

Abstract
Stimulation of bradykinin (BK) B2 receptor (BK2R) has been shown to increase renal Na+ excretion. The aim of the present study is to explore the role of BK2R in regulating Kir4.1 and Na-Cl cotransporter (NCC) in the distal convoluted tubule (DCT). Immunohistochemical studies demonstrated that BK2R was highly expressed in both apical and lateral membrane of Kir4.1 positive tubules such as DCT. Patch-clamp experiments demonstrated that BK inhibited the basolateral 40 pS K+ channel (a Kir4.1/5.1 heterotetramer) in the DCT and this effect was blocked by BK2R antagonist but not by BK1R antagonist. Whole-cell recordings also demonstrated that BK decreased the basolateral K+ conductance of the DCT and depolarized the membrane. Renal clearance experiments showed that BK increased urinary Na+ and K+ excretion. However, the BK-induced natriuretic effect was completely abolished in kidney-specific conditional Kir4.1 knockout mice, suggesting that Kir4.1 activity is required for BK-induced natriuresis. The continuous infusion of BK with osmotic pump for 3 days decreased the basolateral K+ conductance and the negativity of the DCT membrane. Western blot showed that infusion of BK decreased the expression of total NCC and phosphorylated NCC. Renal clearance experiments demonstrated that thiazide-induced natriuresis was blunted in the mice receiving BK infusion, suggesting that BK inhibited NCC function. Consequently, mice receiving BK infusion for 3 days were hypokalemic. We conclude that stimulation of BK2R inhibits NCC activity, increases urinary K+ excretion and causes mice hypokalemic and that Kir4.1 is required for BK2R-mediated stimulation of urinary Na+ and K+ excretion.
Keywords: Kcnj10, Kcnj16, bradykinin B2receptor, Na-Cl-Cotranspotor (NCC)
Introduction
Increasing dietary K+ intake or infusion of high potassium solution has been shown to augment urinary excretion of kallikrein, a serine protease which cleaves kinogen and produces kinins such as bradykinin (BK) in the kidney (1). Also, these interventions increase bradykinin B2 receptor (BK2R) density and mRNA levels in the kidney (2-4). Previous studies reported that the overexpression of BK2R increased renal blood flow, glomerular filtration rate and urine flow in the BK2R transgenic mice (1;5). Conversely, the inhibition of BK2R or renal kallikrein has been shown to decrease urinary volume and Na+ excretion in rats (6;7). BK2R is expressed in the proximal tubules, thick ascending limb (TAL) and aldosterone-sensitive distal nephron (ASDN) including DCT and collecting duct (8;9). However, the role of BK2R in the regulation of membrane transport in the DCT is not understood.
The DCT is responsible for the absorption of 5% filtered Na+ load by a thiazide-sensitive Na-Cl cotransporter (NCC). It is well established that high K+ (HK) intake inhibits NCC expression and activity thereby contributing to HK-intake-induced natriuresis and anti-hypertensive effect. We have previously demonstrated that HK-intake-induced inhibition of NCC requires the presence of inwardly-rectifying K+ channel (Kir 4.1) since the deletion of Kir4.1 in the DCT abolished the effect of HK intake on NCC expression (10). Kir4.1 is expressed in the basolateral membrane of the late TAL, DCT and CCD (11-14). Kir4.1 interacts with Kir5.1 to form a 40 pS basolateral K+ channel (13;15). Moreover, Kir4.1/Kir5.1 heterotetramer is the only type of K+ channel expressed in the basolateral membrane of the DCT (15). Furthermore, we demonstrated that HK-intake-induced inhibition of the basolateral Kir4.1/Kir5.1 K+ channels is essential for the inhibition of NCC in the DCT (10). Also, since HK intake stimulated bradykinin production and BK2R expression (3), it raises the possibility that bradykinin and BK2R may play a role in the regulation of Kir4.1 and NCC in the DCT during increased dietary K+ intake. Thus, the aim of the present study is to test whether the stimulation of BK2R regulates Kir4.1 and NCC in the DCT.
Methods
The authors declare that all supporting data and detailed methods including animal preparation, electrophysiology, western blot and renal clearance method are available within the article (and its online supplementary file.
Animals
C57BL/6 mice (either sex, 12 weeks old) and kidney-specific conditional Kir4.1 knockout (KS-Kir4.1 KO) mice were used in the present study. C57/BL/6 mice were purchased from the Second Hospital of Harbin Medical University or Jackson Laboratory (Bar Harbor, ME). KS-Kir4.1 KO mice were bred in New York Medical College for the experiments and the procedure for generating KS-Kir4.1 KO mice are described in the on-line supplemental material. The mice were fed with normal K+ diet (1% KCl) or high K+ diet (5%) for 7 days as indicated and had free access to water. The preparation of DCT for the patch-clamp experiments has been described in detail in the supplemental material.
Electrophysiology
A Narishige electrode puller (Narishige, Japan) was used to manufacture the patch-clamp pipettes from Borosilicate glass (1.7-mm OD). The resistance of the pipette was 5 MΩ (for single channel recording) or 2 MΩ (for whole cell-recording) when it was filled with solution containing (in mmol/L) 140 KCl, 1.8 MgCl2 and 10 HEPES (titrated with KOH to pH 7.4). The details for the single channel and whole-cell recordings are described in on-line supplemental material.
Immnoblotting and Immunohistochemistry
The details for immunoblotting are described in on-line supplemental material. For immunohistochemistry, renal slices were fixed in Bouin’s fixative and processed as described (9;16). The localization of BK2R and Kir4.1 was assessed by double immunolabeling in the same tissue sections. Briefly, the first antibody immunostaining (Kir4.1) (1:1200, Alomone Labs, Israel) was developed with diaminobenzidine-hydrogen peroxide to give a brown color, whereas BK2R immunostaining was developed with Vector SG substrate to give a blue color (1:1000, Santa Cruz). The sections were observed and photographed on a Nikon Eclipse 600 microscope with a Nikon DS-Ri1 digital camera.
Material and statistical analysis
All chemicals including hydrochlorothiazide (HCTZ), phorbol 12-myristate-13 acetate (PMA), bradykinin, HOE140 and Lys-(des-Arg9, Leu8)-bradykinin were purchased from Sigma Chemicals (St Louis, MO). Polyclone antibodies for NCC, pNCC and NKCC2 were purchased from EMD Millipore, Phosphosolutions and Abcam, respectively. Data were analyzed using student t test for comparisons between two groups or one-way ANOVA for comparisons among more than 2 groups. P-values <0.05 were considered statistically significant. Data are presented as the mean ± SEM.
Results
We first used immunohistochemistry to examine whether BK2R was expressed in the DCT using Kir4.1 as a marker. Fig. 1A shows an image of double staining with low magnification and it demonstrates that BK2R is expressed in Kir4.1-positive tubules. Two areas of the image (indicated by a square) in Fig. 1B are enlarged and demonstrate a detailed view of BK2R staining (Fig. 1C and 1D). It is apparent that BK2R is expressed in the DCT as evidenced by strong basolateral Kir4.1 staining and convoluted tubule appearance. Moreover, BK2R staining is visible not only in the apical membrane but also in the lateral membrane of the DCT (indicated by a red arrow).
Fig. 1. BK2R is expressed in Kir4.1-positive distal tubules.
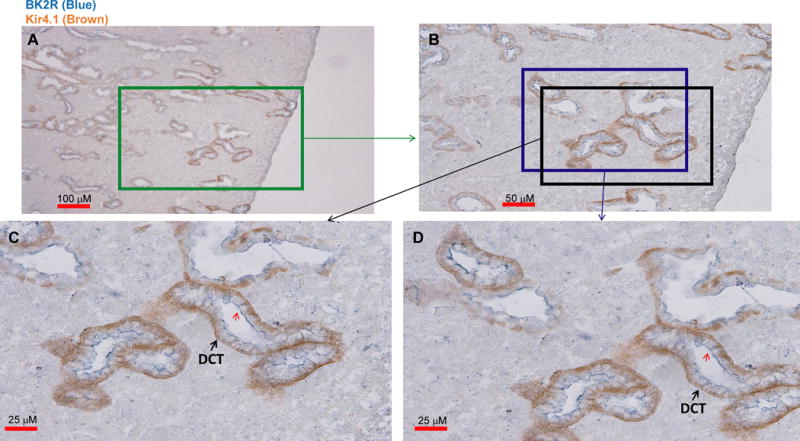
A double staining image shows the expression of Kir4.1 (brown) and BK2R (blue) with low magnification (A). Areas marked by two squares (B) are enlarged, demonstrating detailed view of BK2R staining in Fig. 1C and 1D, respectively. The distal convoluted tubules (DCT) are indicated by arrows. A red arrow indicates BK2R staining in the lateral membrane of the DCT.
Since BK2R is expressed in both apical and lateral membrane of the DCT, we next used the patch-clamp technique to examine whether BK regulated the basolateral 40 pS K+ channel (a Kir4.1/Kir5.1 heterotetramer) in the DCT. Fig. 2A is a channel recording showing the effect of BK on the basolateral 40 pS K+ channel activity in the DCT. Adding 1 μM BK decreased K+ channel activity and reduced NPo of the 40 pS K+ channels to 1.45±0.3, although this effect was not significant (Fig. 2B). However, 10 μM BK significantly inhibited the basolateral 40 pS K+ channel. Results from six experiments are summarized in Fig. 2B demonstrating that BK (10 μM) significantly decreased the channel activity defined by NPo from 1.9±0.25 to 0.54±0.07. The effect of BK on the basolateral K+ channels was reversible since the washout was able to partially restore the channel activity. Moreover, the inhibitory effect of BK on the basolateral K+ channel was mediated by protein kinase C (PKC) pathway since calphostin C (1 μM) abolished the inhibitory effect of BK (10 μM) on the basolateral K+ channels in the DCT (NPo =1.8±0.25, n=6)(Fig. 2B). The notion that PKC mediated the effect of BK on the basolateral K+ channels was also supported by the observation that the stimulation of PKC with PMA (10 μM) also inhibited the basolateral K+ channel activity and reduced NPo to 0.91±0.14 (n=6).
Fig. 2. Bradykinin (BK) inhibits the basolateral 40 pS K+ channels in the DCT.
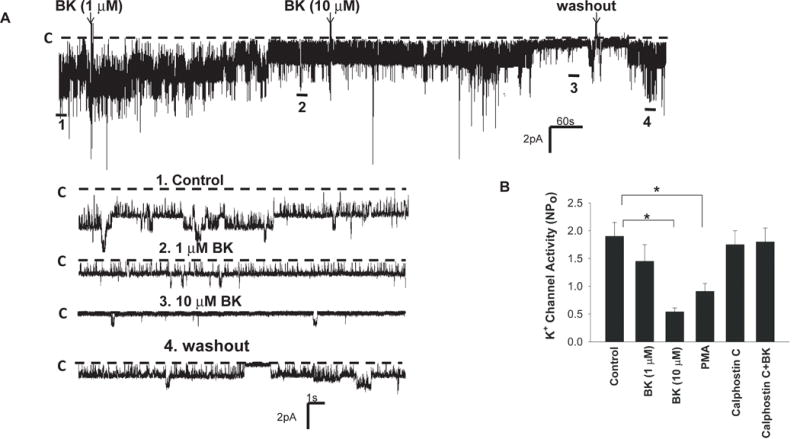
(A) A single channel recording shows the effects of 1 and 10 μM BK on the basolateral K+ channels in the DCT. The top trace shows the time course of the experiments and 4 parts of the record indicated by numbers are extended to demonstrate the fast time resolution. The holding potential was 0 mV and the channel closed level is indicated by a dotted line and “C”. The experiments were performed in cell-attached patches with 140 mmol/L K+ in the pipette and 140 mmol/L Na+/5 mmol/L K+ in the bath solution. (B) A bar graph summarizes the results of experiments in which the effects of BK, PMA (10 μM), calphostin C (1μM) and BK+calphostin C on the basolateral 40 pS K+ channel were examined (n=6).
We have also used the whole-cell recording to measure the Ba2+-sensitive K+ currents under control conditions and in the presence of 10 μM BK in the early portion of the DCT (DCT1). Since Kir4.1/Kir5.1 heterotetramer is the only type of K+ channel in the DCT1, the whole-cell K+ currents represent the activity of whole population of Kir4.1/Kir5.1 in the DCT. Fig. 3A is a whole-cell recording showing the Ba2+-sensitive K+ currents measured from −60 mV to 60 mV at a 20 mV step and Fig. 3B is a recording demonstrating the whole-cell K+ currents measured with RAMP protocol from −100 mV to 100 mV. It is apparent that BK decreased the Ba2+-sensitive whole-cell K+ currents. Fig. 3C is a bar graph summarizing the results of six experiments in which Ba2+-sensitive K+ currents were measured at −60 mV using step protocol (as shown in Fig 3A), demonstrating that BK treatment decreased the K+ currents from 1280±60 pA to 860±50 pA (n=6). Because the basolateral Kir4.1/Kir5.1 channels participate in generating the negative membrane potential, BK-induced inhibition of the basolateral K+ channels is expected to decrease the negativity of the DCT membrane (depolarization). This speculation is confirmed by measuring K+-current (IK) reversal potential, an index of the membrane potential, with perforated whole-cell recording. From the inspection of Fig. 3D it is apparent that BK treatment shifted the IK reversal potential of the DCT to a positive range (depolarization). Results summarized in Fig. 3C show that IK reversal potential was −66±3 mV under control conditions and it was −55±3 mV after acute BK treatment. Thus, BK decreases the basolateral Kir4.1 activity and depolarizes the DCT membrane. Moreover, the inhibitory effect of BK on the basolateral K+ conductance was enhanced in the DCT of the mice on a HK diet for 7 days. Fig.s1A is a whole-cell recording showing the Ba2+-sensitive K+ currents measured from −60 mV to 60 mV at a 20 mV step and Fig.s1B is a recoding demonstrating the whole-cell K+ currents measured with RAMP protocol from −100 mV to 100 mV. Like under control conditions, BK inhibited the basolateral Kir4.1/Kir5.1 in the DCT of the mice on HK diet. Fig.s1C is a bar graph summarizing the results of seven experiments showing that BK treatment decreased the K+ currents from 690±60 pA to 330±40 pA. Thus, HK intake enhanced the inhibitory effect of BK on the basolateral K+ channels since BK-induced inhibition of basolateral K+ conductance was significantly larger in the mice on HK diet (51±2%) than in the mice on normal K+ diet (32±2%) (Fig. s1D).
Fig. 3. BK decreases the basolateral K+ conductance of the DCT and depolarizes the membrane.
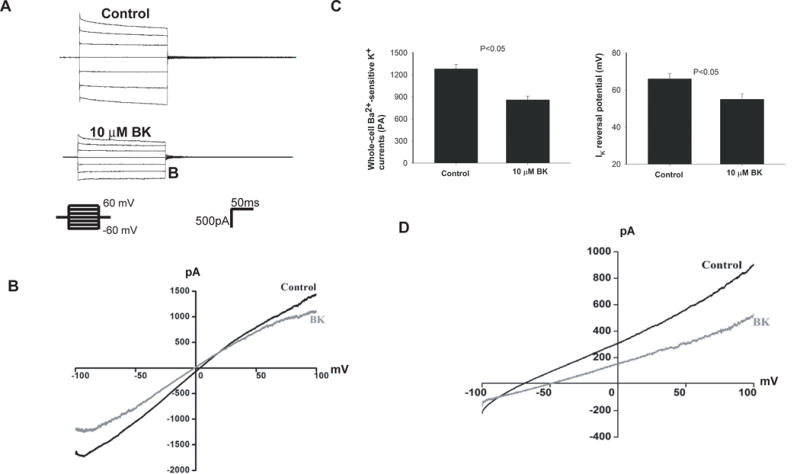
A whole-cell recording shows Ba2+ -sensitive K+ currents in the DCT treated with 10 μM BK and the K+ currents were measured with a step protocol from −60 to 60 mV at a 20 mV step (A) or with Ramp protocol from −100 to 100 mV (B). Symmetrical 140 mmol/L KCl solution in the bath and pipette was used for the measurement. Results of six experiments are summarized in a bar graph (C). A whole-cell recording shows the effect of 10 μM BK on K+-current (IK) reversal potential of the DCT (D). The bath solution contains (in mmol/L) 140 NaCl and 5 KCl while the pipette solution has 140 KCl. Results of five experiments are summarized in a bar graph (left panel of Fig. 3C).
To further examine whether the inhibitory effect of BK on the basolateral K+ channels was mediated by BK2R, we examined the effect of BK on the basolateral K+ channels in the presence of HOE140, a specific BK2R antagonist (7). Fig. 4A is a channel recording showing the effect of BK on the basolateral 40 pS K+ channel activity in the DCT treated with HOE140 (1 μM). Results from six similar experiments are summarized in Fig. 4B showing that HOE140 abolished the inhibitory effect of BK on the basolateral K+ channel activity in the DCT (NPo =1.7±0.14), although BK2R antagonist, per se, had no significant effect on the channel activity (NPo =1.7±0.14). In contrast, BK was still able to inhibit the basolateral K+ channel activity in the presence of 1 μM Lys-(des-Arg9, Leu8)-bradykinin (BK1R antagonist) (Fig. 4C) and decreased NPo to 0.8±0.1 (n=6) (Fig. 4B). Thus, the results suggest that BK-induced inhibition of basolateral K+ channels in the DCT is mediated by BK2R and PKC-dependent pathway.
Fig. 4. BK2R mediates BK-induced inhibition of the basolateral 40 pS K+ channels in the DCT.
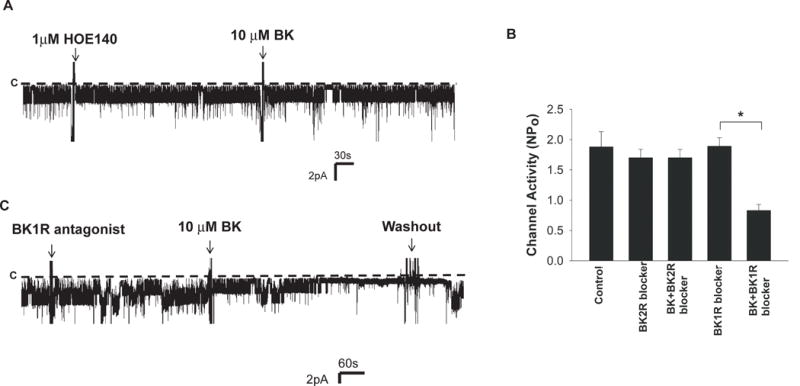
A single channel recording demonstrates the effect of 10 μM BK on the basolateral K+ channels in the DCT in the presence of 1 μM HOE (BK2R antagonist) (A) or in the presence of 1μM Lys-(des-Arg9, Leu8)-bradykinin (BK1R antagonist) (C). The holding potential was 0 mV and the channel closed level is indicated by a dotted line and “C”. (B) Bar graph summarizes the results of experiments in which the effects of HOE140 (1μM), BK (10 μM)+HOE140, BK1R antagonist (1μM) and BK+BK1R antagonist on the basolateral 40 pS K channel were examined (n=6).
Previous studies have demonstrated that the basolateral K+ channel activity in the DCT plays a key role in regulating NCC activity such that a stimulation of the basolateral Kir4.1 activity increases whereas an inhibition of the basolateral K+ channel activity decreases NCC activity (17). Because BK inhibits the basolateral Kir4.1 activity in the DCT and depolarizes the membrane, it is conceivable that BK might increase Na+ excretion by inhibiting NCC. This hypothesis was tested with renal clearance experiments to examine the effect of BK on renal Na+ excretion (ENa) by one time infusion of BK (0.5 ng/kg BW within 60 seconds). For the clearance study, the mice were intravenously perfused with isotonic saline for 4 hr (0.3 ml/1 hr) and urine collections started one hr after saline infusion. Results from four experiments are summarized in Fig. 5A demonstrating that acute BK infusion increased ENa from 0.76±0.06 to 1.48±0.12 μEq/min/100g BW. Also, Fig. s2 shows the time course of BK-infusion-induced changes in urine volume. It is apparent that BK-infusion-induced urine volume changes appear in the 2nd (60 min after injection) and 3rd collections (90 min after injection). The BK infusion-induced stimulation of ENa was, at least in part, due to the inhibition of NCC because BK failed to increase ENa in kidney-specific Kir4.1 knockout mice (KS-Kir4.1 KO) in which NCC activity was inhibited (10;17). Results summarized in Fig 5B show that the deletion of Kir4.1 not only increased the basal level of renal Na+ excretion (1.56±0.12 μEq/min/100g BW) but also abolished the effect of BK infusion on ENa (1.58±0.12 μEq/min/100g BW). We have also examined the effect of BK infusion on renal K+ excretion (EK) with renal clearance methods. Fig. 5C is a line graph demonstrating the results of each experiment and Fig. 5D summarizes the results of four experiments demonstrating that BK infusion increased EK from 0.61±0.04 to 0.9±0.05 μEq/min/100g BW. The deletion of Kir4.1 not only caused K+ wasting (1.06±0.05 μEq/min/100g BW) but also abolished the effect of BK on renal K+ excretion (1.03±0.05 μEq/min/100g BW). Thus, BK-infusion-induced stimulation of both ENa and EK depends on the presence of Kir4.1.
Fig. 5. BK infusion stimulates renal Na+ and K+ excretion.
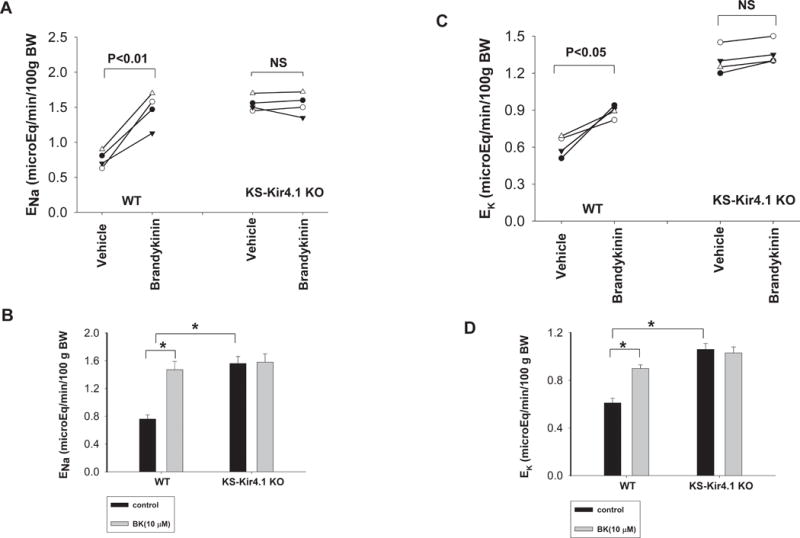
(A) A line graph shows the results of each experiment in which urinary Na+ excretion (ENa) was measured before and after BK infusion in WT mice and KS-Kir4.1 KO mice. (B) The mean value and statistical information are shown in a bar graph. The basal level of ENa of KS-Kir4.1 KO mice is significantly different in comparison to WT mice. (C) A line graph shows the results of each experiment in which urinary K+ excretion (EK) was measured before and after BK infusion in WT mice and KS-Kir4.1 KO mice. (D) The mean value of EK and statistical information are shown in a bar graph. The basal level of EK of KS-Kir4.1 KO mice is significantly different in comparison to WT mice.
To further test whether BK-infusion induced stimulation of renal Na+ excretion was due to the inhibition of Kir4.1 and NCC in the DCT, we examined the basolateral K+ channel activity in the DCT and NCC activity in the mice receiving BK infusion through an osmotic pump (4 μg/min/kg BW). Figure 6A is a recording showing Ba2+ -sensitive K+ currents of DCT cells clamped from −60 mV to 60 mV at a 20 mV step from untreated control and BK-treated mice. As summarized in Fig. s3a, the mean whole-cell K+ current at −60 mV was −1250±60 pA (n=6) in the control mice and it was −520±30 pA (n=6) in BK-treated mice. Because basolateral K+ channels participate in generating the membrane potential of DCT cells, an inhibition of the basolateral K+ channels should depolarize DCT membrane. Thus, we used the whole-cell recording to measure the IK reversal potential in the control (vehicle) and BK-treated mice. Fig. 6B is a perforated whole-cell recording showing current/voltage curve of the DCT with 140 mmol/L K+ in the pipette (intracellular solution) and 140 mmol/L Na+/5 mmol/L K+ in the bath from the control and BK-treated mice. The results from seven experiments are summarized in Fig. s3B showing that BK treatment depolarized DCT membrane and decreased IK reversal potential of the DCT from −65±4 mV (control) to −42±5 mV (BK infusion). Thus, patch-clamp experiments confirm that the BK inhibited the basolateral K+ channel activity in the DCT and depolarized the membrane. We have also used the renal clearance method to examine the effect of HCTZ on ENa in the control mice and in the mice receiving BK infusion for 3 days. Figure 6C summarizes results of four experiments showing that HCTZ-induced natriuretic effect (from 1.96±0.23 to 3.20±0.13 μEq/min/100g BW) in BK-treated mice was significantly smaller than those in the untreated group (from 0.84±0.13 to 2.68±0.16 μEq/min/100g BW). Moreover, the fact that basal level of ENa was higher in BK-treated mice (1.96±0.23 μEq/min/100g BW) than that of the control animals (0.84±0.13 μEq/min/100g BW) also indicates that BK inhibits Na+ transporters such as NCC. The notion that BK-infusion inhibited NCC activity was also supported by the western blot analysis. Figure 6D is a western blot showing the effect of BK infusion (3 days) on the expression of phosphor-NCC at Thr53 (pNCC) and total NCC (tNCC). The normalized band intensity for pNCC and tNCC (n=6) is summarized in Fig. 6E. BK infusion for 3 days significantly decreased the abundance of pNCC (36±4% of the control) and tNCC (55±5% of the control). Thus, western blot data are consistent with the results of the renal clearance study, suggesting that BK infusion inhibits the NCC activity in the DCT. The effect of BK infusion on NCC was specific because BK infusion for 3 days had no effect on the expression of NKCC2 and full-length ENaC-α subunit (Fig. s4). However, from the inspection of Fig s4B, it is apparent that BK treatment decreased the expression of cleaved ENaC-a subunit (65±5% of the control), suggesting that BK treatment inhibits both ENaC and NCC. Since BK-induced inhibition of NCC should increase the flow-stimulated K+ secretion by increasing Na+ and volume delivery to the late portion of ASDN, the mice receiving BK infusion for 3 days were hypokalemic (Fig. 6F). This strongly suggests the role of BK2R in regulating Na+ and K+ transport in ASDN.
Fig. 6. BK infusion inhibits Kir4.1 and NCC.
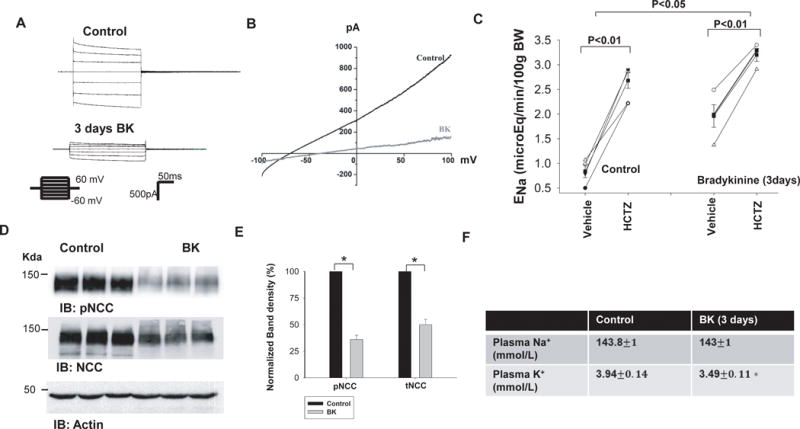
(A) A whole-cell recording shows Ba2+ -sensitive K+ currents in the DCT of mice treated with vehicle (control) or BK infusion for 3 days. K+ currents were measured with a step protocol from −60 to 60 mV at a 20 mV step. (B) A whole-cell recording shows K+-current (IK) reversal potential of the DCT in mice treated with vehicle (control) or BK infusion for 3 days. (C) A line graph shows the results of experiments in which urinary sodium excretion (ENa) was measured before and after a single dose of HCTZ (25 mg/kg BW) in control and BK-treated mice. BK was delivered for three days through an osmatic pump. (D) A western blot shows the expression of pNCC and tNCC in control and BK-treated mice. (E) A bar graph summarizing the normalized band density of pNCC and tNCC from tissues obtained in the control and BK-treated mice (n=6). (F) A table shows the plasm Na+ and K+ concentrations in the control and BK-treated mice (n=7). Asterisk indicates a significant difference between two groups.
Discussion
In the present study, we demonstrate that BK inhibits the basolateral 40 pS K+ channel activity in the DCT. Moreover, the observation that PKC inhibitor abolished while PKC stimulator mimicked the effect of BK on the K+ channels strongly suggests that the effect of BK on the basolateral K+ channels in the DCT was mediated by PKC. Moreover, two lines of evidence suggest that BK2R is responsible for the effect of BK on the basolateral 40 pS K+ channels: 1) Immunostaining shows positive BK2R staining in the DCT; and 2) HOE140 (a specific BK2R antagonist) but not BK1R antagonist abolished the effect of BK on the basolateral 40 pS K+ channels in the DCT. Thus, BK2R plays a role in tonic regulation of the basolateral 40 pS K+ channels in the DCT.
The basolateral 40 pS K+ channel in the DCT is composed of Kir4.1 (encoded by Kcnj10) and Kir5.1 (encoded by Kcnj16). Moreover, Kir4.1 is a pore-forming component for the Kir4.1/Kir5.1 heterotetramer because the deletion of Kcnj10 completely eliminates the basolateral K+ conductance in the DCT (13;17;18). Consequently, the Ks-Kir4.1 KO mice recapitulated the Gitelman’s syndrome including modest Na+ wasting, high aldosterone, hypokalemia and metabolic alkalosis (17). Since this 40 pS K+ channel is the only type of K+ channel expressed in the basolateral membrane of the DCT (17), it is conceivable that BK2R-mediated inhibition of the 40 pS K+ channel activity should have a significant effect on the basolateral K+ conductance and the membrane potentials. Indeed, we observed that BK treatment not only decreased the basolateral K+ conductance but also depolarized the DCT membrane. This suggests that the bradykinin-BK2R pathway is involved in tonic regulation of the membrane potential in the DCT.
Not only inhibiting the basolateral K+ channels in the DCT, BK infusion also increased urine Na+ excretion. Three lines of evidence suggest that BK-induced natriuresis was at least in part mediated by inhibiting NCC. First, renal clearance study showed that BK-induced natriuresis was absent in KS-Kir4.1 KO mice in which NCC function was inhibited (17). Second, BK infusion decreased the expression of both tNCC and pNCC in the kidney. Third, HCTZ-induced natriuretic effect, an indication of NCC function, was smaller in the mice treated with BK than untreated control animals, suggesting that BK suppressed NCC. A large body of evidence has demonstrated that the stimulation of BK-BK2R increases renal Na+ excretion by mechanisms of increasing renal blood flow or inhibiting Na+ transport in distal nephron (6;19;20). Our present study has further suggested that BK-induced inhibition of NCC should contribute to kallicrein-kinin system-induced natriuresis. Thus, the results of the present study have suggested that bradykinin-BK2R should play a role in the regulation of DCT function by inhibiting both Kir4.1 and NCC.
BK2R is also highly expressed in the vascular structure and BK is a powerful vasodilator (1), it is conceivable that BK infusion may increase glomerular filtration rate (GFR) thereby increasing Na+ excretion. However, BK-induced hemodynamic changes may have a minor role in stimulating Na+ excretion in our experiment settings. This speculation is supported by two pieces of the observations. First, the largest increase in urinary volume after BK infusion was appeared in the second (60 min after injection) and third collection (90 min after injection) rather than the first collection (30 min after injection) (Fig.s2). Second, the effect of BK on urinary Na+ excretion and urine volume was absent in Ks-Kir4.1 KO mice. These findings suggest that BK-induced hemodynamic change in GFR may not play a major role in increasing urinary Na+ excretion.
Also, BK2R is highly expressed in the CNT and CCD and previous elegant study by Zaika et al has shown that BK inhibits ENaC in the CCD (20). Our present study also shows that BK treatment decreased the expression of cleaved ENaC-α isoform. Thus, it is conceivable that bradykinin-mediated inhibition of ENaC should contribute to the bradykinin-infusion-induced increase in urinary Na+ excretion. Moreover, bradykinin-induced increase in urinary volume delivery to the collecting duct should stimulate flow-dependent K+ excretion thereby enhancing urinary K+ excretion. However, the observation that BK infusion did not increase urinary Na+ excretion in Kir4.1 KO mice despite of the upregulation of ENaC activity in the collecting duct (21), suggesting that BK-induced inhibition of ENaC activity may be blunted in Kir4.1 KO mice. One possibility is that the volume-depletion in Ks-Kir4.1 KO mice may suppress the inhibitory effect of BK on ENaC. Further experiments are needed to explore this possibility. However, BK-induced inhibition of NCC and ENaC should have a synergistic effect on overall renal Na+ excretion under physiological conditions.
Two lines of evidence suggest the possibility that Kir4.1 activity is essential for the effect of BK on NCC, although we could not completely rule out the direct effect of BK on NCC. First, BK-induced natriuresis was largely abolished in KS-Kir4.1 KO mice. Second, BK-induced inhibition of NCC was closely correlated with BK-induced depolarization of DCT membrane. A large body of evidence has suggested the membrane potential in the DCT plays an important role in the regulation of NCC activity such that an increase in the membrane negativity (hyperpolarization) stimulates, whereas a decrease in membrane negativity (depolarization) inhibits NCC activity (10;17;22;23). Our previous studies have demonstrated that the membrane potential is linked to NCC expression through the modulation of Cl−-sensitive with-no-lysine kinase (WNK) which is suppressed by high intracellular Cl− levels and stimulated by decreased intracellular Cl− levels (22;24;25). Thus, the BK-induced inhibition of the basolateral Kir4.1 is expected to increase the intracellular Cl− levels thereby inhibiting WNK. Because WNK activity determines the activity of both Ste20-Proline-and-Alanine-rich Kinase (SPAK) and Oxidative-Sensitive Responsive Kinase (OSR), the inhibition of WNK should suppress SPAK and OSR activity thereby inhibiting NCC (26-30). Thus, it is conceivable that the BK-induced inhibition of the basolateral Kir4.1 in the DCT is involved for BK-induced decrease in NCC expression and activity.
Recent developments in the field of renal K+ excretion have indicated that NCC activity in the DCT plays an important role in the regulation of K+ excretion and K+ homeostasis (22;31;32). For instance, an increase in dietary K+ intake suppresses the NCC activity thereby increasing Na+ and volume delivery to the late ASDN, whereas a decrease in dietary K+ intake stimulates the NCC activity thereby decreasing Na+ and volume delivery to the late ASDN (33). Genetic and clinical studies have also shown that abnormal NCC activity is associated with hyperkalemia or hypokalemia. For instance, hyperkalemia in patients with pseudohypoaldosteronism type II (PHAII) is the result of high NCC activity (28;34;35), whereas hypokalemia and K+ wasting are two main phenotypes in patients with Gitelman syndrome in which genetic mutations cause the inhibition of NCC (36). Indeed, our present study has also demonstrated that the application of BK-induced inhibition of NCC was associated with increasing urinary K+ excretion. Consequently, the mice treated with BK were hypokalemic. This finding suggests the role of BK in the stimulation of K+ excretion in ASDN and in maintaining K+ homeostasis. In this regard, increased dietary K+ intake has been reported to augment urinary kallikrein excretion and to stimulate BK2R expression (3;4), suggesting that BK may play an important role in stimulating K+ excretion during high dietary K+ intake. This notion is also suggested by our finding that BK-induced inhibition of the basolateral K+ channel activity was enhanced in the mice on HK diet. Our previous study has shown that increasing dietary K+ intake inhibited the basolateral K+ channel activity and that HK-induced inhibition of Kir4.1 was essential for the effect of HK intake on NCC (10). Since BK2R regulates basolateral Kir4.1 in the DCT, it is possible that the stimulation of bradykinine-BK2R pathway is an important mechanism by which HK intake stimulates renal K+ excretion.
Perspective
The main finding of the present study is to demonstrate that the stimulation of BK-BK2R inhibited the basolateral Kir4.1 in the DCT and increased renal Na+ and K+ excretion by inhibiting NCC. Although we and others have demonstrated that BK-BK2R pathway plays a role in increasing urinary Na+ excretion (6;7;19), animals deficient in BK2R are normotensive under control conditions (37-39). This suggests that BK-BK2R pathway may not be essential for maintaining overall Na+ homeostasis and controlling normal blood pressure under physiological conditions. However, the finding that BK stimulates urinary K+ excretion and causes hypokalemia suggests the possible role of BK2R in maintaining K+ homeostasis. In this regard, the previous study has also shown that the mice with over-expression of BK2R increased urinary K+ excretion (5). Thus, it is possible that BK2R is involved in stimulating renal K+ excretion through suppressing Kir4.1 and NCC in the DCT. Our previous experiments have also demonstrated that type II angiotensin II receptor (AT2R) plays a role in regulating urinary K+ excretion (40). Thus, both AT2R and BK2R may play a role in regulating urinary K+ excretion during increased dietary K+ intake.
Supplementary Material
Novelty and Significance.
1) What is New?
Bradykinin (BK) inhibits the basolateral Kir4.1 in the DCT by activating BK2R and depolarizes DCT membrane.
BK application increases renal Na+ excretion and inhibits NCC activity. This natriuretic effect of BK depends on the presence of Kir4.1 in the DCT.
BK stimulates renal K+ excretion and causes mice hypokalemic.
2) What is relevant?
NCC plays a key role in regulating renal K+ excretion and maintaining K+ homeostasis while Kir.4.1 plays a key role in controlling NCC activity. The finding that BK2R is involved in the regulation of Kir4.1 and NCC is relevant for understanding an integrated mechanism of regulating K+ homeostasis.
The finding that BK inhibits NCC is highly relevant for understanding the mechanism of BK2R-induced natriuretic effect.
Summary.
BK2R plays a role in inhibiting NCC activity by suppressing the basolateral Kir4.1 activity in the DCT and that BK2R plays a role in stimulating renal K+ excretion and K+ homeostasis.
Acknowledgments
Authors thank Ms.Gail Anderson for her assistance in preparing the manuscript.
Source of Funding:
The work is supported by Chinese National Natural Science Foundation #31671196 (RMG), # 31400993(PW), PIF#YJSCX2015-10HYD(DDZ), NIH grant DK 54983 (WHW), DK 115366 (LDH). Dr. C. Vio es supported by a Grant CONICYT PFB-12/2007 and SQM.
Footnotes
Conflict(s) of Interest/Disclosure:
None.
References
- 1.Rhaleb N-E, Yang X-P, Carretero OA. The kallikrein-kinin system as a regulator of cardiovascular and renal function. Compr Physiol. 2011;1:971–993. doi: 10.1002/cphy.c100053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vio CP, Figueroa CD. Evidence for a stimulatory effect of high potassium diet on renal kallikrein. Kidney Internat. 1987;31(6):1327–1334. doi: 10.1038/ki.1987.146. [DOI] [PubMed] [Google Scholar]
- 3.Jin L, Chao L, Chao J. Potassium supplement upregulates the expression of renal kallikrein and bradykinin B2 receptor in SHR. Am J Physiol Renal Physiol. 1999;276:F476–F484. doi: 10.1152/ajprenal.1999.276.3.F476. [DOI] [PubMed] [Google Scholar]
- 4.Suzuki T, Katori M, Fujita T, Kumagai Y, Majima M. Involvement of the renal kallikrein-kinin system in K(+)-induced diuresis and natriuresis in anesthetized rats. Eur J Pharmacol. 2000;399:223–227. doi: 10.1016/s0014-2999(00)00382-4. [DOI] [PubMed] [Google Scholar]
- 5.Wang D, Yoshida H, Song Q, Chao L, Chao J. Enhanced renal function in bradykinin B2 receptor transgenic mice. American Journal of Physiology-Renal Physiology. 2000;278(3):F484–F491. doi: 10.1152/ajprenal.2000.278.3.F484. [DOI] [PubMed] [Google Scholar]
- 6.Saitoh S, Scicli AG, Peterson E, Carretero OA. Effect of Inhibiting Renal Kallikrein on Prostaglandin E2, Water, and Sodium Excretion. Hypertension. 1995;25(5):1008. doi: 10.1161/01.hyp.25.5.1008. [DOI] [PubMed] [Google Scholar]
- 7.Madeddu P, Anania V, Demontis MP, Varoni MV, Pisanu G, Troffa C, Tonolo G, Glorioso N, Parpaglia PP. Effects of Hoe 140, a bradykinin B2-receptor antagonist, on renal function in conscious normotensive rats. Br J Pharmacol. 1992;106:380–386. doi: 10.1111/j.1476-5381.1992.tb14344.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rodriguez JA, Vio CP, Pedraza PL, McGiff JC, Ferreri NR. Bradykinin Regulates Cyclooxygenase-2 in Rat Renal Thick Ascending Limb Cells. Hypertension. 2004;44(2):230–235. doi: 10.1161/01.HYP.0000136751.04336.e9. [DOI] [PubMed] [Google Scholar]
- 9.Vio CP, Velarde V, Mueller-Esterl W. Cellular distribution and fate of the bradykinin antagonist HOE 140 in the rat kidney. Colocalization with the bradykinin B2 receptor. Immunopharmacology. 1996;33(1):146–150. doi: 10.1016/0162-3109(96)00031-8. [DOI] [PubMed] [Google Scholar]
- 10.Wang MX, Cuevas-Gallardo C, Su XT, Wu P, Gao Z-X, Lin DH, McCormick JA, Yang CL, Wang WH, Ellison DH. Potassium (K+) intake modulates NCC activity via the K+ channel. Kir4.1. Kid Int. 2018;93:893–902. doi: 10.1016/j.kint.2017.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zaika OL, Mamenko M, Palygin O, Boukelmoune N, Staruschenko A, Pochynyuk O. Direct inhibition of basolateral Kir4.1/5.1 and Kir4.1 channels in the cortical collecting duct by dopamine. American Journal of Physiology - Renal Physiology. 2013;305(9):F1277–F1287. doi: 10.1152/ajprenal.00363.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lachheb S, Cluzeaud F, Bens M, Genete M, Hibino H, Lourdel S, Kurachi Y, Vandewalle A, Teulon J, Paulais M. Kir4.1/Kir5.1 channel forms the major K+ channel in the basolateral membrane of mouse renal collecting duct principal cells. AJP - Renal Physiology. 2008;294(6):F1398–F1407. doi: 10.1152/ajprenal.00288.2007. [DOI] [PubMed] [Google Scholar]
- 13.Lourdel S, Paulais M, Cluzeaud F, Bens M, Tanemoto M, Kurachi Y, Vandewalle A, Teulon J. An inward rectifier K+ channel at the basolateral membrane of the mouse distal convoluted tubule: similarities with Kir4-Kir5.1 heteromeric channels. J Physiol. 2002;538(Pt 2):391–404. doi: 10.1113/jphysiol.2001.012961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang C, Wang L, Su XT, Lin DH, Wang WH. KCNJ10 (Kir4.1) is expressed in the basolateral membrane of the cortical thick ascending limb. American Journal of Physiology - Renal Physiology. 2015;308:F1288–F1296. doi: 10.1152/ajprenal.00687.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang C, Wang L, Thomas S, Wang K, Lin DH, Rinehart J, Wang WH. Src-family protein tyrosine kinase regulates the basolateral K channel in the distal convoluted tubule (DCT) by phosphorylation of KCNJ10. J Biol Chem. 2013;288:26135–26146. doi: 10.1074/jbc.M113.478453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vio CP, AN SJ, CeSPEDES CARL, McGiff JC, Ferreri NR. Induction of Cyclooxygenase-2 in Thick Ascending Limb Cells by Adrenalectomy. J Am Soc Nephrol. 2001;12(4):649–658. doi: 10.1681/ASN.V124649. [DOI] [PubMed] [Google Scholar]
- 17.Cuevas CA, Su XT, Wang MX, Terker AS, Lin DH, McCormick JA, Yang C-L, Ellison DH, Wang WH. Potassium sensing by renal distal tubules requires Kir4.1. J Am Soc Nephrol. 2017;28:1814–1825. doi: 10.1681/ASN.2016090935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pessia M, Tucker SJ, Lee K, Bond CT, Adelman JP. Subunit positional effects revealed by novel heteromeric inwardly rectifying K+ channels. EMBO J. 1996;15(12):2980–2987. [PMC free article] [PubMed] [Google Scholar]
- 19.Willis L, Ludens JH, Hook JB, Williamsom H. Mechanism of natriuretic action of bradykinin. Am J Pathol. 1969;217:1–5. doi: 10.1152/ajplegacy.1969.217.1.1. [DOI] [PubMed] [Google Scholar]
- 20.Zaika O, Mamenko M, O’Neil RG, Pochynyuk O. Bradykinin acutely inhibits activity of the epithelial Na+ channel in mammalian aldosterone-sensitive distal nephron. American Journal of Physiology-Renal Physiology. 2011;300(5):F1105–F1115. doi: 10.1152/ajprenal.00606.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Su XT, Zhang C, Wang L, Gu R, Lin DH, Wang WH. Disruption of KCNJ10 (Kir4.1) stimulates the expression of ENaC in the collecting duct. American Journal of Physiology - Renal Physiology. 2016;310(10):F985–F993. doi: 10.1152/ajprenal.00584.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Terker A-S, Zhang C, McCormick J-A, Lazelle R-A, Zhang C, Meermeier N-P, Siler D-A, Park H-J, Fu Y, Cohen D-M, Weinstein A-M, Wang WH, Yang CL, Ellison D-H. Potassium Modulates Electrolyte Balance and Blood Pressure through Effects on Distal Cell Voltage and Chloride. Cell Metabolism. 2015;21(1):39–50. doi: 10.1016/j.cmet.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang C, Wang L, Zhang J, Su X-T, Lin DH, Scholl UI, Giebisch G, Lifton RP, Wang WH. KCNJ10 determines the expression of the apical Na-Cl cotransporter (NCC) in the early distal convoluted tubule (DCT1) Proc Natl Acad Sci USA. 2014;111:11864–11869. doi: 10.1073/pnas.1411705111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Piala AT, Moon TM, Akella R, He HX, Cobb MH, Goldsmith EJ. Chloride sensing by WNK1 involves inhibition of autophosphosphorylation. Sciencesignaling. 2014;7(324):ra41. doi: 10.1126/scisignal.2005050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bazua-Valenti S, Chavez-Canales M, Rojas-Vega L, Gonzalez-Rodriguez X, Vazquez N, Rodriguez-Gama A, Argaiz ER, Melo Z, Plata C, Ellison DH, Garcia-Valdes J, Hadchouel J, Gamba G. The Effect of WNK4 on the Na+Cl Cotransporter Is Modulated by Intracellular Chloride. J Am Soc Nephrol. 2014;26:1781–1786. doi: 10.1681/ASN.2014050470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McCormick JA, Mutig K, Nelson JH, Saritas T, Hoorn EJ, Yang C-L, Rogers S, Curry J, Delpire E, Bachmann S, Ellison DH. A SPAK isoform switch modulates renal salt transport and blood pressure. Cell Metabolism. 2011;14:352–364. doi: 10.1016/j.cmet.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang SS, Lo YF, Wu CC, Lin SW, Yeh CJ, Chu P, Sytwu HK, Uchida S, Sasaki S, Lin SH. SPAK-Knockout Mice Manifest Gitelman Syndrome and Impaired Vasoconstriction. Journal of the American Society of nephrology. 2010;21(11):1868–1877. doi: 10.1681/ASN.2009121295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lalioti MD, Zhang J, Volkman HM, Kahle KT, Hoffmann KE, Toka HR, Nelson-Williams C, Ellison DH, Flavell R, Booth CJ, Lu Y, Geller DS, Lifton RP. Wnk4 controls blood pressure and potassium homeostasis via regulation of mass and activity of the distal convoluted tubule. Nat Genet. 2006;38(10):1124–1132. doi: 10.1038/ng1877. [DOI] [PubMed] [Google Scholar]
- 29.Piechotta K, Lu J, Delpire E. Cation Chloride Cotransporters Interact with the Stress-related Kinases Ste20-related Proline-Alanine-rich Kinase (SPAK) and Oxidative Stress Response 1 (OSR1) J Biol Chem. 2002;277(52):50812–50819. doi: 10.1074/jbc.M208108200. [DOI] [PubMed] [Google Scholar]
- 30.Liu Z, Xie J, Wu T, Truong T, Auchus RJ, Huang CL. Downregulation of NCC and NKCC2 cotransporters by kidney-specific WNK1 revealed by gene disruption and transgenic mouse models. Human Molecular Genetics. 2011;20(5):855–866. doi: 10.1093/hmg/ddq525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van der Lubbe N, Moes AD, Rosenbaek LL, Schoep S, Meima ME, Danser AHJ, Fenton RA, Zietse R, Hoorn EJ. K+ -induced natriuresis is preserved during Na+ depletion and accompanied by inhibition of the Na+ -Cl− cotransporter. American Journal of Physiology - Renal Physiology. 2013;305(8):F1177–F1188. doi: 10.1152/ajprenal.00201.2013. [DOI] [PubMed] [Google Scholar]
- 32.Sorensen MV, Grossmann S, Roesinger M, Gresko N, Todkar AP, Barmettler G, Ziegler U, Odermatt A, Loffing-Cueni D, Loffing J. Rapid dephosphorylation of the renal sodium chloride cotransporter in response to oral potassium intake in mice. Kidney Int. 2013;83(5):811–824. doi: 10.1038/ki.2013.14. [DOI] [PubMed] [Google Scholar]
- 33.Ellison DH, Terker AS, Gamba G. Potassium and Its Discontents: New Insight, New Treatments. J Am Soc Nephrol. 2015;27:981–989. doi: 10.1681/ASN.2015070751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Take C, Ikeda K, Kurasawa T, Kurokawa K. Increased Chloride Reabsorption as an Inherited Renal Tubular Defect in Familial Type II Pseudohypoaldosteronism. N Engl J Med. 1991;324(7):472–476. doi: 10.1056/NEJM199102143240707. [DOI] [PubMed] [Google Scholar]
- 35.Shibata S, Zhang J, Puthumana J, Stone KL, Lifton RP. Kelch-like 3 and Cullin 3 regulate electrolyte homeostasis via ubiquitination and degradation of WNK4. Proceedings of the National Academy of Sciences. 2013;110(19):7838–7843. doi: 10.1073/pnas.1304592110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Simon DB, Nelson-Williams C, Bia MJ, Ellison D, Karet FE, Molina AM, Vaara I, Iwata F, Cushner HM, Koolen M, Gainza FJ, Gitleman HJ, Lifton RP. Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nature Genetics. 1996;12(1):24–30. doi: 10.1038/ng0196-24. [DOI] [PubMed] [Google Scholar]
- 37.Milia AF, Gross V, Plehm R, De Silva JA, Bader M, Luft FC. Normal Blood Pressure and Renal Function in Mice Lacking the Bradykinin B2 Receptor. Hypertension. 2001;37(6):1473–1479. doi: 10.1161/01.hyp.37.6.1473. [DOI] [PubMed] [Google Scholar]
- 38.Cervenka L, Maly J, Karasova L, Simova M, Vitko S, Hellerova S, Heller J, El-Dahr SS. Angiotensin II-Induced Hypertension in Bradykinin B2 Receptor Knockout Mice. Hypertension. 2001;37(3):967–973. doi: 10.1161/01.hyp.37.3.967. [DOI] [PubMed] [Google Scholar]
- 39.Alfie ME, Sigmon DH, Pomposiello SI, Carretero OA. Effect of High Salt Intake in Mutant Mice Lacking Bradykinin-B2 Receptors. Hypertension. 1997;29(1):483–487. doi: 10.1161/01.hyp.29.1.483. [DOI] [PubMed] [Google Scholar]
- 40.Wu P, Gao Z-X, Duan X, Su X-T, Wang MX, Lin D-H, Gu RM, Wang WH. AT2R-mediated regulation of Na-Cl cotransporter (NCC) and renal K excretion depends on the K channel, Kir4.1. Hypertension. 2018;71:622–630. doi: 10.1161/HYPERTENSIONAHA.117.10471. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.