

Summary
Sepsis is a life-threatening condition caused by pathogen infection and associated with pyroptosis. Pyroptosis occurs upon activation of proinflammatory caspases and their subsequent cleavage of gasdermin D (GSDMD), resulting in GSDMD N-terminal fragments that form membrane pores to induce cell lysis. Here, we show that antioxidant defense enzyme glutathione peroxidase 4 (GPX4) and its ability to decrease lipid peroxidation, negatively regulate macrophage pyroptosis, and septic lethality in mice. Conditional Gpx4 knockout in myeloid lineage cells increases lipid peroxidation-dependent caspase-11 activation and GSDMD cleavage. The resultant N-terminal GSDMD fragments then trigger macrophage pyroptotic cell death in a phospholipase C gamma 1 (PLCG1)-dependent fashion. Administration of the antioxidant vitamin E that reduces lipid peroxidation, chemical inhibition of PLCG1, or genetic Caspase-11 or Gsdmd inactivation prevents polymicrobial sepsis in Gpx4−/− mice. Collectively, this study suggests that lipid peroxidation drives GSDMD-mediated pyroptosis and hence constitutes a potential therapeutic target for lethal infection.
eTOC Blurb
Kang et al demonstrates that glutathione peroxidase 4 (GPX4) serves a protective role in mice undergoing septic shock. Myeloid-specific deficiency of GPX4 coordinates lipid peroxidation-dependent caspase-11 activation and GasderminD-mediated pyroptosis during polymicrobial sepsis. Vitamin E administration reverses Gpx4Mye−/−-susceptibility, thereby revealing a potential target for therapeutic intervention for lethal infection.
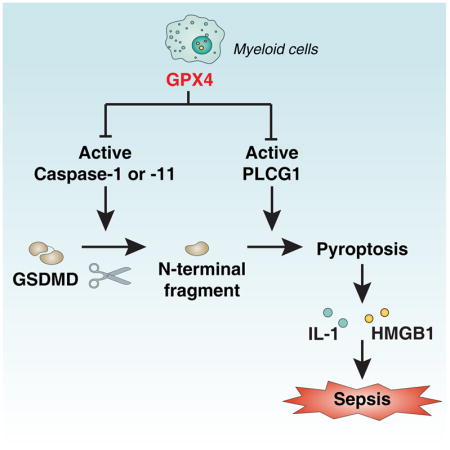
Introduction
Sepsis and septic shock are frequently lethal complications of bacterial infections, accounting for approximately 250,000 deaths per year in the US alone (Singer et al., 2016). Although infection-elicited inflammatory responses are crucial for host defense against invading microbes (Jorgensen et al., 2017), excessive activation of innate immune cells such as macrophages may contribute to tissue damage and multiple organ failure. Pyroptosis, an inflammatory form of regulated necrosis, occurs as a consequence of the activation of caspase-1 and caspase-11 in macrophages (Man and Kanneganti, 2016). It has recently been shown that the cleavage of gasdermin D (GSDMD) by caspases-1 and -11 constitutes a critical event in the lethal cascade leading to infection-associated pyroptotic cell death (Kayagaki et al., 2015; Shi et al., 2015). Indeed, the N-terminal fragment of GSDMD (GSDMD-N) can bind to inner membrane lipids, forming pores in the plasma membrane that ultimately result in cellular lysis (Kayagaki et al., 2015; Shi et al., 2015).
Cytoplasmic membrane lipid peroxidation, an auto-oxidative process initiated by the attack of free radicals, contributes to the progression of various types of regulated cell death (Gaschler and Stockwell, 2017). However, it remains unknown whether lipid peroxidation contributes to GSDMD-mediated pyroptosis during lethal infection. Here, we demonstrate that glutathione peroxidase 4 (GPX4), an antioxidant defense enzyme active in repairing oxidative damage to lipids, is an important negative regulator of macrophage pyroptosis. Genetic inactivation of GPX4 increases lipid peroxidation, thus exacerbating GSDMD-mediated pyroptosis in macrophages as well as septic lethality in mice. Thus, our findings uncover a critical mechanism that controls lipid peroxidation in the context of infection-induced lethality.
Results
Gpx4 deficiency in myeloid cells results in increased susceptibility to polymicrobial infection
Mammalian glutathione peroxidases (GPXs) including GPX1-8 are functional antioxidant defense enzymes that protect cells from oxidative damage (Brigelius-Flohe and Maiorino, 2013). To understand the roles of GPXs in polymicrobial infections, we quantified the expression of Gpx1-8 in peritoneal macrophages (PMs) and peripheral blood mononuclear cells (PBMCs) from C57BL/6 mice subjected to cecal ligation and puncture (CLP), a well-established animal model of polymicrobial infection. Unlike most other Gpx family members, Gpx4 mRNA was time-dependently upregulated in PMs and PBMCs from septic mice (Fig. 1A). In contrast, experimental sepsis failed to significantly alter Gpx4 mRNA levels in the heart, liver, kidney, and lung (Fig. 1B). Of note, the mRNA levels of Gpx4 (but not Gpx1, Gpx2, Gpx3, Gpx5, Gpx6, Gpx7, and Gpx8) were also upregulated in PBMCs from septic patients compared to the healthy control group (Fig. 1C). Consistently, GPX4 protein expression was similarly increased in mouse PMs (Fig. 1D) and human PBMCs (Fig. 1E) under septic conditions. These findings indicate that Gpx4 is specifically upregulated in innate immune cells during the course of experimental and clinical sepsis.
Fig. 1. Gpx4 deficiency in myeloid cells results in increased susceptibility to polymicrobial infection.

(A) Q-PCR analysis of Gpx1-8 mRNA in PMs or PBMCs from septic mice at 24–72 hours (n=3 mice/group; data are expressed as means ± SD, *, P<0.05 versus control group, t test). (A) Q-PCR analysis of Gpx1-8 mRNA in the indicated tissues from septic mice at 24–72 hours (n=3 mice/group; data are expressed as means ± SD, *, P<0.05 versus control group, t test). (C) Box plots comparing Gpx1-8 mRNA levels in PBMC samples from septic patients (n=16) and healthy controls (n=16). The mRNAs are presented as median value (black line), interquartile range (box), and minimum and maximum of all data (black line). *, P<0.05 versus control group, t test. (D) Western blot analysis of GPX4 protein expression in mouse PMs from control group or septic mice at 72 hours (n=3 mice/group; data expressed as means ± SD, *, P<0.05, t test). (E) Western blot analysis of GPX4 protein expression in human PBMCs or septic patients at hospital admission (n=3 patient/group; data expressed as means ± SD, *, P<0.05, t test). (F) Genotype identification of transgenic mice based on reverse transcription polymerase chain reaction. (G) Western blot analysis of GPX4 expression in PMs and hepatocytes from Gpx4 flox/flox, Gpx4Hep−/−, or Gpx4Mye−/− mice with or without CLP for 72 hours. (H) Mice with the indicated genotypes were subjected to CLP with syringe needles with gauges ranging from 27 (A, “low-grade sepsis”), 22 (B, “middle-grade sepsis”), to 17 (C, “high-grade sepsis”). Animal survival was assayed (n=20 mice/group; *, P<0.05, Kaplan-Meier survival analysis).
To define the role of GPX4 in polymicrobial infection, we generated myeloid cell-specific (LysM-Cre;Gpx4 flox/flox, termed Gpx4Mye−/− mice) and hepatocyte-specific Gpx4-knockout mice (Alb-Cre;Gpx4 flox/flox, termed Gpx4Hep−/− mice) (Fig. 1F). Immunoblot analysis confirmed a diminished GPX4 protein expression in PMs and hepatocytes from Gpx4Mye−/− and Gpx4Hep−/− mice, respectively (Fig. 1G). Remarkably, Gpx4Mye−/− mice, but not Gpx4Hep−/− mice, were significantly more susceptible to death induced by low-, middle-, and high-grade polymicrobial sepsis than control Gpx4 flox/flox mice (Fig. 1H). Thus, Gpx4 expressed in the myeloid lineage cells, but not in hepatocytes, functions as an endogenous inhibitor of lethal sepsis.
Vitamin E and caspase inhibitor protect against sepsis in mice
To define the molecular mechanisms underlying GPX4-mediated protection against polymicrobial infection, we treated mice with the antioxidant vitamin E and examined the contribution of oxidative injury to septic shock. Although vitamin E protected against CLP-induced animal lethality in both Gpx4flox/flox and Gpx4Mye−/− mice, vitamin E exhibited a greater increase in animal survival for Gpx4Mye−/− mice than Gpx4flox/flox mice (50% versus 30%, Fig. 2A). The serum levels of lipid peroxidation products (e.g., malondialdehyde [MDA] and 4-hydroxynonenal [4-HNE]) and markers indicating organ dysfunction (e.g., creatine kinase [CK], blood urea nitrogen [BUN], and alanine aminotransferase [ALT]) were all increased in septic Gpx4Mye−/− mice compared to septic Gpx4flox/flox mice (Fig. 2B). Vitamin E administration significantly attenuated the sepsis-induced elevation of serum MDA, 4-HNE, CK, BUN, and ALT levels (Fig. 2B), supporting the notion that oxidative injury contributes to septic lethality. As expected, there were no significant phenotype differences between Gpx4flox/flox and Gpx4Mye−/− mice after sham surgery (Fig. 2A and 2B).
Fig. 2. Effects of vitamin E and cell death inhibitors on sepsis in mice.

(A) Analysis of animal survival in mice with or without repeated intraperitoneal administration of vitamin E (500 mg/kg) at three, 24, 48, and 72 hours after CLP (22-gauge needle)-induced sepsis (n=5–10 mice/group; *, P<0.05, Kaplan-Meier survival analysis). (B) In parallel, serum levels of MDA, 4-HNE, CK, BUN, and ALT were assayed at 72 hours after CLP (n=5 mice/group; *, P<0.05, ANOVA LSD test). Data are presented as median value (black line), interquartile range (box), and minimum and maximum of all data (black line). (C) Analysis of animal survival in mice with or without repeated intraperitoneal administration of Z-VAD-FMK (“ZVF”, 5 mg/kg), wedelolactone (“Wed”, 20 mg/kg), ferrostatin-1 (“Fer”, 10 mg/kg), or necrostatin-1 (“Nec”, 5 mg/kg) at three, 24, 48, and 72 hours after CLP (22-gauge needle)-induced sepsis (n=10 mice/group; *, P<0.05, Kaplan-Meier survival analysis). (D) In parallel, serum levels of IL-1β, IL-18, HMGB1, IL-6, IL-10, IL-17, and IL-12 were assayed at 72 hours after CLP (n=5 mice/group; *, P<0.05, ANOVA LSD test). Data are presented as median value (black line), interquartile range (box), and minimum and maximum of all data (black line).
Gpx4 deficiency-induced lipid peroxidation has been implicated in the progression of regulated cell death, including apoptosis (Ran et al., 2003; Seiler et al., 2008), necroptosis (Canli et al., 2016), and ferroptosis (Friedmann Angeli et al., 2014; Yang et al., 2014b). Importantly, administration of a specific inhibitor for ferroptosis (ferrostatin-1) or necroptosis (necrostatin-1) failed to affect CLP-induced septic lethality in Gpx4Mye−/− mice (Fig. 2C). In contrast, administration of Z-VAD-FMK (a pan caspase inhibitor) or wedelolactone (an inhibitor of caspase-11) to Gpx4Mye−/− mice conferred significant protection against lethal sepsis (Fig. 2C). Similarly, Z-VAD-FMK also partly protected against CLP-induced septic death in Gpx4flox/flox mice (Fig. 2C). Furthermore, the sepsis-induced elevation of serum levels of inflammasome-dependent cytokines (e.g., IL-1β, IL-18, and HMGB1), but not inflammasome-independent cytokines (e.g., IL-6, IL-10, IL-17, and IL-12), was attenuated by the administration of Z-VAD-FMK or wedelolactone, but not by ferrostatin-1 or necrostatin-1 (Fig. 2D). Collectively, these findings suggest a major role for pyroptosis in the pathogenesis of lethal sepsis.
Caspase-11-dependent pyroptosis mediates septic death in Gpx4Mye−/− mice
It has been shown that global knockout of caspase-11 has no impact on CLP-induced sepsis (Vanden Berghe et al., 2014), but protects against lethal endotoxemia (Hagar et al., 2013). Moreover, conditional knockout of caspase-11 in endothelial cells confers similar protection against LPS- and CLP-induced acute lung jury and lethality in mice (Cheng et al., 2017), suggesting a cell type-dependent role of caspase-11 in sepsis.
To test whether caspase-11-dependent pyroptosis is required for lethal inflammation in septic Gpx4Mye−/− mice, we generated Caspase-11 and Gpx4 double knockout mice using myeloid lineage cells (LysM-Cre;Gpx4 flox/flox;Casp11flox/flox, termed Gpx4Mye−/−Casp11Mye−/− mice). As compared to Gpx4 flox/flox controls, Casp11Mye−/− or Gpx4Mye−/− Casp11Mye−/− mice exhibited increased survival (Fig. 3A) with decreased serum levels of IL-1β, IL-18, HMGB1, CK, BUN, and ALT, but not IL-6 and IL-10, during experimental sepsis (Fig. 3B), supporting an essential role for caspase-11 in mediating lethal inflammation and tissue injury.
Fig. 3. Caspase-11-dependent pyroptosis mediates septic death in Gpx4Mye−/− mice.

(A) Mice with the indicated genotypes were subjected to CLP with 22-gauge syringe needles (middle-grade sepsis) and animal survival was monitored (n=20 mice/group; *, P<0.05, Kaplan-Meier survival analysis). Gpx4Mye−/− mice data was the same control as Fig. 1H (middle-grade sepsis). (B) Analysis of serum levels of IL-1β, IL-18, HMGB1, IL-6, IL-10, CK, BUN, and ALT in middle grade septic mice at 72 hours (n=5 mice/group; *, P<0.05, ANOVA LSD test). Data are presented as median value (black line), interquartile range (box), and minimum and maximum of all data (black line). (C) Analysis of bacterial loading in middle grade septic mice at 72 hours (n=5 mice/group; *, P<0.05, ANOVA LSD test). Data are presented as median value (black line), interquartile range (box), and minimum and maximum of all data (black line).
Although pyroptotic macrophages may have the ability to clear bacteria (Jorgensen et al., 2016), the number of bacterial colony-forming units recovered from the blood and peritoneal lavage after CLP did not significantly differ between Gpx4Mye−/− and Gpx4Mye−/−Casp11Mye−/− mice (Fig. 3C). In light of the tremendous impact of altered microbiota on the outcome of CLP sepsis (Wilmore et al., 2018), it will be interesting to examine whether Gpx4 deficiency causes microbiome variation in independent studies.
GPX4 blocks GSDMD cleavage during inflammasome activation
The cleavage and formation of activated N-terminal domain of GSDMD (GSDMD-N) by inflammatory caspases is a recently identified executioner of pyroptosis (Aglietti et al., 2016; Ding et al., 2016; Liu et al., 2016; Sborgi et al., 2016). We next investigated whether, in lieu of suppressing bacterial elimination, GPX4 regulated the cleavage of GSDMD following inflammasome activation. Caspase-11-mediated non-canonical inflammasome activation requires at least two signals (Kayagaki et al., 2015; Shi et al., 2015). The first signal (signal 1), also known as the priming signal, is triggered by microbial products or proinflammatory cytokines such as interferon-γ (IFN-γ) that are recognized by macrophage pattern recognition and cytokine receptors (Kayagaki et al., 2015; Rathinam et al., 2012). Signal 1 activates the NF-κ B pathway, leading to upregulation of pro-IL-1β and pro-caspase-11 protein levels (Kayagaki et al., 2015). However, the loss of Gpx4 failed to affect TLR ligands [e.g., lipopolysaccharide (LPS), poly(I:C), and Pam3CSK4], and IFN-γ-induced pro-IL-1β and pro-caspase-11 protein (Fig. 4A) and mRNA (Fig. 4B) expression in bone-marrow-derived macrophages (BMDMs). The second signal (Signal 2) is mediated by cytosolic LPS or bacterial infection, resulting in caspase-11-dependent GSDMD cleavage to generate activated GSDMD-N (Kayagaki et al., 2015; Shi et al., 2015). We observed that the loss of Gpx4 increased GSDMD-N formation, proteolytic IL-1β maturation (p17), and caspase-11 (p26) activation in BMDMs following LPS electroporation or Escherichia coli infection (Fig. 4C). This process was blocked by caspase-11 depletion (Fig. 4C). Consistently, the LPS electroporation- or Escherichia coli infection-induced cytotoxicity and associated extracellular release of lactate dehydrogenase [LDH], IL-1β, and HMGB1 was increased in Gpx4−/−, but not in Casp11−/− or Gpx4−/−Casp11−/− BMDMs (Fig. 4D).
Fig. 4. GPX4 blocks GSDMD cleavage during inflammasome activation.

(A) Western blot analysis of IL-1β and caspase-11 protein expression in BMDMs following treatment with LPS (500 ng/ml), poly (I:C) (5 μg/ml), Pam3CSK4 (1 μg/ml), or IFN-γ (10/ng/ml) for six hours. (B) Q-PCR analysis of IL-1β and caspase-11 mRNA expression in BMDMs following treatment with LPS (500 ng/ml), poly (I:C) (5 μg/ml), Pam3CSK4 (1 μg/ml), or IFN-γ (10/ng/ml) for six hours. (C) Western blot analysis of indicated proteins in BMDMs recovered from mice with the indicated genotypes following LPS electroporation or E. coli (MOI=25) infection for 16 hours. Sup= supernatants. (D) In parallel, cytotoxicity (LDH release) and levels of IL-1β and HMGB1 in the supernatant were assayed (n=3 wells/group; data expressed as means ± SD, *, P<0.05, ANOVA LSD test). (E) Western blot analysis of LPS (500 ng/ml)-primed indicated BMDMs following treatment with ATP (5 mM) for one hour. Sup=supernatants. (F) In parallel, cytotoxicity and levels of IL-1β and HMGB1 in the supernatant were assayed (n=3 wells/group; data expressed as means ± SD, *, P<0.05, ANOVA LSD test).
In addition to the non-canonical (caspase-11-dependent) inflammasome, the canonical (caspase-1-dependent) NLRP3 inflammasome contributes to GSDMD cleavage under certain conditions (He et al., 2015; Kayagaki et al., 2015; Shi et al., 2015). Indeed, adenosine triphosphate (ATP), a classical NLRP3 inflammasome activator, induced GSDMD-N formation (Fig. 4E), caspase-1 (p20) activation (Fig. 4E), cytotoxicity (Fig. 4F), and consequent release of matured IL-1β (p17) (Fig. 4F) and HMGB1 (Fig. 4F) in LPS-primed Gpx4−/−, Casp11−/−, or Gpx4−/−Casp11−/−, but not in Nlrp3−/−, Casp1−/−Casp11−/−, Gpx4−/−Nlrp3−/− or Gpx4−/−Casp1−/−Casp11−/− BMDMs. Consistent with previous studies (Kayagaki et al., 2011; Py et al., 2014), ATP failed to induce caspase-11 activation in macrophages (Fig. 4E). Collectively, these findings indicate that GPX4 functions as an endogenous repressor of caspase-11- or caspase-1-mediated cleavage of GSDMD in activated macrophages.
Oxidation of phospholipids promotes GSDMD-N-mediated pyroptosis
We next investigated the impact of GPX4 on GSDMD-N-induced pyroptosis. Consistent with a previous study (Shi et al., 2015), transfection with GSDMD-N (1–276), but not GSDMD-C (277–487), GSDMD cleavage mutant (D275A), or full-length FL-GSDMD (1–487), induced cytotoxicity to BMDMs (Fig. 5A). The GSDMD-N-mediated cytotoxicity was further increased in Gpx4−/− BMDMs (Fig. 5A), but was reversed by vitamin E supplementation or enforced expression of Gpx4 (Fig. 5A). In contrast, GSDMD-N-induced cytotoxicity was not reversed by the depletion of Caspase-1 and Caspase-11 or enforced expression of a catalytically inactive Gpx4 cDNA (U46S mutant) (Mannes et al., 2011) in Gpx4−/− BMDMs (Fig. 5A), indicating that caspase-1 and caspase-11 are not essential for GSDMD-N-mediated cytotoxicity despite their involvement in the generation of GSDMD-N.
Fig. 5. Oxidation of phospholipids promote GSDMD-N-mediated pyroptosis.

(A) WT and Gpx4−/− BMDMs were pretreated with vitamin E (100 μM) for three hours before transfection with GSDMD-C, GSDMD-N, GSDMD-D275A, or GSDMD-FL expression constructs. Cell viability was assayed within 24 hours of transfection (n=3 wells/group; means ± SD, *, P<0.05, t test). (B) WT and Gpx4−/− BMDMs were pretreated with indicated lipid components (20 μM) for three hours, and then transfected with GSDMD-N. Cell viability was assayed within 24 hours of transfection (n=3 wells/group; means ± SD, *, P<0.05 versus GSDMD-N group, t test). (C) WT and Gpx4−/− BMDMs were pretreated with indicated lipid components (20 μM) for three hours and then transfected with GSDMD-N. Intracellular MDA level was assayed within 24 hours of transfection (n=3 wells/group; means ± SD, *, P<0.05 versus GSDMD-N group, t test).
The plasma membrane is a complex mixture of multiple lipids including fatty acids, phospholipids, sterols, and sphingolipids (van Meer et al., 2008). Treatment with several types of phospholipids (e.g., phosphatidylinositol 4-phosphate [PI4P], phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2], diacylglycerol [DAG], and phosphatidic acid [PA]) sensitized BMDMs to GSDMD-N-induced cytotoxicity (Fig. 5B), which was associated with increased MDA production (Fig. 5C). In contrast, this process was not affected by the administration of linoleic acid (a polyunsaturated fatty acid), ceramide (a sphingolipid), or cholesterol (a sterol) (Fig. 5B and 5C). Together, these findings suggest that lipid peroxides generated by the oxidation of phospholipids promote GSDMD-N-mediated pyroptosis in BMDMs.
PLC activation is required for N-terminal GSDMD-mediated pyroptosis
Phospholipase C (PLC) mediates the hydrolysis of phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2) into diacyl glycerol (DAG) and inositol 1,4,5-trisphosphate (IP3), which in turn triggers the activation of calcium signal (Kadamur and Ross, 2013). We therefore investigated whether the increased GSDMD-N-mediated cytotoxicity observed in Gpx4−/− BMDMs was associated with PLC activation. Indeed, the phosphorylation of PLC gamma 1 (PLCG1), but not that of PLC beta 3 (PLCB3), was increased in Gpx4−/− BMDMs in response to GSDMD-N-mediated pyroptosis (Fig. 6A). The GSDMD-N-induced PLCG1 phosphorylation in BMDMs was significantly inhibited by vitamin E (Fig. 6A), suggesting that oxidative stress induces PLCG1 activation. Notably, shRNA-mediated depletion of Plcg1 and addition of the pharmacological PLC inhibitor U73122 similarly inhibited GSDMD-N-induced pyroptosis, which was associated with a decreased production of DAG and IP3, as well as a reduced intracellular calcium influx (Fig. 6B). The calcium chelator BAPTA-AM blocked GSDMD-N-induced pyroptosis and calcium influx, but not DAG and IP3 production (Fig. 6B). These findings indicate that GSDMD-N-mediated activation of calcium signaling is downstream of PLCG1-mediated DAG and IP3 production (Hayashi et al., 2007; Pocock and Bates, 2001).
Fig. 6. PLC activation contributes to GSDMD-mediated pyroptosis.

(A) WT and Gpx4−/− BMDMs were pretreated with or without vitamin E (100 μM) for three hours, and then transfected with GSDMD-N. Protein expression was assayed using western blot. (B) Analysis of GSDMD-N-mediated cytotoxicity, DAG, IP3, and calcium production at 24 hours in WT and Gpx4−/− BMDMs in the absence or presence of Plcg1-shRNA, U73122 (10 μM), or BAPTA-AM (10 μM) (n=3 wells/group; data expressed as means ± SD, *, P<0.05 versus GSDMD-N group, t test). (C) Analysis of GSDMD-N-mediated cytotoxicity at 24 hours in THP1, HL-60, and HeLa cell lines (n=3 wells/group; data expressed as means ± SD, *, P<0.05, t test). (D) WT BMDMs were pretreated with indicated lipid components (20 μM) for three hours, and then transfected with GSDMD-N for 24 hours. Protein expression was assayed using western blot. (E) Analysis of GSDMD-N/PI4P- or GSDMD-N/PI(4,5)P2-mediated cytotoxicity at 24 hours in BMDMs in the absence or presence of Plcg1-shRNA, U73122 (10 μM), or BAPTA-AM (10 μM) (n=3 wells/group; data expressed as means ± SD, *, P<0.05 versus control group, t test). (F) Analysis of LPS electroporation- or E. coli (MOI=25) infection-mediated cytotoxicity at 16 hours in BMDMs in the absence or presence of Plcg1-shRNA or U73122 (10 μM) (n=3 wells/group; data expressed as means ± SD, *, P<0.05 versus control group, t test). (G) Analysis of LPS electroporation- or E. coli (MOI=25) infection-mediated protein expression at 16 hours in BMDMs in the absence or presence of Plcg1-shRNA or U73122 (10 μM).
Knockdown of PLCG1 also blocked GSDMD-N-mediated cytotoxicity in THP1 (human monocytic leukemia cell line), HL-60 (human promyelocytic leukemia cell line), and HeLa (human cervical cancer) cells (Fig. 6C). Moreover, exogenous PI4P, but not linoleic acid, ceramide, or cholesterol, enhanced GSDMD-N-induced PLCG1 phosphorylation (Fig. 6D), suggesting a possible reciprocal relationship between PLCG1 activation and PIP2 metabolism during GSDMD-N-induced pyroptosis. The knockdown of Plcg1 and administration of U73122 or BAPTA-AM similarly inhibited GSDMD-N-induced cell death in the presence of phospholipids (PI4P and PI(4,5)P2) (Fig. 6E), supporting a possible association between Ca2+ elevation and lipid peroxidation during GSDMD-N-induced pyroptosis (Aglietti et al., 2016; Liu et al., 2016).
Of note, genetic or pharmacologic inhibition of Plcg1 by shRNA or using U73122 similarly attenuated the LPS electroporation- or Escherichia coli infection-induced cytotoxicity (Fig. 6F) without affecting the production of active caspase-11 (p26), GSDMD-N, and matured IL-1β (p17) (Fig. 6G). Collectively, these findings suggest that phospholipid peroxidation-mediated PLCG1 activation may contribute to GSDMD-N-mediated cytotoxicity without affecting GSDMD-N production.
PLC activation is required for N-terminal GSDMD-mediated septic death
To determine whether GSDMD activation contributes to septic death of Gpx4Mye−/− mice, we used backcrossed GsdmdI105N/I105N mice (which bear a GSDMD cleavage site mutation that renders the protein resistant to proteolytic activation by caspase-1 or -11) in the Gpx4Mye−/− background to generate LysM-Cre;Gpx4 flox/flox;GsdmdI105N/I105N (in short Gpx4Mye−/−Gsdmd−/−) mice. Such GSDMD cleavage mutant mice lacking Gpx4 in their myeloid cells were protected against CLP-induced lethal sepsis (Fig. 7A) with decreased serum levels of CK, BUN, and ALT (Fig. 7B) compared to Gpx4Mye−/− mice expressing activatable wild-type (WT) GSDMD. Compared to WT control mice, the PLC inhibitor U73122 exhibited greater protection against CLP-induced organ damage and animal lethality in Gpx4Mye−/− mice compared to control WT animals (Fig. 7A and 7B). Collectively, these animal studies indicate that the caspase-11-GSDMD-PLC pathway contributes to multi-organ failure in septic mice, especially in Gpx4Mye−/− mice.
Fig. 7. PLC activation contributes to GSDMD-mediated septic death.

(A) Analysis of animal survival in mice with or without repeated intraperitoneal administration of U73122 (30 mg/kg) at three, 24, 48, and 72 hours after CLP (22-gauge needle)-induced sepsis (n=10–20 mice/group; *, P<0.05, Kaplan-Meier survival analysis). Gpx4Mye−/− mice data was the same control as Fig. 1H (middle-grade sepsis). (B) In parallel, quantitation of serum CK, BUN, and ALT in middle grade septic mice at 72 hours (n=5–8 mice/group; *, P<0.05, ANOVA LSD test). Data are presented as median value (black line), interquartile range (box), and minimum and maximum of all data (black line).
Discussion
Oxidative stress occupies a critical role in the regulation of various biological processes including cell death and innate immunity (Reuter et al., 2010; Ryter et al., 2007). Here, we demonstrate a potential involvement of GPX4 in coordinating lipid peroxidation, inflammasome activation, and pyroptosis during polymicrobial infection. In response to infectious insults, GPX4 is upregulated in innate immune cells to counter-regulate GSDMD-N mediated pyroptotic cell death, thereby preventing lethal systemic inflammation. These findings therefore establish GPX4 as an essential negative regulator of pyroptosis in lethal inflammation.
In conjunction with antioxidants (e.g., vitamin E), GPX4 catalyzes the reduction of phospholipid hydroperoxides within membranes and lipoproteins to inhibit lipid peroxidation, thereby exerting protection against oxidative stress (Imai and Nakagawa, 2003). Global knockout of Gpx4 results in early embryonic lethality, while conditional knockout of Gpx4 in specific organs (e.g., myeloid cells) is compatible with survival into adulthood. Conditional knockout models revealed that Gpx4 protects against experimental renal failure (Friedmann Angeli et al., 2014), neurodegenerative diseases (Chen et al., 2015), viral and parasitic infections (Matsushita et al., 2015), male subfertility (Brutsch et al., 2016), atherogenesis (Guo et al., 2008), anemia (Canli et al., 2016), and thrombus formation (Wortmann et al., 2013). Gpx4 depletion resulted in a marked elevation of apoptosis (Ran et al., 2003; Seiler et al., 2008), necroptosis (Canli et al., 2016), and ferroptosis (Friedmann Angeli et al., 2014; Yang et al., 2014b). The results from the present study indicate that GPX4 also plays a major role in reducing excessive macrophage pyroptosis in the context of sepsis.
We propose that GPX4 serves as a negative regulator of GSDMD cleavage and activation. The generation of GSDMD-N enables its oligomerization to form membrane-damaging pores and to initiate the lethal phase of pyroptosis. The production of GSDMD-N is catalyzed by inflammatory caspases such as caspase-1 and -11 (Aglietti et al., 2016; Ding et al., 2016; He et al., 2015; Kayagaki et al., 2015; Liu et al., 2016; Sborgi et al., 2016; Shi et al., 2015). In contrast to caspase-1-dependent pyroptosis, caspase-11-mediated pyroptosis is triggered by the intracellular invasion of bacteria (Aachoui et al., 2013; Hagar et al., 2013; Kayagaki et al., 2011; Kayagaki et al., 2013). Live Escherichia coli and cytosolic LPS require caspase-11, but not caspase-1, to trigger pyroptosis in macrophages (Kayagaki et al., 2011). Specifically, caspase-11 recognizes cytosolic LPS via binding to the lipid A moiety, which results in caspase-11 oligomerization and GSDMD cleavage (Shi et al., 2014). In the present study, we demonstrated that myeloid lineage cell-specific Gpx4 depletion caused a marked increase in caspase-11- and caspase-1-mediated GSDMD cleavage, suggesting that lipid peroxidation may serve as an accelerator of inflammasome activation and pyroptosis.
It is well-known that GSDMD-N induces pyroptosis only when delivered to the cytosol by transfection, but not when directly added extracellularly, supporting the notion that GSDMD is an endogenous pore-forming protein that inserts itself into the inner leaflet of the plasma membrane (Ding et al., 2016; Liu et al., 2016). The formation of membrane GSDMD pores disrupts the distribution of phospholipids, causing cell swelling (oncosis) and eventual lysis (Aglietti et al., 2016; Ding et al., 2016; Liu et al., 2016; Sborgi et al., 2016). While ferroptosis involves the oxidation of polyunsaturated fatty acids within cell membranes (Kagan et al., 2017; Yuan et al., 2016), our study indicates that oxidation of phospholipids may be involved in GSDMD-N-induced pyroptosis.
Specifically, our data suggest that lipid peroxidation specifically induced the activation of PLCG1 in myeloid cells and that PLCG1 contributes to GSDMD-N-mediated cytotoxicity in a calcium-dependent manner. PLCG1 is a phosphatidylinositol-specific PLC, a class of membrane-associated enzymes that cleaves PIP2 into DAG and IP3, resulting in the mobilization of intracellular calcium stores that may contribute to cell death and inflammatory reactions (Kadamur and Ross, 2013). Importantly, the PLC inhibitor U73122 and the calcium chelator BAPTA-AM protect against GSDMD-N cytotoxicity in macrophages or against lethal infection in mice.
In summary, we demonstrated that Gpx4 expressed by cells from the myeloid lineage plays a major role in attenuating lipid peroxidation, inflammasome activation, and pyroptotic cell death in the context of sepsis. The depletion of Gpx4 results in increased septic lethality, which is at least partly mediated through caspase-11-mediated GSDMD cleavage and PLCG1-dependent GSDMD-N activation in macrophages. Genetic or pharmacologic inhibition of this pathway reversed the lethal consequences of excessive pyroptosis in the context of sepsis.
STAR METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal [EPNCIR144] to GPX4 | Abcam | ab125066, RRID:AB_10973901 |
| Rabbit polyclonal to caspase-1 | Abcam | ab1872, RRID:AB_302644 |
| Goat polyclonal to IL-1β | R&D Systems | AF-401-NA, RRID:AB_416684 |
| Rabbit polyclonal to GSDMD | Sigma Aldrich | G7422, RRID:AB_1850381 |
| Mouse monoclonal to caspase-1 (p20) | AdipoGen | AG-20B-0042, RRID:AB_2490248 |
| Mouse monoclonal [A-7] to GSDMD | Santa Cruz Biotechnology | Sc-393656, RRID: AB_2728694 |
| Mouse monoclonal [64-Y] to GSDMD | Santa Cruz Biotechnology | Sc-81868, RRID: RRID:AB_2263768 |
| Rabbit polyclonal to PLCG1 | Cell Signaling Technology | 2822, RRID:AB_10691689 |
| Rabbit monoclonal [D6M9S] to phospho-PLCG1 (Tyr783) | Cell Signaling Technology | 14008, RRID: AB_2728690 |
| Rabbit monoclonal [D9D6S] to PLCB3 | Cell Signaling Technology | 14247, RRID: AB_2728691 |
| Rabbit monoclonal [D8K2R] to phospho-PLCB3 (Ser537) | Cell Signaling Technology | 29021, RRID: AB_2728692 |
| Mouse monoclonal [8H10D10] to actin | Cell Signaling Technology | 3700, RRID:AB_2242334 |
| Rat monoclonal [17D9] to caspase-11 (p20) | Cell Signaling Technology | 14340, RRID: AB_2728693 |
| Rabbit polyclonal to HMGB1 | Cell Signaling Technology | 2822, RRID:AB_10691689 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Escherichia coli LPS 0111:B4 | Sigma Aldrich | L4391 |
| Vitamin E | Sigma Aldrich | 258024 |
| Necrostatin-1 | Sigma Aldrich | N9037 |
| Z-VAD-FMK | Sigma Aldrich | V116 |
| Wedelolactone | Sigma Aldrich | W4016 |
| PA | Sigma Aldrich | P9511 |
| DAG | Sigma Aldrich | D8394 |
| Linoleic acid | Sigma Aldrich | L1376 |
| Ceramide | Sigma Aldrich | 22244 |
| Cholesterol | Sigma Aldrich | C8667 |
| U73122 | Sigma Aldrich | 662035 |
| Gentamicin | Sigma Aldrich | G1397 |
| Ferrostatin-1 | Selleck Chemicals | S7243 |
| ATP | InvivoGen | tlrl-atp |
| Poly(I:C) | InvivoGen | 31852-29-6 |
| Pam3CSK4 | InvivoGen | tlrl-pms |
| PI4P | Echelon Biosciences Incorporated | P-4004 |
| PI(4,5)P2 | Echelon Biosciences Incorporated | P-4508 |
| BAPTA-AM | Calbiochem | 126150-97-8 |
| Lipofectamine 3000 | Invitrogen | L3000-015 |
| Puromycin | InvivoGen | ant-pr-1 |
| SuperSignal™ West Pico Chemiluminescent Substrate | Thermo Fisher Scientific | 34080 |
| SuperSignal™ West Femto Maximum Sensitivity Substrate | Thermo Fisher Scientific | 34095 |
| Critical Commercial Assays | ||
| Cell Counting Kit-8 (CCK-8) | Dojindo Laboratories | CK04 |
| HMGB1 ELISA Kit | Shino Test Corporation | ST51011 |
| IL-1β ELISA Kit | R&D Systems | MLB00C |
| IL-18 ELISA Kit | R&D Systems | 7625 |
| IL-12 ELISA Kit | R&D Systems | DY419 |
| IL-17 ELISA Kit | R&D Systems | DY421 |
| LDH Assay Kit | Abcam | ab102526 |
| MDA Assay Kit | Abcam | ab118970 |
| 4-HNE Assay Kit | MyBioSource | MBS702101 |
| DAG Assay Kit | Cell Biolabs Inc. | MET-5028 |
| IP3 Assay Kit | LifeSpan Biosciences, Inc. | LS-F10644-1 |
| CK single-slide test | IDEXX | 98-11073-01 |
| BUN single-slide test | IDEXX | 98-11070-01 |
| ALT single-slide test | IDEXX | 98-11067-01 |
| iScript cDNA Synthesis Kit | Bio-Rad | 1708890 |
| 4–12% Criterion XT Bis-Tris gels | Bio-Rad | 3450124 |
| PVDF membranes | Bio-Rad | 1620233 |
| Experimental Models: Cell Lines | ||
| THP1 | ATCC | TIB-202 |
| HL-60 | ATCC | CCL-240 |
| HeLa | ATCC | CCL-2 |
| Escherichia coli | ATCC | 11775 |
| Experimental Models: Organisms/Strains | ||
| GsdmdI105N/I105N mice | (Kayagaki et al., 2015) | N/A |
| Casp11 flox/flox mice | This study | N/A |
| Gpx4 flox/flox mice | (Yoo et al., 2012) | N/A |
| LysM-Cre mice | The Jackson Laboratory | 004781 |
| Alb-Cre mice | The Jackson Laboratory | 003574 |
| Nalp3−/− mice | The Jackson Laboratory | 021302 |
| Casp1−/−Casp11−/− mice | The Jackson Laboratory | 016621 |
| Oligonucleotides | ||
| Mouse Plcg1-shRNA-1 (Sequence:CCGGGCCAGCTTGTAGCACTCAATTCTCGAGAATTGAGTGCTACAAGCTGGCTTTTT) | Sigma Aldrich | TRCN0000024974 |
| Mouse Plcg1-shRNA-2 (Sequence:CCGGCCAACTTTCAAGTGTGCAGTACTCGAGTACTGCACACTTGAAAGTTGGTTTTT) | Sigma Aldrich | TRCN0000024976 |
| Human Plcg1-shRNA (Sequence:GTACCGGAGAAGTTCCTTCAGTACAATCCTCGAGGATTGTACTGAAGGAACTTCTTTTTTTG) | Sigma Aldrich | TRCN0000218478 |
| See Table S2 for primers used for qPCR | This paper | Table S2 |
| Software and Algorithms | ||
| Image Lab™ software | Bio-Rad | 1709691 |
| GraphPad Prism 7 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be obtained from the lead contact, Daolin Tang (Email: tangd2@upmc.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
THP1 (#TIB-202, male), HL-60 (#CCL-240, female), and HeLa (#CCL-2, female) cell lines were obtained from American Type Culture Collection (ATCC). BMDMs from male mice were obtained using 30% L929-cell conditioned medium as a source of granulocyte/macrophage colony stimulating factor (Marim et al., 2010). Mouse PMs were isolated from male mice as previously described (Andujar et al., 2012). Primary PBMCs were isolated from whole blood of male or female using the Ficoll-Paque method (Boyum, 1968). These cells were cultured in Dulbecco’s Modified Eagle’s Medium (#11995073, ThermoFisher Scientific) supplemented with 10% heat-inactivated fetal bovine serum (#TMS-013-B, Millipore) and 1% penicillin and streptomycin (#15070-063, ThermoFisher Scientific) at 37 °C, 95% humidity, and 5% CO2. All cells used were authenticated using STR profiling and mycoplasma testing was negative.
Bacterial infection
Escherichia coli (#11775) were obtained from ATCC and then added to cells at a multiplicity of infection (MOI) of 25 in media without antibiotics. After 30 min, cells were washed and incubated for 1.5 h at 37°C in fresh medium supplemented with gentamicin (100 μg/ml, #G1397, Sigma Aldrich) to kill extracellular bacteria.
Animal model of sepsis
We conducted all animal care and experiments in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care guidelines (http://www.aaalac.org) and with approval from our Institutional Animal Care and Use Committee. Mice were housed with their littermates in groups of four or five animals per cage and kept on a regular 12 hr light and dark cycle in a specific pathogen-free barrier facility.
GsdmdI105N/I105N mice (C57BL/6) were a gift from Dr. Vishva M. Dixit (Genentech Inc., South San Francisco, CA, USA). Casp11 flox/flox mice (C57BL/6) were a gift from Dr. Timothy R. Billiar (University of Pittsburgh). Gpx4 flox/flox mice (C57BL/6) were a gift from Dr. Qitao Ran (University of Texas Health Science Center). LysM-Cre (#004781), Alb-Cre (#003574), Nalp3−/− (#021302), and Casp1−/−Casp11−/− (#016621) mice were obtained from The Jackson Laboratory. These mice were used to generate indicated transgenic mice as described in the text.
Sepsis was induced in male or female C57BL/6 mice (eight- to 10-weeks old, 22 to 26 g body weight) using a surgical procedure termed cecal ligation and puncture (CLP) as previously described (Xie et al., 2016; Yang et al., 2014a). Briefly, anesthesia was induced with ketamine (80–100 mg/kg/i.p., #1867-66-9, Cayman Chemical) and xylazine (10–12.5 mg/kg/i.p., #S2516, Selleck Chemicals). A small midline abdominal incision was made and the cecum was exteriorized and ligated with 4–0 silk immediately distal to the ileocecal valve without causing intestinal obstruction. The cecum was then punctured twice with a 17–27-gauge needle. The abdomen was closed in two layers and mice were injected subcutaneously with 1 ml Ringer’s solution.
Blood was collected at indicated time points, allowed to clot for two hours at room temperature, and then centrifuged for 15 minutes at 1,500×g. Serum samples were stored at −80°C before analysis. Mortality was recorded for up to 10 days after the onset of lethal sepsis to ensure that no additional late deaths occurred.
Patient samples
PBMCs from patients with sepsis (n=16) at hospital admission and healthy controls (n=16) were collected from Daping Hospital and Xiangya Hospital by a density gradient centrifugation method using Ficoll Histopaque. The subjects were not involved in previous procedures and test naïve. Clinical characteristics of sepsis patients and healthy control individuals including sex and age are shown in our previous publication (Table S1) (Zeng et al., 2017). Collection of samples was approved by the Institutional Review Board. Sepsis was identified according to The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3) (Singer et al., 2016). No statistical methods were used to pre-determine sample sizes. We did not analysis of the influence (or association) of sex, gender identity, or both on the results of the study because a small sample size for each subgroup.
METHOD DETAILS
Biochemical assay
Commercially available enzyme linked immunosorbant assay (ELISA) kits were used to measure the concentrations of HMGB1 (#ST51011, Shino Test Corporation), IL-1β (#MLB00C, R&D Systems), IL-18 (#7625, R&D Systems), IL-12 (#DY419, R&D Systems), IL-17 (#DY421, R&D Systems), LDH (#ab102526, Abcam), MDA (#ab118970, Abcam), 4-HNE (#MBS702101, MyBioSource), DAG (#MET-5028, Cell Biolabs Inc.), and IP3 (LS-F10644-1, LifeSpan Biosciences Inc.) in serum, culture medium, or whole cell lysate according to the manufacturer’s instructions. Measurement of serum tissue enzymes (CK, BUN, and ALT) were performed using the IDEXX Catalyst Dx® Chemistry Analyzer according to the manufacturer’s protocol.
Calcium measurement
The intracellular Ca2+ concentration was detected using the fluorescent probe Fura-2 (#F1201) from Thermo Fisher Scientific according to the manufacturer’s protocol. Briefly, intracellular calcium concentrations were calculated from the ratio of background corrected Fura-2 emission (520 nm) at two excitation wavelengths (340nm/380nm) using a microplate reader (Cytation™ 5 Cell Imaging Multi-Mode Reader, BioTek, USA).
LPS transfection
To stimulate caspase-11 noncanonical inflammasome activation, LPS were electroporated into indicated cells using the Neon Transfection System (Thermo Fisher Scientific) according to the manufacturer’s protocol. Briefly, 1×106 BMDMs were electroporated LPS (1–3 μg) in buffer R (#MPK10025, Thermo Fisher Scientific) under pulse voltage 1400V, pulse width 10ms, and pulse number 2.
RNAi and gene transfection
Mouse PLCG1-shRNA (#TRCN0000024974 and #TRCN0000024976), human PLCG1-shRNA (#TRCN0000218478), and control-shRNA (#SHC001) were obtained from Sigma (St. Louis, MO, USA). GSDMD-N (1–276), GSDMD-C (277–487), GSDMD cleavage mutant (D275A), and full length WT GSDMD plasmids were a kind gift from Dr. Feng Shao (National Institute of Biological Sciences in China) (Ding et al., 2016). The Gpx4 cDNA expression construct was a gift from Dr. Qitao Ran (University of Texas Health Science Center). Inactive Gpx4 cDNA (U46S mutant) was generated as previously described (Mannes et al., 2011). Transfection was performed using lentiviral vector or Lipofectamine 3000 (#L3000-015, Invitrogen, Grand Island, NY, USA,) according to the manufacturer’s instructions (Xie et al., 2017b). Puromycin (#ant-pr-1; InvivoGen) was used to generate stable knockdown cell lines.
Western blot analysis
Western blot was used to analyze protein expression as described previously (Sun et al., 2016; Tang et al., 2010; Xie et al., 2017a). In brief, after extraction, proteins in cell lysates were first resolved by 4–12% Criterion XT Bis-Tris gel electrophoresis and then transferred to polyvinylidene difluoride membrane and subsequently incubated with the primary antibody (1:500–1:1000). After incubation with peroxidase-conjugated secondary antibodies (1:1000–1:2000), the signals were visualized by enhanced chemiluminescence (#32106, Thermo Fisher Scientific) according to the manufacturer’s instructions.
Quantitative real-time polymerase chain reaction
Total RNA was extracted using TRI reagent (#93289, Sigma Aldrich) according to the manufacturer’s instructions. First-strand cDNA was synthesized from 1 μg of RNA using the iScript cDNA Synthesis kit (#1708890, Bio-Rad). cDNA from various cell samples was amplified using real-time quantitative PCR with specific primers (Table S2). The data were normalized to 18S RNA and the fold change was calculated via the 2−ΔΔCt method (Song et al., 2018; Zhu et al., 2017). Relative concentrations of mRNA were expressed in arbitrary units based on the untreated group, which was assigned a value of 1.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis
Data are presented as mean ± SD. All data meet the assumptions of the tests (e.g., normal distribution). Unpaired Student’s t tests were used to compare the means of two groups. One-way Analysis of Variance (ANOVA) was used for comparison among the different groups. When ANOVA was significant, post hoc testing of differences between groups was performed using the Least Significant Difference (LSD) test. The Kaplan-Meier method was used to compare differences in mortality rates between groups. A p-value < 0.05 was considered statistically significant. The exact value of n within figures was indicated in figure legends. We did not exclude samples or animals. For every figure, statistical tests are justified as appropriate. All data meet the assumptions of the tests. No statistical methods were used to pre-determine sample sizes, but our sample sizes are similar to those generally employed in the field.
Supplementary Material
Highlights.
Gpx4 deficiency in myeloid cells increases severity of polymicrobial sepsis in mice
Caspase-11-dependent pyroptosis mediates septic death in Gpx4Mye−/− mice
GPX4 decreases lipid peroxidation to block PLCG1-mediated GSDMD activity and pyroptosis
Antioxidant activity of Vitamin E promotes survival during sepsis in Gpx4Mye−/− mice
Acknowledgments
We thank Christine Heiner (Department of Surgery, University of Pittsburgh) for her critical reading of the manuscript. This work was supported by grants from the US National Institutes of Health (R01GM115366, R01CA160417, R01AT005076, R01GM063075, R01GM044100, and R01GM050441), the Natural Science Foundation of Guangdong Province (2016A030308011), the American Cancer Society (Research Scholar Grant RSG-16-014-01-CDD), the National Natural Science Foundation of China (31671435, 81400132, and 81772508), Guangdong Province Universities and Colleges Pearl River Scholar Funded Scheme (2017), Lin He’s Academician Workstation of New Medicine and Clinical Translation (2017), and International Scientific and Technology Cooperation Program of China (2015DFA31490). This project partly utilized University of Pittsburgh Cancer Institute shared resources supported by award P30CA047904. GK is supported by the Ligue contre le Cancer Comité de Charente-Maritime (équipe labelisée); Agence National de la Recherche (ANR) – Projets blancs; ANR under the frame of E-Rare-2, the ERA-Net for Research on Rare Diseases; Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; Chancelerie des universités de Paris (Legs Poix), Fondation pour la Recherche Médicale (FRM); the European Commission (ArtForce); the European Research Council (ERC); Fondation Carrefour; Institut National du Cancer (INCa); Inserm (HTE); Institut Universitaire de France; LeDucq Foundation; the LabEx Immuno-Oncology; the RHU Torino Lumière, the Searave Foundation; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); the SIRIC Cancer Research and Personalized Medicine (CARPEM); and the Paris Alliance of Cancer Research Institutes (PACRI).
Footnotes
Author Contributions
D.T. and J. J. designed the experiments. R.K., S.Z., Y.X., L.Z., J.L., Q.W., L.C., M.X., and D.T conducted the experiments. D.T. wrote the paper. Q.R., H.W., and T.R.B. provided important reagents and samples. G.K., H.W., and T.R.B. edited and commented on the manuscript.
Declaration of Interests
The authors declare no conflicts of interest or financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, Tan MH, Cotter PA, Vance RE, Aderem A, et al. Caspase-11 protects against bacteria that escape the vacuole. Science. 2013;339:975–978. doi: 10.1126/science.1230751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, Ciferri C, Dixit VM, Dueber EC. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci U S A. 2016;113:7858–7863. doi: 10.1073/pnas.1607769113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andujar I, Rios JL, Giner RM, Miguel Cerda J, del Recio MC. Beneficial effect of shikonin on experimental colitis induced by dextran sulfate sodium in BALB/c mice. Evid Based Complement Alternat Med. 2012;2012:271606. doi: 10.1155/2012/271606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyum A. Isolation of mononuclear cells and granulocytes from human blood. Isolation of monuclear cells by one centrifugation, and of granulocytes by combining centrifugation and sedimentation at 1 g. Scand J Clin Lab Invest Suppl. 1968;97:77–89. [PubMed] [Google Scholar]
- Brigelius-Flohe R, Maiorino M. Glutathione peroxidases. Biochimica et biophysica acta. 2013;1830:3289–3303. doi: 10.1016/j.bbagen.2012.11.020. [DOI] [PubMed] [Google Scholar]
- Brutsch SH, Rademacher M, Roth SR, Muller K, Eder S, Viertel D, Franz C, Kuhn H, Borchert A. Male Subfertility Induced by Heterozygous Expression of Catalytically Inactive Glutathione Peroxidase 4 Is Rescued in Vivo by Systemic Inactivation of the Alox15 Gene. The Journal of biological chemistry. 2016;291:23578–23588. doi: 10.1074/jbc.M116.738930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canli O, Alankus YB, Grootjans S, Vegi N, Hultner L, Hoppe PS, Schroeder T, Vandenabeele P, Bornkamm GW, Greten FR. Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood. 2016;127:139–148. doi: 10.1182/blood-2015-06-654194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Hambright WS, Na R, Ran Q. Ablation of the Ferroptosis Inhibitor Glutathione Peroxidase 4 in Neurons Results in Rapid Motor Neuron Degeneration and Paralysis. The Journal of biological chemistry. 2015;290:28097–28106. doi: 10.1074/jbc.M115.680090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng KT, Xiong S, Ye Z, Hong Z, Di A, Tsang KM, Gao X, An S, Mittal M, Vogel SM, et al. Caspase-11-mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J Clin Invest. 2017;127:4124–4135. doi: 10.1172/JCI94495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang DC, Shao F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111–116. doi: 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
- Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch A, Eggenhofer E, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nature cell biology. 2014;16:1180–1191. doi: 10.1038/ncb3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaschler MM, Stockwell BR. Lipid peroxidation in cell death. Biochemical and biophysical research communications. 2017;482:419–425. doi: 10.1016/j.bbrc.2016.10.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Ran Q, Roberts LJ, 2nd, Zhou L, Richardson A, Sharan C, Wu D, Yang H. Suppression of atherogenesis by overexpression of glutathione peroxidase-4 in apolipoprotein E-deficient mice. Free radical biology & medicine. 2008;44:343–352. doi: 10.1016/j.freeradbiomed.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science. 2013;341:1250–1253. doi: 10.1126/science.1240988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Mo JH, Gong X, Rossetto C, Jang A, Beck L, Elliott GI, Kufareva I, Abagyan R, Broide DH, et al. 3-Hydroxyanthranilic acid inhibits PDK1 activation and suppresses experimental asthma by inducing T cell apoptosis. Proc Natl Acad Sci U S A. 2007;104:18619–18624. doi: 10.1073/pnas.0709261104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang ZH, Zhong CQ, Han J. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell research. 2015;25:1285–1298. doi: 10.1038/cr.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai H, Nakagawa Y. Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free radical biology & medicine. 2003;34:145–169. doi: 10.1016/s0891-5849(02)01197-8. [DOI] [PubMed] [Google Scholar]
- Jorgensen I, Rayamajhi M, Miao EA. Programmed cell death as a defence against infection. Nature reviews Immunology. 2017;17:151–164. doi: 10.1038/nri.2016.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen I, Zhang Y, Krantz BA, Miao EA. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. The Journal of experimental medicine. 2016;213:2113–2128. doi: 10.1084/jem.20151613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadamur G, Ross EM. Mammalian phospholipase C. Annual review of physiology. 2013;75:127–154. doi: 10.1146/annurev-physiol-030212-183750. [DOI] [PubMed] [Google Scholar]
- Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13:81–90. doi: 10.1038/nchembio.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–671. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–121. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, Miyake K, Zhang J, Lee WP, Muszynski A, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 2013;341:1246–1249. doi: 10.1126/science.1240248. [DOI] [PubMed] [Google Scholar]
- Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, Lieberman J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–158. doi: 10.1038/nature18629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man SM, Kanneganti TD. Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nature reviews Immunology. 2016;16:7–21. doi: 10.1038/nri.2015.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannes AM, Seiler A, Bosello V, Maiorino M, Conrad M. Cysteine mutant of mammalian GPx4 rescues cell death induced by disruption of the wild-type selenoenzyme. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2011;25:2135–2144. doi: 10.1096/fj.10-177147. [DOI] [PubMed] [Google Scholar]
- Marim FM, Silveira TN, Lima DS, Jr, Zamboni DS. A method for generation of bone marrow-derived macrophages from cryopreserved mouse bone marrow cells. PLoS One. 2010;5:e15263. doi: 10.1371/journal.pone.0015263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita M, Freigang S, Schneider C, Conrad M, Bornkamm GW, Kopf M. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. The Journal of experimental medicine. 2015;212:555–568. doi: 10.1084/jem.20140857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pocock TM, Bates DO. In vivo mechanisms of vascular endothelial growth factor-mediated increased hydraulic conductivity of Rana capillaries. J Physiol. 2001;534:479–488. doi: 10.1111/j.1469-7793.2001.00479.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Py BF, Jin M, Desai BN, Penumaka A, Zhu H, Kober M, Dietrich A, Lipinski MM, Henry T, Clapham DE, et al. Caspase-11 controls interleukin-1beta release through degradation of TRPC1. Cell Rep. 2014;6:1122–1128. doi: 10.1016/j.celrep.2014.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran Q, Van Remmen H, Gu M, Qi W, Roberts LJ, 2nd, Prolla T, Richardson A. Embryonic fibroblasts from Gpx4+/− mice: a novel model for studying the role of membrane peroxidation in biological processes. Free radical biology & medicine. 2003;35:1101–1109. doi: 10.1016/s0891-5849(03)00466-0. [DOI] [PubMed] [Google Scholar]
- Rathinam VA, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, Leong JM, Fitzgerald KA. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell. 2012;150:606–619. doi: 10.1016/j.cell.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med. 2010;49:1603–1616. doi: 10.1016/j.freeradbiomed.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryter SW, Kim HP, Hoetzel A, Park JW, Nakahira K, Wang X, Choi AM. Mechanisms of cell death in oxidative stress. Antioxid Redox Signal. 2007;9:49–89. doi: 10.1089/ars.2007.9.49. [DOI] [PubMed] [Google Scholar]
- Sborgi L, Ruhl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, Farady CJ, Muller DJ, Broz P, Hiller S. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. Embo J. 2016;35:1766–1778. doi: 10.15252/embj.201694696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler A, Schneider M, Forster H, Roth S, Wirth EK, Culmsee C, Plesnila N, Kremmer E, Radmark O, Wurst W, et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008;8:237–248. doi: 10.1016/j.cmet.2008.07.005. [DOI] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–665. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514:187–192. doi: 10.1038/nature13683. [DOI] [PubMed] [Google Scholar]
- Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3) Jama. 2016;315:801–810. doi: 10.1001/jama.2016.0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Zhu S, Xie Y, Liu J, Sun L, Zeng D, Wang P, Ma X, Kroemer G, Bartlett DL, et al. JTC801 Induces pH-dependent Death Specifically in Cancer Cells and Slows Growth of Tumors in Mice. Gastroenterology. 2018;154:1480–1493. doi: 10.1053/j.gastro.2017.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, Tang D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63:173–184. doi: 10.1002/hep.28251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D, Kang R, Livesey KM, Cheh CW, Farkas A, Loughran P, Hoppe G, Bianchi ME, Tracey KJ, Zeh HJ, 3rd, et al. Endogenous HMGB1 regulates autophagy. The Journal of cell biology. 2010;190:881–892. doi: 10.1083/jcb.200911078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 2008;9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Berghe T, Demon D, Bogaert P, Vandendriessche B, Goethals A, Depuydt B, Vuylsteke M, Roelandt R, Van Wonterghem E, Vandenbroecke J, et al. Simultaneous targeting of IL-1 and IL-18 is required for protection against inflammatory and septic shock. Am J Respir Crit Care Med. 2014;189:282–291. doi: 10.1164/rccm.201308-1535OC. [DOI] [PubMed] [Google Scholar]
- Wilmore JR, Gaudette BT, Gomez Atria D, Hashemi T, Jones DD, Gardner CA, Cole SD, Misic AM, Beiting DP, Allman D. Commensal Microbes Induce Serum IgA Responses that Protect against Polymicrobial Sepsis. Cell Host Microbe. 2018;23:302–311 e303. doi: 10.1016/j.chom.2018.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wortmann M, Schneider M, Pircher J, Hellfritsch J, Aichler M, Vegi N, Kolle P, Kuhlencordt P, Walch A, Pohl U, et al. Combined deficiency in glutathione peroxidase 4 and vitamin E causes multiorgan thrombus formation and early death in mice. Circulation research. 2013;113:408–417. doi: 10.1161/CIRCRESAHA.113.279984. [DOI] [PubMed] [Google Scholar]
- Xie M, Yu Y, Kang R, Zhu S, Yang L, Zeng L, Sun X, Yang M, Billiar TR, Wang H, et al. PKM2-dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat Commun. 2016;7:13280. doi: 10.1038/ncomms13280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J, Zhong M, Yuan H, Zhang L, Billiar TR, et al. The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep. 2017a;20:1692–1704. doi: 10.1016/j.celrep.2017.07.055. [DOI] [PubMed] [Google Scholar]
- Xie Y, Zhu S, Zhong M, Yang M, Sun X, Liu J, Kroemer G, Lotze M, Zeh HJ, 3rd, Kang R, et al. Inhibition of Aurora Kinase A Induces Necroptosis in Pancreatic Carcinoma. Gastroenterology. 2017b;153:1429–1443. e1425. doi: 10.1053/j.gastro.2017.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Xie M, Yang M, Yu Y, Zhu S, Hou W, Kang R, Lotze MT, Billiar TR, Wang H, et al. PKM2 regulates the Warburg effect and promotes HMGB1 release in sepsis. Nat Commun. 2014a;5:4436. doi: 10.1038/ncomms5436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014b;156:317–331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo SE, Chen L, Na R, Liu Y, Rios C, Van Remmen H, Richardson A, Ran Q. Gpx4 ablation in adult mice results in a lethal phenotype accompanied by neuronal loss in brain. Free Radic Biol Med. 2012;52:1820–1827. doi: 10.1016/j.freeradbiomed.2012.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Li X, Zhang X, Kang R, Tang D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochemical and biophysical research communications. 2016;478:1338–1343. doi: 10.1016/j.bbrc.2016.08.124. [DOI] [PubMed] [Google Scholar]
- Zeng L, Kang R, Zhu S, Wang X, Cao L, Wang H, Billiar TR, Jiang J, Tang D. ALK is a therapeutic target for lethal sepsis. Sci Transl Med. 2017:9. doi: 10.1126/scitranslmed.aan5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Zhang Q, Sun X, Zeh HJ, 3rd, Lotze MT, Kang R, Tang D. HSPA5 Regulates Ferroptotic Cell Death in Cancer Cells. Cancer Res. 2017;77:2064–2077. doi: 10.1158/0008-5472.CAN-16-1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.