of PDAC progression.
Keywords: Pancreatic cancer, pancreatitis, acinar-to-ductal metaplasia, glycogen synthase kinase-3β, KRas, S6K
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is among the most deadly human malignancies[1–3]. Oncogenic KRas mutation represents the most frequent and earliest genetic alteration in PDAC patients, highlighting its role as a driver of PDAC[4, 5]. Indeed, genetically engineered mouse models expressing activated mutant KRas (G12D) in the pancreas develop pancreatic cancer with a long latency, which can be substantially accelerated by experimentally inducing chronic pancreatitis through the administration of the CCK agonist caerulein[6–8]. Several studies have shown that inflammation and aberrant growth factor signaling can drive acinar cells to dedifferentiate into metaplastic duct-like cells, a process termed acinar-to-ductal metaplasia (ADM)[8–10]. Interestingly, lineage-tracing experiments in these models have demonstrated that preneoplastic pancreatic intraepithelial neoplasia (PanIN) lesions are mainly derived from acinar cells undergoing ADM[4, 11–13]. Although ADM is a benign lesion and a reversible process[14, 15], the presence of mutant KRas ‘locks’ the metaplastic cells in a duct-like state, suggesting that ADM might be an early event that interacts with mutant KRas to promote PDAC development.
Glycogen synthase kinase-3 (GSK3) α and β are highly conserved serine-threonine kinases involved in several cellular functions including glycogen metabolism, Wnt/β-catenin signaling, immune regulation, and maintenance of stem cell identity[16, 17]. Not surprisingly, these two kinases participate in the pathogenesis of various human diseases, such as diabetes, inflammatory and neurological disorders, as well as cancer[18, 19]. Reports from ours and other groups have indicated that GSK-3β is progressively overexpressed from PanIN to advanced PDAC, and becomes nuclear accumulated in most moderately and poorly differentiated tumors[20, 21]. Significantly, GSK-3β overexpression contributes to PDAC cell proliferation and survival, whereas GSK-3 inhibition reduces pancreatic cancer cell viability in vitro and suppresses tumor xenograft growth in vivo[22, 23]. Lastly, oncogenic KRas signaling regulates GSK-3β gene expression, suggesting that GSK-3β may be important for KRas-driven tumor-promoting pathways during the development of PDAC[24].
Although most studies have shown functional redundancy between the two homologs of GSK-3 as exemplified by their regulation of Wnt/β-catenin signaling, they also exhibit tissue-specific physiologically important functions that may have varying impact on human cancer etiology[25, 26]. We have previously demonstrated that the two GSK-3 isoforms exhibit differential activity toward TRAIL- or TNFα-induced apoptosis in PDAC cells[27]. Significantly, addition of a GSK-3 inhibitor concomitantly with caerulein prevented the onset of ADM in a dose-dependent manner[28]. However, the role of GSK-3β in early PDAC, especially pancreatitis-induced ADM, remains unknown.
Herein, we provide evidence that GSK-3β is necessary for oncogenic KRas-mediated formation of duct-like structures in vitro. Moreover, conditional deletion of GSK-3β in Pdx1-cre;LSL-KRasG12D mice abrogated caerulein-induced ADM, PanIN lesion formation, inflammation and fibrosis. Significantly, GSK-3β deletion resulted in a substantial decrease in the number of proliferating cells within ADM and PanIN areas. Mechanistically, we find enhanced activation of S6 kinase (S6K) resulting in the elevated phosphorylation of S6 in ADM and PanIN epithelial cells, which is substantially reduced in the absence of GSK-3β. As ADM is an early event contributing to the development of PDAC, our data suggest that pharmacologic inhibition of GSK-3 may be an effective strategy to prevent early changes that are required for further development to PDAC.
MATERIALS AND METHODS
Antibodies
Antibodies to GSK-3α/β, GSK-3β, phospho-GSK-3β (ser9), Ki-67, S6 ribosomal protein, phospho-S6 ribosomal protein (ser235, 236), phospho-S6 ribosomal protein (ser240, 244) were obtained from Cell Signaling Technologies (Beverly, MA), β-catenin was purchased from BD Transduction Lab (San Jose, CA), β-actin, α-amylase, and αSMA were from Sigma (St. Louis, MO), cytokeratin 19 was from DSHB (University of Iowa, Iowa). For immunohistochemical and immunofluorescence staining, the following primary antibodies were used: mouse anti-β-catenin (1:200), mouse anti-αSMA (1:200), rabbit anti-amylase (1:400), rat anti-CK19 (TROMAIII, 1:300), rabbit anti-GSK-3β (1:100), rabbit anti-phospho-S6 ribosomal protein (ser235, 236) at 1:300 dilution, and rabbit anti-Ki67 (1:200). For immunofluorescence staining, Alexa Fluor 488 donkey anti-mouse IgG, Alexa Fluor 568 donkey anti-rabbit IgG (Life Technologies), and Alexa Fluor 633 donkey anti-rat IgG (Jackson ImmunoResearch Laboratories) were used as secondary antibodies at a 1:300 dilution.
Reagents, Cell culture and drug treatment
Caerulein, DMSO and all other chemicals unless specified were obtained from Sigma (St. Louis, MO). The GSK-3 inhibitor 9-ING-41 (GSK-3i) was obtained from Actuate Therapeutics (Fort Worth, TX). Recombinant human TGF-α was obtained from R&D Systems (Minneapolis, MN). Soybean trypsin inhibitor was obtained from Affymetrix (Santa Clara, CA). Rat-tail collagen I was obtained from BD (Franklin Lakes, NJ). AR42J cells were passaged in Ham’s F-12K (Kaighn’s) medium supplemented with 15% fetal bovine serum (FBS), 2 mM L-glutamine and penicillin/streptomycin. For caerulein stimulation experiments, cells were serum starved for 16 hours and pre-treated with 9ING41 (500 nM) for 2 hours, then 10 nM of caerulein for 2 or 6 hours. Stable shGSK-3β or scramble AR42J cells were fasted overnight prior to caerulein (10 nM) stimulation. Cells were harvested 2, 6, and 12 hours post caerulein stimulation.
Lentiviral packaging, transduction and selection of stable cells
Lentivirus packaging, cell infection and selection of pLKO-shRNA stable cells were performed as previously described following institutional biosafety regulations[27]. AR42J cells were infected with appropriate amounts of lentiviral particles containing medium. Twenty-four hours later, virus-containing medium was removed and replaced with fresh medium supplemented with 2 μg/ml of puromycin. Pooled resistant clones were used for experiments.
Animals and acute pancreatitis model
Experimental animals were generated by crossing Pdx1-Cre;LSL-KRasG12D mice with GSK-3βF/F mice. To induce pancreatitis, 6-week old male sibling littermates from each genotype were selected for treatment. Mice were starved for 12 h and allowed water ad libitum one day prior to the experiment. Animals were injected intraperitoneally into the right lower quadrant with a 50 μg/kg/bw (body weight) of caerulein dissolved in 0.9% saline in a volume of 100 μl. Injections were given at hourly intervals up to 8 times. Pancreata were harvested 2 and 7 days after caerulein treatment. All animal experiments were approved by the Mayo Clinic Institutional Animal Care and Use Committee and were performed in accordance with relevant institutional and national guidelines and regulations.
Isolation of primary pancreatic acinar cells for 3D explant culture in vitro
Primary pancreatic acinar cells were isolated as previously described[29, 30]. Briefly, the pancreas was washed in cold serum-free Hanks Balanced Salt Solution, cut into 1–2 mm pieces and digested with collagenase I. Small clusters of acinar cells were filtered through a 100 μm cell strainer and further purified with density gradient centrifugation, and resuspended in complete Waymouth medium (10% FBS, 0.1 mg/ml trypsin inhibitor, and 1 μg/ml dexamethasone). Freshly isolated acinar cells were then mixed with an equal volume of collagen solution and seeded in cell culture plates pre-coated with collagen I in Waymouth medium. Following 15–30 minutes incubation at 37°C, another layer of Waymouth complete medium with or without TGF-α, or GSK-3 inhibitors was added. To express proteins using lentiviral expression system, acinar cells were infected with appropriate amount of packaged viral particles and incubated for 3–5 h before embedding in the collagen I/medium mixture. At day 6 or 7 (dependent on time course of duct formation) the number of ducts was counted under a microscope.
IHC, EdU labeling and Immunofluorescence
Following caerulein treatment, mice were anesthetized using isoflurane (Nova Plus Pharmaceuticals), followed by cervical dislocation. The whole pancreas was quickly removed and fixed overnight in 4% PFA with gentle shaking, embedded in paraffin, cut into 5 μm-thick sections. Sections were subjected to H&E, IHC and immunofluorescence staining as described[31]. For EdU labeling, mice were injected with EdU at a concentration of 50 mg/kg/bw in saline 2 hours before sacrifice. Staining was performed by Click-iT® EdU Alexa Fluor® 488 Imaging Kit following manufacturer’s instructions (Thermofisher scientific, USA). Confocal images were collected with an LSM-710 laser scanning confocal microscope with a 63× water Plan-Apochromat objective lens using ZEN 2009 software (Carl Zeiss, Oberkochen, Germany).
Western Blot analysis, quantitative RT-PCR and Qiagen RT2 PCR array
Snap frozen pancreata from mice of desired genotypes were homogenized in RIPA buffer (Abcam, Cambridge, MA). Protein extracts were prepared, separated by SDS-PAGE, transferred to PVDF membrane and immunoblotted with specific antibodies as described [24]. Protein bands of interest were quantified by calculating an integrated density value for each band using ImageJ (National Institutes of Health, Bethesda, MD). Pancreatic total RNA was isolated using Trizol and further purified with an RNeasy Mini Kit (Qiagen, Valencia, CA, USA). Reverse transcription was performed with the Superscript III RT-PCR Kit (Invitrogen). Quantitative PCR was performed with the SYBR Green PCR Master Mix using the ABI StepOnePlus Sequence Detection System (Applied Biosystems, Carlsbad, CA). GAPDH and RPLP0 were used as housekeeping genes for normalization of gene expression. CCND1 and Myc expression was analyzed using the Qiagen RT2 PCR array kit (PAMM-225Z, Qiagen, Valencia, CA, USA). The Qiagen RT2 PCR array kit contains five internal controls for gene expression. The double delta Ct method was used to analyze gene expression. Experiments were performed in triplicate using three independent cDNAs. Primer sequences are provided in Supplementary Table S1.
Statistical analysis
Data are expressed as mean ± SEM and analyzed by repeated measures analysis of variance, unpaired Student’s t-test using GraphPad Prism software (GraphPad Software Inc., La Jolla, CA). A value of p <0.05 denotes statistical significance.
RESULTS
GSK-3β expression is increased following caerulein-induced pancreatitis in mice
Pancreatic tissues sections from control- or caerulein-treated Pdx1-cre and Pdx1-cre;LSL-KRasG12D mice were stained for GSK-3β expression following caerulein-induced pancreatitis. GSK-3β expression is weakly cytoplasmic in saline-treated Pdx1-cre mice, but is increased in the areas of ADM by two days post caerulein treatment (Fig. 1A). Similar to what we have previously shown, GSK-3β expression is elevated in Pdx1-cre;LSL-KRasG12D mice and is further increased upon caerulein treatment (Fig. 1A). In addition, protein extracts at selected time points post caerulein treatment demonstrate increased levels GSK-3β compared to control mice and slightly increased levels of GSK-3β following the administration of caerulein (Fig. 1B, C). We also noted increased levels of GSK-3α in control and Pdx1-cre;LSL-KRasG12D mice following caerulein treatment, suggesting that this kinase may also be linked to oncogenic KRasG12D signaling (Fig. 1B, C). Overall, these data indicate that the expression levels of both GSK-3 kinase isoforms are increased in response to caerulein-induced pancreatitis and oncogenic KRasG12D signaling.
Figure 1. GSK-3β protein expression is elevated following caerulein-induced pancreatitis in mice.
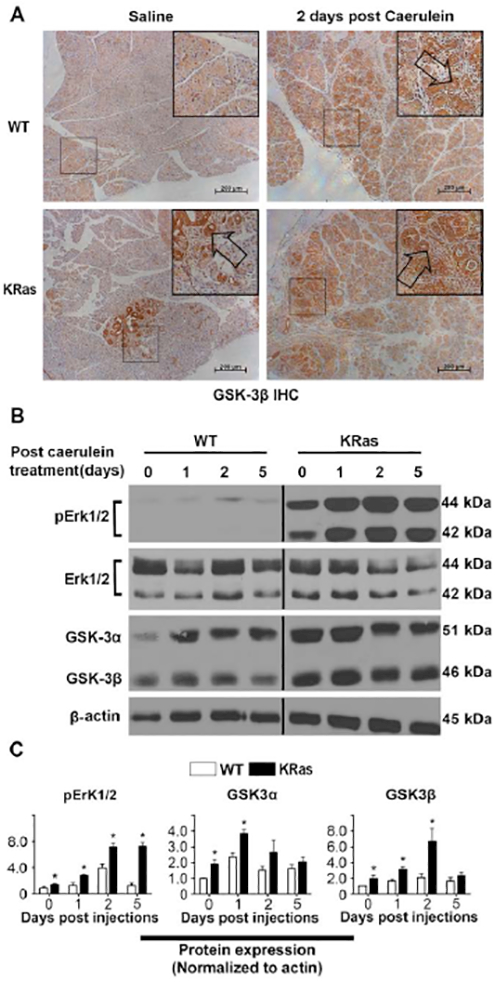
(A) Representative IHC images of GSK-3β in pancreatic sections from Pdx1-cre wild-type (WT) (upper panel) and Pdx1-cre;LSL-KRasG12D (KRas) mice (lower panel) treated with or without caerulein. Arrows point to the abnormal pancreas area. Bars, 200 μm. (B) Representative immunoblot analysis for phosphorylated Erk1/2, total Erk1/2, GSK-3α and GSK-3β in pancreatic tissue of WT and KRas mice before and 1, 2, 5 days post caerulein treatment. β-actin was used as loading controls. The black vertical line in (B) denotes where an additional time point was removed. Samples were developed from the same membrane. Shown are representative results from 3 experiments. (C) The average signal intensity of phospho-Erk1/2, GSK-3α, GSK-3β was quantified and expressed as mean ± SEM. n=6. *P<0.05 KRas versus WT mice.
GSK-3β promotes TGF-α-induced ADM
To investigate whether GSK-3 kinases are involved in ADM, we cultured isolated mouse pancreatic acinar cells in collagen. This ex vivo explant model for ADM is based on a previously established cell culture model in which growth factors such as TGF-α that activate the EGF receptor ErbB1 have been shown to induce ADM within 6–8 days[30]. Consistent with this, TGF-α stimulation of primary acinar cells resulted in a 3-fold increase in ADM events (Fig.2 A, B, and supplemental Fig. S1). Significantly, we found that addition of a GSK-3 inhibitor (GSK-3i) slightly reduced the number of ADM events in unstimulated cells, but substantially impaired TGF-α induced ductal formation, showing a six-fold decrease (Fig. 2A, B, and supplemental Fig. S1). Taken together, these data indicate that GSK-3 kinases are involved in the process of ADM in vitro.
Figure 2. GSK-3β promotes TGF-α-induced ADM.
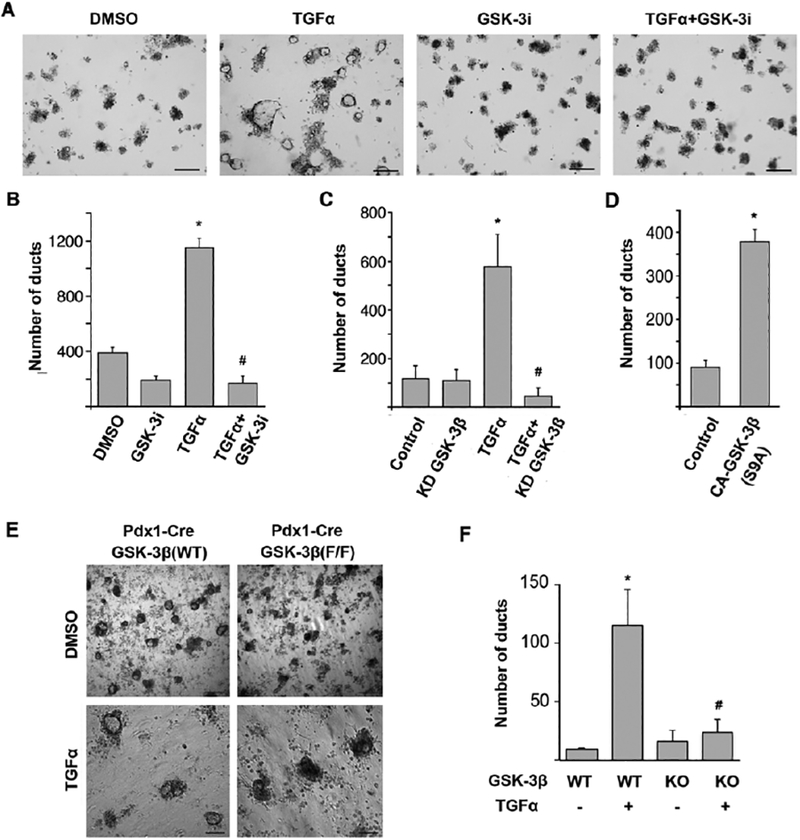
(A) Freshly isolated primary mouse acinar cells were embedded in a 3D collagen culture. Images show acinar cell clusters or ducts on day 6 following treatment with DMSO, 50 ng/ml TGF-α, 1 μM GSK-3i or 50 ng/ml TGF-α combined with 1 μM GSK-3i. Bars, 250 μm. (B) Effect of GSK-3 inhibition on TGF-α-induced ADM in vitro. Number of ducts was quantified by counting. *P<0.05 TGF-α versus DMSO. #P<0.05 TGF-α combined with GSK-3i versus TGF-α only. (C) Primary mouse pancreatic acinar cells were isolated, transduced with GSK-3β kinase-dead (KD) lentiviral particles and seeded in collagen with or without TGF-α (50 ng/ml). The number of ducts formed was enumerated on day 6 post stimulation. (D) Primary mouse pancreatic acinar cells were isolated, transduced with constitutively active GSK-3β (S9A) lentiviral particles and seeded in collagen. The number of ducts formed was enumerated on day 6 post stimulation. *P<0.05 TGF-α or constitutively active GSK-3β (S9A) versus Control. #P<0.05 TGF-α combined with kinase-dead (KD) mutant versus TGF-α only. (E) Primary acinar cells from Pdx1-cre; GSK-3β (WT) and Pdx1-cre;GSK-3βF/F (KO) mice were isolated and cultured for 6 days in collagen in the presence of DMSO or TGF-α 50 ng/ml. Representative images of ductal-like structures are shown. Bars, 250 μm. (F) Quantification of ducts formed following 6-days of stimulation. *P<0.05 TGF-α treated versus untreated WT acinar cells. #P<0.05 TGF-α treated KO acinar cells versus WT acinar cells. All experiments were repeated using acinar cells derived from at least three different mice. Data were analyzed and expressed as mean ± SEM. n = 15.
Since the GSK-3i can inhibit both GSK-3 kinases, we next sought to determine whether GSK-3β was necessary for TGF-α-induced ADM. First, using an shRNA lentivirus targeting GSK-3β, we infected isolated acinar cells and then left them unstimulated or stimulated them with TGF-α for 6 days. Significantly, knockdown of GSK-3β resulted in a 12-fold decrease in ductal formation compared to scrambled control (Fig. 2C). Furthermore, ectopic expression of a constitutively active GSK-3β (S9A) promoted ductal formation even in the absence of TGF-α (Fig. 2D). Lastly, we isolated acinar cells from Pdx1-Cre and Pdx1-Cre;GSK-3βF/F mice and examined TGF-α-induced ADM ex vivo. As shown in Figure 2E and F, acinar cells from GSK-3β knockout animals had significantly diminished duct forming ability under TGF-α stimulation compared to WT acinar cells. Overall, these data suggest that GSK-3β is an important signaling molecule involved in TGF-α-induced ADM in vitro.
Generation of Pdx1-cre;LSL-KRasG12D;GSK-3βF/F compound mouse strain
To further define the role of GSK-3β in the process of ADM in the context of the oncogenic KRas, we generated pancreas-specific GSK-3β knockout mice harboring oncogenic KRas (Pdx1-cre;LSL-KRasG12D;GSK-3βF/F). To confirm the successful deletion of GSK-3β gene in this model, we isolated protein from the pancreas of pairs of Pdx1-cre (WT), Pdx1-cre;LSL-KRasG12D (KRas), Pdx1-cre;GSK-3βF/F (KO), and Pdx1-cre;LSL-KRasG12D;GSK-3βF/F (RKO) mice. As shown in Figure 3A,B, protein extracts from both KO and RKO animals showed a substantial loss of both phosphorylated and total GSK-3β as compared to WT and KRas mice. Consistent with a prior study [32], we did not observe an upregulation of GSK-3α or β-catenin protein level upon loss of GSK-3β (Fig. 3A). Furthermore, KO and RKO animals showed neither nuclear translocation of β-catenin nor induced expression of two well known target genes (myc or CCND1) compared to WT and KRas animals (Supplemental Fig. S2A, B). IHC staining confirmed that GSK-3β was widely expressed in both endocrine and exocrine cells within the pancreas. Similar to the immunoblot results, GSK-3β staining was reduced in the KO and RKO animals (Fig. 3C). Consistent with our prior data shown in Figure 1A, we found aberrant accumulation of GSK-3β in pancreatic cancer precursor lesions in KRas mice (Figure 3C, KRas panel). Thus, we used this model to evaluate the contribution of GSK-3β to the process of ADM in vivo.
Figure 3. Deletion of GSK-3β in the pancreas of genetically engineered mice.
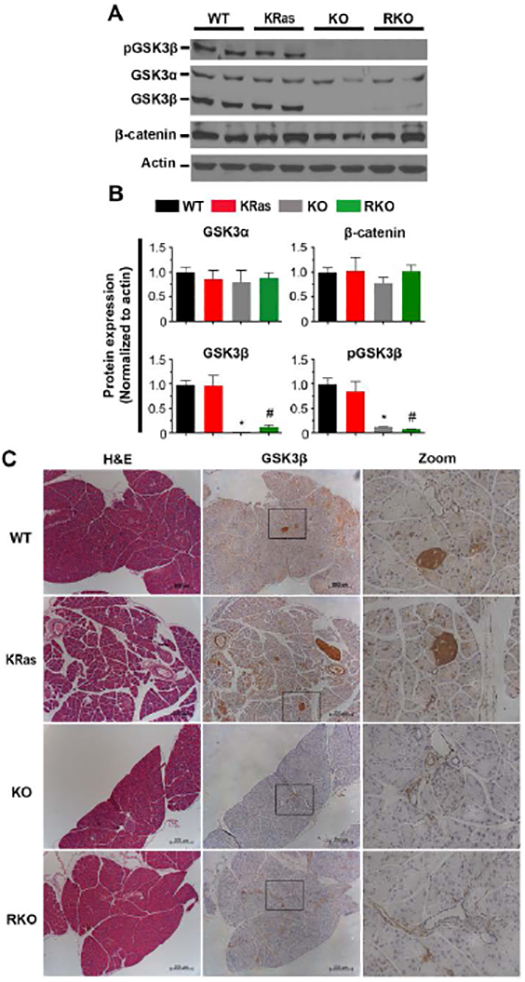
(A) Pdx1-cre, LSL-KRasG12D, GSK-3βF/F mice were used to generate mice of desired genotypes as described in the Materials and Methods. The expression of phospho-GSK-3β (Ser9), GSK-3α/β and β-catenin in pancreatic tissues was examined by immnoblot. β-actin was used as a loading control. Shown are representative results from 6 experiments. (B) The average signal intensity of phospho-GSK-3β (Ser9), GSK-3α/β, β-catenin were quantified and expressed as mean ± SEM. n=6. *P<0.05 KO versus WT mice. #P<0.05 RKO versus KRas mice. (C) H&E and IHC staining for detection of GSK-3β expression in pancreas sections of WT, KRas, KO, and RKO mice. Representative images were taken under low and high magnification lens. Bars, 200 μm.
GSK-3β is necessary for KRas-mediated ADM in vivo
Caerulein has been used to induce acute pancreatitis in various animal models harboring oncogenic KRasG12D[33]. Interestingly, in addition to preventing conversion of metaplastic ductal cells back to acinar cells following the resolution of pancreatitis, mice harboring oncogenic KRas develop more advanced PanIN lesions and ultimately PDAC[6, 33]. To test the impact of GSK-3β deletion alone or in the context of oncogenic KRas on acinar cell recovery and PanIN progression, we injected saline or caerulein as indicated in Figure 4A and harvested pancreata on day 2 and 7. Saline injected mice showed no gross pathological changes in any genotype (Fig. 4B, top panel). However, compared to WT mice, gross pathological examination revealed hallmarks of severe pancreatitis in caerulein-treated KRas mice, including a gelatinous change of the pancreas, edema and swelling by day 2 and formation of a dense desmoplastic reaction by day 7 (Fig. 4B). In contrast, there were no significant differences between KO and WT mice that had been treated with caerulein. Interestingly, the grade of pancreatitis was macroscopically reduced in RKO mice with less edema, swelling and desmoplasia at 2 and 7 days post caerulein injection as compared to KRas mice (Fig. 4B). Histologically, WT and KO mice had limited areas of pancreatitis at day 2-post caerulein injection (Fig. 4C and supplemental Fig. S3A), which mostly resolved by day 7. In contrast, KRas mice showed extensive areas of pancreatitis at day 2 and further development of ADM and PanIN regions by day 7 along with extensive desmoplasia (Fig. 4C, Ras panel). Alcian Blue staining further confirmed the reduction of ADM formation and PanIN progression in RKO mice (Supplemental Fig. S3B).
Figure 4. GSK-3β is necessary for KRas-initiated ADM in vivo.
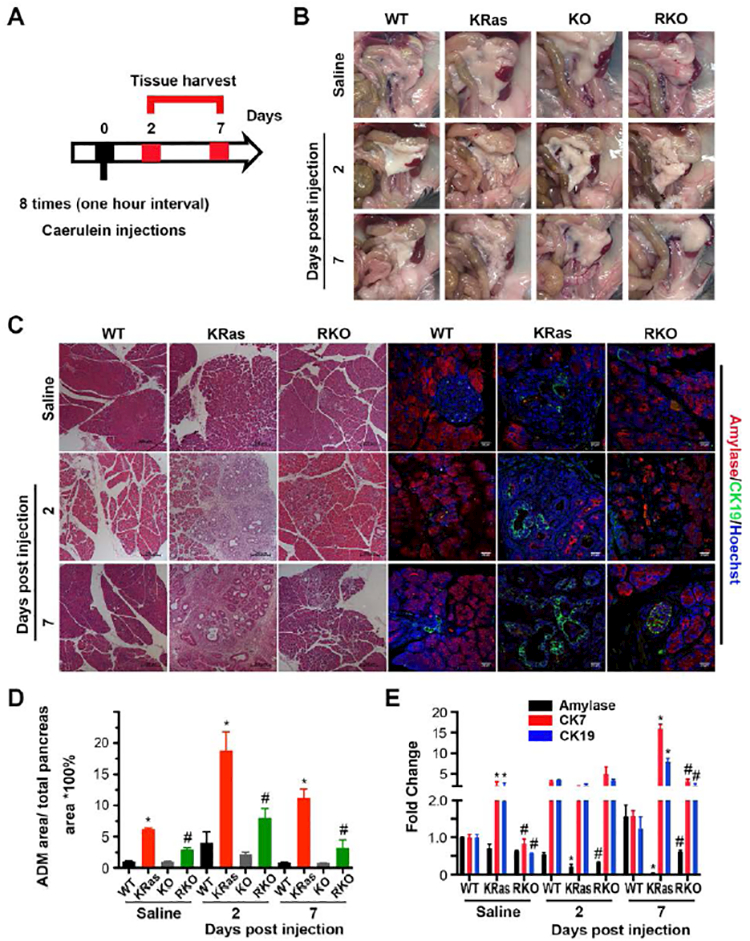
(A) Scheme for caerulein-induced acute pancreatitis model and analysis. (B) Gross pathology of pancreas and adjacent tissues from transgenic mice of indicated genotypes treated with saline or 2 and 7 days post caerulein injection. (C) H&E stained pancreatic sections from WT, KRas or RKO mice (left panel) and immunofluorescence staining of amylase and CK19 (right panel) of mice treated with saline or 2 and 7 days post caerulein injection. Nuclei were counter-stained with Hoechst (blue). (D) H&E stained tissue samples from WT (black), KRas (red), KO (grey) and RKO (green) mice, respectively, were evaluated and quantitatively analyzed for ADM area as percentage to the total area. Data were analyzed and expressed as mean ± SEM. n = 25. *P<0.05 KRas versus WT mice. #P<0.05 RKO versus KRas mice. (E) Real-time PCR quantification of pancreatic gene expression for amylase (black), CK7 (red) and CK19 (blue) from WT, KRas or RKO mice treated with saline or 2, 7 days post caerulein injection. RPLP0 and GAPDH were used as internal housekeeping gene controls. Data were analyzed and expressed as mean ± SEM. n = 5. *P<0.05 KRas versus WT mice. #P<0.05 RKO versus KRas mice.
Amylase and CK19 are two commonly used markers of acinar and ductal cells in the pancreas, respectively. The co-existence of their staining within the same area indicates the loss of acinar identity and transdifferentiation into ductal cells[11]. As determined by immunofluorescence and the expression of the ductal markers CK7 and CK19, WT and KO mice have very little to no areas of spontaneous ADM, which increases modestly in response to caerulein treatment and is mostly resolved by day 7 (Fig. 4C-E and supplemental Fig. S3A). Consistent with previous reports, KRas mice showed higher levels of spontaneous ADM in saline-treated animals, as well as a dramatic increase in ADM and desmoplasia by day 2, which continued into day 7 (Fig. 4C,D). The decrease in ADM area by day 7 in KRas mice is accounted for by the increase in PanIN lesion formation (Supplemental Figure S3C). The expression of CK7 and CK19 levels also remained elevated at day 7 consistent with the increase in ADM and PanIN areas in KRas mice (Fig. 4E). In contrast, the pancreas in saline-treated RKO mice was comprised of mostly acinar cells and importantly, fewer spontaneous ADM areas (3.0 ± 0.3 versus 6.2 ± 0.2 percentage of ADM area/total pancreas; Fig. 4C). Significantly, upon caerulein exposure, ADM areas with abnormal ductal structures were reduced in RKO mice as compared to Ras mice (7.7 ± 1.6 versus 18.8 ± 5.2 percentage of ADM area/total pancreas) (Fig. 4C,D and supplemental Fig. S3D). Seven days post treatment, ADM progression was greatly limited in RKO mice compared to KRas mice (3.2 ± 2.2 versus 11.2 ± 2.5) and more amylase positive acinar cells remained compared to CK19 and CK7-expressing ductal cells (Fig. 4C-E). Taken together, these data reveal an important role for GSK-3β in the maintenance/progression of ADM to PanIN lesion formation.
GSK-3β contributes to cell proliferation within ADM and PanIN lesions.
Normally, alpha-smooth muscle actin (αSMA) is associated with blood vessels but not ductal structures in WT mice (supplemental Figure 4A). However, αSMA becomes associated with the areas of metaplastic ducts and PanIN lesions (supplemental Figure 4A)[34]. Previous studies have shown that in the WT pancreas acinar and ductal cells are proliferatively quiescent[11, 35]. During ADM, KRas mutation activates a proliferation program resulting in a subset of cells progressing toward a duct cell phenotype. Significantly, we could observe αSMA+/Ki-67+/CK19+ ductal cells within areas of ADM from KRas mice, but not in CK19+/ αSMA- normal ductal cells (supplemental Figure 4B). As shown in Figure 5A, several Ki-67 positive cells were detected in ADM areas in KRas mice 2 days after caerulein injection with a dramatic accumulation of Ki-67 positive cells in PanIN lesions 7 days after caerulein injection. In contrast, RKO mice showed fewer Ki-67 positive cells in ADM and PanIN lesions (Fig. 5A). The percentage of Ki-67 positive ADM cells, as determined by co-staining with CK19, is significantly reduced in RKO mice compared to KRas mice at both day 2 and day 7 (Fig. 5B,C). In addition, EdU labeling, which marks cells actively synthesizing DNA, showed a significant decrease in S-phase in RKO versus KRas mice at both day 2- and day 7-post caerulein treatment (supplemental Fig. S4C, D). We did not observe an increase in the number of apoptotic cells in the ADM or PanIN lesions in RKO mice at either time point following caerulein injection (data not shown). Taken together, these data suggest that GSK-3β is required for oncogenic KRas-driven cell proliferation within ADM and PanIN regions.
Figure 5. GSK-3β contributes to proliferation of ADM cells.
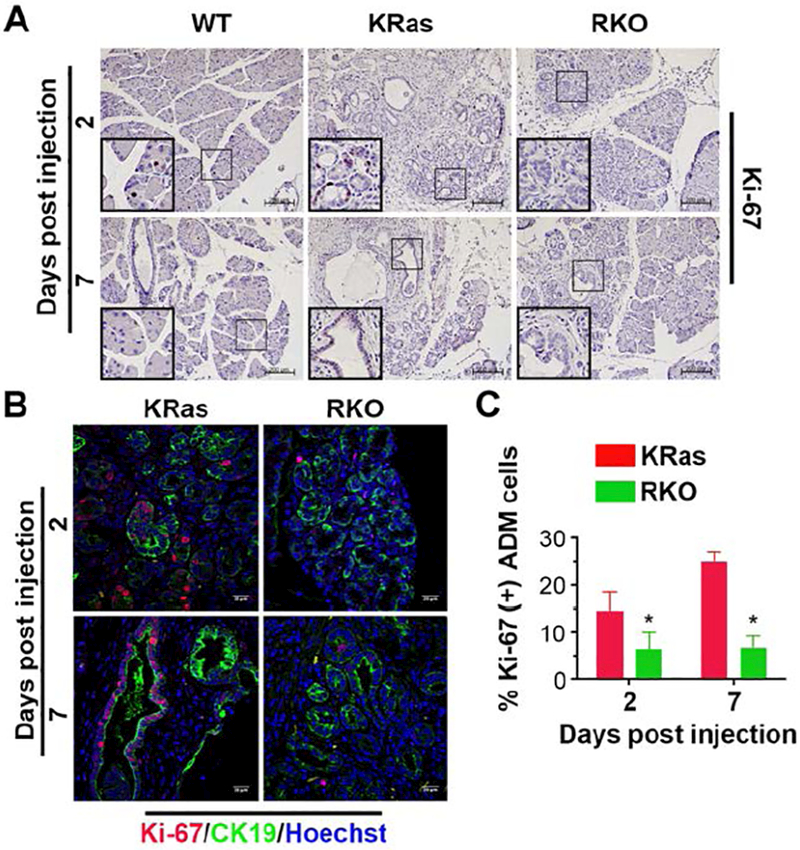
(A) IHC staining and analysis of WT, KRas and RKO mice for Ki-67 expression in the pancreas. Representative images were taken under 10× magnification lens. Bars, 200 μm. (B) Double-labeling of pancreatic sections from KRas and RKO mice was performed 2 or 7 days post caerulein injection using Ki-67 (red) and CK19 (green) antibodies. Nuclei were counter-stained with Hoechst (blue). (C) Quantification of the percentage of Ki-67 positive ADM cells in KRas and RKO mice 2 or 7 days post caerulein injection. Data were analyzed and expressed as mean ± SEM. n = 6. *P<0.05 RKO versus KRas mice.
GSK-3β facilitates proliferation through mTOR
Previous studies in breast cancer cell lines have shown that GSK-3β cooperates with mammalian target of rapamycin (mTOR) to regulate the activity of ribosomal protein S6 kinase (S6K), an important regulator of cell proliferation and growth[36]. Significantly, we observed a substantial increase in the phosphorylation of the S6K substrate S6 (pS6) from day 2 to day 7 post caerulein treatment in KRas mice compared to RKO mice (Fig. 6A). Interestingly, more of the pS6 positive cells in the KRas mice are also CK19 positive as compared to those in the RKO mice (supplemental Fig. S5A). In contrast, while pS6 and CK19 double positive staining was observed in RKO mice at day 2 and 7, the majority of pS6 positive cells were negative for CK19 and reside outside the ADM area (Fig. 6A and supplemental Fig. S5A). These data indicate that deletion of GSK-3β affects S6K activity within the KRas-driven metaplastic duct cells.
Figure 6. GSK-3β deletion decreases the levels of phospho-S6.
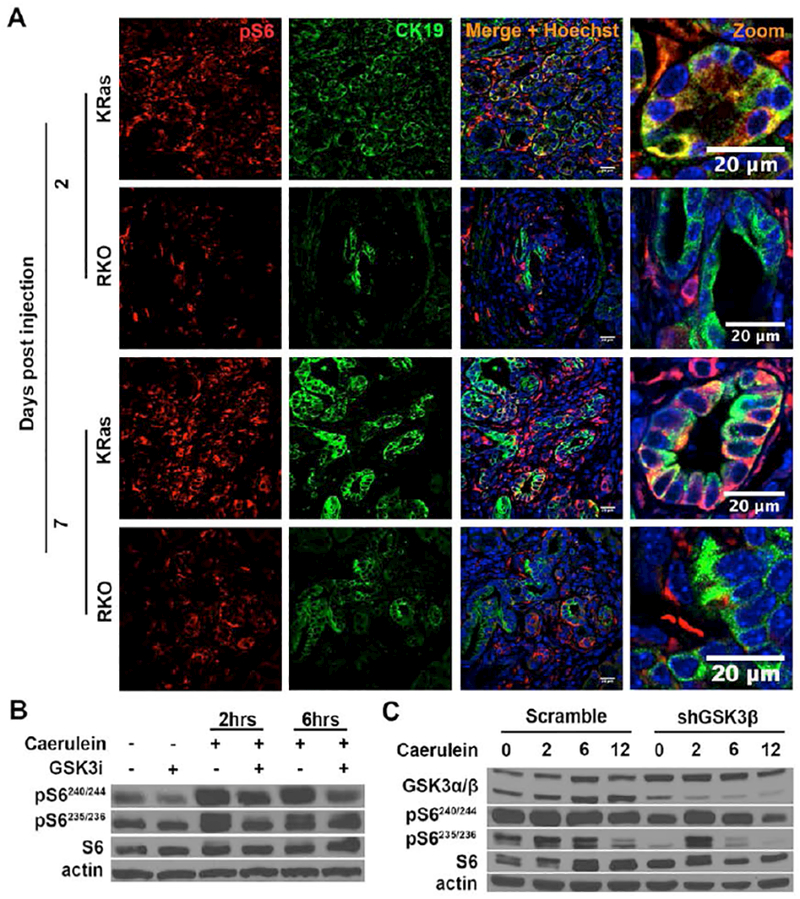
(A) Double-labeling of the pancreatic sections for pS6 (red) and CK19 (green). The pancreatic sections were derived from KRas and RKO mice 2 or 7 days post caerulein injection. Nuclei were counter-stained with Hoechst (blue). (B) AR42J cells were pre-treated with diluent (DMSO) or GSK-3i for 2 hours. They were then treated with caerulein for an additional 2 or 6 hours. Cell lysates were prepared and probed with the indicated antibodies. (C) Stable control and shGSK-3β AR42J cells were treated with caerulein for 2, 6 and 12 hours. Cell lysates were prepared and probed with the indicated antibodies. Shown are representative results from 6 independent experiments.
To further examine the role of GSK-3β in regulating S6K activity, we treated AR42J pancreatic acinar tumor cells with the GSK3i in the presence and absence of caerulein stimulation. We found greatly increased S6 phosphorylation on all four phosphorylation sites in the control group after caerulein stimulation for 2 or 6 hours. Interestingly, we detected a decrease of S6 phosphorylation after GSK-3i treatment (Fig. 6B and supplemental Fig. S5B). Similarly, knockdown of GSK-3β in AR42J cells led to reduced phosphorylation of S6 after caerulein stimulation (Fig. 6C and supplemental Fig. S5C). Taken together, these data suggest that GSK-3β regulates the activity of S6K in response to caerulein stimulation.
Discussion
The present study demonstrates a critical role for GSK-3β in pancreatitis-induced ADM and PanIN lesion formation. This general conclusion is supported by the following distinct observations: (1) The expression of GSK-3β is increased in caerulein-induced ADM areas and PanIN lesions; (2) Acinar cells from GSK-3β knockout mice form fewer ductal structures after TGF-α stimulation relative to WT acinar cells in vitro; (3) GSK-3β ablation results in a reduction of caerulein-induced ADM development and PanIN progression in KRasG12D mice; (4) GSK-3β deletion inhibits the proliferation of ADM cells driven by oncogenic KRas; and (5) GSK-3β could in part stimulate proliferation of metaplastic ductal-like cells through activation of S6K. These results highlight the role of GSK-3β in the process of ADM and suggest that GSK-3 inhibitors may prove beneficial in abrogating these early events leading to the development of pancreatic cancer.
We have previously demonstrated that GSK-3β becomes progressively over-expressed in PanIN lesions and PDAC patient samples. Consistent with a role for this kinase in PDAC, others and we have shown that genetic or pharmacologic inhibition of GSK-3β abrogates the growth of PDAC cell lines in vitro and in vivo[20, 22]. Significantly, we have previously reported that oncogenic KRas, which is present in more than 90% of PDAC cases, increases GSK-3β gene expression[24]. Since oncogenic KRas has been shown to drive ADM both in vivo and ex vivo, we reasoned that GSK-3β might be involved in altering the plasticity of pancreas cells during the process of ADM. Indeed, using the 3D explant ex vivo cell culture model we show that TGF-α-induced ADM is impaired by either pharmacologic inhibition of GSK-3, specific knockdown of GSK-3β or when acinar cells from GSK-3β conditional KO mice are used. Furthermore, we show in vivo, that GSK-3β deletion reduces the extent of ADM and PanIN lesion progression in mice expressing oncogenic KRas. These results highlight an important role for GSK-3β signaling downstream of oncogenic KRas during ADM and the formation of these preneoplastic lesions. As deletion of GSK-3β did not completely abolish ADM and PanIN lesions formation in the presence of KRas, it remains possible that GSK-3α might also be involved, but will require further experimentation.
We have previously shown that co-injection of a GSK-3 inhibitor along with caerulein in oncogenic KRas mutant mice results in a dose-dependent abrogation of pancreatitis and ADM formation[28]. GSK-3β has multiple intracellular targets including several inflammatory transcription factors including NFκB and NFATs. Importantly, induced NFAT expression during ADM contributes to the development of PDAC by driving pathways involved in inflammation, proliferation and transcription factors involved in stemness including Sox2 and Sox9[37–39], the latter factor being linked to PanIN lesion formation and the development of PDAC in the presence of oncogenic KRas. Whether GSK-3β is involved in the regulation of SOX9 during the process of ADM remains to be determined. In addition, as the role of GSK-3β in driving inflammation is well known, it remains possible that decreased expression of inflammatory signature genes in the absence of GSK-3β could impact sustained ADM development an the extent of inflammation in our model. In fact, previous studies have shown that GSK-3 inhibitors diminished inflammatory responses in experimentally induced colitis in rats[40] as well as arthritis and peritonitis in mice[41]. Future experiments aimed at identifying the transcriptional networks regulated by GSK-3β during ADM development will provide valuable insight into the role of this kinase in supporting pancreatitis and the development of PDAC.
Another feature of GSK-3β loss in our model is the significant decrease in the proliferation of duct-like cells within the ADM areas and PanIN lesions. Interestingly, while many of the Ki-67+ cells in the KRas mice are also CK19+, this is not the case in RKO mice, suggesting a critical role of GSK-3β in driving the proliferation of the metaplastic duct-like cells in the presence of oncogenic KRas. Mechanistically, we provide evidence that GSK-3β affects the activity of S6K. S6K is known to stimulate mRNA translation, protein synthesis and proliferation, but GSK-3β regulation of S6K is controversial. Inoki et al. showed that GSK-3β negatively regulated phosphorylation of S6K at T389 by activating TSC, an upstream negative regulator of mTOR[42]. However, Sejeong et al. established that GSK-3β directly phosphorylates S6K1 at S371 resulting in increased phosphorylation of its downstream substrate, S6[36]. Consistent with this, we found increased phosphorylation of S6 in CK19+ cells within the ADM and PanIN lesions of KRas mice, which was significantly lost in the RKO mice. Moreover, while pS6 positive cells were observed in the RKO mice, most of the cells containing the highest levels of pS6 were not CK19+, suggesting that GSK-3β is important to maintaining S6K activity in metaplastic duct-like cells.
In summary, our study provides evidence that GSK-3β is necessary to mediate TGF-α- and KRas mutation-induced pancreatic cellular plasticity leading to ADM and the formation of PanIN lesions. In addition, GSK-3β contributes to the proliferation of these metaplastic duct-like cells in part through the regulation of S6K activity. Thus, targeting GSK-3β pharmacologically might have clinical benefit in removing or eliminating these precursor cells during different diseased states.
Supplementary Material
Acknowledgement
This work was supported by grants from NCI Pancreatic Cancer SPORE grant CA102701 to DDB, National Natural Science Foundation of China [grant number 81472601] to JSZ and NCI CA200572 to PS. The rat pancreatic acinar tumor AR42J cells were a gift from Dr. Raul Urrutia (Mayo Clinic). Pdx1-Cre;LSL-KRasG12D mice were obtained from Dr. Baoan Ji (Mayo Clinic) and GSK-3βF/F mice were obtained from Dr. James Woodgett (Mount Sinai Hospital, Toronto, Canada).
Footnotes
Statement of author contributions
L.D.: study concept and design, data acquisition, analysis and interpretation, drafting the manuscript and critical revision of the manuscript for important intellectual content; GY.L., P.S.: contribute to data acquisition, analysis and interpretation, technical and material support; J-S.Z.: study concept and design, data acquisition, analysis and interpretation, technical support, study supervision, drafting the manuscript and critical revision of the manuscript; D.M.S.: provided key reagents, technical and material support, editing of the manuscript; D.D.B.: study concept and design, data acquisition, analysis and interpretation, drafting the manuscript, critical revision of the manuscript for important intellectual content, obtainment of funding, technical support, study supervision and guarantor of this work.
Financial disclosures: D.M.S. is CEO of Actuate Therapeutics Inc. and has ownership interest in Actuate Therapeutics Inc. D.D.B. has ownership interest in Actuate Therapeutics Inc.
The other authors disclosed no potential conflicts of interest.
References
- 1.Rahib L, Smith BD, Aizenberg R, et al. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 2014; 74: 2913–2921. [DOI] [PubMed] [Google Scholar]
- 2.Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med 2014; 371: 2140–2141. [DOI] [PubMed] [Google Scholar]
- 3.Wolfgang CL, Herman JM, Laheru DA, et al. Recent progress in pancreatic cancer. CA Cancer J Clin 2013; 63: 318–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morris JPt Wang SC, Hebrok M. KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat Rev Cancer 2010; 10: 683–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kanda M, Matthaei H, Wu J, et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology 2012; 142: 730–733 e739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guerra C, Schuhmacher AJ, Canamero M, et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 2007; 11: 291–302. [DOI] [PubMed] [Google Scholar]
- 7.Reichert M, Rustgi AK. Pancreatic ductal cells in development, regeneration, and neoplasia. J Clin Invest 2011; 121: 4572–4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hingorani SR, Petricoin EF, Maitra A, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003; 4: 437–450. [DOI] [PubMed] [Google Scholar]
- 9.Wagner M, Luhrs H, Kloppel G, et al. Malignant transformation of duct-like cells originating from acini in transforming growth factor transgenic mice. Gastroenterology 1998; 115: 1254–1262. [DOI] [PubMed] [Google Scholar]
- 10.Carriere C, Seeley ES, Goetze T, et al. The Nestin progenitor lineage is the compartment of origin for pancreatic intraepithelial neoplasia. Proc Natl Acad Sci U S A 2007; 104: 4437–4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu L, Shi G, Schmidt CM, et al. Acinar cells contribute to the molecular heterogeneity of pancreatic intraepithelial neoplasia. Am J Pathol 2007; 171: 263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gidekel Friedlander SY, Chu GC, Snyder EL, et al. Context-dependent transformation of adult pancreatic cells by oncogenic K-Ras. Cancer Cell 2009; 16: 379–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Strobel O, Dor Y, Alsina J, et al. In vivo lineage tracing defines the role of acinar-to-ductal transdifferentiation in inflammatory ductal metaplasia. Gastroenterology 2007; 133: 1999–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cano DA, Hebrok M, Zenker M. Pancreatic development and disease. Gastroenterology 2007; 132: 745–762. [DOI] [PubMed] [Google Scholar]
- 15.Collins MA, Bednar F, Zhang Y, et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest 2012; 122: 639–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jope RS, Yuskaitis CJ, Beurel E. Glycogen synthase kinase-3 (GSK3): inflammation, diseases, and therapeutics. Neurochem Res 2007; 32: 577–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci 2003; 116: 1175–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCubrey JA, Steelman LS, Bertrand FE, et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget 2014; 5: 2881–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takahashi-Yanaga F Activator or inhibitor? GSK-3 as a new drug target. Biochem Pharmacol 2013; 86: 191–199. [DOI] [PubMed] [Google Scholar]
- 20.Ougolkov AV, Fernandez-Zapico ME, Bilim VN, et al. Aberrant nuclear accumulation of glycogen synthase kinase-3beta in human pancreatic cancer: association with kinase activity and tumor dedifferentiation. Clin Cancer Res 2006; 12: 5074–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ougolkov AV, Fernandez-Zapico ME, Savoy DN, et al. Glycogen synthase kinase-3beta participates in nuclear factor kappaB-mediated gene transcription and cell survival in pancreatic cancer cells. Cancer Res 2005; 65: 2076–2081. [DOI] [PubMed] [Google Scholar]
- 22.Garcea G, Manson MM, Neal CP, et al. Glycogen synthase kinase-3 beta; a new target in pancreatic cancer? Curr Cancer Drug Targets 2007; 7: 209–215. [DOI] [PubMed] [Google Scholar]
- 23.Mishra R Glycogen synthase kinase 3 beta: can it be a target for oral cancer. Mol Cancer 2010; 9: 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang JS, Koenig A, Harrison A, et al. Mutant K-Ras increases GSK-3beta gene expression via an ETS-p300 transcriptional complex in pancreatic cancer. Oncogene 2011; 30: 3705–3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoeflich KP, Luo J, Rubie EA, et al. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 2000; 406: 86–90. [DOI] [PubMed] [Google Scholar]
- 26.MacAulay K, Doble BW, Patel S, et al. Glycogen synthase kinase 3alpha-specific regulation of murine hepatic glycogen metabolism. Cell Metab 2007; 6: 329–337. [DOI] [PubMed] [Google Scholar]
- 27.Zhang JS, Herreros-Villanueva M, Koenig A, et al. Differential activity of GSK-3 isoforms regulates NF-kappaB and TRAIL- or TNFalpha induced apoptosis in pancreatic cancer cells. Cell Death Dis 2014; 5: e1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baumgart S, Chen NM, Zhang JS, et al. GSK-3beta Governs Inflammation-Induced NFATc2 Signaling Hubs to Promote Pancreatic Cancer Progression. Mol Cancer Ther 2016; 15: 491–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liou GY, Doppler H, Necela B, et al. Macrophage-secreted cytokines drive pancreatic acinar-to-ductal metaplasia through NF-kappaB and MMPs. J Cell Biol 2013; 202: 563–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Means AL, Meszoely IM, Suzuki K, et al. Pancreatic epithelial plasticity mediated by acinar cell transdifferentiation and generation of nestin-positive intermediates. Development 2005; 132: 3767–3776. [DOI] [PubMed] [Google Scholar]
- 31.Ding L, Yin Y, Han L, et al. TSC1-mTOR signaling determines the differentiation of islet cells. J Endocrinol 2016. [DOI] [PubMed] [Google Scholar]
- 32.Doble BW, Patel S, Wood GA, et al. Functional redundancy of GSK-3alpha and GSK-3beta in Wnt/beta-catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev Cell 2007; 12: 957–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carriere C, Young AL, Gunn JR, et al. Acute pancreatitis markedly accelerates pancreatic cancer progression in mice expressing oncogenic Kras. Biochem Biophys Res Commun 2009; 382: 561–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi C, Washington MK, Chaturvedi R, et al. Fibrogenesis in pancreatic cancer is a dynamic process regulated by macrophage-stellate cell interaction. Lab Invest 2014; 94: 409–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuroki H, Hayashi H, Okabe H, et al. EZH2 is associated with malignant behavior in pancreatic IPMN via p27Kip1 downregulation. PLoS One 2014; 9: e100904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shin S, Wolgamott L, Yu Y, et al. Glycogen synthase kinase (GSK)-3 promotes p70 ribosomal protein S6 kinase (p70S6K) activity and cell proliferation. Proc Natl Acad Sci U S A 2011; 108: E1204–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hessmann E, Zhang JS, Chen NM, et al. NFATc4 Regulates Sox9 Gene Expression in Acinar Cell Plasticity and Pancreatic Cancer Initiation . Stem Cells Int 2016; 2016: 5272498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Singh SK, Chen NM, Hessmann E, et al. Antithetical NFATc1-Sox2 and p53-miR200 signaling networks govern pancreatic cancer cell plasticity. EMBO J 2015; 34: 517–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen NM, Singh G, Koenig A, et al. NFATc1 Links EGFR Signaling to Induction of Sox9 Transcription and Acinar-Ductal Transdifferentiation in the Pancreas. Gastroenterology 2015; 148: 1024–1034 e1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hofmann C, Dunger N, Scholmerich J, et al. Glycogen synthase kinase 3-beta: a master regulator of toll-like receptor-mediated chronic intestinal inflammation. Inflamm Bowel Dis 2010; 16: 1850–1858. [DOI] [PubMed] [Google Scholar]
- 41.Hu X, Paik PK, Chen J, et al. IFN-gamma suppresses IL-10 production and synergizes with TLR2 by regulating GSK3 and CREB/AP-1 proteins. Immunity 2006; 24: 563–574. [DOI] [PubMed] [Google Scholar]
- 42.Inoki K, Ouyang H, Zhu T, et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 2006; 126: 955–968. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.