

Abstract
A dearomative reduction of simple arenes has been developed which employs a visible-light-mediated cycloaddition of arenes with an N-N-arenophile and in situ diimide reduction. Subsequent cycloreversion or fragmentation of the arenophile moiety affords 1,3-cyclohexadienes or 1,4-diaminocyclohex-2-enes, compounds that are not synthetically accessible using existing dearomatization reactions. Importantly, this strategy also provides numerous opportunities for further derivatization as well as site-selective functionalization of polynuclear arenes.
Keywords: arenes, arenophiles, dearomatization, functionalization, photochemistry
The dearomative strategies that convert arenes to compounds of broader utility are of fundamental importance for organic synthesis.[1] Despite significant advances that have been made in recent years, mainly involving heteroarenes and phenols,[2,3] there are only limited options available for the dearomatization of non-activated benzene derivatives.[4] For example, in the area of reductive dearomatization, the most common transformations are dissolving-metal (Birch) reduction and dearomative hydrogenation (Scheme 1a).[5,6] The resulting 1,4-cyclohexadiene and cyclohexane products have been extensively used in the preparation of natural products and pharmaceuticals. In addition, osmium-arene η2-complexes are known to undergo chemoselective partial hydrogenation of the arene ligand;[7] however, the high toxicity and cost have significantly restricted their widespread use. Finally, a microbial reductive process involving benzoyl-coenzyme A reductases exists,[8] but its translation to organic synthesis has not yet been realized. Accordingly, to the best of our knowledge, there are no known general methods for the preparation of 1,3-dienes from arenes, even though these highly versatile building blocks are only one reduction level away from the parent aromatic material.[9] Herein, we report a reductive dearomatization strategy (Scheme 1b), which delivers 1,3-cyclohexadienes as well as cis-1,4-diaminocyclohex-2-enes.
Scheme 1.
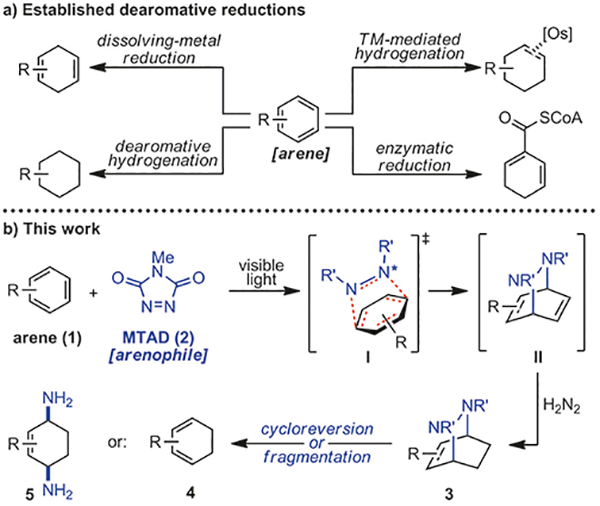
Reductive dearomatizations, including this work.
Recently, we introduced a new dearomative platform based on the application of small organic molecules, termed arenophiles (Scheme 1b).[10] The underlying feature of this approach is the ability of visible-light-excited arenophiles[11] to undergo para-cycloaddition with arenes (1 + 2 → I) to provide bicycles II that are amenable to further transformations. Formally, reactions with arenophiles could be seen as isolation of π-bonds within an aromatic starting material and have the potential to connect olefin chemistry to the area of dearomative functionalizations. Accordingly, this strategy could be used for dearomative reductions, as it would provide products that are complementary to those obtained by known dearomative processes. Described reactions are conducted using operationally simple conditions, under ambient atmosphere and have been performed on gram-scale. Moreover, the utility of this method is demonstrated by the dearomative diversification of 1-phenylheptane, as well as the rapid conversion of naphthalene to naturally occurring γ-hydroxytetralone.
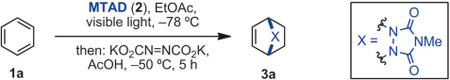
Our studies commenced with exploring the photochemical reaction of benzene (1a) with N-methyl-1,2,4-triazoline-3,5-dione (2, MTAD) and subsequent in situ reduction. Initial experiments evaluated metal-catalyzed homogenous or heterogeneous hydrogenation; however, no reduced products were observed at cryogenic conditions with cycloadducts of type II. Next, diimide reduction was examined, as this reagent is capable of engaging olefins at low temperatures and has a broad functional group compatibility.[12] Accordingly, we identified that the cycloaddition in EtOAc at −78 °C followed by reduction with diimide, generated from potassium azodicarboxylate and acetic acid, at −50 °C provided the best result and gave product 3 a in 65% yield (Table 1, entry 1). Several aspects observed during optimization are noteworthy: 1) keeping the temperature at −78 °C throughout the sequence resulted in significantly lower yields of reduced bicycle (entry 2). 2) As shown in entries 3–5, several other solvents were suitable for this reaction, with CH2Cl2 being comparable to EtOAc. 3) Direct, hydrazine-based diimide generation was not as effective (entry 6).[13] 4) Use of stronger acids, as well as lower stoichiometry of acetic acid or azodicarboxylate salt had a detrimental effect on the reaction yield (entries 7–10).
Table 1:
Optimization of dearomative cycloaddition/diimide reduction.[a]
| Entry | Deviation from standard conditions | Yield [%][b] |
|---|---|---|
| 1 | none | 65[c] |
| 2 | diimide reduction at −78°C | 36 |
| 3 | CH2CI2 was used instead of EtOAc | 61[c] |
| 4 | acetone was used instead of EtOAc | 31[c] |
| 5 | EtCN was used instead of EtOAc | 55[c] |
| 6 | o-NO2-C6H4-SO2CI and N2H4 H2O were used instead of KO2CN=NCO2K and AcOH | 10 |
| 7 | HCO2H was used instead of AcOH | 61 |
| 8 | TFA was used instead of AcOH | 16 |
| 9 | with 10 equiv of AcOH | 26 |
| 10 | with 2.0 equiv of KO2CN=NCO2K | 48 |
Reaction conditions: benzene (1a, 5.0 mmol, 10 equiv), MTAD (2, 0.5 mmol, 1.0 equiv), EtOAc (0.1 m), visible light LEDs, −78°C; then KO2CN=NCO2K (3.0 equiv), AcOH (15 equiv), −50°C, 5 h.
Determined by GC relative to an internal standard.
Isolated yields after purification by flash chromatography.
Having identified the optimal reaction conditions for the cycloaddition and subsequent in situ reduction, we explored the substrate scope of this process with mononuclear arenes (Scheme 2). Thus, on a preparative scale (1.0 mmol), the model reaction with benzene (1a) successfully furnished product 3a in 68% yield. Furthermore, a range of monosubstituted arenes bearing ester (3f, 3h, 3i, 3j, 3o, 3q), halogen (3d, 3n, 3p), amide (3g), acetal (3k), ketal (31), and orthoester (3m) functional groups delivered the corresponding reduced bicycles. Most notably, benzylic heteroatomcontaining functional groups (3i–3n) were well tolerated in this process. High regioselectivity of cycloaddition and chemoselectivity of reduction were observed in most cases; only reaction with phenyltrimethylsilane (1e) resulted in a nearly equal mixture of constitutional isomers 3e and 3e′. Moreover, MTAD cycloaddition with methyl 2-phenylbenzoate (1q) and subsequent reduction delivered products 3q and the corresponding column-separable 3,6-constitutional isomer in a 1.9:1 ratio. Importantly, no over-reduction was observed in any case.[14] All reactions were run under air and using commercial grade solvents and reagents, making this cycloaddition/reduction protocol practical to perform. Finally, this process can be performed on a multigram scale as demonstrated in preparation of 3f (2.0 g, 17.7 mmol of MTAD used, 68% yield, 10:1).
Scheme 2.
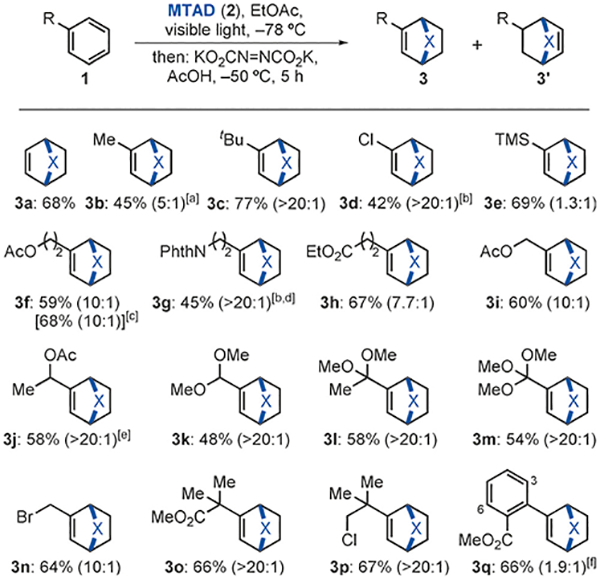
Scope of the dearomative cycloaddition/diimide reduction of mononuclear arenes. All reactions were carried out on 1.0 mmol scale under the standard conditions (see Table 1, entry 1). Yield of the isolated product after purification by chromatography. Ratios of 3 and 3′ were determined by 1H NMR of the crude reaction mixtures and is shown in parenthesis. [a] MeOH was added with AcOH. [b] Reaction was conducted in CH2CI2. [c] Run on gram-scale. [d] 3.0 equiv of 1g used. [e] 1.7:1 d.r. [f] Mixture with 3,6-constitutional isomer.
We next investigated the conversion of reduced bicycles 3 to the corresponding 1,3-cyclohexadienes 4 (Scheme 3, left side) using one-pot hydrolysis followed by in situ oxidation of the resulting cyclic hydrazines with CuCl2.[15] While esters were hydrolyzed under the basic reaction conditions to alcohols (4f, 4i, 4j) and acids (4h, 4o, 4q), other functional groups were tolerated, including acetal (4k), ketal (41), orthoester (4m), alkyl chloride (4p), and silyl (4e and 4e′) as well as alkyl substituents (4b and 4c). Only allyl bromide 3n was converted to the corresponding isopropoxy ether 4n. For the cycloreversion of phthalimide 3g, we modified this one-pot sequence by using hydrazine and amino group reprotection to obtain Boc-protected aminodiene 4g. Finally, conducting this transformation on a sevenfold larger scale proceeded smoothly and without significant decrease in yield as exemplified with diene 4f (0.88 g, 3.15 mmol of 3f used, 77 % yield).
Scheme 3.
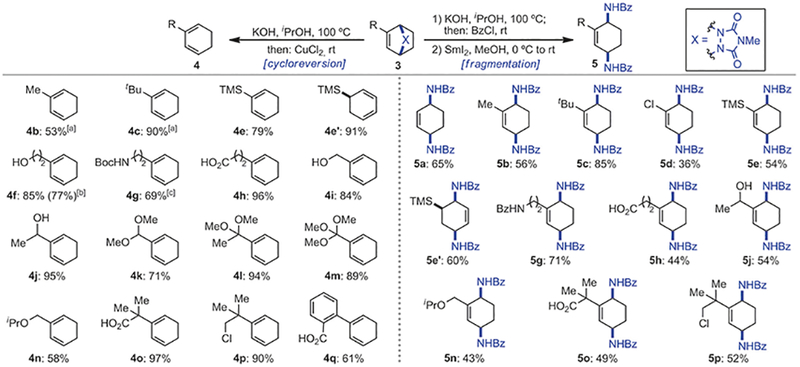
Cycloreversion and fragmentation of cycloadducts 3. Conditions for cycloreversion: 3 (0.4 mmol, 1.0 equiv), KOH (4.0 mmol, 10 equiv), iPrOH (0.2 m), 100°C, 12–24 h; then pH 7, CuCl2, (0.4 mmol, 1.0 equiv), 25°C, 1 min. Conditions for fragmentation: 1) 3 (0.2 mmol, 1.0 equiv), KOH (2.0 mmol, 10 equiv), iPrOH (0.3 m), 100°C, 12–24 h; then BzCl (1.0 mmol, 5.0 equiv), rt, 4–8 h. 2) Sml2 (0.5 mmol, 2.5 equiv), MeOH (0.1 m), 0°C to rt. Yields of isolated product after purification by flash chromatography. [a] Determined by ‘H NMR using internal standard. [b] Run on 3.15 mmol scale. [c] N2H4 was used instead of KOH followed by in situ protection with Boc2O.
In a similar fashion, we examined the fragmentation of the arenophile moiety within the reduced bicycles 3 to deliver 1,4-diaminocyclohex-2-ene derivatives 5 (Scheme 3, right side). These products formally represent the dearomative bis-1,4-hydroamination of the parent arenes, a functionalization method for which no synthetic or biological equivalent exists. Thus, a two-step protocol, involving urazole hydrolysis/hydrazine benzoylation and subsequent SmI2-mediated N-N reduction furnished the desired diamine products protected as a benzamides.[16] Again, a variety of functionalities were tolerated with the exception of esters and allylic bromide, which delivered alcohol 5j, acids 5h and 5o, and ether 5n. While free alcohols did not undergo re-protection, the primary amino group was benzoylated during the first step (5g). Noteworthy, substrates bearing vinyl and aliphatic chlorides 5d and 5p were compatible with the reductive conditions, although a slight decrease in yield was observed with vinyl chloride 5d.
In addition to benzene derivatives, the arenophile-mediated dearomative bis-1,4-hydroamination strategy was also applied to polynuclear arenes (Scheme 4). In all cases, the cycloaddition proceeded with exclusive site-selectivity for the terminal aromatic moiety. The fragmentation step was accomplished by a one-pot hydrazinolysis[17] and Ni-catalyzed reductive cleavage.[18] Overall, this protocol proved efficient for several naphthalene derivatives (8a–8c)[19] and phenanthrene (8d),[20] as well as heteroarenes such as quinoline (8e), quinoxaline (8f), acridine (8g), and benzo[h]quinoline (8h). Sensitive arene substituents, such as benzylic ketal (8b) and bromide (8e) were tolerated under the described reductive conditions. Moreover, dearomatization of polynuclear substrates can be successfully performed on gram-scale, as demonstrated with the conversion of naphthalene (6a) to 7a (1.13 g, 10.0 mmol of MTAD used, 82% yield).
Scheme 4.
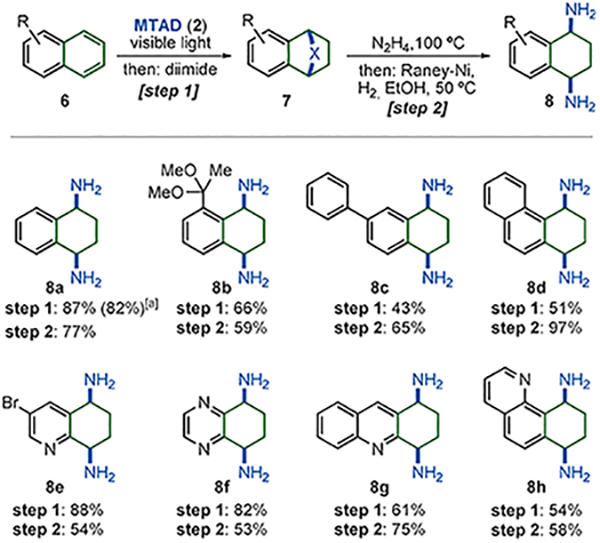
Dearomative bis-1,4-hydroamination of polynuclear arenes. Conditions for cycloaddition/reduction: see Table 1 for reaction conditions, 6 (2.0 mmol, 2.0 equiv), MTAD (2, 1.0 mmol, 1.0 equiv). Conditions for cycloreversion: N2H4 (20 equiv), 100°C, 12 h, then Raney-Ni (50 mol%), EtOH (0.2 m), H2 (1 atm), 50°C, 8 h. Yields are of isolated product after purification by flash chromatography. [a] Run on gram-scale.
This arenophile-mediated dearomatization can provide an expedient entry to a range of functionally diverse compounds. In addition to 1,3-dienes, which are extensively used in organic synthesis, dearomatized products 5 are versatile entities that can be subjected to further synthetic manipulations (Scheme 5). For example, 1-phenylheptane (1r) was converted to the corresponding partially unsaturated diamine derivative 5r (37% over three steps), which was oxidized to epoxide 9 or diol 10. Moreover, Mukaiyama hydration[21] delivered tertiary alcohol 11, and reductive olefin coupling[22] with methyl acrylate installed a quaternary center in carbocyle 12, obtained as a readily separable mixture of diastereoisomers. Finally, substrate-directed, catalytic hydrogenation afforded saturated analog 13.
Scheme 5.
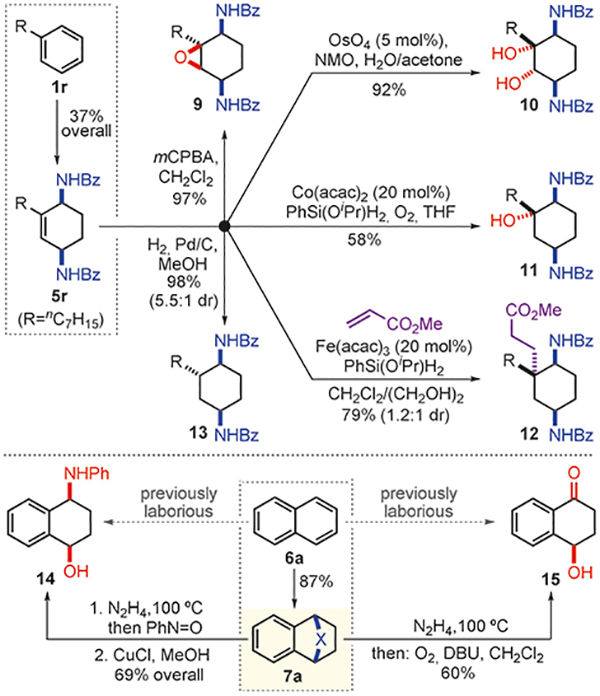
Elaboration of dearomatized products.
Furthermore, the reduced cycloadducts based on polynuclear arenes could also be elaborated to different compounds by manipulating the cycloreversion step. For instance, the conversion of naphthalene derivative 7a to aminoalcohol 14 was achieved through a one-pot cycloreversion/nitroso Diels–Alder reaction, and subsequent N–O bond cleavage. Similarly, the urazole moiety was replaced with an endoper oxide that underwent an in situ Kornblum-DeLaMare rearrangement[23] to deliver γ-hydroxytetralone 15. This reaction sequence exemplifies the production of an expensive bioactive natural product 15[24] ($577 g−1, Aldrich) from a cheap feedstock naphthalene (6a, $0.03 g−1) in two one-pot chemical operations.
In summary, we have developed a new strategy for dearomative reduction of simple arenes to deliver 1,3-cyclohexadienes and 1,4-diaminocyclohex-2-ene derivatives containing a variety of pre-installed functional groups that have been previously incompatible with reductive dearomatizations. The obtained products are orthogonal to those accessible through known dearomatization chemistry and are amenable to further functionalizations. From a practical standpoint, these reactions are operationally simple and have been conducted on gram-scale. Studies to expand the scope as well as additional applications of the versatile products are currently ongoing and will be reported in due course.
Supplementary Material
Acknowledgements
We acknowledge support from the University of Illinois and the National Science Foundation for CAREER Award No. CHE-1654110. M.O. thanks the Honjo International Scholarship Foundation and J.P. acknowledges the German Research Foundation (DFG) for a postdoctoral fellowship.
Footnotes
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under http://dx.doi.org/10.1002/anie.201609686.
References
- [1].a) Roche SP, Porco JA, Angew. Chem. Int. Ed 2011, 50, 4068–4093; Angew. Chem 2011, 123, 4154–4179; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhuo C-X, Zhang W, You S-L, Angew. Chem. Int. Ed 2012, 51, 12662–12686; Angew. Chem 2012, 124, 12834–12858; [DOI] [PubMed] [Google Scholar]; c) Pouysegu L, Deffieux D, Quideau S, Tetrahedron 2010, 66, 2235–2261; [Google Scholar]; d) Lewis SE, Chem. Commun 2014, 50, 2821–2830. [DOI] [PubMed] [Google Scholar]
- [2].For recent examples involving heteroarenes, see: DiPoto MC, Hughes RP, Wu J, J. Am. Chem. Soc 2015, 137, 14861–14864; Acharya A, Anumandla D, Jeffrey CS, J. Am. Chem. Soc 2015, 137, 14858–14860;Zhuo C-X, Zhou Y, Cheng Q, Huang L, You S-L, Angew. Chem. Int. Ed 2015, 54, 14146–14149; Angew. Chem 2015, 127, 14352–14355;Liu C, Yi J-C, Zheng Z-B, Tang Y, Dai L-X, You S-L, Angew. Chem. Int. Ed 2016, 55, 751–754; Angew. Chem 2016, 128, 761–764;Dudnik AS, Weidner VL, Motta A, Delferro M, Marks TJ, Nat. Chem 2014, 6, 1100–1107;Yang Z-P, Wu Q-F, Shao W, You S-L, J. Am. Chem. Soc 2015, 137, 15899–15906.
- [3].For recent examples involving phenols, see: Cheng Q, Wang Y, You S-L, Angew. Chem. Int. Ed 2016, 55, 3496–3499; Angew. Chem 2016, 128, 3557–3560;Wu Q-F, Liu W-B, Zhuo C-X, Rong Z-Q, Ye K-Y, You S-L, Angew. Chem. Int. Ed 2011, 50, 4455–4458; Angew. Chem 2011, 123, 4547–4550;Grenning AJ, Boyce JH, Porco JA Jr., J. Am. Chem. Soc 2014, 136, 11799–11804;Dong S, Zhu J, Porco JA Jr., J. Am. Chem. Soc 2008, 130, 2738–2739;Uyanik M, Yasui T, Ishihara K, Angew. Chem. Int. Ed 2013, 52, 9215–9218; Angew. Chem 2013, 125, 9385–9388;Nan J, Liu J, Zheng H, Zuo Z, Hou L, Hu H, Wang Y, Luan X, Angew. Chem. Int. Ed 2015, 54, 2356–2360; Angew. Chem 2015, 127, 2386–2390.
- [4].For recent examples involving nitroarenes, see: Lee S, Chataigner I, Piettre SR, Angew. Chem. Int. Ed 2011, 50, 472–476; Angew. Chem 2011, 123, 492–496;Trost BM, Ehmke V, O’Keefe BM, Bringley DA, J. Am. Chem. Soc 2014, 136, 8213–8216.
- [5].a) Birch AJ, Pure Appl. Chem 1996, 68, 553–556; [Google Scholar]; b) Rabideau PW, Marcinow Z, Org. React 1992, 42, 1–334. [Google Scholar]
- [6].a) Foubelo F, Yus M in Arene Chemistry: Reaction Mechanisms and Methods for Aromatic Compounds, (Ed.: Mortier J), Wiley, Hoboken, 2015, pp. 337–364; [Google Scholar]; b) Stanislaus A, Cooper BH, Catal. Rev. Sci. Eng 1994, 36, 75–123. [Google Scholar]
- [7].a) Harman WD, Taube H, J. Am. Chem. Soc 1988, 110, 7906–7907; [Google Scholar]; b) Keane JM, Harman WD, Organometallics 2005, 24, 1786–1798. [Google Scholar]
- [8].Boll M, J. Mol Microbiol Biotechnol 2005, 10, 132–142. [DOI] [PubMed] [Google Scholar]
- [9].For recent examples involving 1,3-cyclohexadiene syntheses, see:; a) Kulkarni AA, Diver ST, J. Am. Chem. Soc 2004, 126, 8110–8111; [DOI] [PubMed] [Google Scholar]; b) Hilt G, Paul A, Harms K, J. Org. Chem 2008, 73, 5187–5190; [DOI] [PubMed] [Google Scholar]; c) Ryan SJ, Candish L, Lupton DW, J. Am. Chem. Soc 2011, 133, 4694–4697; [DOI] [PubMed] [Google Scholar]; d) Nagasawa S, Sasano Y, Iwabuchi Y, Angew. Chem. Int. Ed 2016, 55, 13189–13194; Angew. Chem 2016, 128, 13383–13388. [DOI] [PubMed] [Google Scholar]
- [10].Southgate EH, Pospech J, Fu J, Holycross DR, Sarlah D, Nat. Chem 2016, 8, 922–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hamrock SJ, Sheridan RS, J. Am. Chem. Soc 1989, 111, 9247–9249. [Google Scholar]
- [12].a) Pasto DJ, Taylor RT, Org. React 1991, 40, 91–155; [Google Scholar]; b) Adam W, Eggelte HJ, J. Org. Chem 1977, 42, 3987–3988. [DOI] [PubMed] [Google Scholar]
- [13].Marsh BJ, Carbery DR, J. Org. Chem 2009, 74, 3186–3188. [DOI] [PubMed] [Google Scholar]
- [14].A limitation in scope at the current level of development is the lack of reactivity between MTAD and polysubstituted benzene derivatives. In addition, electron-deficient arenes do not react and some electron-rich arenes undergo undesired side reactions with MTAD. See the Supporting Information for details.
- [15].Nelsen SF, Trieber DA, Wolff JJ, Powell DR, Rogers-Crowley S, J. Am. Chem. Soc 1997, 119, 6873–6882. [Google Scholar]
- [16].Burk MJ, Feaster JE, J. Am. Chem. Soc 1992, 114, 6266–6267. [Google Scholar]
- [17].Adam W, Arias LA, De Lucchi O, Synthesis 1981, 543–545. [Google Scholar]
- [18].Egli M, Hoesch L, Dreiding AS, Helv. Chim. Acta 1985, 68, 220–230. [Google Scholar]
- [19].a) Kjell DP, Sheridan RS, J. Am. Chem. Soc 1984, 106, 5368–5370; [Google Scholar]; b) Breton GW, Newton KA, J. Org. Chem 2000, 65, 2863–2869. [DOI] [PubMed] [Google Scholar]
- [20].Hamrock SJ, Sheridan RS, Tetrahedron Lett 1988, 29, 5509–5512. [Google Scholar]
- [21].a) Isayama S, Mukaiyama T, Chem. Lett 1989, 18, 1071–1074; [Google Scholar]; b) Mukaiyama T, Isayama S, Inoki S, Kato K, Yamada T, Takai T, Chem. Lett 1989, 18, 449–452. [Google Scholar]
- [22].a) Lo JC, Yabe Y, Baran PS, J. Am. Chem. Soc 2014, 136, 1304–1307; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Obradors C, Martinez RM, Shenvi RA, J. Am. Chem. Soc 2016, 138, 4962–4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kornblum N, DeLaMare HE, J. Am. Chem. Soc 1951, 73, 880–881. [Google Scholar]
- [24].a) Fournet A, Barrios A, Munoz V, Hocquemiller R, Roblot F, Cave A, Planta Med 1994, 60, 8–12; [DOI] [PubMed] [Google Scholar]; b) Dwivedi GR, Upadhyay HC, Yadav DK, Singh V, Srivastava SK, Khan F, Darmwal NS, Darokar MP, Chem. Biol. Drug Des 2014, 83, 482–492. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.