

Abstract
Mitochondria are integral to cellular energy metabolism and ATP production, and are involved in regulating many cellular processes. Mitochondria produce reactive oxygen species (ROS) that can damage cellular components but that also participate in signal transduction. The kinase ATM, which is mutated in the neurodegenerative, autosomal recessive disease Ataxia-Telangiectasia (A-T), is a key player in the nuclear DNA-damage response. However, ATM also performs a redox-sensing function mediated by its ROS-dependent formation of disulfide-linked dimers. Here, we found that mitochondria-derived hydrogen peroxide promoted ATM redox dimerization. In HeLa cells, ATM dimers were localized to the nucleus and inhibited by the redox regulatory protein thioredoxin 1 (TRX1), suggesting the existence of a ROS-mediated, stress-signaling relay from mitochondria to the nucleus. ATM dimer formation did not affect its association with chromatin in the absence or presence of nuclear DNA damage, consistent with separation of its redox and DNA-damage signaling functions. Comparative analysis of U2OS cells expressing either wild-type ATM or the redox-sensing-deficient C2991L mutant revealed that one function of ATM redox-sensing is to promote glucose flux through the pentose phosphate pathway (PPP) by increasing the expression and activity of glucose-6-phosphate dehydrogenase (G6PD), thereby increasing cellular antioxidant capacity. The PPP produces the coenzyme NADPH needed for a robust antioxidant response, including the regeneration of TRX1, indicating the existence of a regulatory feedback loop involving ATM and TRX1. We propose that loss of the mitochondrial ROS-sensing function of ATM may cause cellular ROS accumulation and oxidative stress in A-T.
Introduction
Ataxia-Telangiectasia (A-T) is an autosomal recessive disease, pathology of which includes cerebellar neurodegeneration, cancer susceptibility, hypersensitivity to ionizing radiation and immunodeficiency (1). A-T is caused by loss-of-function mutations in the gene encoding ATM (A-T mutated), a serine/threonine kinase belonging to the family of phosphatidylinositol 3-kinase-related kinases (PIKKs) (1, 2). ATM mediates nuclear DNA-damage signaling and DNA double-strand break (DSB) repair and can promote cell cycle arrest and apoptosis (1, 2). These activities are mediated by its phosphorylation of a large number of substrates, including the histone variant H2AX, the transcription factor p53, and Checkpoint kinase 2 (CHK2) (1–3). When activated by DNA damage, ATM is converted from a noncovalent, inactive dimer into monomers that are recruited to DSBs by the MRE11/RAD50/NBS1 (MRN) complex (4). In addition to defective nuclear DNA damage responses, A-T is also associated with oxidative stress that is thought to contribute to disease pathology (1, 5, 6). For example, higher levels of reactive oxygen species (ROS) contribute to increased lipid oxidation (1) and defects in neuronal cell proliferation (8, 9), hematopoietic stem cell (HSC) exhaustion (10), and impaired B and T cell function (11, 12). The cause of the oxidative stress in A-T remains uncertain, but could be related to the redox-sensing function of ATM that was initially identified by Paull and colleagues (13) and is conserved in yeast (14). When activated by oxidative stress (such as in the presence of increased hydrogen peroxide), ATM forms disulfide-linked dimers that can phosphorylate some of its canonical substrates (such as CHK2), but signals in a manner distinct from the DNA-damage response (13, 15) and important for other downstream functions, including proteostasis (16). There are multiple disulfide linkages in the redox-sensing ATM dimer and mutation of one of the cysteines involved to a leucine (C2991L) prevents redox signaling without affecting DNA-damage signaling (13, 16). Thus, analysis of this mutant provides the ability to dissect downstream consequences of the redox-sensing function of ATM (16), which we took advantage of in this study.
The pentose phosphate pathway (PPP) is alternate route of glucose oxidation from glycolysis that generates nicotinamide adenine dinucleotide phosphate (NADPH) and ribose-5-phosphate. This pathway is essential for cellular redox homeostasis and DNA repair because NADPH is a reducing agent critical for maintaining the active, reduced forms of glutathione and thioredoxin needed for cellular antioxidant defenses and ribose-5-phosphate is a precursor for nucleotide biosynthesis needed for DNA repair (17). ATM is linked to the PPP through activation of the rate-limiting enzyme, glucose-6-phosphate dehydrogenase (G6PD). This occurs by ATM-mediated phosphorylation of heat shock protein 27 (HSP27), which binds to and activates G6PD (18). Whether the redox-sensing function of ATM is related to its activation of cellular antioxidant defenses is an open question, but if so this could represent a cellular antioxidant defense mechanism triggered by ROS signaling.
Multiple lines of evidence point to dysfunction of mitochondria as a component of A-T. As sites of the electron transport chain and other redox reactions (19), mitochondria are a major source of ROS and hence likely relevant to the oxidative stress in A-T. The initial observations of mitochondrial defects in A-T patient cells were aberrant mitochondrial morphology and respiration in lymphoblastoid cells (20) and mitochondrial DNA (mtDNA) instability, downstream of defects in ribonucleotide reductase, in primary fibroblasts (21). Subsequently, A-T thymocytes were shown to have inefficient mitophagy, which in turn results in oxidative stress and ATP depletion due to accumulation of defective mitochondria (22). ATM also induces mitochondrial biogenesis through adenine-monophosphate-activated protein kinase (AMPK) in response to etoposide-induced DNA damage (23). Antioxidants have beneficial effects on cancer development and HSC defects in the Atm-null mouse models of A-T (24–26), and we previously showed that reducing mitochondrial hydrogen peroxide, by targeting the antioxidant enzyme catalase to mitochondria (mCAT), prolongs mean survival and partially rescues HSC and other immune system defects (27). Finally, we have also previously shown that reduced signaling through the nutrient-sensing mechanistic target of rapamycin complex 1 (mTORC1) pathway leads to mitochondrial ROS-mediated changes in nuclear gene expression that are dependent on Tel1p and Rad53p, the yeast homologs of ATM and CHK2, but independently of their role in the nuclear DNA damage response (14). Activation of this pathway resulted in an adaptive response that led to increased stress resistance and longevity, which is fully recapitulated by transient exposure to low doses of the redox-cycling compound menadione that increases mitochondrial superoxide (28). This is one salient example of mitochondria as signaling organelles with ROS as the proximal messenger, which has been observed in many other physiological contexts (29, 30). In this study, we examined the potential role for mammalian ATM in mitochondrial ROS signaling and cellular antioxidant responses.
Results
ATM senses mitochondrial hydrogen peroxide
Using low doses of the redox-cycling chemical menadione to generate superoxide in mitochondria, we showed previously that the yeast homolog of ATM (Tel1p) is responsive to mitochondrial ROS and induces adaptive changes in nuclear gene expression that increase longevity (14, 28). This response was separate from the role of Tel1p in nuclear DNA damage signaling. Mammalian ATM senses ROS by forming disulfide-linked dimers, but the cellular sources of ROS to which it is responsive have not been elucidated. Thus, to test for the involvement of mitochondrial ROS, we induced their production in HeLa cells using menadione. This resulted in increased mitochondrial ROS, as judged by MitoSOX staining (Figure 1A) and promoted ATM dimer formation, which was probed in non-reducing, SDS-PAGE gels (Figure 1B, lane 2). Mitochondrial ROS and ATM dimer formation under these conditions were inhibited by the addition of the mitochondrial antioxidant Mito-TEMPO (Figures 1A and 1B), consistent with mitochondrial ROS sensing. Menadione also induced cellular ROS and ATM dimer formation in primary mouse embryonic fibroblasts (MEFs), and this was inhibited in MEFs isolated from mice that have the hydrogen peroxide-scavenging enzyme catalase targeted to mitochondria (Figure 1C and 1D). These data indicate the menadione-generated mitochondrial superoxide is converted to hydrogen peroxide that can exit mitochondria and promote ATM dimerization.
Figure 1. Mitochondrial ROS promote redox-dependent ATM dimerization.
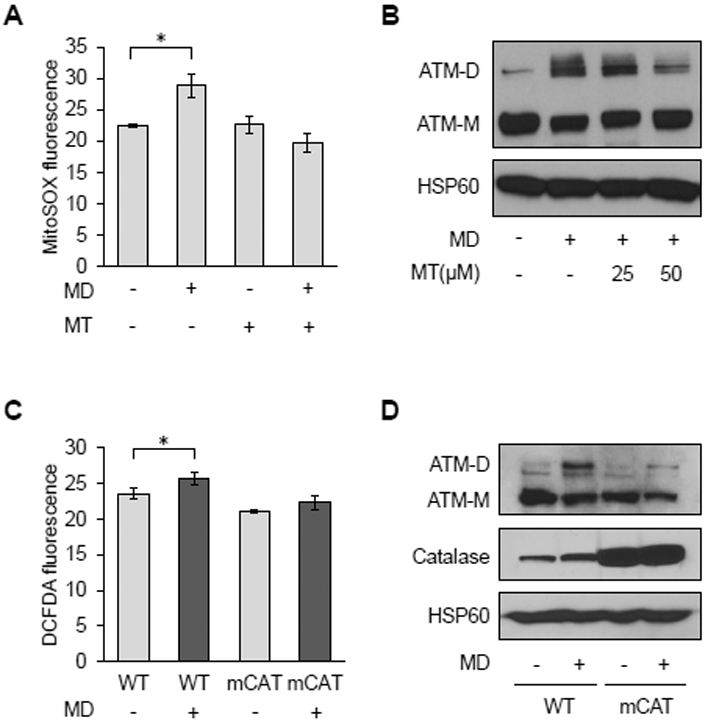
A, MitoSOX staining by flow cytometry in HeLa cells treated with vehicle (−) or 50 μM Mito-Tempo (MT; +) for 24 hours prior to being treated with vehicle (−) or 20 μM menadione (MD; +) for 30 minutes. Data are mean fluorescence intensities ± SD of biological triplicates. * p<0.05 by Student’s t-test. B, Western blot analysis of HeLa cells treated with vehicle (−) or 25 or 50 μM Mito-Tempo (MT;+) for 2 hours before treatment with vehicle (−) or 20 μM MD (+) for 30 minutes. ATM monomers (ATM-M) and disulfide-linked dimers (ATM-D) were resolved by non-reducing SDS-PAGE. Mitochondrial HSP60 was probed as a loading control. C, Primary MEFs from wild-type (WT) or mitochondrial catalase overexpressing (mCAT) mice treated with vehicle or 20 μM MD for 30 minutes were analyzed for cellular ROS using DCFDA and flow cytometry as described in A. D, Western blots of ATM and HSP60 of MEFs treated as in C were performed as described in B, except catalase was also probed to demonstrate its overexpression. Blots are representative of three experiments.
Inhibition of mitochondrial respiration can also lead to increased mitochondrial ROS production. However, treating HeLa cells with electron transport chain inhibitors, rotenone or antimycin A, at concentrations that increased mitochondrial superoxide to similar extent as menadione (Figure 2A), did not result in ATM dimerization (Figure 2C). Using MitoPY1, a fluorescent probe that senses mitochondrial hydrogen peroxide (31), we observed that menadione increased mitochondrial hydrogen peroxide whereas rotenone and antimycin A did not (Figure 2B). Peroxiredoxin 1 (PRDX1) is an antioxidant enzyme localized in the cytosol and nucleus that reacts with hydrogen peroxide (32, 33). Similar to the MitoPY1 results, we found that PRDX1 was further oxidized to disulfide bond-linked dimers by menadione, but not rotenone or antimycin A (Figure 2C). These results demonstrate that mitochondrial superoxide must be converted to hydrogen peroxide in order to gain access to the cytosol and/or nucleus to dimerize ATM and other redox reactive proteins.
Figure 2. Conversion of mitochondrial superoxide to membrane-permeable hydrogen peroxide is necessary for ATM dimerization.
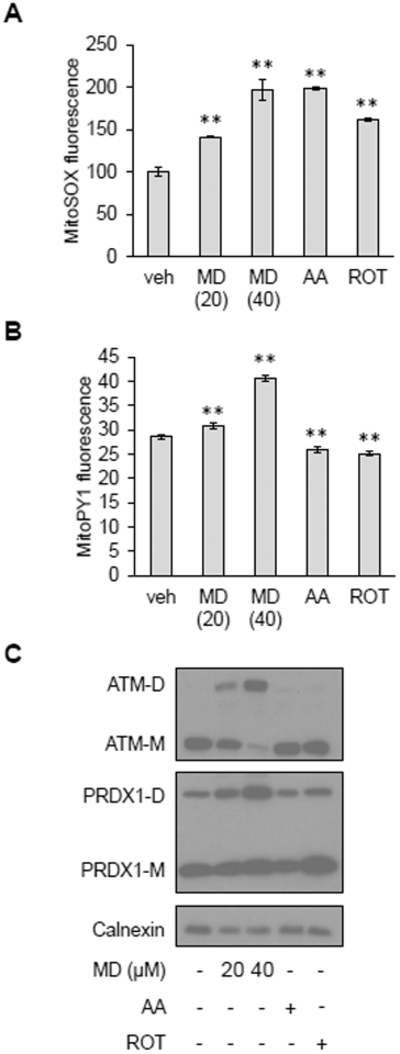
A, MitoSOX staining in HeLa cells treated with vehicle (veh), 20 or 40 μM MD, 2 μM antimycin A (AA) or 0.4 μM rotenone (ROT) for 30 minutes. Data are mean fluorescence intensities ± SD of biological triplicates. ** p<0.01 by Student’s t-test. B, MitoPY1 staining in HeLa cells treated with vehicle (veh), 20 or 40 μM MD, 2 μM antimycin A (AA) or 0.4 μM rotenone (ROT) for 30 minutes. Plotted is the mean fluorescence intensity ± SD of biological triplicates. ** as in (A). C, Western blot analysis of HeLa cells treated with vehicle (−), 20 or 40 μM MD, 2 μM antimycin A (AA) or 0.4 μM rotenone (ROT) for 30 minutes. ATM monomers (ATM-M) and dimers (ATM-D), PRDX1 monomers (PRDX1-M) and dimers (PRDX1-M) were resolved by non-reducing SDS-PAGE. Calnexin was probed as a loading control. Blots are representative of three experiments.
ATM redox dimers are localized to the nucleus, but do not inhibit DNA-damage signaling.
Paull and colleagues have shown in several contexts that the redox and DNA-damage sensing functions of ATM are distinct (13, 16). Consistent with this, in HeLa cells, menadione increased ATM-dependent phosphorylation of CHK2, but not γH2AX (Figure 3A), indicating ATM activation by mitochondrial ROS is not the result of nuclear DNA DSBs. Conversely, inducing nuclear DNA damage with topotecan, a topoisomerase I inhibitor, led to phosphorylation of CHK2 and H2AX but not dimerization of ATM (Fig. 3A). We next investigated the cellular localization of ATM and ATM dimers. In different cell types, ATM has been reported to localize to the cytoplasm, mitochondria and peroxisomes (22, 34–36). In HeLa cells, we found that both ATM monomers and dimers resided primarily in the nucleus, based on biochemical fractionation studies (Figure 3B). Since nuclear ATM is recruited to chromatin after DNA damage and we found ATM dimers in the nucleus, we hypothesized that ATM dimerization might interfere with its recruitment to chromatin. However, when we treated HeLa cells with menadione and topotecan to induce mitochondrial ROS and DNA damage simultaneously, we observed that ATM dimerization did not affect the ability of ATM to bind to chromatin basally or in the presence of DNA damage (Figure 3C). This again indicates that redox signaling and DNA-damage signaling by ATM are largely independent and that mitochondrial hydrogen peroxide signaling can promote ATM dimerization in the nucleus.
Figure 3. Mitochondrial hydrogen-peroxide-induced ATM dimers are in the nucleus, but do not interfere with ATM chromatin association.
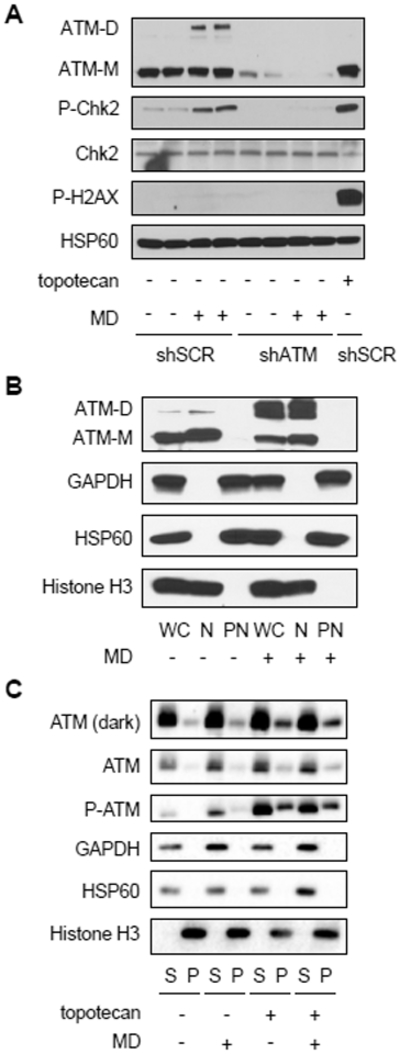
A, Western blot analysis of HeLa cells expressing shRNA targeting a scrambled sequence (shSCR) or ATM (shATM) treated (+) with 20 μM menadione (MD) or vehicle (ethanol, −) for 30 minutes or 0.75 μM topotecan for 1 hour as indicated. ATM dimer (ATM-D), ATM monomer (ATM-M), CHK2, phosphorylated CHK2 Thr68 (P-CHK2) and phosphorylated H2AX Ser139 (P-H2AX) were probed, along with HSP60 as a loading control. B, HeLa cells treated with vehicle (ethanol, −) or 30 μM MD (+) for 1 hour were separated into nuclear (N) and post-nuclear (PN) fractions which were analyzed by western blot along with a cell-equivalent amount of whole-cell (WC) lysate. ATM monomer (ATM-M), ATM dimer (ATM-D), histone H3, GAPDH and HSP60 were analyzed. C, HeLa cells treated with vehicle (−) or 20 μM MD (+) for 30 minutes and/or 0.75 μM topotecan for 1 hour were subjected to salt extraction of chromatin-bound proteins. An equivalent volume of the supernatant fraction (S) and the pellet (P) fraction (containing chromatin-bound proteins) was loaded for western blot. ATM, phosphorylated ATM Ser1981 (P-ATM), histone H3, GAPDH and HSP60 were analyzed. Blots are representative of two experiments.
Thioredoxin 1 (TRX1) reduces ATM dimers
TRX1 is a redox-responsive enzyme found in the nucleus and cytoplasm that reduces protein disulfide bonds (37). Since we found that ATM dimers are mostly in the nucleus in HeLa cells, and that their formation can be mediated by mitochondrial hydrogen peroxide, we asked if TRX1 is involved in regulating this pathway. TRX1 knock-down in HeLa cells led to increased ATM dimerization basally (Figure 4A) and greater dimer formation at lower concentrations of menadione (Figure 4A). In the absence of menadione, TRX1 knock-down had no effect on cellular ROS abundance but did exacerbate menadione-induced ROS production (Figure 4B). To better characterize the effect of TRX1 on ATM dimers, we examined their stability in the presence and absence of TRX1 knock-down. To do this we induced the formation of dimers with menadione, then changed the media to that without menadione and monitored their rate of decay. In the presence of TRX1, ATM dimers exhibited a half-life of ∼30 minutes, whereas TRX1 knock-down significantly increased the half-life of ATM dimers (Figure 4C). In this experiment, menadione-induced ROS returned to baseline abundance within 30 minutes after the media change in both control and TRX1-knock-down cells (Figure S1A). In addition, oxidized PRDX1 and PRDX2 are reported to promote protein oxidation through redox relays (38, 39), leading us to examine their potential role in ATM dimerization. Knocking down PRDX1 and PRDX2 by siRNA (Figure 4D) or knocking them out using CRISPR (Figure S1B) failed to prevent ATM dimerization induced by menadione, suggesting that PRDX1 and PRDX2 are not required.
Figure 4. Thioredoxin 1 negatively regulates ATM redox dimerization.
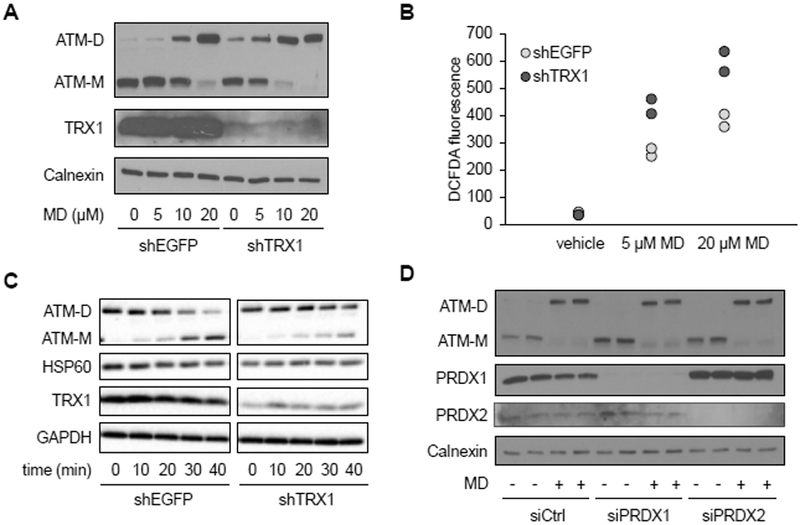
A, Western blot analysis of HeLa cells expressing shRNA targeting EGFP (shEGFP) as a negative control or thioredoxin 1 (shTRX1) treated with vehicle or the indicated concentration of MD for 30 minutes. Blot was probed for ATM monomer (ATM-M), ATM dimer (ATM-D), and TRX1, with calnexin as a loading control. Blots are representative of two experiments. B, DCFDA staining by flow cytometry, in cells treated with vehicle, 5 μM or 20 μM MD as described in A. Plot is representative of three experiments utilizing biological duplicate samples in each. C, Western blot of HeLa cell treated with 20 μM MD for 30 minutes, after which the media was changed and cells were allowed to recover for 0, 10, 20, 30, 40 minutes as indicated. Blot was probed for ATM monomer (ATM-M), ATM dimer (ATM-D) and TRX1, with HSP60 and GAPDH as loading controls. Blots are representative of three experiments. D, Western blot of HeLa cells transfected with control or siRNA targeting PRDX1 and PRDX2 for 72 hours and treated with vehicle (−) or 20 μM MD (+) for 30 minutes. Blot was probed for ATM monomer (ATM-M), ATM dimer (ATM-D), PRDX1 and PRDX2, with calnexin as a loading control. Blots are representative of three experiments.
ATM redox signaling activates the pentose phosphate pathway through increased expression of glucose-6-phophate dehydrogenase
We next wondered why ATM might sense mitochondrial ROS and reasoned that it might be part of a cellular antioxidant response. The pentose phosphate pathway (PPP) is an alternate route of glucose metabolism that generates NADPH needed to maintain cellular antioxidant responses and to supply ribose-5-phosphate for nucleotide synthesis and DNA repair (17). ATM is known to activate the PPP under DNA damage conditions through its phosphorylation of HSP27, a positive regulator glucose-6-phosphate dehydrogenase (G6PD) (18), the rate-limiting enzyme in the pathway. Thus, we hypothesized that the redox-sensing function of ATM might also be involved in regulating the PPP, perhaps by a similar mechanism. To test this, we used a U2OS cell system devised by Paull and colleagues (16), that enabled comparison of cells expressing either wild-type (WT) ATM or the C2991L mutant that is deficient in redox signaling (13). In this system, endogenous ATM is knocked down by shRNA and replaced by the WT or mutant protein (for further details, see the Materials and Methods). In these cells, we observed robust knock-down of endogenous ATM and similar expression of WT and C2991L ATM protein expressed ectopically (Figure S2A). Under these conditions, relative to cells expressing the C2991L mutant, those expressing WT ATM had significantly lower DCFDA staining in the presence and absence of menadione (Figure 5A) and maintained lower MitoSOX staining in the presence of menadione (Figure 5B). These data indicate that ATM redox signaling is needed to maintain lower cellular and mitochondrial ROS abundance. We next asked whether this was due to lack of HSP27 phosphorylation; however, this was not the case (Figure S2B). Instead, we observed higher G6PD activity in cells expressing WT ATM compared to the C2991L mutant (Figure 5C), that correlated with higher levels of G6PD mRNA and protein (Figure 5, D and E).
Figure 5. Redox sensing by ATM upregulates antioxidant capacity and G6PD expression.
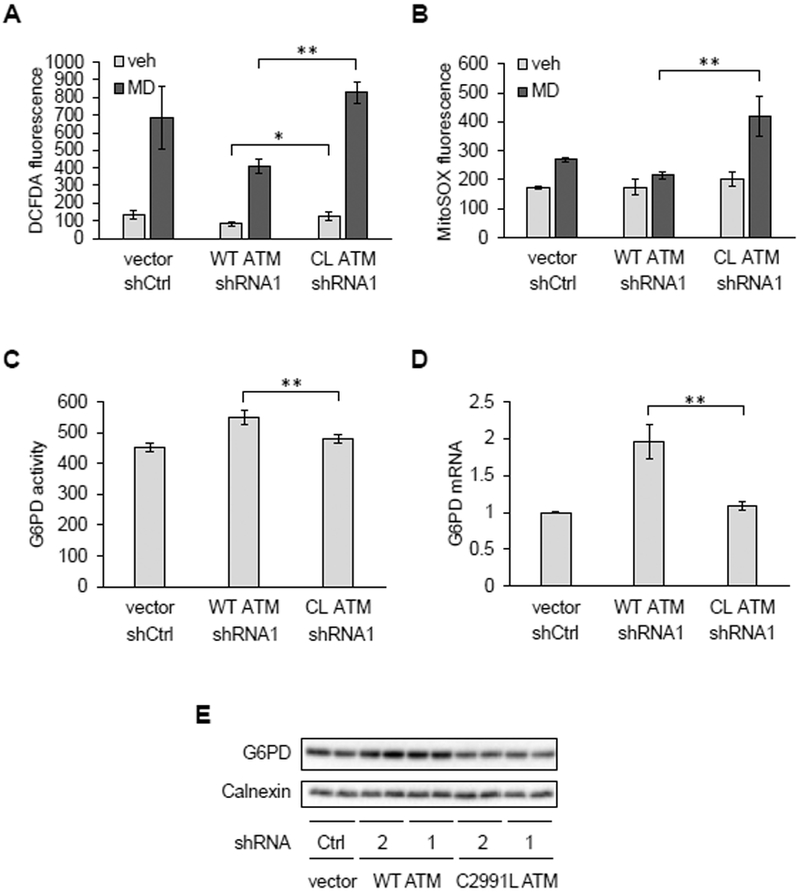
A and B, Analysis of U2OS cells transfected with empty vector (vector) and control shRNA (shCtrl) or overexpressing wild-type (WT) or the C2991L (CL) ATM mutant while having their endogenous ATM knocked down by shRNA targeting ATM (shATM1) were treated with vehicle (veh) or 20 μM MD for 30 minutes. Cellular and mitochondrial ROS were then measured by DCFDA (A) or MitoSOX (B) staining and flow cytometry. Plotted is the mean fluorescence intensity ± SD of biological triplicates. * p<0.05 and ** p<0.01 by Student’s t-tests. C, Glucose-6-phosphate dehydrogenase (G6PD) activity. Plotted is the mean ± SD of technical quadruplicates from a representative of three experiments. ** as in (A). D, G6PD mRNA abundance by qPCR. Plotted is the mean ± SD of biological triplicates. ** as in (A). E, Western blot of G6PD protein. Here, endogenous ATM was knocked down by one of two shRNAs (1 and 2). Calnexin was probed as a loading control. Blots are representative of three experiments.
To further validate this new mode of PPP regulation by ATM, we performed metabolic tracing experiments with 13C1,2,3-glucose in U2OS cells expressing WT or C2991L mutant ATM. This approach can distinguish metabolic flux through either glycolysis (also known as the Embden-Meyerhoff-Parnas pathway, or EMP) or the PPP. Increased relative flux through the PPP is expected to generate more hexose and pentose phosphate intermediates and byproducts of the PPP (Figure 6A). At 6 and 24 hours after adding 13C1,2,3-glucose, more light and heavy (M+2) ribose phosphate (and pentose phosphate isobaric isomers) as well as higher 6-phosphogluconate/glucose-6-phosphate ratio were observed in the U2OS cells expressing WT ATM compared to the C2991L mutant (Figure 6B–C), indicating more flux through the PPP in the WT cells.
Figure 6. Cell deficient in ATM redox signaling flux less glucose through the PPP and have increased total glutathione.
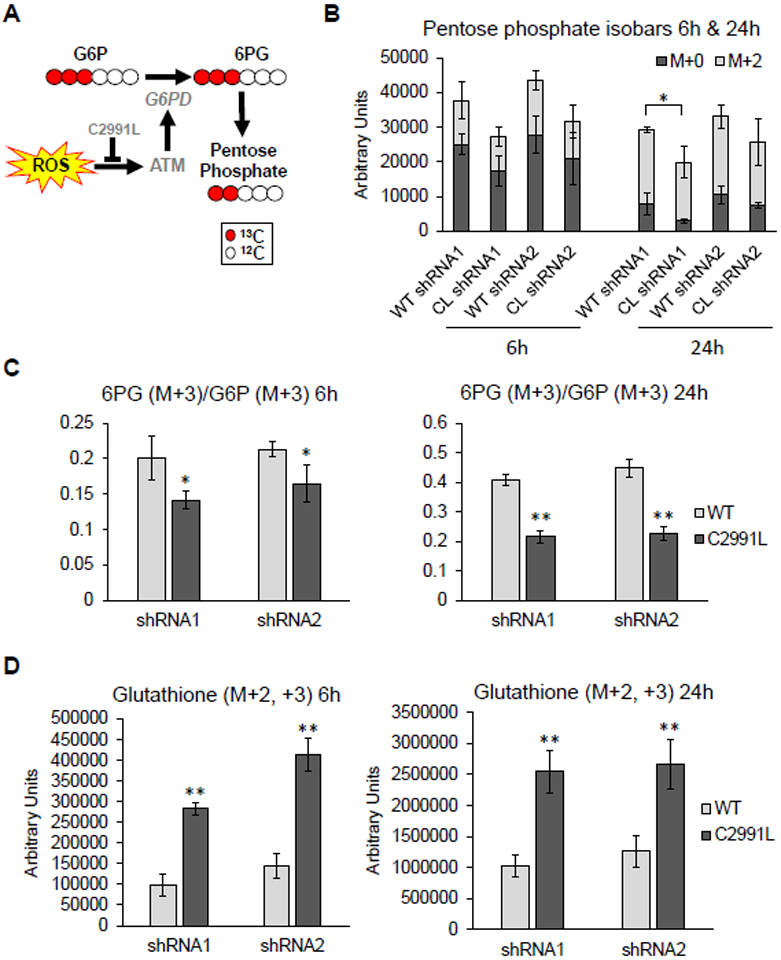
A, G6PD activity limits metabolic fluxes through the PPP, which generates pentose phosphate compounds. Generation of these compounds was greater in cells expressing the WT allele of ATM. B, Amount of light (M+0) and heavy (M+2) pentose phosphate isobars 6 and 24 hours after cells were labeled with heavy glucose in U2OS cells expressing either WT or C2991L mutant (CL) ATM after endogenous ATM knockdown by each of two shRNAs (1 and 2). Plotted is the mean ± SD of biological triplicates. C, Ratio of the G6PD substrate [glucose 6-phosphate (G6P)] and its downstream product 6-phosphogluconate (6PG) were measured and plotted as described in (B). D, Amount of heavy glutathione plotted as described in (B). * p<0.05 and ** p<0.01 by Student’s t-tests.
Catabolism of 13C1,2,3-glucose generates distinct isotopologues of lactate, either M+2 and M+3, depending on whether glucose oxidation preferentially occurs through the PPP or the EMP glycolytic pathway (Figure S3A). We observed increased glucose consumption and lactate generation through glycolysis in the C2991L mutant cells (Figure S3B–C). These cells also exhibited increased shuttling of glucose-derived heavy carbons into the TCA cycle (Figure S4). Lastly, our metabolic flux analysis revealed that C2991L cells produced more heavy glutathione than WT ATM cells (Figure 6D), suggesting an increased rate of glutathione synthesis. This was confirmed by the higher steady-state levels of glutathione in the C2991L cells (Figure S5A). However, despite the higher total level of glutathione, there was no difference in the GSH/GSSG ratio (Figure S5B). Altogether, these data demonstrate a role of ATM redox sensing in the shunting of glucose catabolism towards the PPP for redox and anabolic purposes and that there is a disruption of glutathione homeostasis when this function of ATM is lost.
Discussion
In addition to its role in nuclear DNA DSB sensing and repair, ATM has a redox-signaling function that is initiated by ROS-mediated disulfide-linked dimer formation (2, 13). Here, we found that mitochondria are a physiological source of hydrogen peroxide that can elicit this signaling mode. The primarily nuclear localization of ATM in HeLa cells, as well as the requirement for induction of cellular hydrogen peroxide, indicates that the signaling mechanism could be direct oxidation of ATM by mitochondrial hydrogen peroxide that gains access to the cytoplasm and/or nucleus (40). This could occur from perinuclear mitochondria, which have been implicated in redox signaling via diffusion of hydrogen peroxide to the nucleus (41). Alternatively, redox signaling via hydrogen peroxide can also be facilitated by enzymes that chemically modify specific cysteine residues in target proteins or that regulate disulfide bond formation (42). For example, PRDX2 facilitates hydrogen peroxide-mediated disulfide bond formation in the nuclear transcription factor STAT3 (38). However, while PRDX1 and PRDX2 appear not to have a role dimerization of ATM based on our results, TRX1 is a negative regulator of this process. The involvement of TRX1 might provide a link between mitochondrial hydrogen peroxide in the cytoplasm and dimer formation in the nucleus since it is located in both compartments (43). Thus, mitochondrial hydrogen peroxide could result in reduced nuclear accumulation of TRX1 to allow formation of ATM dimers. Alternatively, a negative regulator of TRX1 could enter the nucleus in response to mitochondrial hydrogen peroxide to inhibit nuclear TRX1 and induce ATM dimers. For example, TXNIP is a negative regulator of TRX1 (and TRX2) that appears to shuttle between the nucleus, cytoplasm and mitochondria (44, 45). Thus, altered TXNIP localization dynamics could be involved in regulation of nuclear TRX1 and hence ATM dimerization.
Despite the independence of DNA-damage signaling and redox-sensing functions of ATM, the redox-sensing role of ATM does make teleological sense with its ultimate goal of protecting the genome. That is, we propose that ATM likely senses an increase in mitochondrial ROS as a potential threat to DNA (nuclear and mitochondrial) and enacts preemptive signaling to detoxify ROS and prime pathways needed to repair the genome. ATM regulates the expression or activity of several antioxidant enzymes including superoxide dismutase 1 and 2, catalase, glutathione peroxidase 1 (46), and the rate-limiting enzyme in the PPP, glucose-6-phosphate dehydrogenase (G6PD) (18). Ionizing radiation activates the PPP through ATM-dependent phosphorylation of HSP27, which promotes its binding to and activation of G6PD (18). The PPP provides NADPH needed for many antioxidant pathways and produces ribose-5-phosphate for nucleotide synthesis needed for DNA damage repair (17). However, our results show the redox-sensing function of ATM activates the PPP via expression of G6PD instead of by modulating HSP27 phosphorylation. While the precise mechanism of gene activation was not elucidated, transcription factors that regulate G6PD transcription, including Nrf2, SP1, CREB and SREBP1 (47–50), could be targets of ATM redox sensing. Likewise, the regulation of histone methylation by ATM (51) might activate the G6PD promoter. It is noteworthy that a major downstream consequence of PPP activation and increased cellular NADPH is enhanced regeneration of reduced thioredoxins. Since TRX1 reduces ATM dimers, a negative feedback loop might exist to modulate redox signaling by ATM. Our metabolic tracing experiments confirmed increased glucose flux through the PPP in WT ATM versus the C2991L mutant cells and the WT cells also showed less shunting of glucose-derived heavy carbon atoms into glycolysis and the TCA cycle. This places ATM at critical juncture to regulate central carbohydrate metabolism via its ability to sense mitochondrial hydrogen peroxide or other cellular sources of ROS. We also observed increased glutathione synthesis in the ATM C2991L mutant cells that we speculate is being upregulated in an attempt to compensate for the lower flux through the PPP and hence less NADPH available to recycle reduced glutathione. There are several points where glutathione synthesis might be activated in the C2991L mutant cells. For example, the rate-limiting enzyme of glutathione synthesis, glutamate cysteine ligase (GCL), is activated by oxidative stress at the transcriptional and post-translational level (52). The expression of the catalytic and modifier subunit of glutathione cysteine ligase (GCLC and GCLM) is under control of the ROS-responsive transcription factor Nrf2 (52). In addition, oxidative stress promotes the interaction of GCLC and GCLM, which increases enzyme activity (52, 53).
Mitochondrial dysfunction and oxidative stress are now recognized as features of A-T (1, 54, 55), the disease caused by loss-of-function mutations in ATM. Our results strongly suggest that these phenotypes are linked through the loss of the redox-sensing function of ATM. In the absence of this function, we propose that cells are unable to convey a homeostatic ROS signal from mitochondria to ATM to upregulate antioxidant defenses, including activation of the PPP through G6PD gene expression. This, in turn, would lead to accumulation of ROS and oxidative damage to mitochondria and ot likely enhance the oxidative stress phenotype in A-T. We propose the oxidative stress in A-T is precipitated initially by loss of the mitochondrial hydrogen peroxide signaling function of likely enhance the oxidative stress phenotype in A-T. We propose the oxidative stress in A-T is precipitated initially by loss of the mitochondrial hydrogen peroxide signaling function of ATM that normally would have quelled toxic ROS accumulation and prevented cellular oxidative stress. Reducing mitochondrial hydrogen peroxide in mice can reduce cancer lethality and partially rescue immune phenotypes in the mouse model of A-T (27). Thus, in the absence of gene therapy for the disease, potential therapeutic strategies might be to target mitochondrial ROS production directly, enhance cellular antioxidant capacity by increasing G6PD activity or PPP flux, or mitigating other downstream consequences resulting from lack of ATM redox signaling.
Materials and Methods
Reagents:
Menadione, N-ethylmaleimide (NEM), rotenone, and antimycin A were from Sigma-Aldrich. Topotecan and Mito-TEMPO were from Enzo Life Sciences. MitoSOX and CM-DCFDA were from Invitrogen. MitoPY1 was from Tocris (4428).
Cell culture, siRNA and shRNA transfection:
HeLa cells and primary mouse embryonic fibroblasts (prepared from day 13.5 embryos) from wild-type and mCAT mice were propagated in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). ATM knock-down in HeLa cells was done by infecting cells with retrovirus encoding shRNA targeting ATM (sequence 1, shATM, a gift from David Stern), or a scrambled (SCR) shRNA. TRX1 knock-down in HeLa cells was done through infecting cells with lentiviruses containing shRNA targeting TRX1 (CCGGATCAAGCCTTTCTTTCATTCCCTCGAGGGAATGAAAGAAAGGCTTGATTTTTTG) or EGFP as a control (target sequence: GCAAGCTGACCCTGAAGTTCAT). In both cases infected cells were selected in 0.3 μg/mL puromycin. PRDX1 and PRDX2 knock-down was done by transfecting 20 nM siRNA (IDT, reference #: 130748509 and 130748512) with Lipofectamine RNAiMAX transfection reagent for 72 hours.
The U2OS cell system, in which endogenous ATM is knocked-down by shRNA and replaced with inducible, vector-born wild-type or C2991L alleles, was described previously by Paull and colleagues (16). U2OS cells containing control vector (T-Rex FLP-in) or expressing wild-type or C2991L ATM alleles were cultured in DMEM supplemented with 10% FBS containing 15 μg/mL Blasticidin (A1113903, Life Technology), and 200 μg/mL Hygromycin (400052–50 mL, Life Technologies). Depletion of endogenous ATM was performed by incubating cells with lentivirus containing shRNA targeting ATM (sc-29761-SH, Santa Cruz Biotechnology) overnight and selected in medium containing 1 μg/mL puromycin (Invitrogen) for 5–7 days. To induce wild-type or mutant ATM expression, media was supplemented with 1 μg/mL doxycycline (Sigma) for 3 days. Approximately 16 hours before analysis, the culture medium was changed to DMEM with 10% FBS and 1 μg/mL doxycycline, without puromycin, blasticidin or hygromycin.
Generation of PRDX1 and PRDX2 knock-out Hela cells:
PRDX1 and PRDX2 knock-out HeLa cells were generated using the CRISPR-Cas9 system. Guide RNA (gRNA) design was based on the CRISPR design tool developed by the Zhang lab (http://crispr.mit.edu) (Table S1). The sgRNA-encoding sequences targeting PRDX1 or PRDX2 genes were inserted into the Bbs I site of the p2Tol-U6-sgPal1-HygR vector (sgPal1; Addgene plasmid # 71483) (56). Briefly, 2 × 106 cells were transfected with sgRNA construct plasmid (1 μg), p2to12cbhCas9-Blasticidin plasmid (1 μg), and p2tol2Transponsase plasmid (1 μg) by using Neon™ Transfection System (Invitrogen) according to the manufacturer’s protocol (the Cas9 and Transponsase plasmids were gifts from Dr. Richard Sherwood). Three days later, cells were treated with Hygromycin B (300 μg/ml) and Blasticidin S (10 μg/ml) for three days, and then isolated using a cell sorter (BD FACSARIA Special Order 11 color sorter) into 96-well plates and allowed to expand for 3 weeks. Several clones per transfection were assessed by Western blot analysis and sequencing of DNA fragments covering the sgRNA target region.
Measurements of reactive oxygen species:
To detect mitochondrial superoxide, cells were grown in 12-well plates and incubated with MitoSOX in DMEM + 10% FBS at a final concentration of 2.5 or 5 μM for 30 min at 37°C. Cells were then trypsinized, washed once with PBS, and resuspended in PBS plus 0.1% FBS prior to analyzing MitoSOX fluorescence using Stratedigm 13 in the PE 580/30 channel. To measure levels of cellular or mitochondrial hydrogen peroxide, cells were stained with 5 μM CM-DCFDA in DMEM + 10% FBS for 30 min or 10 μM MitoPY1 in serum-free DMEM for 50 min, respectively, after which cells were trypsinized, washed and pelleted. Cell pellets were resuspended in PBS + 0.1% FBS and immediately measured for fluorescence using Stratedigm 13 in the FITC 530/55 channel. For all FACS experiments, FloJo software was used to determine the mean fluorescence intensity for each sample.
Protein extraction and western blotting:
For standard western blots to detect proteins, cells were lysed with RIPA buffer (50 mM Tris-HCl pH 8.5, 150 mM NaCl, 1.0% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 1X protease inhibitor (Roche 11836170001), 1X phosphatase inhibitor (Roche 04906837001)). Cell lysates were then sonicated in a Bioruptor sonicator (Diagenode) on high power (7 pulses, 10 seconds on, 10 seconds off). Samples were pelleted in a microcentrifuge at 14,000 rpm for 10 min, and supernatants (20 μg) were loaded for western blot.
For non-reducing western blots to detect ATM or PRDX1 dimerization, protein extraction was done in two ways which gave consistent results. For Figures 1 and 2, cells were plated at least 16 h prior to each experiment. After treatment, cells were scraped in PBS and proteins were precipitated with 10% trichloroacetic acid on ice for 10 min. Protein pellets were collected by centrifugation at 2000 × g for 5 min. To prevent post-lysis oxidation of protein thiols, pellets were washed in 100% acetone and then resuspended in SDS buffer containing NEM (100 mM Tris-HCl pH 6.8, 2% SDS, 5 mg/mL NEM) for 30 min at 25 °C. Samples were then sonicated in a Bioruptor sonicator (Diagenode) on high power (5 pulses, 10 seconds on, 10 seconds off). Samples were pelleted in a microcentrifuge at 14,000 rpm for 10 min, and supernatants were analyzed by western blot. The ATM dimer (∼700 kDa) co-migrates with the dimeric 660-kDa form of thyroglobulin (not shown). An improvement in this method was used in Figures 3, 4 and S1. After drug treatment, cells were washed once with PBS and then incubated in 100 mM NEM in PBS for 5 min at 37 °C or 10 min on ice and scraped and pelleted. Cells were resuspended in lysis buffer (100 mM NaCl, 20 mM Tris-Cl pH 7.5, 0.5% NP-40, 5mM CaCl2, 1X protease inhibitor (Roche)) and incubated on ice for 10 min. Nuclear DNA was digested with 30 U/mL micrococcal nuclease for 8 min at 37 °C and the reaction was terminated by adding 5 mM EGTA (pH 8.0). The nuclei were then lysed by adding SDS to 1% and incubating on ice for 10 min. Samples were spun in a microcentrifuge at 14,000 rpm for 10 min at 4 °C, and supernatants were analyzed by western blot. For either protein extraction method, SDS sample buffer without 2-mercaptoethanol (BME) was added to samples and 20 μg of protein were loaded into each lane of a 6% polyacrylamide gel to resolve ATM dimers and monomers. Transfer was done overnight at 4°C at 30 V using transfer buffer without methanol.
Antibodies used were ATM (Sigma-Aldrich A-1106, 1:7500), phospho-ATM (Ser1981, Abcam ab81292, 1:5000), phospho-CHK2 (Thr68, Cell Signaling Technology 2197, 1:1000), CHK2 (Santa Cruz Biotechnology sc5278, 1:200), phospho-H2AX (Ser139, Millipore 05–636, 1:1000), HSP60 (Santa Cruz Biotechnology sc1052, 1:20000), catalase (Sigma-Aldrich C-0979, 1:5000), calnexin (Santa Cruz Biotechnology sc11397, 1:20000), TRX1 (Abcam ab26320, 1:15000), GAPDH (Ambion AM4300, 1:10000), G6PD (Cell Signaling Technology 12263, 1:1000), phospho-HSP27 (Ser78, Cell Signaling Technology 2405, 1:1000), histone H3 (Abcam ab1791, 1:2000), PRDX1 (Santa Cruz Biotechnology sc-7381, 1:2000), PRDX2 (Santa Cruz Biotechnology sc-23967, 1:500, and Abcam ab109367, 1:1000).
Nuclear Fractionation:
HeLa cells were grown to ∼80% confluence on two 15-cm plates, treated with 30 μM menadione for 1 hour, washed with PBS, and pelleted by centrifugation. The cell pellet was resuspended in 1 mL of buffer containing 0.1 M Tris-HCl pH 6.8, 10 mg/mL NEM, and protease inhibitors (Roche). NEM modification proceeded for 5 min at room temperature after which 300 μL of the sample was removed and saved as whole cell extract (WC). To the remaining 700 μL, Triton X-100 was added to a final concentration of 0.5% and cells were incubated on ice for 5 min until visually broken and intact nuclei could be seen. Nuclei were pelleted at 720 × g for 5 min and the supernatant, representing the post-nuclear extract (PN) was saved. Nuclei (N) were washed 3 times with NEM Buffer with 0.5% Triton X-100 and once with NEM Buffer without Triton X-100. Nuclei were resuspended in 700 μL NEM Buffer with 1% SDS and lysed for 10 min on ice. The PN fraction was clarified by three sequential centrifugation steps at 720 × g for 5 min, discarding the pellet each time. To WC, N, and PN fractions, Triton X-100 was added to a final concentration of 1%, and SDS was added to a final concentration of 0.5%. Samples were then sonicated in a Bioruptor sonicator (Diagenode) on high power (5 pulses, 30-second on, 30 off) and clarified by centrifugation at 14,000 rpm at 4 °C for 10 min. Equal volumes (25 μL) of the supernatants were loaded per lane on a 6% SDS-PAGE gel.
Salt extraction of chromatin bound proteins:
The protocol used was modified from a previously described method (57). HeLa cells (2 × 106) were plated in a 10-cm dish ∼16 hours prior to the experiment. After drug treatment, cells were washed once with 5 mL of PBS, scraped in PBS and pelleted at 350 × g for 5 min at 4 °C. To lyse cells, pellets were resuspended in 50 μL NETN buffer (100 mM NaCl, 20 mM Tris-Cl pH 7.5, 0.5% NP-40, 2 mM EDTA) for every 5 × 105 cells and incubated for 15 min on ice, during which they were vortexed twice. The nuclei were pelleted in a microcentrifuge at 14000 rpm for 5 min. The supernatant was moved to a new tube, and after another centrifugation at 14000 rpm for 5 min, was saved as the cytoplasmic/soluble nuclear protein fraction (“S”). The pellet was washed once with 50 μL NETN buffer, resuspended in 50 μL nuclear digestion buffer (20 mM Tris-Cl pH7.5, 150 mM NaCl, 5 mM CaCl2, 200 U/mL micrococcal nuclease) and incubated for 8 min at 37 °C. This pellet (“P”) was saved as the chromatin fraction. After adding 10 μL of 6 X SDS sample buffer with BME, the samples were boiled for 5 min, clarified in a microcentrifuge at 13,000 rpm for 10 min at room temperature, and 15 μL loaded for SDS-PAGE and western blot.
Quantitative RT-PCR:
RNA was isolated with TRIzol reagent (Life Technologies15596–026) and converted to cDNA using the High Capacity cDNA Reverse Transcription Kit (Agilent) according to the manufacturer’s instructions. cDNA was diluted 1:50, and each 10 μL reaction contained 4.5 μL diluted cDNA, 0.5 μL primer mix (from a 10 μM stock), and 5 μL Applied Biosystems Fast SYBR Green Master Mix (Life Technologies). PCR was performed on a Bio-Rad CFX96 Real Time PCR machine in 96-well plates using fast ramp speed. Data were analyzed according to the ΔΔCt method using GAPDH as a reference gene. Primers used were as following: G6PD_F: ACCGCATCGACCACTACCT, G6PD_R: TGGGGCCGAAGATCCTGTT, GAPDH_F: ACAACTTTGGTATCGTGGAAGG, GAPDH_R: GCCATCACGCCACAGTTTC
G6PD activity assay:
The protocol used was modified from a previously described method (58). U2OS cells were plated at 1.5 × 106 cells per 10-cm dish ∼16 hours before the experiment. Cells were washed with 5 mL PBS, scraped in 1 mL PBS and pelleted. Cell pellets were lysed with 75 μL NP-40 buffer (100 mM NaCl, 20 mM Tris-Cl pH 7.5, 0.5% NP-40, 1X protease inhibitor) and incubated for 10 min on ice. Cell lysate was clarified in a microcentrifuge at 14000 rpm for 10 min at 4 °C. The supernatant was diluted to 1 μg/μL protein. In a 96-well plate with black sides and clear bottom, 20 μL cell lysate and 180 μL reaction mix (50 mM Tris-Cl pH 8, 1 mM MgCl2, 200 μM glucose 6-phosphate (Sigma G7879), 100 μM NADP+ (Sigma N0505)) were added to each well. To measure NADPH production, fluorescence was measured immediately using excitation at 340 nm and emission at 460 nm on a plate reader (measured at 1-min intervals for 10 minutes). The fluorescence intensity was plotted versus time and the slope was calculated. Data points plotted in Figure 5C are the averaged slope of four technical replicates.
Metabolomics analysis:
Metabolomics analyses and tracing experiments with 13C1,2,3-glucose were performed as previously reported (59, 60). All metabolite standard reagents were purchased from Sigma Aldrich (St. Louis, MO) or Cambridge Isotope Laboratories, Inc., Tewksbury, MA, unless otherwise stated. U2OS cells in which endogenous ATM was replaced by WT or C2991L alleles of ATM (see details above) were cultured in DMEM without glucose and supplemented with 25 mM 13C1,2,3-glucose (no. CLM-4673-PK – Cambridge Isotopes, Tewksbury, MA). Longitudinal sampling of cells and supernatants was performed at 0, 1, 6 and 24 hours from incubation with stable heavy isotope-labeled glucose. Cells (1 million) and supernatants (20 μL) were extracted in 1 mL of ice-cold lysis/extraction buffer (methanol: acetonitrile: water 5:3:2 – triplicate experiment). Samples were then agitated at 4 °C for 30 min followed by centrifugation at 10,000 g for 15 min at 4 °C. Protein pellets were discarded, while supernatants were stored at −80°C prior to metabolomics analyses. Extracts (10 μL) were injected into an UHPLC system (Ultimate 3000, Thermo, San Jose, CA, USA) and run on a Kinetex XB-C18 column (150 × 2.1 mm i.d., 1.7 μm particle size – Phenomenex, Torrance, CA, USA), as previously reported (59). Technical mixes were generated by pooling ten microliters of all the cells and supernatants, separately. Pooled reference samples were run every 10 analytical runs to control for technical variability, determined as coefficients of variation (CV). CV were determined by calculating the ratios of standard deviations divided by mean measurements for compounds of interest (e.g. all isotopologues of lactate) across all technical mix runs. The Q Exactive mass spectrometer (Thermo, San Jose, CA, USA) was operated in Full MS mode (2 μscans) at 70,000 resolution in the 60–900 m/z range, 4kV spray voltage, 15 sheath gas and 5 auxiliary gas, either in negative or positive ion mode. Metabolite assignments and isotopologue distributions were determined using the software Maven (Princeton, NJ, USA) (61) and assignments confirmed against 700 standards (IROATech, Sigma Aldrich, St. Louis, MO, USA).
Glutathione measurements:
Total glutathione and GSH/GSSG ratio were measured using the GSH/GSSG-Glo assay kit (Promega V6611) according to manufacturer instructions.
Statistical analysis:
Unless indicated otherwise, statistical significance was calculated using Student’s t-test, with * denoting p<0.05 and ** denoting p<0.01. Error bars represent the standard deviation.
Supplementary Material
Acknowledgments.
The authors wish to thank Dr. Laura Newman for help troubleshooting the use of redox-sensitive dyes and Dr. David Stern for providing the human ATM shRNA.
Funding. This work was supported by NIH R01 AG047632 and R33 ES025636, a grant from the A-T Children’s Project, the Audrey Geisel Chair Fund and the Joseph A. and Lucille K. Chair fund to G.S.S, and NIH R01 GM065204 to V.N.G. B.E.C was supported by NIH NRSA NS077723, E.A.S. by NIH F31AG043242, Y.Z. by the China Scholarship Counsel, and A.D. by the Webb-Waring Early Career Award from the Boettcher Foundation.
Footnotes
Author contributions. Y.Z., performed experiments and wrote manuscript; J.H., provided critical reagents and data analysis; T.J.P., provided critical reagents, data analysis, and funding; S.G., performed experiments and data analysis; A.D., performed data analysis, wrote manuscript, provided funding; Q.D., provided critical reagents; V.N.G., provided critical reagents and funding; E.A.S, performed experiments; S.K.S, performed experiments; B.E.C., performed experiments, wrote manuscript, provided funding; G.S.S. wrote manuscript, performed data analysis, and provided funding.
Competing interests. The authors have no competing interests to declare pertaining to this work.
Data and Materials Availability. Transfer of reagents used in this work may require an MTA. All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
References and Notes
- 1.Shiloh Y, Ziv Y, The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 14, 197–210 (2013). [PubMed] [Google Scholar]
- 2.Paull TT, Mechanisms of ATM Activation. Annu Rev Biochem 84, 711–738 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, Shiloh Y, Gygi SP, Elledge SJ, ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316, 1160–1166 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Bakkenist CJ, Kastan MB, DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421, 499–506 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Ditch S, Paull TT, The ATM protein kinase and cellular redox signaling: beyond the DNA damage response. Trends in Biochemical Sciences 37, 15–22 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barzilai A, Rotman G, Shiloh Y, ATM deficiency and oxidative stress: a new dimension of defective response to DNA damage. DNA Repair (Amst) 1, 3–25 (2002). [DOI] [PubMed] [Google Scholar]
- 7.Barlow C, Dennery PA, Shigenaga MK, Smith MA, Morrow JD, Roberts LJ 2nd, Wynshaw-Boris A, Levine RL, Loss of the ataxia-telangiectasia gene product causes oxidative damage in target organs. Proc Natl Acad Sci U S A 96, 9915–9919 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim J, Wong PK, Loss of ATM impairs proliferation of neural stem cells through oxidative stress-mediated p38 MAPK signaling. Stem Cells 27, 1987–1998 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Kim J, Wong PK, Oxidative stress is linked to ERK1/2-p16 signaling-mediated growth defect in ATM-deficient astrocytes. J Biol Chem 284, 14396–14404 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ito K, Hirao A, Arai F, Takubo K, Matsuoka S, Miyamoto K, Ohmura M, Naka K, Hosokawa K, Ikeda Y, Suda T, Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med 12, 446–451 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Bagley J, Singh G, Iacomini J, Regulation of oxidative stress responses by ataxia- telangiectasia mutated is required for T cell proliferation. J Immunol 178, 4757–4763 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Ito K, Takubo K, Arai F, Satoh H, Matsuoka S, Ohmura M, Naka K, Azuma M, Miyamoto K, Hosokawa K, Ikeda Y, Mak TW, Suda T, Hirao A, Regulation of reactive oxygen species by Atm is essential for proper response to DNA double-strand breaks in lymphocytes. J Immunol 178, 103–110 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT, ATM activation by oxidative stress. Science 330, 517–521 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Schroeder EA, Raimundo N, Shadel GS, Epigenetic silencing mediates mitochondria stress-induced longevity. Cell Metab 17, 954–964 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo Z, Deshpande R, Paull TT, ATM activation in the presence of oxidative stress. Cell Cycle 9, 4805–4811 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee JH, Mand MR, Kao CH, Zhou Y, Ryu SW, Richards AL, Coon JJ, Paull TT, ATM directs DNA damage responses and proteostasis via genetically separable pathways. Sci Signal 11, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patra KC, Hay N, The pentose phosphate pathway and cancer. Trends Biochem Sci 39, 347–354 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cosentino C, Grieco D, Costanzo V, ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J 30, 546–555 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brand MD, The sites and topology of mitochondrial superoxide production. Exp Gerontol 45, 466–472 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ambrose M, Goldstine JV, Gatti RA, Intrinsic mitochondrial dysfunction in ATM-deficient lymphoblastoid cells. Hum Mol Genet 16, 2154–2164 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Eaton JS, Lin ZP, Sartorelli AC, Bonawitz ND, Shadel GS, Ataxia-telangiectasia mutated kinase regulates ribonucleotide reductase and mitochondrial homeostasis. J Clin Invest 117, 2723–2734 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Valentin-Vega YA, Maclean KH, Tait-Mulder J, Milasta S, Steeves M, Dorsey FC, Cleveland JL, Green DR, Kastan MB, Mitochondrial dysfunction in ataxia-telangiectasia. Blood 119, 1490–1500 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fu X, Wan S, Lyu YL, Liu LF, Qi H, Etoposide induces ATM-dependent mitochondrial biogenesis through AMPK activation. PLoS One 3, e2009 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ito K, Hirao A, Arai F, Matsuoka S, Takubo K, Hamaguchi I, Nomiyama K, Hosokawa K, Sakurada K, Nakagata N, Ikeda Y, Mak TW, Suda T, Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature 431, 997–1002 (2004). [DOI] [PubMed] [Google Scholar]
- 25.Schubert R, Erker L, Barlow C, Yakushiji H, Larson D, Russo A, Mitchell JB, Wynshaw-Boris A, Cancer chemoprevention by the antioxidant tempol in Atm-deficient mice. Hum Mol Genet 13, 1793–1802 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Reliene R, Fischer E, Schiestl RH, Effect of N-acetyl cysteine on oxidative DNA damage and the frequency of DNA deletions in atm-deficient mice. Cancer Res 64, 5148–5153 (2004). [DOI] [PubMed] [Google Scholar]
- 27.D’Souza AD, Parish IA, Krause DS, Kaech SM, Shadel GS, Reducing mitochondrial ROS improves disease-related pathology in a mouse model of ataxia-telangiectasia. Mol Ther 21, 42–48 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pan Y, Shadel GS, Extension of chronological life span by reduced TOR signaling requires down-regulation of Sch9p and involves increased mitochondrial OXPHOS complex density. Aging (Albany NY) 1, 131–145 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sena LA, Chandel NS, Physiological roles of mitochondrial reactive oxygen species. Mol Cell 48, 158–167 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shadel GS, Horvath TL, Mitochondrial ROS signaling in organismal homeostasis. Cell 163, 560–569 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dickinson BC, Lin VS, Chang CJ, Preparation and use of MitoPY1 for imaging hydrogen peroxide in mitochondria of live cells. Nat Protoc 8, 1249–1259 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee KW, Lee DJ, Lee JY, Kang DH, Kwon J, Kang SW, Peroxiredoxin II restrains DNA damage-induced death in cancer cells by positively regulating JNK-dependent DNA repair. J Biol Chem 286, 8394–8404 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang X, He S, Sun JM, Delcuve GP, Davie JR, Selective association of peroxiredoxin 1 with genomic DNA and COX-2 upstream promoter elements in estrogen receptor negative breast cancer cells. Mol Biol Cell 21, 2987–2995 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alexander A, Cai SL, Kim J, Nanez A, Sahin M, MacLean KH, Inoki K, Guan KL, Shen J, Person MD, Kusewitt D, Mills GB, Kastan MB, Walker CL, ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci USA 107, 4153–4158 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang J, Tripathi DN, Jing J, Alexander A, Kim J, Powell RT, Dere R, Tait-Mulder J, Lee JH, Paull TT, Pandita RK, Charaka VK, Pandita TK, Kastan MB, Walker CL, ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat Cell Biol 17, 1259–1269 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watters D, Kedar P, Spring K, Bjorkman J, Chen P, Gatei M, Birrell G, Garrone B, Srinivasa P, Crane DI, Lavin MF, Localization of a portion of extranuclear ATM to peroxisomes. J Biol Chem 274, 34277–34282 (1999). [DOI] [PubMed] [Google Scholar]
- 37.Watson WH, Jones DP, Oxidation of nuclear thioredoxin during oxidative stress. FEBS Lett 543, 144–147 (2003). [DOI] [PubMed] [Google Scholar]
- 38.Sobotta MC, Liou W, Stocker S, Talwar D, Oehler M, Ruppert T, Scharf AN, Dick TP, Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat Chem Biol 11, 64–70 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Jarvis RM, Hughes SM, Ledgerwood EC, Peroxiredoxin 1 functions as a signal peroxidase to receive, transduce, and transmit peroxide signals in mammalian cells. Free Radic Biol Med 53, 1522–1530 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Murphy MP, Modulating mitochondrial intracellular location as a redox signal. Sci Signal 5, pe39 (2012). [DOI] [PubMed] [Google Scholar]
- 41.Al-Mehdi AB, Pastukh VM, Swiger BM, Reed DJ, Patel MR, Bardwell GC, Pastukh VV, Alexeyev MF, Gillespie MN, Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci Signal 5, ra47 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reczek CR, Chandel NS, ROS-dependent signal transduction. Curr Opin Cell Biol 33, 8–13 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Go YM, Jones DP, Redox compartmentalization in eukaryotic cells. Biochim Biophys Acta 1780, 1273–1290 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saxena G, Chen J, Shalev A, Intracellular shuttling and mitochondrial function of thioredoxin-interacting protein. J Biol Chem 285, 3997–4005 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.World C, Spindel ON, Berk BC, Thioredoxin-interacting protein mediates TRX1 translocation to the plasma membrane in response to tumor necrosis factor-alpha: a key mechanism for vascular endothelial growth factor receptor-2 transactivation by reactive oxygen species. Arterioscler Thromb Vasc Biol 31, 1890–1897 (2011). [DOI] [PubMed] [Google Scholar]
- 46.Yalcin S, Zhang X, Luciano JP, Mungamuri SK, Marinkovic D, Vercherat C, Sarkar A, Grisotto M, Taneja R, Ghaffari S, Foxo3 is essential for the regulation of ataxia telangiectasia mutated and oxidative stress-mediated homeostasis of hematopoietic stem cells. J Biol Chem 283, 25692–25705 (2008). [DOI] [PubMed] [Google Scholar]
- 47.Liu B, Fang M, He Z, Cui D, Jia S, Lin X, Xu X, Zhou T, Liu W, Hepatitis B virus stimulates G6PD expression through HBx-mediated Nrf2 activation. Cell Death Dis 6, e1980 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leopold JA, Dam A, Maron BA, Scribner AW, Liao R, Handy DE, Stanton RC, Pitt B, Loscalzo J, Aldosterone impairs vascular reactivity by decreasing glucose-6-phosphate dehydrogenase activity. Nat Med 13, 189–197 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, Vander Heiden MG, MacKeigan JP, Finan PM, Clish CB, Murphy LO, Manning BD, Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell 39, 171–183 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Makarona K, Caputo VS, Costa JR, Liu B, O’Connor D, Iskander D, Roper D, Robertson L, Bhatnagar N, Terpos E, Georgiou E, Papaioannou M, Layton DM, Luzzatto L, Roberts I, Karadimitris A, Transcriptional and epigenetic basis for restoration of G6PD enzymatic activity in human G6PD-deficient cells. Blood 124, 134–141 (2014). [DOI] [PubMed] [Google Scholar]
- 51.Ayrapetov MK, Gursoy-Yuzugullu O, Xu C, Xu Y, Price BD, DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proc Natl Acad Sci U S A 111, 9169–9174 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lu SC, Glutathione synthesis. Biochim Biophys Acta 1830, 3143–3153 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krejsa CM, Franklin CC, White CC, Ledbetter JA, Schieven GL, Kavanagh TJ, Rapid activation of glutamate cysteine ligase following oxidative stress. J Biol Chem 285, 16116–16124 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guleria A, Chandna S, ATM kinase: Much more than a DNA damage responsive protein. DNA Repair (Amst) 39, 1–20 (2016). [DOI] [PubMed] [Google Scholar]
- 55.Shadel GS, Expression and maintenance of mitochondrial DNA: new insights into human disease pathology. Am J Pathol 172, 1445–1456 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Arbab M, Srinivasan S, Hashimoto T, Geijsen N, Sherwood RI, Cloning-free CRISPR. Stem Cell Reports 5, 908–917 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bian C, Chen Q, Yu X, The zinc finger proteins ZNF644 and WIZ regulate the G9a/GLP complex for gene repression. Elife 4, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tian WN, Braunstein LD, Pang J, Stuhlmeier KM, Xi QC, Tian X, Stanton RC, Importance of glucose-6-phosphate dehydrogenase activity for cell growth. J Biol Chem 273, 10609–10617 (1998). [DOI] [PubMed] [Google Scholar]
- 59.D’Alessandro A, Nemkov T, Yoshida T, Bordbar A, Palsson BO, Hansen KC, Citrate metabolism in red blood cells stored in additive solution-3. Transfusion 57, 325–336 (2017). [DOI] [PubMed] [Google Scholar]
- 60.Reisz JA, Wither MJ, Dzieciatkowska M, Nemkov T, Issaian A, Yoshida T, Dunham AJ, Hill RC, Hansen KC, D’Alessandro A, Oxidative modifications of glyceraldehyde 3-phosphate dehydrogenase regulate metabolic reprogramming of stored red blood cells. Blood 128, e32–42 (2016). [DOI] [PubMed] [Google Scholar]
- 61.Clasquin MF, Melamud E, Rabinowitz JD, LC-MS data processing with MAVEN: a metabolomic analysis and visualization engine. Curr Protoc Bioinformatics Chapter 14, Unit14 11 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.