

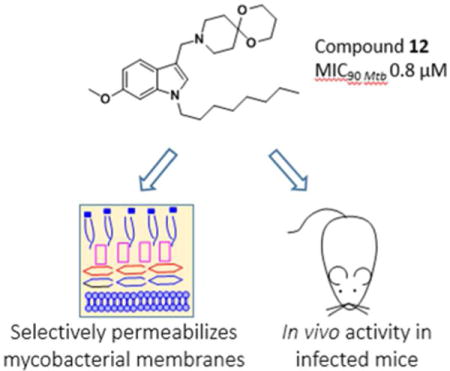
Abstract
The inclusion of an azaspiroketal Mannich base in the membrane targeting anti-tubercular 6- methoxy-1-n-octyl-1H-indole scaffold resulted in analogs with improved selectivity and submicromolar activity against Mycobacterium tuberculosis H37Rv. The potency enhancing properties of the spiro-ring fused motif was affirmed by SAR and validated in a mouse model of tuberculosis. As expected for membrane inserting agents, the indolyl azaspiroketal Mannich bases perturbed phospholipid vesicles, permeabilized bacterial cells and induced the mycobacterial cell envelope stress reporter promoter piniBAC. Surprisingly, their membrane disruptive effects did not appear to be associated with bacterial membrane depolarization. This profile was not uniquely associated with azaspiroketal Mannich bases but was characteristic of indolyl Mannich bases as a class. Whereas resistant mycobacteria could not be isolated for a less potent indolyl Mannich base, the more potent azaspiroketal analog displayed low spontaneous resistance mutation frequency of 10−8/CFU. This may indicate involvement of an additional envelope-related target in its mechanism of action.
Graphical abstract
INTRODUCTION
Tuberculosis (TB) has been a scourge for most of history.1 In spite of advances in our understanding of the pathogenic organism Mycobacterium tuberculosis (Mtb), TB remains frustratingly difficult to treat and continues to extract a high toll of human lives. In 2016, TB surpassed HIV-AIDS and malaria as the leading cause of death due to a single infectious agent.2 The past decade has witnessed continuing resistance to first line anti-TB drugs isoniazid and rifampicin (multi-drug resistance), additional resistance to fluoroquinolones and second-line injectables (extensive drug resistance), and more recently, cases of programmatically incurable TB where treatment regimens constructed with available drugs completely fail.3 Most drugs used today were discovered decades ago and the global TB drug pipeline, notwithstanding recent additions like bedaquiline, remains thin.4 While the urgency for new drugs cannot be overemphasized, a more compelling need is for agents that kill both replicating and phenotypically drug tolerant nonreplicating populations of mycobacteria. Disrupting the mycobacterial membrane has been posited as a viable means of achieving these objectives.5 First, the loss of an architecturally intact and functional membrane is fatal to both growing and quiescent bacteria.5 Second, the ensuing perturbation of multiple membrane-embedded targets would further delay resistance development among surviving organisms.5 The feasibility of this approach is corroborated by membrane-active small molecules (verapamil,6 boromycin,7 amphiphilic xanthones,8 propyltriphenylphosphonium phenothiazine conjugates,9 1-octylindolyl Mannich bases10) that have antimycobacterial activity. Involvement of the mycobacterial membrane was inferred from the ability of these compounds to permeabilize mycobacterial membranes,7,8,10 disrupt transmembrane potential6,7 and/or deplete intracellular ATP.7,8 Tellingly, conjugation of the membrane targeting triphenylphosphonium (TPP) moiety to antimycobacterial phenothiazines led to significant improvements in activity, likely due to enhanced localization within the mycobacterial membrane.9 Relatably, grafting TPP onto an inactive indole scaffold resulted in mycobactericidal cationic amphiphilic TPP indoles that rapidly depolarized the membrane of M. bovis BCG.11 1-Octylindolyl Mannich bases embedded with a cationic amphiphilic motif were similarly mycobactericidal and additionally, more potent and selective than the TPP indoles.10,11 Among the more potent indolyl Mannich bases, compound 1 (8-[(6-methoxy-1-octyl-1H-indolyl-3-yl)methyl]-1,4-dioxa-8-azaspiro[4,5]decane) stood out as the only member with an azaspiroketal side chain (Figure 1).10 We noted that a structurally similar motif was present in the preclinical TB candidate BTZ043 (2, Figure 1).4,12 Mechanistically, the azaspiroketal has not been implicated in the potent activity of BTZ043 which was attributed to inhibition of the oxidoreductase DprE1, a key enzyme involved in mycobacterial cell wall synthesis.13,14 The contribution of the azaspiroketal to the anti-TB activity of BTZ043 remains elusive as structural variations have yielded mixed results. Notably, the antimycobacterial activity of 3 which does not possess a spirocyclic side chain, was comparable to BTZ043 (Figure 1)15 whereas replacing the ketal oxygens in BTZ043 with sulfur resulted in a more active analog 4 (Figure 1).16 Spirocyclic motifs are widely perceived to possess attractive attributes from a drug design perspective due to their unique dimensionality and complexity.17 Evidence from literature has shown that the inclusion of spirocycles in molecules led to tighter and more selective binding to desired targets, improved solubility and greater metabolic stability.18,19 Viewed in this context and as part of our efforts to optimize the antimycobacterial activity of indolyl Mannich bases, we explored the azaspirocyclic motif with two goals in mind: First, to assess its potential as a potency-enhancing chemotype, and second, to determine if it is associated with distinctive mechanistic effects that set it apart from other Mannich bases.
Figure 1.
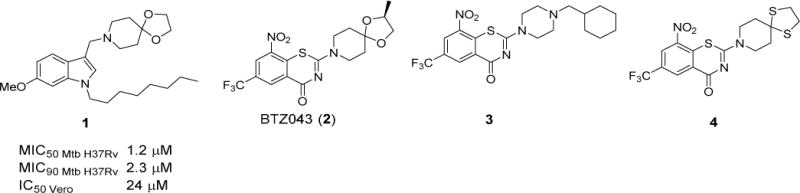
Structures of indolyl Mannich base 1, BTZ043 (2) and its benzothiazinone congeners 3 and 4
Chemistry
Three series of indolyl Mannich bases were synthesized to interrogate the structure-activity contribution of the spirocyclic motif. In Series 1, diverse azaspirocycles were introduced as the basic moiety at position 3 of the 1-n-octyl-6-methoxyindolyl scaffold. We then sought to optimize activity of the more potent Series 1 members by replacing n-octyl with ring bearing side chains (Series 2), and introducing alternative substituents (nitro, cyano, propoxy, isopropoxy, benzyloxy) in place of methoxy (Series 3). Also prepared were 1-octylindoles in which the azaspirocycle was not part of the Mannich base motif but attached to indole via an alkoxy linker (Series 4).
Series 1-3 were synthesized by methods previously reported for indolyl Mannich bases.10 Scheme 1 outlines the synthesis of Series 1 which involves reacting 6-methoxy-1H-indole with 1-bromooctane in the presence of sodium hydride to give the 1-(n-octyl) analog 5, followed by the insertion of the basic 3-substituted aminomethyl side chain by two approaches. In the first, the indole-3-carbaldehyde 6 was prepared by a Vilsmeier-Haack reaction and then subjected to reductive amination to give compounds 21, 22, 28 – 32, 35. In the second approach, 5 was reacted with an amine and formaldehyde in the presence of an acid catalyst (acetic acid or zinc chloride) to give the remaining Series 1 compounds.
Scheme 1.
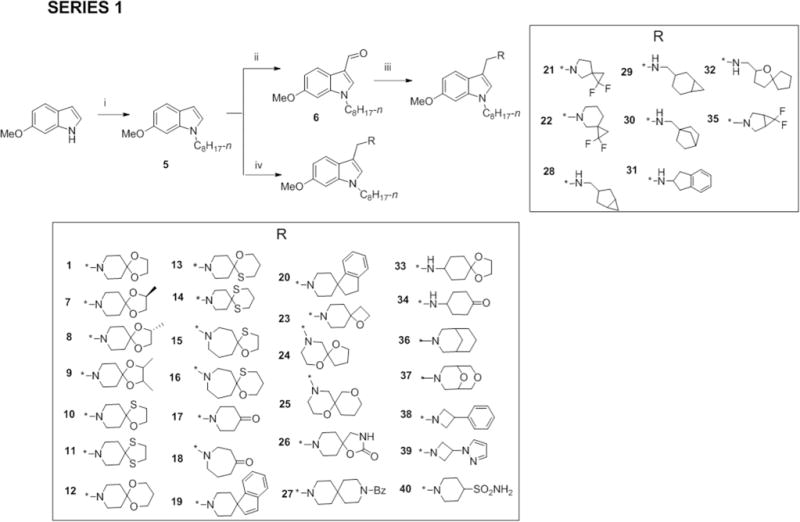
Reagents and conditions: (i) NaH, CH3(CH2)6CH2Br, anhydrous DMF, 0°C to RT, 2 – 8 h (ii) POCl3, DMF, 0°C – 40°C, 2 h (iii) CH3COOH, appropriate amines, Na(AcO)3BH, anhydrous THF, RT, 24 – 48 h (iv) CH2O (aq., 36%), appropriate amines, CH3COOH, RT or ZnCl2, EtOH, RT, 3 – 24 h.
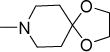
Most of the amines indicated in Scheme 1 were commercially available but those required for 1, 7-16 were synthesized. These were the azaspirocycles 1,4-dioxa-8-azaspiro[4.5]decane (1a), (S)-2-methyl-1,4-dioxa-8-azaspiro[4.5]decane (7a), (R)-2-methyl-1,4-dioxa-8-azaspiro[4.5]decane (8a), 2,3-dimethyl-1,4-dioxa-8-azaspiro[4.5]decane (9a), 1-oxa-4-thia-8-azaspiro [4.5]decane (10a), 1,4-dithia-8-azaspiro[4.5]decane (11a), 1,5-dioxa-9-azaspiro[5.5]undecane (12a), 1-oxa-5-thia-9-azaspiro[5.5]undecane (13a), 1,5-dithia-9-azaspiro[5.5]undecane (14a), 1-oxa-4-thia-8-azaspiro[4.6]undecane (15a) and 1-oxa-5-thia-9-azaspiro[5.6]dodecane (16a). These amines were prepared by reacting piperidin-4-one or azepan-4-one with the relevant alkanediols, alkanedithiols or mercaptoalkanols in the presence of catalytic amounts of p-toluenesulfonic acid.
In Series 2, we replaced the n-octyl side chain of 1, 12 and 6-methoxy-1-octyl-3-(piperidin-1- ylmethyl)-1H-indole (52) with various ring-bearing side chains, namely 3-phenoxypropyl, fluoro/methoxy/methyl-substituted 3-phenoxypropyl, 4-phenoxybutyl, 3-benzyloxypropyl, cinnamyl, 3-phenylpropyl and 4-phenylbutyl. Of these side chains, phenoxypropyl bromide (53) and 1-(3-bromopropoxy)-4-methoxybenzene (54) were prepared by reacting phenol/4- methoxyphenol with 1,3-dibromopropane in the presence of potassium carbonate in acetonitrile (Supporting Information, Scheme S1). Scheme 2 outlines the synthesis of Series 2 which involved firstly, N-alkylation of 6-methoxy-1H-indole as described in Scheme 1 to give the 1-ring substituted 6-methoxyindoles (41-51), followed by the Mannich reaction with bases 1a, 12a and piperidine to give the desired compounds (41a,b,c – 50a,b,c; 51a,b). The 1-octylindolyl Mannich base 52 was synthesized by a similar method.10
Scheme 2.
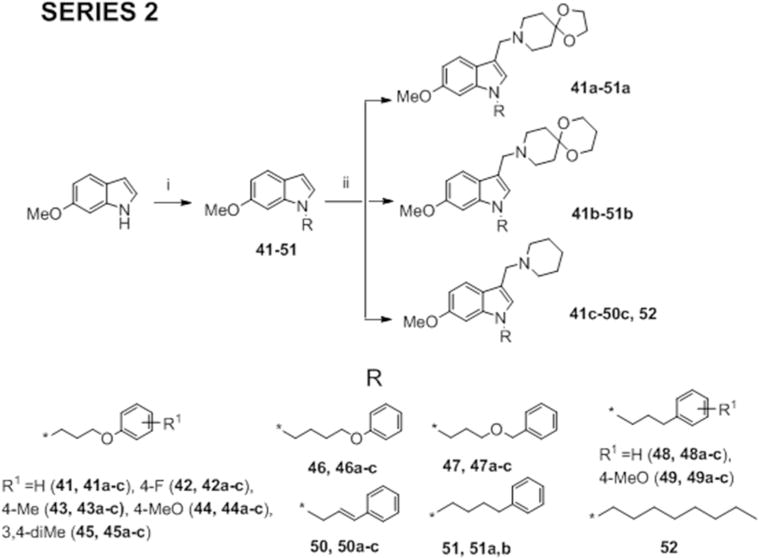
Reagents and conditions: (i) NaH, appropriate n-octyl, phenalkyl, phenoxyalkyl, benzyloxyalkyl or cinnamyl bromide, anhydrous DMF, 0°C to RT, 2 – 8 h (ii) CH2O (aq., 36%), amines (1a, 12a or piperidine), CH3COOH, RT or ZnCl2, EtOH, RT, 3 – 24 h.
In Series 3, other functionalities (nitro, cyano, propoxy, isopropoxy, benzyloxy) were introduced in place of methoxy on the indole ring while retaining n-octyl and selected Mannich bases at positions 1 and 3 respectively. The 6-isopropoxy (55a), 6-propoxy (55b) and 6-benzyoxyindoles (55c) were synthesized from 6-hydroxy-1H-indole and the corresponding alkyl/benzyl halide in the presence of an inorganic base (cesium carbonate Cs2CO3 or potassium carbonate K2CO3) in DMF. The regioisomeric nitroindoles and 6-cyanoindoles were purchased. The substituted indoles were then alkylated with 1-bromooctane and reacted via a Mannich reaction to give the final compounds 57a-h, 58a-e and 59a-c (Scheme 3).
Scheme 3.
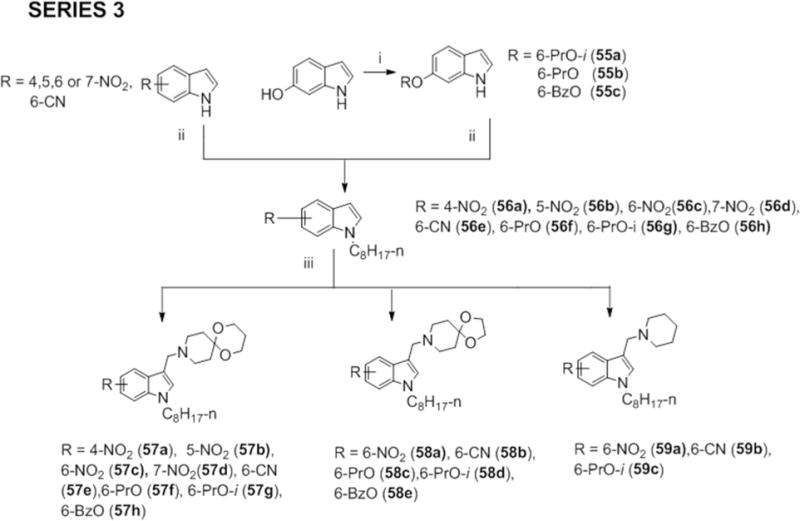
Reagents and conditions: (i) 1-iodopropane; 2-iodopropane; or benzyl bromide, K2CO3 or Cs2CO3, anhydrous DMF, 80°C, 3 – 4 h (ii) NaH, CH3(CH2)6CH2Br, anhydrous DMF, 0°C to RT, 2 – 8 h (iii) CH2O (aq., 36%), amines (1a, 12a or piperidine), CH3COOH, RT or ZnCl2, EtOH, RT, 3 – 24 h.
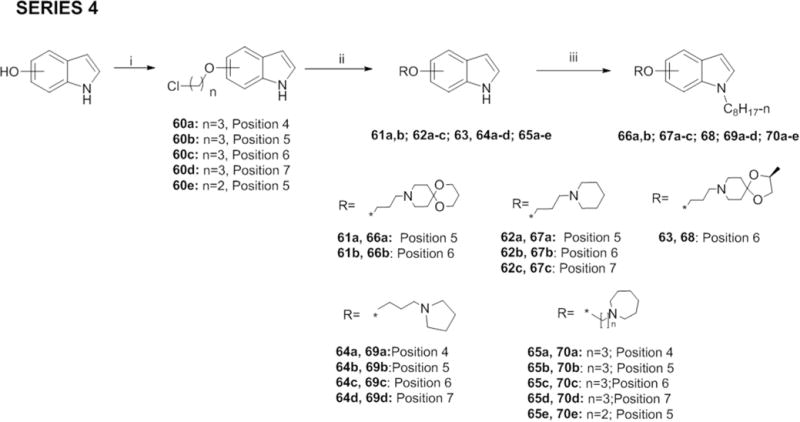
To obtain Series 4, we first reacted regioisomeric 4-, 5-, 6- and 7-hydroxy-1H-indoles with 1- bromo-3-chloropropane or 1-bromo-2-chloroethane in the presence of K2CO3 in ethanol to obtain the chloropropoxyindole (60a-d) or 5-(2-chloroethoxy)-1H-indole (60e). Displacement of the terminal chlorine with iodine followed by reaction with amines azepane, 1,5-dioxa-9-azaspiro [5.5]undecane (12a), piperidine, (S)-2-methyl-1,4-dioxa-8-azaspiro[4.5]decane (7a), and pyrrolidine in the presence of K2CO3 or direct reaction of 60a-e with the appropriate amine and Cs2CO3 gave 61a-b, 62a-c, 63, 64a-d, 65a-e. The latter were then alkylated as before to give 66a-b, 67a-c, 68, 69a-d, 70a-e.
RESULTS AND DISCUSSION
Series 1 compounds with azaspiroketal and isosterically related sidechains have submicromolar potencies and outstanding selectivity
The minimum inhibitory concentrations (MIC50, MIC90) of Series 1 were determined by turbidity measurements on M. bovis BCG, an attenuated tubercule bacillus and mycobacterial model organism (Table 1). The more potent analogs (MIC50 < 5 μM) were assessed on mammalian African Green Monkey kidney epithelial Vero cells for selective cytotoxicities. Also assessed were the hemolytic potential of some analogs on human red blood cells (RBCs). This was in view of the cationic amphiphilic nature of these compounds which would predispose them to non-selective membrane disruptive effects.
Table 1.
Antimycobacterial activity (M. bovis BCG), mammalian Vero cell cytotoxicity (IC50) and hemolytic activity (HC50, human RBC) of Series 1 compounds.
| |||||||
|---|---|---|---|---|---|---|---|
| No | R | MIC (BCG) (μM)a | Vero IC50 (μM)b |
SIC | HC50 (μM)d |
LogPe | |
| MIC50 | MIC90 | ||||||
| 1 |
|
2 | 4 | 46 | 23 | 75 | 5.01 |
| 7 |
|
0.5 | 0.9 | 34 | 68 | 51 | 5.33 |
| 8 |
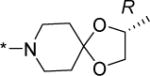
|
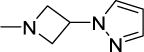
0.6 | 0.8 | 37 | 62 | 75 | 5.33 |
| 9 |
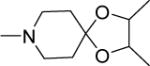
|
0.7 | 1.6 | 34 | 49 | 76 | 5.65 |
| 10 |

|
0.6 | 0.8 | 64 | 107 | >300 | 5.73 |
| 11 |
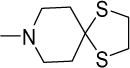
|
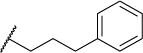
0.5 | 0.8 | >200 | >400 | >300 | 6.46 |
| 12 |
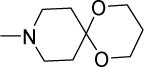
|
0.3 | 0.8 | 29 | 97 | 64 | 5.12 |
| 13 |
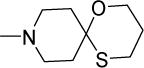
|
0.4 | 0.8 | 39 | 98 | >300 | 5.84 |
| 14 |
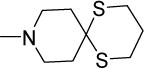
|
0.4 | 0.8 | 152 | 380 | >300 | 6.56 |
| 15 |
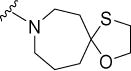
|
0.7 | 1.2 | 35 | 50 | >300 | 5.70 |
| 16 |
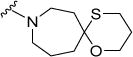
|
0.5 | 1 | 34 | 68 | 132 | 5.95 |
| 17 |
|
17 | 24 | NDf | NDf | NDf | 4.47 |
| 18 |
|
17 | 42 | NDf | NDf | NDf | 4.75 |
| 19 |
|
1 | 2 | 44 | 44 | >300 | 6.93 |
| 20 |
|
1 | 2 | 31 | 31 | >300 | 7.25 |
| 21 |
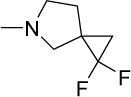
|
13 | 18 | NDf | NDf | NDf | 5.17 |
| 22 |
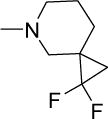
|
13 | 25 | NDf | NDf | NDf | 5.58 |
| 23 |
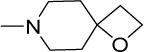
|
2 | 4 | 31 | 16 | 88 | 4.07 |
| 24 |
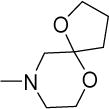
|
15 | 46 | NDf | NDf | NDf | 4.85 |
| 25 |
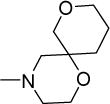
|
1.8 | 6.3 | >500 | >278 | >300 | 4.16 |
| 26 |
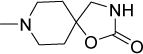
|
22 | 43 | NDf | NDf | NDf | 3.74 |
| 27 |
|
2.3 | 4.4 | 14 | 6 | 69 | 7.03 |
| 28 |
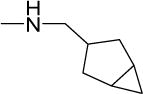
|
2 | 4 | 10 | 5 | 33 | 5.46 |
| 29 |
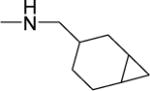
|
1.6 | 2.3 | 20 | 13 | NDf | 5.88 |
| 30 |
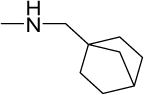
|
1.1 | 1.9 | 10 | 9 | 19 | 6.09 |
| 31 |
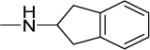
|
4 | 6 | 24 | 6 | NDf | 6.13 |
| 32 |
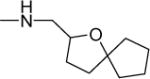
|
1.8 | 4.6 | 21 | 12 | NDf | 5.31 |
| 33 |
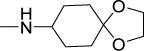
|
3.6 | 6.3 | 22 | 6 | 207 | 4.83 |
| 34 |
|
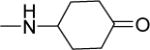
15 | 24 | NDf | NDf | NDf | 4.63 |
| 35 |
|
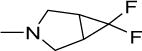
21 | 50 | NDf | NDf | NDf | 4.92 |
| 36 |
|
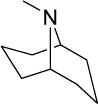
3.8 | 7.6 | 31 | 8 | 109 | 5.96 |
| 37 |
|
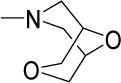
17 | 30 | NDf | NDf | NDf | 3.70 |
| 38 |
|
2 | 4 | 44 | 22 | >300 | 6.07 |
| 39 |
|
6 | 15 | NDf | NDf | NDf | 4.40 |
| 40 |
|
8 | 22 | NDf | NDf | NDf | 3.18 |
Minimum Inhibitory Concentrations required to reduce growth by 50% (MIC50) or 90% (MIC90) compared to untreated controls. Average of 2 or more determinations.
Concentration required to reduce growth of Vero cells by 50% compared to untreated controls. Average of 2 or more determinations.
Selective Index = IC50 Vero/MIC50 BCG.
Concentration required to hemolyze 50% of human RBCs, compared to 2% Triton X-100 (100% hemolysis).
Log P values were determined on ChemDraw Professional Version 15.1.0.144.
ND = Not Determined
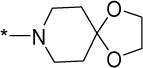
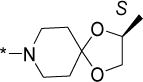
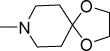
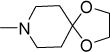
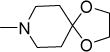
Structurally diverse Mannich bases were explored at R in Series 1 (Table 1). These were (i) azaspiroketals (7-9) and congeners in which one or both ketal oxygen atoms were replaced by sulfur (10-16); (ii) azaspirocycles without the ketal moiety (19-27); (iii) secondary amines substituted with fused/bridged bicyclic rings (28-31), the spirocycle 1-oxaspiro[4.4]nonane (32) and the spiroketal 1,4-dioxaspiro[4.5]decane (33); (iv) azabicycles (35-37) and (v) substituted azetidines 38,39 and piperidine 40. Of these, only the azaspiroketals and their sulfur isosteres (7-16, MIC50 0.3-0.7 μM) surpassed the original lead 1 (MIC50 2 μM). These significant gains in potency which also led to better selective cytotoxicities, were achieved by modifying the ketal or piperidine ring of 1 in the following ways: (i) methyl or dimethyl substitution of the ketal ring; (ii) replacing one or both ketal oxygen atoms with isosteric sulfur to give 10 (MIC50 0.6 μM) and 11 (MIC50 0.5 μM); (iii) expanding the 5-membered ketal bearing ring to a 6-membered homolog (12, MIC50 0.3 μM); (iv) submitting 12 to oxygen-sulfur isosteric substitution to give 13 and 14 (MIC50 0.4 μM for both) and (v) homologation of the piperidine ring in 11 and 13 to the 7- membered azepanyl homologs 15 (MIC50 0.7 μM) and 16 (MIC50 0.5 μM) respectively.
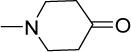
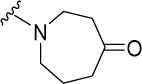
Interestingly, these modifications resulted in compounds that were largely equipotent. We then asked if the narrow variation in MICs could be due to hydrolysis of the spirocyclic side chain to a common potent intermediate. Hydrolysis would yield the piperidinone 17 or azepanone 18 and if the hypothesis holds, these compounds should be as active as their spirocyclic precursors. That both 17 and 18 had only moderate activities (MIC50 17 μM) led us to conclude otherwise.
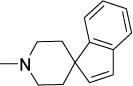
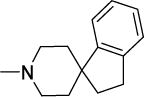
The remaining compounds (19-40) in Series 1 were less potent than 1 but provided useful insight into the structural requirements for activity. First, the diminished activities of azaspirocycles that were not ketals (19-27) emphasized the importance of the intact azaspiroketal motif. Tellingly, the non-ketal azaspirocycles 24 and 25 displayed mediocre activity (MIC50 15 μM, 1.8 μM), in spite of being regioisomers of the azaspiroketals 1 and 12 respectively. Second, positioning the basic nitrogen outside the spirocycle or bicycle resulted in analogs (28-33) that were moderately potent (MIC50 1.1-4 μM) but with strikingly poor selective cytotoxicities (SI 5-13). The comparison of 1 (MIC50 2 μM, SI =23) with 33 (MIC50 3.6 μM, SI =6) was particularly compelling in this regard. As before, hydrolysis of the spiroketal side chain in 33 was dismissed in view of the limited activity of the N-cyclohexanone analog 34 (MIC50 15 μM). Third, there was no apparent advantage in deploying rigid scaffolds like those in the azabicycles 35-37 or conformationally more flexible azacycles such as 38-40.
We also collated the log P values of the Series 1 analogs. Lipophilicities varied over a wide 104 range, from 3.2 (40) to 7.3 (20) (Table 1). A cursory inspection showed that the least active compounds (MIC50 > 10 μM) had low log P values (3.2 – 5.6), an indication that activity may be driven by lipophilicity. Indeed, we found a significant inverse relationship beween MIC50 and log P (Spearman ρ correlation coefficient −0.567, p = 0.01, 2-tailed), althought the potent azaspiroketal Mannich bases (7-16), with an average log P of 5.8, were only moderately lipophilic.
In all, these results pointed to considerable tolerance for structurally diverse Mannich bases at R, most of which have low micromolar activities (MIC50 1-10 μM). However, only azaspiroketals and their sulfur isosteres (7-16) were uniquely associated with highly selective and submicromolar potencies.
Series 2 analogs with ring bearing side chains at the indole N were less potent than n- octyl counterparts
Series 2 was designed to interrogate substitution at the indole N. Here, ring-bearing phenoxyalkyl, phenylalkyl and phenylalkenyl side chains were introduced in place of n-octyl. As shown in Table 2, the R1 substitution was explored in three 6-methoxyindolyl Mannich bases, namely the early hit 1, the potent submicromolar hit 12 and a previously reported piperidinylmethyl analog 52.10
Table 2.
Antimycobacterial activity (M. bovis BCG), mammalian Vero cell cytotoxicity (IC50) and hemolytic activity (HC50, human RBC) of Series 2 compounds.
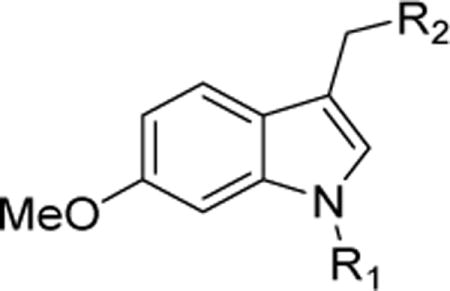
| |||||||
|---|---|---|---|---|---|---|---|
| No | R1 | R2 | MIC (BCG) (μM)a | Vero IC50b |
HC50 (μM)c |
LogPd | |
| MIC50 | MIC90 | ||||||
| 41a |
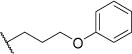
|
|
3 | 10 | NDe | NDe | 3.87 |
| 41b |
|
1.3 | 3.1 | NDe | >300 | 3.98 | |
| 41c |
|
30 | 50 | NDe | NDe | 4.13 | |
| 42a |
|
|
5 | 11 | NDe | NDe | 4.03 |
| 42b |
|
1.5 | 2.6 | NDe | >300 | 4.14 | |
| 42c |
|
20 | 43 | NDe | NDe | 4.29 | |
| 43a |
|
|
4.8 | 11 | NDe | NDe | 4.36 |
| 43b |
|
1.3 | 2.6 | NDe | >300 | 4.46 | |
| 43c |
|
19 | 41 | NDe | NDe | 4.62 | |
| 44a |
|
|
6 | 13 | NDe | NDe | 3.75 |
| 44b |
|
1.5 | 3.5 | 46 | >300 | 3.85 | |
| 44c |
|
34 | >50 | NDe | NDe | 4.00 | |
| 45a |
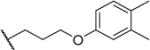
|
|
4.6 | 11 | NDe | NDe | 4.85 |
| 45b |
|
1.7 | 2.7 | NDe | >300 | 4.95 | |
| 45c |
|
18 | 25 | NDe | NDe | 5.10 | |
| 46a |
|
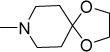
|
20 | 41 | NDe | NDe | 4.33 |
| 46b |
|
11 | 25 | NDe | NDe | 4.43 | |
| 46c |
|
16 | 41 | NDe | NDe | 4.58 | |
| 47a |
|
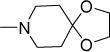
|
16 | 40 | NDe | NDe | 3.78 |
| 47b |
|
2.0 | 6 | 54 | NDe | 3.89 | |
| 47c |
|
46 | >50 | NDe | NDe | 4.04 | |
| 48a |
|
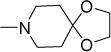
|
6 | 18 | 35 | NDe | 4.53 |
| 48b |
|
2.6 | 9.8 | NDe | NDe | 4.64 | |
| 48c |
|
36 | >50 | 43 | NDe | 4.79 | |
| 49a |
|
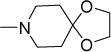
|
7.4 | 17 | 31 | NDe | 4.41 |
| 49b |
|
4.1 | 9.3 | NDe | NDe | 4.51 | |
| 49c |
|
18 | 39 | NDe | NDe | 4.66 | |
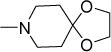
| 50a |
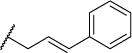
|
|
15 | 31 | NDe | NDe | 4.35 |
| 50b |
|
4.8 | 9 | NDe | NDe | 4.46 | |
| 50c |
|
31 | 49 | NDe | NDe | 4.61 | |
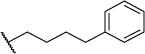
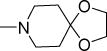
| 51a |
|
|
4.9 | 11.5 | NDe | NDe | 4.95 |
| 51b |
|
1.6 | 3.1 | 21 | >300 | 5.05 | |
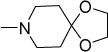
| 1 |
|
|
2 | 4 | 46 | 75 | 5.01 |
| 12 |
|
0.3 | 0.8 | 29 | 64 | 5.12 | |
| 52 |
|
7 | 13 | NDe | NDe | 5.27 | |
As described in footnote of Table 1.
ND = Not Determined.
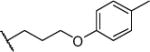
The results largely recapitulated the activity advantage of the R1 n-octyl side chain that was observed in a related series where R1 was varied from ethyl (−C2H5) to dodecyl (−C12H25).10 Here, none of the compounds with ring bearing R1, for the same Mannich base R2, were more potent than their n-octyl counterparts. Tellingly, the ring bearing analogs were also less lipophilic than their n-octyl counterparts with the same R2 group. However, unlike Series 1, Spearman correlation analysis did not reveal any significant relationship between MIC50 and log P in this series. However, it is still likely that the favored status of the n-octyl side chain is due to its lipophilic and less bulky features. As to whether there was an optimal ring bearing R1, we found that this was largely determined by R2. Of the three R2 side chains explored, there was an unambiguous preference for the 6/6 spiro-ring fused 1,5-dioxa-9-azaspiro[5.5]undecane (41b- 51b), followed by its 6/5 homolog 1,4-dioxa-8-azaspiro[4.5]decane (41a-51a) and lastly the monocyclic piperidine (41c-50c). For the 6/5 and 6/6 azaspiroketals, there was a preference for R1 side chains that were phenoxypropyl (41a,b), substituted phenoxypropyl (42a,b-45a,b) and phenylbutyl (51a,b). Benzyloxypropyl at R1 was favored only when paired with 1,5-dioxa-9- azaspiro[5.5]undecane at R2 (47b).
The activity advantage of replacing 6-methoxy with other ring substituents in Series 3 was strongly dependent on the nature of the Mannich base
Series 3 was designed to explore the replacement of 6-methoxy with other groups while retaining the n-octyl chain and Mannich base (1,5-dioxa-9-azaspiro[5.5]undecanylmethyl, 1,4- dioxa-8-azaspiro[4.5]decanylmethyl, piperidinylmethyl) of Series 1 and 2 respectively (Table 3). The electron donating methoxy was replaced by electron withdrawing groups (nitro, cyano) and sterically bulkier/more lipophilic alkoxy groups (n-propoxy, isopropoxy, benzyloxy). To determine if there was a preference for specific positions on the indole ring, we prepared nitro regioisomers of the 1,5-dioxa-9-azaspiro[5.5]undecanyl analog 12 (Table 3). When greater potency was found for the 6-nitro regioisomer (57c, MIC50 0.3 μM), subsequent substitutions were limited to position 6. Here, we found that 6-cyano (57e), 6-propoxy (57f), and 6- isopropoxy (57g) analogs were as potent (MIC50 0.5-0.7 μM) as the 6-nitro 57c. Only the insertion of the bulkier and more lipophilic benzyloxy at position 6 was not favored (57h, MIC50 2.2 μM). Interestingly, none of the compounds were more potent than the 6-methoxy analog 12.
Table 3.
Antimycobacterial activity (M. bovis BCG), mammalian Vero cell cytotoxicity (IC50) and hemolytic activity (HC50, human RBC) of Series 3 compounds.
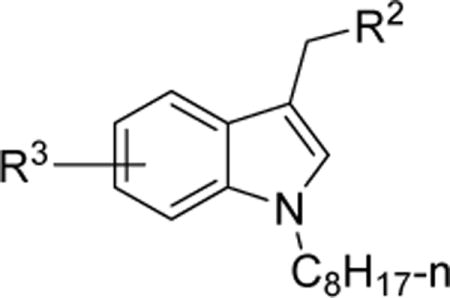
| |||||||
|---|---|---|---|---|---|---|---|
| No | R2 | R3 | MIC (μM)a | Vero IC50 (μM)b |
HC50 (μM)c |
LogPd | |
| MIC50 | MIC90 | ||||||
| 57a |
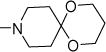
|
4-NO2 | 10 | 25 | NDe | NDe | 5.13 |
| 57b | 5-NO2 | 7 | 18 | NDe | NDe | 4.92 | |
| 57c | 6-NO2 | 0.3 | 0.8 | 28 | >300 | 5.87 | |
| 57d | 7-NO2 | 1.3 | 3.1 | 49 | >300 | 6.06 | |
| 57e | 6-CN | 0.5 | 1.2 | 22 | >300 | 5.53 | |
| 57f | 6-C3H7-n | 0.7 | 1.2 | 29 | 27 | 7.14 | |
| 57g | 6-C3H7-i | 0.6 | 1.6 | 20 | 29 | 6.98 | |
| 57h | 6-BzO | 2.2 | 3.1 | 27 | >300 | 7.42 | |
| 12 | 6-MeO | 0.3 | 0.8 | NDe | NDe | 6.12 | |
| 58a |
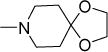
|
6-NO2 | 0.8 | 1.6 | 62 | >300 | 5.67 |
| 58b | 6-CN | 2.5 | 5 | 59 | >300 | 5.33 | |
| 58c | 6-C3H7-n | 3 | 5 | 29 | 31 | 6.94 | |
| 58d | 6-C3H7-i | 3 | 6 | 41 | 56 | 6.78 | |
| 58e | 6-BzO | 4 | 5.5 | 36 | >300 | 7.21 | |
| 1 | 6-MeO | 2 | 4 | NDe | NDe | 5.92 | |
| 59a |
|
6-NO2 | 4 | 5 | 23 | NDe | 6.35 |
| 59b | 6-CN | 2 | 6 | 22 | NDe | 6.01 | |
| 59c | 6-C3H7-i | 4 | 6 | 26 | NDe | 7.46 | |
| 52 | 6-MeO | 7 | 13 | NDe | NDe | 6.60 | |
As described in footnote of Table 1.
Log P values were determined on ACD Labs Physchem History Version 12.5.
ND = Not Determined.
In contrast, selective replacement of 6-methoxy in 1 and 52 resulted in more potent analogs. These were the 6-nitro 58a (MIC50 0.8 μM) and 6-cyano 59b (MIC50 2 μM) which were more potent than 1 and 52 respectively. Other substitutions yielded compounds (58b-e, 59a,c) that were comparable to 1 and 52. Thus, R3 substitution benefited the less potent analogs (1, 52) to a greater exent than the more potent 12. Taken together, two structure-activity trends were evident: First, there was no preference for electron donating or withdrawing R3 groups but size was a limiting factor. Notwithstanding, lipophilicity was not found to significantly influence activity. Second, the optimal R3 in this series was conspicously influenced by the Mannich base at R2, reinforcing the SAR trend observed in Series 2.
Non-Mannich base Series 4 analogs have modest antimycobacterial activities
The indole scaffold in Series 4 comprised an n-octyl side chain at position 1 and a basic spirocyclic or non-spirocyclic moiety attached to the benzenoid carbons of indole via an alkoxy linker. In a significant departure from the preceding series, these compounds retained the cationic amphiphilic signature but were not Mannich bases. Interestingly, this alteration resulted in analogs with mediocre activity (Table 4). Even analogs bearing the highly favored 6/6 spiro ring fused 1,5-dioxa-9-azaspiro[5.5]undecane ring (66a, 66b: MIC50 6 μM, 14 μM), were no more potent than their non-spirocyclic piperidinyl counterparts (67a-c: MIC50 5-13 uM). Clearly the privileged status of the azaspiroketal motif that was so pronounced in the indolyl Mannich bases of Series 1 did not apply to this scaffold. Here, the most potent members were 70b (MIC50 1.9 μM) and 70e (MIC50 2 μM), both of which have azepanylalkoxy side chains. Collectively these results led us to conclude that the topography of the cationic amphiphilic indole was pivotal for activity. To this end, the location of the azaspiroketal moiety was decisive and reconfiguring it to a non-Mannich base was disadvantageous.
Table 4.
MIC50/MIC90 values of Series 4 on M. bovis BCG
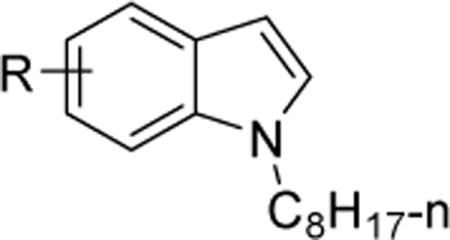
| |||||
|---|---|---|---|---|---|
| No | R | Position on ring | MIC (μM)a | LogPb | |
| MIC50 | MIC90 | ||||
| 66a |
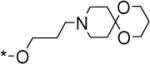
|
5 | 14 | 31 | 5.28 |
| 66b | 6 | 5.6 | 28 | 5.28 | |
| 67a |
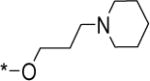
|
5 | 13 | 40 | 5.43 |
| 67b | 6 | 13 | 25 | 5.43 | |
| 67c | 7 | 5 | 13 | 5.43 | |
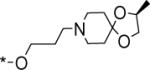
| 68 |
|
6 | 11 | 25 | 5.49 |
| 69a |
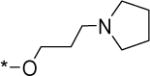
|
4 | 18 | 36 | 5.02 |
| 69b | 5 | 31 | 48 | 5.02 | |
| 69c | 6 | 20 | 42 | 5.02 | |
| 69d | 7 | 10 | 22 | 5.02 | |
| 70a |
|
4 | 9.5 | 22 | 5.85 |
| 70bc | 5 | 1.9 | 8 | 5.85 | |
| 70cd | 6 | 3.9 | 12 | 5.85 | |
| 70d | 7 | 5 | 11 | 5.85 | |
| 70e |
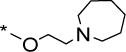
|
5 | 2 | 6 | 5.74 |
As described in footnote of Table 1.
Log P values were determined on ChemDraw Professional Version 15.1.0.144.
Vero cell IC50 34 μM.
Vero cell IC50 33 μM.
Antimycobacterial profiles of azaspiroketals (10-14) with submicromolar potencies
Encouraged by the potent and selective antimycobacterial activities of the azaspiroketals 10-14, we proceeded to investigate their antimycobacterial profiles in greater detail. As shown in Table 5, these compounds retained submicromolar MICs against pathogenic Mtb H37Rv. Both 12 and 13 were cidal against Mtb and achieved 1000-fold kill at 3.2 μM (4 × MIC90). Together with remaining compounds, they were cidal against M. bovis BCG, with MBC99.9 equivalent to 1-2x MIC90 BCG. We also assessed the ability of 12 and 13 to eradicate non-growing, phenotyoically drug tolerant organisms by assessing their cidality on Mtb cultures grown under nutrient deprived conditions (Loebel model). Both compounds were cidal in this model, with LCC99 of 18 μM for 12 and LCC99.9 < 18 μM for 13 (Supporting Information, Figure S1).
Table 5.
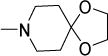
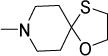
Antimycobacterial profiles of Compounds 1, 10-14
| No | Ra | Mtb H37Rv (μM) | M.bovis BCG (μM) | mic50 (μm) (M.bovis BCG) in 7H9 Mediab | ||||
|---|---|---|---|---|---|---|---|---|
| MIC50b | MIC90b | MBC99.9c | MBC99.9c | Standard Mediumd | Without Glycerol | With 10% FBS | ||
| 1 |
|
1.2 | 2.3 | ND | 3-6 | 1.5 | 1.6 | 1.1 |
| 10 |
|
0.6 | 0.95 | ND | 0.8-1.6 | 0.36 | 0.37 | 0.32 |
| 11 |
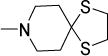
|
0.7 | 1.1 | ND | 0.8-1.6 | 0.29 | 0.27 | 0.28 |
| 12 |
|
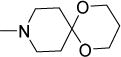
0.44 | 0.76 | 3.2 | 0.8-1.6 | 0.30 | 0.35 | 0.33 |
| 13 |
|
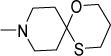
0.5 | 0.8 | 3.2 | 0.4-1.6 | 0.26 | 0.41 | 0.6 |
| 14 |
|
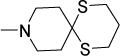
0.5 | 0.78 | ND | 0.8-1.6 | 0.28 | 0.43 | 0.48 |
Refer to general structure given in Table 1
Average of 2 or more separate determinations.
MBC99.9: Minimum bactericidal concentration required to kill 99.9% of bacteria. Average of 2 or more separate determinations. MBC99.9 of 1, 10, 11, 14 on Mtb H37Rv were not determined (ND).
Standard 7H9 medium contains glycerol and does not contain FBS
The antimycobacterial activities of 12, 13 and other analogs (1, 10, 11) were not dependent on media composition. MICs were unchanged in the absence of glycerol or in 7H9 medium containing serum. We also determined the spontaneous resistance mutation frequencies of M. bovis BCG and Mtb H37Rv to 12. A low spontaneous mutation frequency (10−8/CFU) was obtained on Mtb H37Rv exposed to 12 at 4 × and 8 × MIC90. The Mtb H37Rv mutants were confirmed to be resistant to 12, displaying on average 10-fold shifts in MIC50 values (based on analysis of 6 resistant strains). This is in contrast to the indolyl Mannich base 3-(azepan-1- ylmethyl)-4-fluoro-1-octyl-1H indole, 71 (Supporting Information, Figure S2) which we had reported earlier.10 For compound 71 which was not an azaspiroketal Mannich base, the mutation frequency was < 10−9/CFU for M. bovis BCG.10 Here, we confirmed in side-by-side experiments that the spontaneous resistance mutation frequency of 71 was < 10−9/CFU for Mtb H37Rv as well.
The azaspiroketal Mannich bases 10-13 disrupted the melting profiles of dimyristoylphosphatidylglycerol (DMPG) liposomal vesicles
Phosphatidylglycerols are negatively charged phospholipids that are abundant in bacterial plasma membranes but almost absent from membranes derived from eukaryotic cells.20,21 Previously, we employed DMPG liposomal vesicles as surrogates of bacterial membranes and found that indolyl Mannich bases which altered the melting profiles of DMPG vesicles (monitored by differential scanning calorimetry) also permeabilized mycobacterial membranes as seen from greater propidium iodide (PI) uptake by mycobacterial cultures.10 Conversely, antimycobacterial indolylalkyltriphenylphosphonium analogs that did not disrupt the thermotropic profiles of DMPG vesicles failed to increase PI uptake into mycobacteria.11
Here, we found that 10-13 caused pronounced changes in the melting profile of DMPG vesicles (Figure 2). Their presence caused a downward shift (≈ 4°) in the phase transition temperature Tm of the phospholipid and a pronounced 6-9 fold reduction in the calorimetric enthalpy (ΔH) of the gel to liquid transition. Collectively these changes pointed to weakened interactions within the lipid bilayer, conceivably due to the insertion of these compounds within the hydrophobic core of the bilayer. They also closely mimicked those reported earlier for DMPG vesicles containing 1, determined under similar conditions.10
Figure 2.
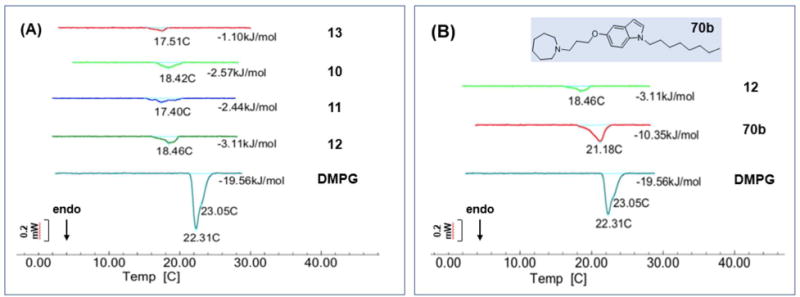
Representative calorimetric curves, in heating mode, of DMPG vesicles in the absence and presence (1 in 10 parts DMPG) of test compounds (A) 10-13, and (B) 70b. The melting transition Tm (°C) and molar enthalpy ΔH (in kJ/mol) for each treatment condition are indicated. Control DMPG vesicles showed a main Tm (22.3°C) and a shoulder at 23.05°C. This appearance has been noted by others.22
Intriguingly, the non-Mannich base 70b of Series 4 had significantly diminished effects on the melting profile of the DMPG vesicles, namely a minimal (1°) shift in Tm and a reduction of ΔH by only half (Figure 2B). Clearly, 70b did not disrupt the DMPG vesicles to the same extent as 12. It would seem that relocating the azaspiroketal in 70b so that it is no longer part of a Mannich base had seriously hampered its interaction with the lipid bilayer.
The azaspiroketal Mannich bases 12 and 13 induced permeabilization of mycobacteria before onset of cell death
In view of the perturbative effects of 10-13 on DMPG vesicles, we posited that they should permeabilize mycobacterial cells. Hence, changes in the permeability of M. bovis BCG cultures treated with representative members 12 and 13 (4× MIC90) were monitored over time (up to 48h) using the nucleic acid dyes SYTO 9 and propidium iodide (PI). Similar experiments were carried out with the less potent analog 1 and isoniazid. Briefly, SYTO 9 is taken up by all cells and emits a green fluorescence whereas PI is selectively concentrated in cells with compromised membranes where it emits a red fluorescence. A decrease in the green/red ratio with time would signify progessive permeabilization of the mycobacterial cultures. The ratios were normalized against values obtained from untreated cultures (no permeabilization) and SDS-treated cultures (complete permeabilization) to give the % permeabilization. To confirm that increased permeability was not an epiphenomenon of cell death, we monitored the viability of the treated cultures by plating and colony counting over the same time period.
Figure 3A shows the time-kill profiles of 12, 13 and isoniazid at 6 h, 12 h, 24 h and 48 h. The onset of cidal activity was more rapid for isoniazid but by 48 h, all three compounds had killed the mycobacterial cultures by more than 100-fold. The time-kill profile of 1 was determined separately and here we found that cultures treated with 1 were still viable at 48h (Figure 3B). A comparison of the time kill profiles with the loss in membrane permeability at the various time points showed that 12 and 13 increased the permeability of bacteria before the onset of massive cell death. This was observed early for 13 (12 h) and later (24 h) for 12 (Figure 3C). 13 induced noticeably less permeabilization at 24 h than 12 but by 48 h, both compounds had permeabilized the cultures to the same extent. The slower acting 1 had a similar time-kill /permeability profile as 12, except that permeabilization did not increase at 48 h (Figure 3D). These differences notwithstanding, the overall profile was one in which increased permeability was incurred early and before the onset of significant cell death, suggesting that increased permeability may have caused cell death. In the case of isoniazid, whose mode of action does not directly involve membrane disruption, both events - cell death and permeabilization - were aligned as would be expected if cell death was causal to the loss of the permeability barrier.
Figure 3.
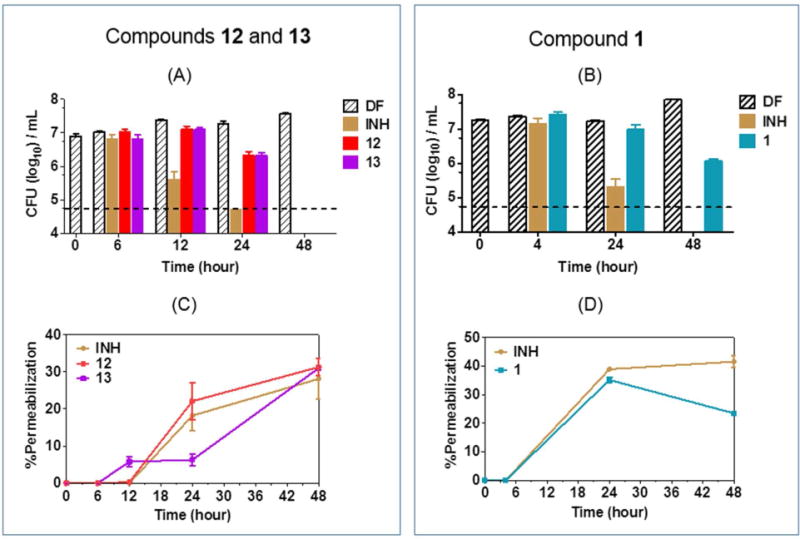
Time-kill kinetics (A, B) and membrane permeabilization (C, D) of M. bovis BCG cultures by test compounds (1,12,13) and isoniazid (INH). Mid log phase cultures were treated with (A) 12, 13 and (B) 1 over 48h at 4× MIC90 for the determination of time-kill profiles. Samples were concurrently withdrawn at the indicated time points for the determination of green/red fluorescence ratios, normalized to drug free (DF) inoculum at the same time point, to give % permeabilization induced by (C) 12, 13 and (D) 1. Dashed lines in (A, B) represent the limit of detection. Experiments were performed 3 times independently. Mean and SD from representative experiments are shown
Compounds 12 and 13 induce the mycobacterial promoter piniBAC
The promoter of the mycobacterial iniBAC operon is upregulated under conditions of cell envelope stress such as those encountered when cell wall biosynthesis is disrupted23.24 or when the mycobacterial membrane is permeabilized.10 To further verify the membrane disruptive effects of the azaspiro ketal Mannich bases, we monitored the fluorescent signal emitted by cultures of M. bovis BCG expressing Red Fluorescent Protein (RFP) under control of piniBAC promoter, that had been treated with various concentrations (0.4–100 μM) of 1, 12, 13 and the positive control isoniazid. Upregulation of transcriptional activity would increase the fluorescent signal of the RFP. Here we found that 12 and 13 increased the transcriptional activity of the piniBAC in a concentration dependent manner (Figure 4). The piniBAC activation profiles of 1, 12 and 13 were broadly similar to that of isoniazid. In all, the ability of 1, 12 and 13 to perturb phospholipid vesicles, permeabilize mycobacterial cultures and upregulate cell envelope stress reporter genes were in accord with their membrane disruptive properties.
Figure 4.
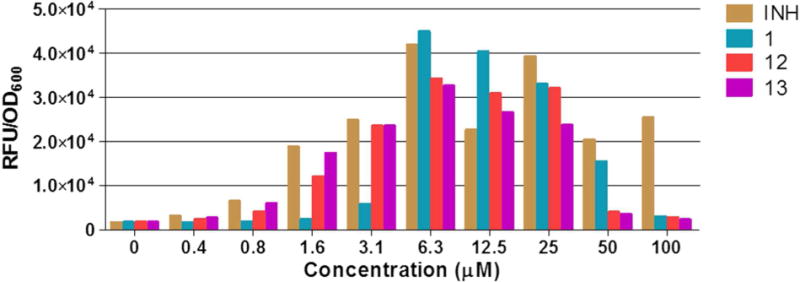
Concentration dependent induction of piniBAC promoter activity by 1, 12, 13 and isoniazid (INH) in a recombinant strain of M. bovis BCG-piniBAC-RFP after 24 h incubation. Experiments were performed in biological replicates, data shown here is from a representative experiment.
Compounds 12 and 13 did not depolarize mycobacterial membranes
We expected the permeabilizing effects of the azaspiroketals on mycobacterial cultures to be accompanied by a loss in membrane potential. Hence, we examined the ability of 12 and 13 to dissipate the membrane potential of M. bovis BCG cultures over time (minutes to days) using the fluorescent membrane permeable dye 3,3-diethyloxacarbocyanine iodide (DiOC2). Briefly, DiOC2 emits red fluorescence in cells with polarized membranes and green fluorescence in cells that are depolarized. A fall in the red/green fluorescence ratio of DiOC2 would indicate a loss in the membrane potential. Here, we monitored time dependent changes in the DiOC2 ratios of M. bovis BCG cultures treated with 1, 12, 13, the membrane depolarizer carbonyl cyanide m-chlorophenylhydrazine (CCCP) and rifampicin.
As shown in Figure 5, the changes in the DiOC2 ratios of CCCP and rifampicin-treated mycobacteria reflected their roles as positive and negative controls respectively. In cultures treated with 12 and 13, the DiOC2 ratios were comparable to drug-free controls over the short time intervals (Figure 5A,B). Only at Days 3-5 did the ratios declined, by which time the losses would be attributed to the dying cultures. A similar observation was made for 1 (Supporting Information, Figure S3). Thus the azaspiroketal Mannich bases had the surprising effect of not depolarizing mycobacterial membranes in spite of their ability to induce permeabilization. There was however precedent to the separation of these closely related membrane effects. First, several antimycobactericidal indolylpentyltriphenylphosphonium analogs were found to rapidly depolarize M. bovis BCG cultures without inducing membrane permeabilization.9 Second, Okano and coworkers reported that peripheral modifications of vancomycin resulted in a potent analog that increased permeability of Enterococcus faecium without inducing membrane depolarization or cell lysis.25 The authors posited that the compound in question had a “specific” mode of action that could have led to membrane perturbation.
Figure 5.
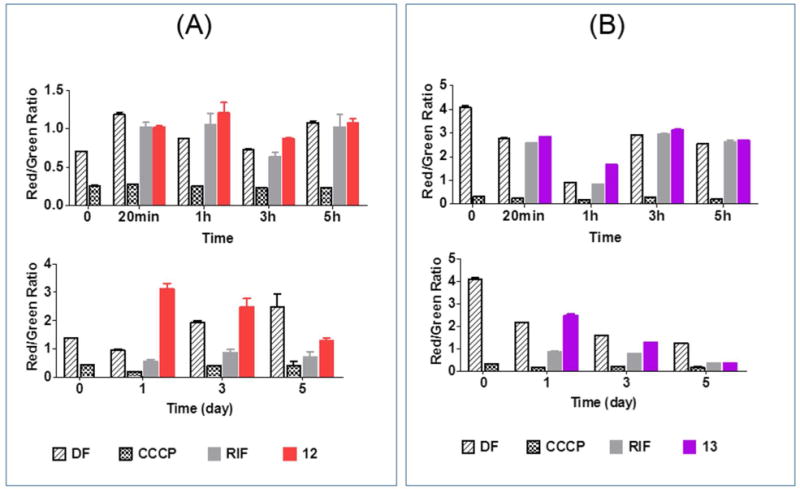
Changes in membrane potential (as reflected by Red/Green fluorescent ratio of DiOC2 of M. bovis BCG cultures treated with (A) 12 and (B) 13 at 4× MIC90 over short time intervals (min – h, upper panel) and 1-5 days (lower panel). Rifampicin (RIF, 0.08 μM) and CCCP (100 μM) were negative and positive controls respectively. DF = Drug Free cultures. Results from 2-3 independent biological replicates.
We had earlier reported that the indolyl Mannich base 71 (Supporting linformation, Figure S2) induced both permeabilization and depolarization of live M. bovis BCG cultures.10 In view of the present findings, we re-examined its effects on the membrane potential, specifically at shorter time intervals (minutes to hours) which were not investigated earlier. Here we found no depolarization at these early time points but were able to replicate the loss in membrane potential after 3 days (Supporting Information, Figure S4). Thus we concluded that the unusual uncoupling of permeabilization and depolarization of mycobacterial membranes was not unique to azaspiroketal Mannich bases (1,12,13) but a shared feature of non-azaspiroketal Mannich bases (as exemplified by 71) as well.
Compounds 12 and 13 have limited electrophilic reactivity to cysteamine
3-Alkylindoles have been flagged out for their ability to generate electrophilic species.26 Mechanistically, a similar reaction may occur with the indolyl Mannich bases. It would involve the displacement of the protonated basic nitrogen in the azaspirocyclic side chain of 12 or 13 to form a stabilized electrophilic carbocation that would react with soft nucleophiles (thiols, amines) in the biological milieu (Figure 6). This would implicate the scaffold as a pan-assay interference compound (PAIN) and hinder its progressability as an antimycobacterial pharmacophore.
Figure 6.
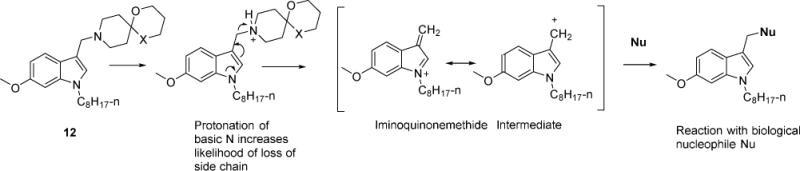
Proposed scheme by which 12 (X = O) or 13 (X = S) generates an electrophilic iminoquinonemethide intermediate. Nu = Nucleophile.
To address this possibility, the reactivity of 12 with the thiol reagent cysteamine was investigated by 1H NMR.27 Here we identified the signal assigned to the C-3 methylene protons of 12 and posited that a reaction with the thiol of the nucleophilic cysteamine would alter the chemical shift and appearance of this signal. The C-3 methylene protons of 12 were detected as a singlet at 3.67 ppm in deuterated DMSO- 1% acetic acid. Downfield from this signal was a multiplet (3.77 ppm) attributed to the 6-methoxy protons and the spirocyclic methylene protons adjacent to the ketal oxygens (Figure 7A). Upon addition of cysteamine to the sample, the C-3 singlet shifted to 3.57 ppm but with no change in its appearance or the chemical shift of the flanking multiplet (Figure 7B). When the spectrum of the same sample was recorded at longer time intervals (30 min, 24 h), the C-3 methylene signal remained unaltered, as were the signals assigned to adjacent multiplet (Figure 7C,D). The cysteamine methylene protons of cysteamine were detected at 2.6-2.8 ppm (multiplet) and remained unchanged over the monitoring period. Similar observations were made for 13 (Supporting Information, Figure S5). Thus, we surmised that the Mannich base side chains of 12 and 13 have limited electrophilic reactivity under the in vitro conditions of these experiments.
Figure 7.
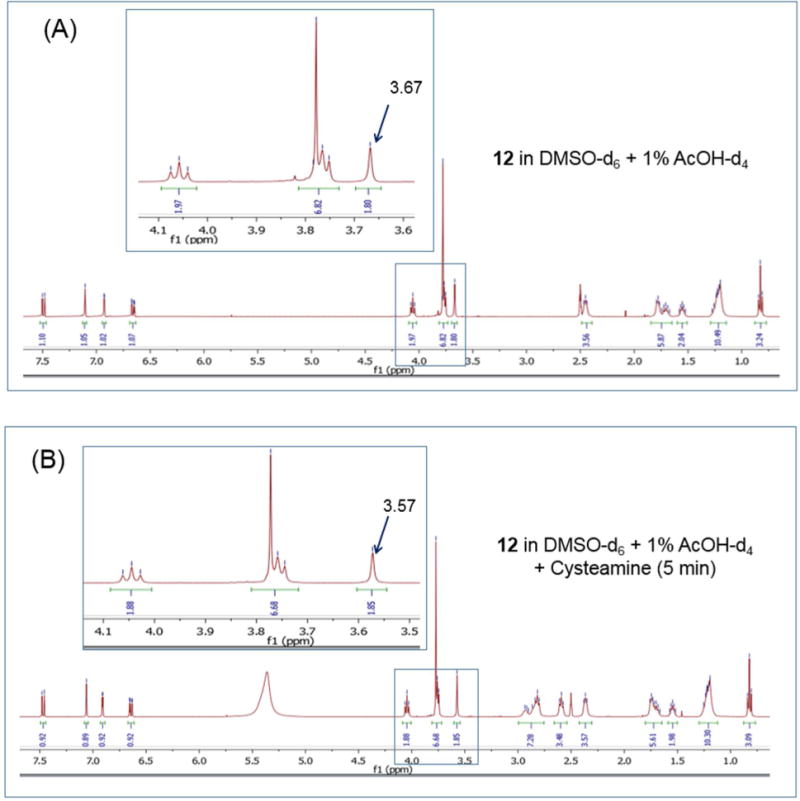
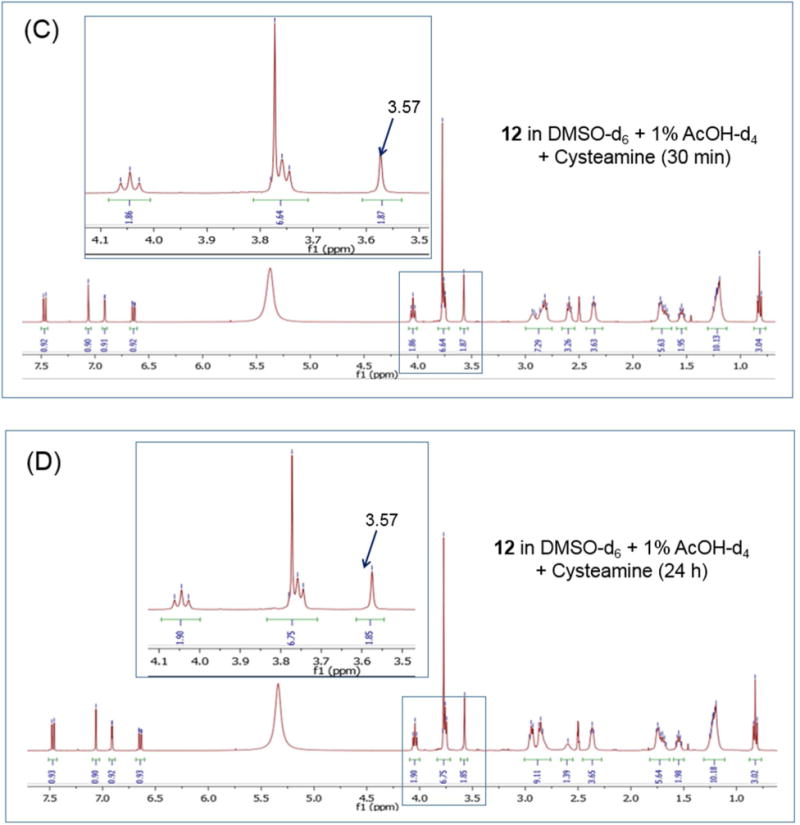
1H NMR spectra of 12 under different conditions: (A) In deuterated DMSO and 1% deuterated acetic acid with spectrum collected immediately (≈ 5 min); (B) As in (A) in the presence of cysteamine and spectrum recorded immediately (≈ 5 min); For (C) and (D), the spectra were recorded after 30 min and 24 h incubation with cysteamine respectively.
“Snapshot” PK of 12 and 13 in mice showed low plasma exposure but high lung partitioning
In view of their attractive mycobactericidal profiles, preliminary PK experiments were planned for 12 and 13. Prior to these experiments, we assessed their PAMPA permeabilities, kinetic solubilities, and in vitro microsomal stabilities (Table 6). Concurrent experiments were carried out with 1. No PAMPA Pe could be derived for 1, 12 and 13 due to their retention in the lecithindodecane lipid layer. In retrospect, this observation affirmed their affinities for lipid bilayers as reflected in the DSC experiments on model membranes. Their aqueous solubilities were largely similar (20-30 μM) but the longer half-lives of 12 and 13 suggested that they may be metabolically more stable to microsomal breakdown than 1.
Table 6.
Physicochemical profiles of 1, 12 and 13
| 1 | 12 | 13 | |
|---|---|---|---|
| Aqueous Solubility (μM) pH7.4a | 23.9 ± 0.8 | 27.5 ± 1.5 | 30.9 ± 1.3 |
| PAMPA Effective Permeability (Pe) cm/sb | Cannot be determined | Cannot be determined | Cannot be determined |
| Half-Life (min), Rat male microsomes | 17.4 ± 0.5 | 27.7 ± 0.6 | 32.9 ± 1.9 |
Multiscreen filter plates (Millipore), 24 h agitation, 25°C, mean ± SD, n=3 determinations
Multiscreen-IP PAMPA assay plates (Millipore), lecithin-dodecane as lipid layer, 24 h agitation, 25°C.
Next, we assessed the PK profiles of 1, 12 and 13 in preliminary “snapshot” experiments carried out in CD1 mice. Briefly, two mice were dosed with the test compound at 25 mg/kg, administered orally in an aqueous solution of 5% dimethylacetamide-60% PEG300. This dose was well tolerated in the mice. Tail bleeds were collected from the treated animals at various time points (30min, 1h, 3h, 5h) and analyzed for their concentrations in plasma. Lung tissue was collected and analyzed at the terminal time point (5h). The analyses revealed that the compounds had low plasma exposures (33 ng/mL - 58 ng/mL) and high levels in lung tissue (468 ng/g - 1596 ng/g) (Table 7). The preferential partitioning of these compounds in the lung may be due to their lipophilic character. However, additional time point determinations are required to confirm the selective lung distribution profiles of these compounds.
Table 7.
Snapshot PK profiles of 1, 12 and 13a
| 1 | 12 | 13 | |
|---|---|---|---|
| Peak plasma concentration (ng/mL) | 42.0 | 64.8 | 60.6 |
| Area under the concentration time curve (AUC) (ng.h/mL) | 173 | 282 | 224 |
| Plasma concentration at 5 h (ng/mL) | 33.0 | 58.5 | 55.2 |
| Lung concentration at 5 h (ng/g) | 467.5 | 1408 | 1596 |
| Ratio of lung concentration to plasma concentration (Lung\Plasma) at 5 h | 14.2 | 24.1 | 28.9 |
1, 12 and 13 were administered at 25 mg/kg (PO) to CD1 mice (n=2 for each compound).
Compound 12 demonstrated in vivo efficacy in an acute TB mouse model
Next, 1 and 12 were evaluated for in vivo efficacy in an acute TB mouse model. In this model, the compound was administered orally to mice infected with Mtb H37Rv at 100mg/kg daily over 4 weeks (6 days per week, total of 24 doses). Concurrent experiments were carried out with the positive control isoniazid at 25 mg/kg. Encouragingly, no adverse effects were detected in animals dosed with 1 or 12 throughout the treatment period, suggesting excellent in vivo tolerability of the compounds. At the end of this time, bacterial loads in the lungs and spleen were determined by plating organ homogenates on agar and subsequent colony counting (Figure 8). 12 significantly inhibited bacterial growth in both organs but 1 only reduced the splenic bacterial load. Although both 1 and 12 preferentially partitioned into lung tissue, we noted that the levels of 12 in lung (~3 μM) exceeded its in vitro MIC90 Mtb (0.8 μM). In the case of 1, lung levels (~1 μM) and MIC90 Mtb (2.3 μM) were nearly equivalent. Speculatively, this might have led to the different efficacy outcomes, although 12, unlike isoniazid, was only bacteriostatic in this infection model.
Figure 8.
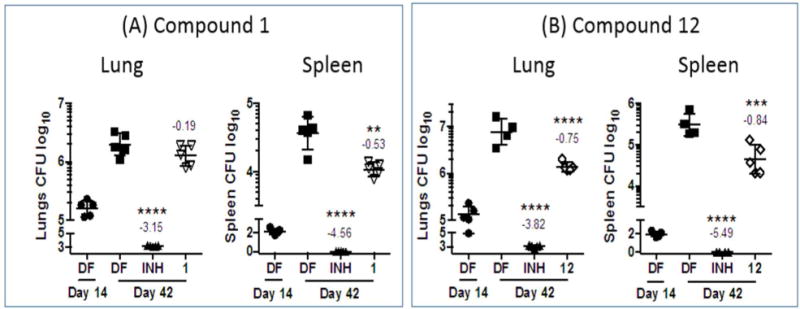
In vivo efficacy of (A) 1 and (B) 12 in a mouse model of acute TB. Mice were dosed with either drug at 100 mg/kg (po) for 6 days per week, × 4 weeks. A similar dosing schedule applied to isoniazid (INH) at 25 mg/kg. CFU analysis of lung and spleen tissues were carried out on treated mice and drug-free (DF) infected mice at Day 14 (start of treatment, Early Control) and Day 42 (end of treatment, Late Control). Statistical analysis was by One-way ANOVA, multi comparison, Bonferroni post test, n=4/5 (4/5 mice per group); **p<0.01; ****p<0.0001. Figure depicts individual data points, means and SD. Numbers above groups reflect difference as compared to the drug free (DF) control at day 42 (Late Control).
CONCLUSIONS
The azaspiroketal is a uniquely rigid and three-dimensional motif that is occasionally found in antimycobacterial agents but whose contribution to activity has not been systematically explored. In this report, we have provided clarity on the potential of this motif as a potency enhancing chemotype in a series of indolyl Mannich bases and affirmed the contribution of selective membrane perturbation to its mode of action.
The distinctive potency advantage of the azaspiroketal motif is predicated on the following evidence. First, it is supported by SAR garnered from Series 1-4. In Series 1, we showed that only specific changes to the 1,4-dioxa-8-azaspiro[4,5]decanyl side chain of the early hit 1 resulted in analogs with submicromolar MICs. These were methylation of the ketal ring, homologation of either ring in the spirocycle, and O to S isosteric replacement. The singularity of these requirements was emphasized by the failure of closely related modifications to achieve the same potency outcomes. Series 2 and 3 highlighted the azaspiroketal motif, notably 1,5- dioxa-9-azaspiro[5.5]undecanyl of 12, as an influential driver of activity, prevailing over other modifications on the indole ring. From Series 4, we noted the striking losses in activity that followed when the azaspiroketal was not part of a Mannich base on the indole scaffold. Clearly it was not the azaspiroketal per se, but its inclusion in a Mannich base, that was pivotal to potency enhancement. The mechanistic implication of the latter remains an intrigue. The likelihood of the Mannich base as a source of reactive electrophilic species was explored but not found to be tenable. Second, the in vivo activity of 12 in an acute mouse model of TB validates the azaspiro ketal Mannich base as a potency enhancing chemotype. The apparent accumulation of 12 in lung tissue, as revealed in preliminary PK studies, may be a point in its favor. The in vivo studies also confirmed the low toxicity potential of the scaffold, as seen from 1 and 12 which were both well tolerated in mice.
The membrane disrupting effects of the indolyl azaspiro ketal Mannich bases were corroborated by their perturbative effects on model membranes, permeabilization of mycobacterial cultures and induction of the mycobacterial cell envelope stress reporter promoter piniBAC. Our investigations on 12 and 13 provided further insight into this phenomenon. First, the membrane disruptive effects were specific to mycobacterial membranes. Both 12 and 13 induced minimal hemolysis of human red blood cells at their mycobactericidal concentrations. They were nearly 100-fold less cytotoxic against mammalian Vero cells and when administered in vivo to mice for PK studies, did not cause obvious adverse effects. Second, the cationic amphiphilicity of 12 and 13 is a necessary but not sufficient requirement for membrane disruption. This is convincingly demonstrated by the limited perturbation elicited by the Series 4 analog 70b on DMPG vesicles. 70b retains the cationic amphiphilic features of 12 but is not a Mannich base. The presence of the latter appears to be essential for membrane disruption. Third, 12 and 13 permeabilized mycobacterial membranes without dissipating the membrane potential. This profile is not uniquely associated with azaspiroketal Mannich bases but characteristic of indolyl Mannich bases, including those without the azaspiroketal side chain as seen with 71. Lastly, whereas resistant mycobacteria could not be isolated for 71, we were able to raise resistant mutants to 12 which was highly unusual as membrane disrupting agents typically have very low mutation resistance frequencies in view of their effects on multiple targets. Perhaps 12 intercepts a membrane target besides perturbing membranes and it is the interplay of these effects that have led to its exceptional mycobactericidal potency. Further investigations would be required to address this question.
EXPERIMENTAL
General chemistry
Reagents were of synthetic grade (or better) and were used without further purification. Reactions were monitored by TLC on aluminium-backed sheets coated with silica gel 60 (Merck) with visualization by UV. Compounds were purified by column chromatography on silica gel 60 (230-400 mesh, Merck). 1H and 13C NMR spectra were recorded at 400 MHz and 101 MHz respectively on a Bruker 400 Ultrashield Plus Instrument (Bruker, Billerica, MA, USA) in CDCl3, DMSO-d6, or MeOH-d4 at room temperature. Chemical shifts were reported in parts per million (ppm) on the δ scale using residual protio-solvent signals (1H NMR: CDCl3 δ7.26, DMSO- d6 δ2.50, MeOH-d4 δ4.78; 13C NMR: CDCl3 δ77.00) as internal references. Coupling constants (J) were reported in Hertz (Hz) and splitting patterns as singlet (s), broad singlet (s, br), doublet (d), triplet (t), quartet (q), quintet (quint), sextet (sext), septet (sept), doublet of doublets (dd), doublet of triplets (dt) or multiplet (m). Nominal mass spectra were captured on a LCMS 2020 LC/MS System (Shimadzu, Singapore) by ESI or dual ion source (ESI, APCI), run in either positive or negative ionization mode. High-resolution mass spectra were recorded on a Bruker micrOTOFQII mass spectrometer (Bruker, Billerica, MA, USA) by ESI (positive ionization mode). Purities of final compounds were determined by reverse-phase HPLC using Zorbax Eclipse XDB-C18 column (5 μM, 150 × 4.6 mm) and found to be > 95%. Spectral and other details of synthesized compounds are given in Supporting Information.
General procedure A for the synthesis of N-substituted indoles in Series 1(5), Series 2 (41-49, 51), Series 3 (56a-56h) and Series 4 (66a, b, 67a-c, 68, 69a-d, 70a-e)
A previously reported method was followed.10 Briefly, the substituted indole (1 eq) was reacted with sodium hydride (NaH, 60 % dispersion in mineral oil; 2 eq.) in anhydrous DMF (20 - 30 mL) in an ice bath for 20 min. 1-Bromooctane, phenylalkyl bromide or phenoxyalkylbromide (2 eq) in DMF (5 mL) was added dropwise to the stirred mixture, after which stirring was continued at room temperature (RT) for 2-8h. Thereafter, isopropanol was added to quench the reaction, followed by methanol (dropwise) and then water. The crude product was extracted with dichloromethane (DCM, 3 × 20 mL), washed with water (2x) and brine (1x). The organic layers were combined, dried (anhydrous Na2SO4) and removed under reduced pressure to give the crude product which was purified by column chromatography using hexane: ethyl acetate (30: 1). Yields for target compounds ranged from 39 - 99%.
1-Cinnamyl-6-methoxy-1H-mdole (50)
A reported method was employed with modifications.28 Briefly, ground potassium hydroxide (KOH; 336 mg, 6.0 mmol) was quickly weighed in a dry round-bottom flask and 10 mL acetonitrile (MeCN) was added with stirring at RT. Commercially available 6-methoxyindole (441 mg, 3.0 mmol) in 2 mL MeCN was added dropwise to the mixture, stirred for 15 min, followed by addition of (E)-cinnamyl bromide (887 mg, 4.5 mmol). Stirring was continued for another 3 h, after which water (20 mL) and 1 M HCl (15 mL) were added in succession and the mixture extracted with ethyl acetate (EA, 3 × 15 mL). The combined organic layer was washed sequentially with water and brine, dried over anhydrous Na2SO4, filtered, and the solvent removed under reduced pressure to give the crude product which was purified by column chromatography (hexane: EA; 20: 1). 50 was obtained as a yellow oil in 57% yield. 1H NMR (400 MHz, CDCl3) δ 7.52 (dd, J = 2.4, 8.5 Hz, 1H), 7.39 – 7.27 (m, 5H), 7.05 (d, J = 3.1 Hz, 1H), 6.90 - 6.76 (m, 2H), 6.58 - 6.43 (m, 2H), 6.40 - 6.29 (m, 1H), 4.82 (ddd, J = 1.5, 5.7, 19.6 Hz, 2H), 3.86 (s, 3H); MS (ESI): Calculated for C18H17NO 263.13, found 264.20 [M+H]+.
General procedures (B, C) for the synthesis of the indolyl Mannich bases in Series 1 (1, 7-20, 23-27, 33, 34 36-40), Series 2 (41a, b, c - 50a, b, c; 51a, b) and Series 3 (57a-h, 58a-e, 59a- c)
Procedure B: To a stirred solution of the appropriate amine (1.2 eq.) in acetic acid (5 mL) was added the N-substituted indole (1 eq.) and formaldehyde (CH2O; 36 % aqueous solution, 1.2 eq).29 The reaction mixture was stirred at RT and monitored periodically by TLC. Upon completion of the reaction (3-24h), the mixture was made alkaline (pH 8) with NaOH (1M, 5-10 mL) and extracted with DCM (3×15 mL). The organic layer was washed with brine, dried (anhydrous Na2SO4) and concentrated under vacuum to give the crude residue which was purified by column chromatography using DCM: MeOH (gradient elution). Target compounds were obtained in yields ranging from 10 – 57%.
Procedure C: The N-substituted indole (1 eq.) was added to a stirred solution or suspension of the amine, formaldehyde (36 % aqueous solution) and ZnCl2 (1.2 eq. of each) in ethanol (3 - 5 mL) at RT.30 The reaction was monitored by TLC and on completion (5-24h), the solvent was removed under reduced pressure and work up was carried out as described earlier. Target compounds were obtained in yields ranging from 10 – 75%.
6-Methoxy-1 -octyl-1H-indole-3-carbaldehyde (6)
A reported method was followed with modifications.31 Briefly, a 50-mL round bottom flask was charged with DMF (15 mL) under an inert atmosphere (N2) in an ice bath. Phosphorus oxychloride (6 mmol) was added dropwise over 30 min. Stirring was continued for 1.5h at 0°C during which time 6-methoxyindole (5, 4 mmol) in DMF (5 mL) was added dropwise. Stirring was continued at 40°C for 2 h, and then cooled to 0°C with crushed ice (50 g). The reaction mixture was adjusted to pH 8 with 1 M NaOH, extracted with DCM (3 × 20 mL) after which the organic layers were washed (brine), dried (anhydrous Na2SO4) and concentrated under reduced pressure to give the crude product which was purified by column chromatography (DCM, 100 %). 6 was obtained as a yellow solid (644 mg, 56% yield), mp 47 – 50°C. 1H NMR (400 MHz, CDCl3) δ 9.91 (s, 1H), 8.17 (d, J = 8.6 Hz, 1H), 7.59 (s, 1H), 6.95 (dd, J = 2.2, 8.6 Hz, 1H), 6.80 (d, J = 2.2 Hz, 1H), 4.08 (t, J = 7.1 Hz, 2H), 3.87 (s, 3H), 1.85 (quint, J = 7.1 Hz, 2H), 1.39 - 1.17 (m, 10H), 0.91 - 0.82 (m, 3H); MS (DUIS): Calculated for C18H25NO2 287.19, found 288.15 [M+H]+.
General Procedure D for the synthesis of Series 1 compounds 21, 22, 28 - 32 and 35
The method by Abdel-Magid et al. was employed.32 Briefly, to a solution of 6 (1 eq.) in tetrahydrofuran (THF, 5 mL) was added the amine (1.2 eq.) and acetic acid (2 eq.) dissolved in THF (2 mL). The reaction mixture was stirred for 30 min, sodium triacetoxyborohydride (3 eq.) was added, and stirring continued for 6 - 12 h or until complete as indicated by TLC (DCM: MeOH; 40: 1). A solution of 1 M NaOH (5 mL) was added to quench the reaction. After stirring for another 30 min, the reaction mixture was extracted with ethyl acetate (2 × 20 mL), washed with water (2 × 20 mL), brine, dried (anhydrous Na2SO4) and concentrated under reduced pressure to give the crude product which was purified by column chromatography (DCM: MeOH, gradient elution). The target compounds were obtained in yields ranging from 18 - 31%.
General procedure E for the synthesis of azaspiro-fused intermediates (1a, 7a - 16a)
The method of Tiwari et al was followed with modifications.33 Briefly, 2 eq of the diol (1,2- ethanediol, 1,3-propanediol, 2-mercaptoethanol, 3-mercapto-1-propanol, 1,2-ethanedithiol or 1,3-propanedithiol) and 4-piperidone monohydrate hydrochloride (1 eq) were dissolved in toluene (20 - 50 mL). p-Toluenesulfonic acid monohydrate (0.05-0.1 eq) was added and the reaction mixture heated to reflux. A Dean-Stark apparatus was attached to the reaction vessel to collect water released during the reaction. When no more water was collected, the mixture was cooled to RT, toluene was removed under reduced pressure and the crude product used as such for the subsequent Mannich reaction without further purification. 4-Azepanone HCl was reacted with 2-mercaptoethanol and 3-mercapto-1-propanol to give 15a and 16a respectively under similar conditions.
General procedures F for the synthesis of (3-chloropropoxy)indoles (60a-d) and 5-(2- chloroethoxy)indole (60e)
The method of Lepri et al. was followed with modifications.34 Briefly, 1-bromo-3-chloropropane (3 eq.) was dissolved in ethanol (20 - 30 mL). K2CO3 (6 eq.) was added with stirring at RT, followed by an ethanolic solution of the commercially available 4-, 5-, 6-, or 7-hydroxyindole (1 eq., 5 mL). The reaction mixture was then refluxed at 80°C for 3 - 5 h. The reaction was monitored by TLC (hexane: ethyl acetate, 2: 1) and cooled to RT on completion. Solvent was removed under reduced pressure and the crude residue extracted with DCM (3 × 20 mL). The organic layer was washed successively with water and brine, dried (anhydrous MgSO4), concentrated under reduced pressure to give the crude product which was purified by column chromatography (hexane: ethyl acetate, 30: 1, or DCM 100%) to give 60a-d in yields ranging from 42 % to 71 %. 1-Bromo-2-chloroethane was reacted with 5-hydroxyindole in the same way to give 60e in 20 % yield.
General procedure G for the synthesis of (3-amino)propoxyindoles (61a, b; 62a-c; 63; 64a-d; 65b-d) and 5-[2-(azepan-1-yl)ethoxy]-1H-indole (65e)
The amine (12a, piperidine, 7a, pyrrolidine or azepane, 3 eq), Cs2CO3 (4 eq) and the 3- chloropropoxyindole (60b-d)/5-(2-chloroethoxy) indole 60e (1 eq) in MeCN (5 - 10 mL) was refluxed with stirring for 24-48 h and cooled to RT on completion. The solvent was removed under vacuum, the residue extracted with DCM (3 × 10 mL) and worked up in the usual way. The crude product was purified by column chromatography (DCM: methanol, 30: 1) to give the desired products (61a, b; 62a - c; 63; 64 a - e; 65b - e) in yields ranging from 15 to 53 %.
4-[2-(Azepan-1-yl)propoxy]-1H-indole (65a)
Potassium iodide (KI, 236 mg, 1.42 mmol) was added to a solution of 4-(3-chloropropoxy)-1H- indole (60a) (150 mg, 0.71 mmol) in MeCN (10 mL). The reaction mixture was heated to reflux for 30-45 min, then cooled to RT, followed by addition of azepane (240 μL, 2.13 mmol) and K2CO3 (392 mg, 2.84 mmol). Heating was resumed for another 24 to 48 h. The reaction was monitored by TLC (DCM: MeOH, 5: 1), and on completion, cooled to RT, extracted with DCM (3 × 10 mL) and worked up in the usual way. The crude residue was purified by column chromatography (DCM: methanol, 30: 1) to give the desired product (65a) as a yellow semisolid in 34 % yield. 1H NMR (400 MHz, CDCl3) δ 8.69 (s, br, 1H), 7.15 - 7.05 (m, 2H), 7.00 (d, J = 8.2 Hz, 1H), 6.67 (ddd, J = 0.8, 2.1, 3.0 Hz, 1H), 6.53 (d, J = 7.6 Hz, 1H), 4.17 (t, J = 6.3 Hz, 2H), 2.83 - 2.68 (m, 6H), 2.15 - 2.03 (m, 2H), 1.77 - 1.57 (m, 8H); MS (ESI): Calculated for C17H24N2O 272.19, found 273.15 [M+H]+.
Bacterial strains and culture conditions
Mycobacterium bovis BCG (ATCC 35734) and Mycobacterium tuberculosis H37Rv (Mtb, ATCC 27294) were grown in complete Middlebrook 7H9 medium (BD Difco, Detroit, Michigan) supplemented with 0.05% (v/v) Tween-80 (Sigma- Aldrich), 0.5% (v/v) glycerol (Fisher Scientific), and 10% (v/v) Middlebrook albumin-dextrose-catalase (BD Difco). To determine if MIC values were subjected to a serum or glycerol shift, Middlebrook 7H9 broth was supplemented with 10% fetal bovine serum or prepared without glycerol.
Colony forming unit (CFU) enumeration
CFU enumeration of M. bovis BCG was performed by plating 10 μL of appropriate dilutions of cultures onto each well of a 12-well plate containing Middlebrook 7H10 agar supplemented with 0.5% (v/v) glycerol and 10% (v/v) oleic acid-albumin- dextrose-catalase (OADC, BD Difco). CFU counting of Mtb H37Rv was carried out by dividing each petri dish into two equal parts and then plating 50 μL of appropriate dilutions of cultures onto each part of the dish containing Middlebrook 7H10 agar.
Minimum inhibitory concentration (MIC) determination
MICs were determined on M. bovis BCG and Mtb H37Rv using the broth dilution method as described earlier.11
Minimum Bactericidal Concentration (MBC) determination
MBC in M. bovis BCG was determined by CFU enumeration on Middlebrook 7H10 agar plates using 12-well plates after exposure to a given concentration of test compound. Bacterial cultures were grown to mid-log phase, adjusted to a final OD600 = 0.05 (approximately 5 × 106 CFU/mL) and then treated with 0.5 x, 1x, 2x or 4x MIC90 of test compound for 5 days at 37°C, 110 rpm. Drug-free cultures were plated at the start of the experiment to determine the bacterial load of the inoculum. After incubation (5 days), the compound-treated cultures were plated to determine CFUs. The same procedure was followed for Mtb H37Rv except that cultures were exposed to test compound for 7days at 37°C, 80 rpm. MBC90, MBC99 and MBC99.9 are defined as the concentrations required to reduce CFU by 10, 100 and 1000-fold respectively as compared to the untreated inoculum at time point zero.
Loebicidal Concentration (LCC)
The method of Gengenbacher et al. was followed.35 Briefly, Mtb H37Rv was grown in 1 L roller bottle containing complete 7H9 broth at 37°C, 2 rpm. Midlog phase bacilli at OD600 = 0.4 - 0.6 were spun down and washed three times with 1 × PBS (Invitrogen, Life Technologies) supplemented with 0.025% Tween-80 and diluted to a final OD600 of 0.1. 50 mL of this suspension was transferred into a 1 L roller bottle and starved for 14 days at 37°C, 2 rpm. The bactericidal activity of 12 or 13 against nutrient-starved non-growing bacilli was determined by exposing 1 mL of OD600 = 0.05 culture to various concentrations of test compound in round-bottom 14 mL snap-cap tubes at 37°C, 80 rpm for 7 days. CFU was determined by plating appropriate dilutions of the cultures on Middlebrook 7H10 agar. LCC90, LCC99 and LCC99.9 of test compound are defined as the Loebicidal concentrations required to bring about 10, 100 and 1000-fold reduction in CFU respectively as compared to the drug free inoculum at time point zero.
Spontaneous resistant mutant selection
Mutant selection in M. bovis BCG was carried out by plating 106 - 109 CFU/mL of mid-log-phase pre-cultures on Middlebrook 7H10 agar plates containing 2 ×, 4 × and 8 × MIC90 of 12 or 71. Plates were incubated at 37°C for 8 weeks. Colonies were picked from selection plates and re-streaked on agar containing the same concentration of 12 or 71 for colony purification and to verify drug resistance.
Mtb H37Rv resistant mutants were isolated by plating CFU (107 - 109) from mid-log phase precultures on Middlebrook 7H10 agar plates containing 2 ×, 4 × and 8 × MIC90 of 12. To select 71 - resistant Mtb mutants, 108 and 109 CFU/mL of mid-log phase Mtb H37Rv pre-cultures were plated onto Middlebrook 7H10 agar containing 1x, 2 × and 4 × MIC90 of 71. Plates were incubated at 37°C for 8 weeks. Colonies were picked from selection plates and re-streaked on agar containing the same concentration of 12 or 71 for colony purification and to verify drug resistance.
Cytotoxicity determinations Vero E6 cells
Cytotoxicity determinations on the African green monkey kidney epithelial cells (48 h) were carried out as previously reported.11
Compound-induced hemolysis of Human Red Blood Cells
Test compounds were tested at various concentrations for hemolysis of human red blood cells (37°C, 1 h, phosphate buffered saline) as described previously.11 The procedure for collection of blood from donors was approved by the Institutional Review Board of the National University of Singapore.
Differential Scanning Calorimetry (DSC)
DSC was employed to determine the effects of selected compounds on DMPG multilamellar vesicles as previously described.10
Membrane Permeability Determination
Mid-log-phase M. bovis BCG cultures at OD600 = 0.1 in complete 7H9 broth were treated with 4-fold MIC90 concentrations of test compounds. Negative control was isoniazid tested at 3.2 μM. At each time point, sufficient volumes of cultures were washed twice and re-suspended into 500 μL of 0.9 % NaCl at OD600 = 0.5. Each 500 pL of sample was incubated with 0.75 μL of SYTO 9 (3.3 mM, Molecular Probes, Invitrogen) and 0.75μL of propidium iodide (PI, 20 mM, Molecular Probes, Invitrogen) in the dark at 37°C for 15 min. Dye-treated cultures were spun down, re-suspended in 500 μL of 0.9% NaCl and dispensed into flat-bottomed 96-well black plates (200 μL/well). Green and red fluorescence were recorded at λex = 488 nm/λem = 530 nm and λex = 488 nm/λem = 630 nm respectively, using Tecan Infinite M200 PRO plate reader. In this assay, the green/red (G/R) fluorescence ratio of drug-free (DF) cultures at each time point was assumed to represent 0% membrane permeabilization and the G/R ratio of 5% (v/v) SDS-treated culture was used to indicate 100% membrane permeabilization. % Membrane permeabilization of drug-treated culture at each time point was calculated as follows:
To correlate changes in membrane permeability to cell viability, the time-kill curves of drug-treated cultures were concurrently monitored by plating diluted cultures on Middlebrook 7H10 agar in 12-well plates for CFU enumeration. Experiments were performed in three independent biological replicates.
piniBAC Cell Envelope Stress Reporter System
Compounds 1,12,13 and isoniazid (0.4 - 100 μM) were incubated with recombinant M. bovis-piniBAC-RFP strain for 24 h to assess their effects on envelope-stress associated promoter activity following a previously described method.11
Determination of Membrane Potential
Membrane potential of treated M. bovis BCG cultures harvested at mid-log phase, and adjusted to OD600 = 0.1 in complete 7H9 medium with test compound (1, 12, 13, 71) at 4-fold MIC90, were measured using the Baclight™ Bacterial Membrane Potential Kit (Life Technologies, CA, USA) as described in an earlier report.11
Solubility Determinations
Solubility determinations were carried out on Multiscreen Solubility filter plates (Millipore- MSSLBPC10) from Millipore Corporation (MA, USA) following the protocol (PC2445EN00) from the manufacturer.
PAMPA Pe Determinations
Determinations were carried out on MultiScreen-IP PAMPA assay (donor) plates (MAIPNTR10) and MultiScreen Receiver Plates (MATRNPS50) from Millipore Corporation (USA) with 1% lecithin (Sigma Aldrich, USA) in dodecane (Reagent Plus®, Sigma Aldrich USA) as the lipid barrier. Pe was determined following the method described by Ramanujulu et al.36
Microsomal Stability
Compounds 1, 12 and 13 were incubated with male rat liver microsomes over 45 min at 37°C for the determination of microsomal stability. Details are given in SI.
Determination of Michael acceptor reactivity by NMR
The method described by Avonto et al was followed with some modifications.27 The 1H NMR spectrum of 12 (0.05 mmol) was recorded in DMSO-d6 (0.5 mL) and deuterated acetic acid (5 μL, 1% v/v). To the same sample in a tightly capped Eppendorf tube was added cysteamine (0.5 mmol, Sigma-Aldrich, USA), sonicated for 10 min, and quickly transferred to an NMR tube for the recording of the spectrum. The 1H NMR spectrum of the same sample was collected again after 30 min and 24 h of standing at RT. 1H NMR spectra were analyzed using Mnova 10.0.2 (Mestrelab Research, CA, USA). The same procedure was followed to determine the reactivity of 13 to cysteamine.
Animals and ethics assurance
Mouse studies were carried out in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health, with approval from the Institutional Animal Care and Use Committee of the New Jersey Medical School, Newark (CD-1 mice), National University of Singapore’s Institutional Animal Care and Use Committee (BALB/c mice). All animals were maintained under specific pathogen-free conditions and fed water and chow ad libitum, and all efforts were made to minimize suffering or discomfort. Studies in M. tuberculosis infected animals were performed in Biosafety Level 3 facilities approved for the containment of M. tuberculosis.
Pharmacokinetic (PK) analyses
Snapshot PK studies were performed in uninfected CD-1 mice after single dose of 25 mg/kg via the oral route (p.o.), as described previously.37 The p.o. formulation was dosed as a solution consisting of of 5%DMA/65% PEG300 (polyethylene glycol)/30% D5W (dextrose 5% in sterile water). Blood was collected at 30 min, 1, 3, and 5h post dose. Plasma was obtained by centrifugation for 10 min at 5,000 rpm and stored at −80°C until analyzed. Lungs were collected at the final time point (5h), weighed and homogenized in approximately 5 volumes of PBS, and stored at -80C until analyzed. Concentrations of 1, 12, and 13 were measured as described below. The PK parameters (area under the curve [AUC0-t and AUC0-24], peak plasma concentration [Cmax], half-life [t1/2], and elimination rate constant [kel]) were calculated from mean concentrations using Microsoft Excel (Office 2010; Microsoft Corp., Redmond, WA).
Analytical Methods
A neat 1mg/mL DMSO stock solution of test compound was first serially diluted in 50/50 acetonitrile water and subsequently serially diluted in drug-free CD1 mouse plasma (K2EDTA, Bioreclamation IVT, NY) to create standard curves and quality control (QC) spiking solutions. 20 μL of standards, QC samples, control plasma, and study samples were extracted by adding 200 μL of acetonitrile/methanol 50/50 protein precipitation solvent containing the internal standard (10 ng/mL verapamil). Extracts were vortexed for 5 minutes and centrifuged at 4000 RPM for 5 minutes. 100 μL of supernatant was transferred for HPLC coupled to tandem mass spectrometry (LC/MS-MS) analysis and diluted with 100 μL of Milli-Q deionized water.
LC/MS-MS quantitative analysis was performed on a AB Sciex Qtrap 6500+ triple-quadrupole mass spectrometer coupled to a Shimadzu 30ACMP HPLC system, and chromatography was performed on an Agilent Zorbax SB-C8 column (2.1×30 mm; particle size, 3.5 μm) using a reverse phase gradient elution. Milli-Q deionized water with 0.1% formic acid was used for the aqueous mobile phase and 0.1% formic acid in acetonitrile for the organic mobile phase. Multiple-reaction monitoring of parent/daughter transitions in electrospray positive-ionization mode was used to quantify all molecules. Data processing was performed using Analyst software (Version 1.6.2; Applied Biosystems Sciex).
Animal tolerability and efficacy experiments
Eight to ten week old female BALB/c mice were maintained in groups of 3 or 4 in individually ventilated cages under specific pathogen free conditions at the National University of Singapore Biosafety Level 3 Core Facility. Food and water were offered ad libitum. Test compounds (1, 12) were formulated in equal volumes of polyethylene glycol 400 and 5% glucose and administered at a dose of 100 mg/kg in a volume of 200 μL by oral gavage. Acute toxicity was assessed by dosing groups of 3 mice on three consecutive days followed by a monitoring period of 7 days. Animals were subsequently euthanized by CO2 to assess gross pathological changes. For in vivo efficacy determination, mice were infected with 100-200 CFU M. tuberculosis H37Rv using a full body inhalation exposure system (GlasCol). After 14 days, drug treatment was initiated for 6 days per week for 4 weeks. Isoniazid at a dose of 25 mg/kg formulated in 0.25% methyl cellulose served as control. Mice were euthanized at designated time points by CO2. Bacterial burden of organs was determined by plating serial dilutions of organ homogenates onto Middlebrook 7H11 agar supplemented with 20 μg/mL ampicillin and 10 μg/mL cycloheximide. Colonies were counted after 3 - 4 weeks of incubation at 37°C.
Supplementary Material
Scheme 4.
Reagents and conditions: (i) 1-Bromo-3-chloropropane or 1-bromo-2-chloroethane, K2CO3, ethanol, reflux, 3 h (ii) KI, K2CO3, appropriate amine, MeCN, reflux, 24 - 48 h; or Cs2CO3, appropriate amine, MeCN, reflux, 24 - 48 h (iii) NaH, CH3(CH2)6CH2Br, anhydrous DMF, RT, 12 - 24 h.
Acknowledgments
This research was supported by the Republic of Singapore Ministry of Education Academic Research Fund R148000234114 to GML and Ministry of Health National Medical Research Council under its TCR Flagship grant NMRC/TCR/011-NUHS/2014 and Centre Grant “MINE”, Research Core #4 NMRC/CG/013/2013 to TD. The TCR Flagship grant and Centre Grant are part of the Singapore Programme of Research Investigating New Approaches to Treatment of Tuberculosis (SPRINT-TB; www.sprinttb.org) managed by Pauline Yoong and led by Nick Paton. TD holds a Toh Chin Chye Visiting Professorship at the Department of Microbiology and Immunology, Yong Loo Lin School of Medicine, National University of Singapore. Research reported in this publication is also supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Number R01AI132374 and by the Cystic Fibrosis Foundation under Award Number DICK17XX00 to TD. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the Cystic Fibrosis Foundation. LM and SAN gratefully acknowledge graduate scholarships from Ministry of Education, Singapore. We thank Jian Liang Low for laboratory management. Our appreciation goes to Dr David Alland, Rutgers New Jersey Medical School for providing the piniBAC reporter plasmid, Dr Yoshiyuki Yamada, National University of Singapore for providing a modified version of the reporter piniBAC-RFP and Dr R. Manjunatha Kini, National University of Singapore for providing human blood for the hemolysis assay.
Abbreviations
- BCG
Bacillus Calmette-Guerin
- CFU
colony forming unit
- DiOC2
3,3-diethyloxacarbocyanine iodide
- DMPG
2-dimyristolyl-sn-glycero-3-phosphatidylglycerol
- DprE1
decaprenylphosphoryl-beta-D-ribose oxidase
- DSC
differential scanning calorimetry
- log P
logarithm to base 10 of partition coefficient P
- MIC
Minimum inhibitory concentration
- MBC
Minimum bactericidal concentration
- MtB
Mycobacterium tuberculosis
- PI
propidium iodide
- RFP
red fluorescent protein
- SI
Selective Index
- TB
tuberculosis
Footnotes
Supporting Information Available
Spectral data of synthesized intermediates and final compounds; Purity data of final compounds; 1H NMR spectra of 6 and 65a; 1H NMR, 13C NMR, HRMS and HPLC spectra of 12, 13; Scheme for synthesis of 52 and 53; Loebicidal activities of 12 and 13 against Mtb H37Rv; Structure and antimycobacterial profile of 71; Membrane depolarization of M. bovis BCG cultures by 1 and 71; Electrophilic reactivity of 13 to cysteamine as evaluated by 1H NMR; Protocol for microsomal stability determinations; Molecular Formula Strings of final compounds. This material is available via the Internet at http://pubs.acs.org.
Conflict of Interest Declaration
The authors declare no competing financial interest.
References
- 1.Daniel TM. The History of Tuberculosis. Resp Med. 2006;100:1862–1870. doi: 10.1016/j.rmed.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization. Global Tuberculosis Report 2017. WHO Press; France: 2017. [Google Scholar]
- 3.Dheda K, Limberis JD, Pietersen E, Phelan J, Esmail A, Lesosky M, Fennelly KP, Te Riele J, Mastrapa B, Streicher EM, Dolby T, Abdallah AM, Ben-Rached F, Simpson J, Smith L, Gumbo T, van Helden P, Sirgel FA, McNerney R, Theron G, Pain A, Clark TG, Warren RM. Outcomes, Infectiousness, and Transmission Dynamics of Patients with Extensively Drug-Resistant Tuberculosis and Home-Discharged Patients with Programmatically Incurable Tuberculosis: A Prospective Cohort Study. Lancet Respiratory Med. 2017;5:269–281. doi: 10.1016/S2213-2600(16)30433-7. [DOI] [PubMed] [Google Scholar]
- 4.Working Group on New TB drugs. https://www.newtbdrugs.org/pipeline/clinical.
- 5.Hurdle JG, O’Neill AJ, Chopra I, Lee RE. Targeting Bacterial Membrane Function: An Underexploited Mechanism for Treating Persistent Infections. Nat Rev Microbiol. 2011;9:62–75. doi: 10.1038/nrmicro2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen C, Gardete S, Jansen RS, Shetty A, Dick T, Dartois V. Verapamil Targets Membrane Energetics in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2018;62:e02107–02117. doi: 10.1128/AAC.02107-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moreira W, Aziz D, Dick T. Boromycin Kills Mycobacterial Persisters without Detectable Resistance. Front Microbiol. 2016;7 doi: 10.3389/fmicb.2016.00199. Article 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koh JJ, Zou H, Mukherjee D, Lin S, Lim F, Tan JK, Tan DZ, Stocker BL, Timmer MSM, Corkran HM, Lakshminarayanan R, Tan DTH, Cao D, Beuerman RW, Dick T, Liu S. Amphiphilic Xanthones as a Potent Chemical Entity of Anti-microbial Agents with Membrane-Targeting Properties. Eur J Med Chem. 2016;123:684–703. doi: 10.1016/j.ejmech.2016.07.068. [DOI] [PubMed] [Google Scholar]
- 9.Dunn EA, Roxburgh M, Larsen L, Smith RAJ, McLellan AD, Heikal A, Murphy MP, Cook GM. Incorporation of Triphenylphosphonium Functionality Improves the Inhibitory Properties of Phenothiazine Derivatives in Mycobacterium tuberculosis. Bioorg Med Chem. 2014;22:5320–5328. doi: 10.1016/j.bmc.2014.07.050. [DOI] [PubMed] [Google Scholar]
- 10.Yang TM, Moreira W, Nyantakyi SA, Chen H, Aziz D, Go ML, Dick T. Amphiphilic Indole Derivatives as Antimycobacterial Agents: Structure-Activity Relationships and Membrane Targeting Properties. J Med Chem. 2017;60:2745–2763. doi: 10.1021/acs.jmedchem.6b01530. [DOI] [PubMed] [Google Scholar]
- 11.Li M, Nyantakyi SA, Gopal P, Aziz DB, Dick T, Go ML. Indolylalkyltriphenyl-phosphonium Analogues are Membrane-Depolarizing Mycobacterial Agents. ACS Med Chem Lett. 2017;8:1165–1170. doi: 10.1021/acsmedchemlett.7b00287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Makarov V, Manina G, Mikusova K, Mollmann U, Ryabova O, Saint-Joanis B, Dhar N, Pasca MR, Buroni S, Lucarelli AP, Milano A, De Rossi E, Belanova M, Bobovska A, Dianiskova P, Kordulakova J, Sala C, Fullam E, Schneider P, McKinney JD, Brodin P, Christophe T, Waddell S, Butcher P, Albrethsen J, Rosenkrands I, Brosch R, Nandi V, Bharath S, Gaonkar S, Shandil RK, Balasubramanian V, Balganesh T, Tyagi S, Grosset J, Riccardi G, Cole ST. Benzathiazinones kill Mycobacterium tuberculosis by Blocking Arabinan Synthesis. Science. 2009;324:801–804. doi: 10.1126/science.1171583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trefzer C, Rengifo-Gonzalez M, Hinner MJ, Schneider P, Makarov V, Cole ST, Johnsson K. Benzothiazinones: Prodrugs that Covalently Modify the Decaprenylphosphoyrl-beta-D-ribofuranose 2′-epimerase DprEI of Mycobacterium tuberculosis. J Am Chem Soc. 2010;132:13663–13665. doi: 10.1021/ja106357w. [DOI] [PubMed] [Google Scholar]
- 14.Trefzer C, Skovierova H, Buroni S, Bobovska A, Nenci S, Molteni E, Pojer F, Pasca MR, Makarov V, Cole ST, Riccardi G, Mikusova K, Johnsson K. Benzothiazinones are Suicide Inhibitors of Mycobacterial Decaprenylphosphoyrl-beta-D- ribofuranose 2′-oxidase DprE1. J Am Chem Soc. 2010;134:912–915. doi: 10.1021/ja211042r. [DOI] [PubMed] [Google Scholar]
- 15.Makarov V, Lechartier B, Zhang M, Neres J, van der Sar AM, Raadsen SA, Hartkoorn RC, Ryabova OB, Vocat A, Decosterd LA, Widmer N, Buclin T, Bitter W, Andries K, Pojer F, Dyson PJ, Cole ST. Towards a New Combination Therapy for Tuberculosis with Next Generation Benzothiazinones. EMBO Mol Med. 2014;6:372–383. doi: 10.1002/emmm.201303575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao C, Ye T, Wang N, Zeng X, Zhang L, Xiong Y, You X, Xia Y, Xu Y, Peng C, Zuo W, Wei Y, Yu L. Synthesis and Structure-Activity Relationships Evaluation of Benzothiazinone Derivatives as Potential Anti-tubercular Agents. Bioorg Med Chem Lett. 2013;23:4919–4922. doi: 10.1016/j.bmcl.2013.06.069. [DOI] [PubMed] [Google Scholar]
- 17.Zheng Y, Tice CM, Singh SB. The Use of Spirocyclic Scaffolds in Drug Discovery. Bioorg Med Chem Lett. 2014;24:3673–3682. doi: 10.1016/j.bmcl.2014.06.081. [DOI] [PubMed] [Google Scholar]
- 18.Tai VWF, Garrido D, Price DJ, Maynard A, Pouliot JJ, Xiong Z, Seal JW, III, Creech KL, Kryn LH, Baughman TM, Peat AJ. Design and Synthesis of Spirocyclic Sompounds as HCV Replication Inhibitors by Targeting Viral NS4B Protein. Bioorg Med Chem Lett. 2014;24:2288–2294. doi: 10.1016/j.bmcl.2014.03.080. [DOI] [PubMed] [Google Scholar]
- 19.Whitney L, Petrilli SB, Hoyt CL, McMasters D, Verras A, Struthers M, Cully D, Wisniewski T, Ren N, Bopp C, Sok A, Chen Q, Li Y, Tung E, Tang W, Salituro G, Knemeyer I, Karanam B, Clemas J, Zhou G, Gibson J, Shipley CA, MacNeil DJ, Duffy R, Tata JR, Ujjainwalla F, Ali A, Xiong Y. Discovery of Spirocyclic Aldosterone Synthase Inhibitors as Potential Treatments for Resistant Hypertension. ACS Med Chem Lett. 2017;8:128–132. doi: 10.1021/acsmedchemlett.6b00455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yeaman MR, Yount NY. Mechanisms of Antimicrobial Peptide Action and Resistance. Pharmacol Rev. 2003;55:27–55. doi: 10.1124/pr.55.1.2. [DOI] [PubMed] [Google Scholar]
- 21.Fischer W. Lipoteichoic Acid and Lipids in the Membrane of Staphylococcus aureus. Med Microbiol Immunol. 1994;183:61–76. doi: 10.1007/BF00277157. [DOI] [PubMed] [Google Scholar]
- 22.Lamy-Freund MT, Riske KA. The Perculiar Thermo-structural Behavior of the Anionic Lipid DMPG. Chem Phys Lipids. 2003;122:19–32. doi: 10.1016/s0009-3084(02)00175-5. [DOI] [PubMed] [Google Scholar]
- 23.Alland D, Steyn AJ, Weisbrod AJ, Aldrich K, Jacobs WR., Jr Characterization of the Mycobacterium tuberculosis iniBAC Promoter, a Promoter that Responds to Cell Wall Biosynthesis Inhibition. J Bacteriol. 2000;182:1802–1811. doi: 10.1128/jb.182.7.1802-1811.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boot M, van Winden VJC, Sparrius M, van de Weerd R, Speer A, Ummels R, Rustad T, Sherman DR, Bitter W. Cell Envelope Stress in Mycobacteria is Regulated by the Novel Signal Transduction ATPase IniR in Response to Trehalose. PLoS Genet. 2017;13:e1007131. doi: 10.1371/journal.pgen.1007131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okano A, Isley NA, Boger DL. Peripheral Modifications of [Ψ[CH2NH]Tpg4] Vancomycin with Added Synergistic Mechanisms of Action Provide Durable and Potent Antibiotics. Proc Natl Acad Sci U S A. 2017;114:E5052–E5061. doi: 10.1073/pnas.1704125114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baell JB, Holloway GA. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for their Exclusion in Bioassays. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 27.Avonto C, Taglialatela-Scafati O, Pollastro F, Minassi A, Di Marzo V, De Petrocellis L, Appendino G. An NMR Spectroscopic Method to Identify and Classify Thiol-Trapping Agents: Revival of Michael Acceptors for Drug Discovery? Angew Chem Int Ed. 2011;50:467–471. doi: 10.1002/anie.201005959. [DOI] [PubMed] [Google Scholar]
- 28.Itahara T, Ikeda M, Sakakibara T. Alkenylation of 1-Acylindoles with Olefins Bearing Electron-withdrawing Substituents and Palladium Acetate. J Chem Soc, Perkin Trans. 1983;1:1361–1363. [Google Scholar]
- 29.Dai H-G, Li J-T, Li T-S. Efficient and Practical Synthesis of Mannich Bases Related to Gramine Mediated by Zinc Chloride. Synth Commun. 2006;36:1829–1835. [Google Scholar]
- 30.Miranda S, López-Alvarado P, Avendaño C, Menéndez JC. Convenient Synthesis of Highly Functionalized, 3,4-Disubstituted Indole Building Blocks. Open Org Chem J. 2007;1:1–12. [Google Scholar]
- 31.Wang Z. Comprehensive Organic Name Reactions and Reagents. John Wiley & Sons, Inc; Hoboken, NJ, USA: 2010. Vilsmeier Formylation; pp. 2872–2879. [Google Scholar]
- 32.Abdel-Magid AF, Carson KG, Harris BD, Maryanoff CA, Shah RD. Reductive Amination of Aldehydes and Ketones with Sodium Triacetoxyborohydride. Studies on Direct and Indirect Reductive Amination Procedures. J Org Chem. 1996;61:3849–3862. doi: 10.1021/jo960057x. [DOI] [PubMed] [Google Scholar]
- 33.Tiwari R, Mollmann U, Cho S, Franzblau SG, Miller PA, Miller MJ. Design and Syntheses of Anti-Tuberculosis Agents Inspired by BTZ043 Using a Scaffold Simplification Strategy. ACS Med Chem Lett. 2014;5:587–591. doi: 10.1021/ml500039g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lepri S, Buonerba F, Goracci L, Velilla I, Ruzziconi R, Schindler BD, Seo SM, Kaatz GW, Cruciani G. Indole Based Weapons to Fight Antibiotic Resistance: A Structure- Activity Relationship Study. J Med Chem. 2016;59:867–891. doi: 10.1021/acs.jmedchem.5b01219. [DOI] [PubMed] [Google Scholar]
- 35.Gengenbacher M, Rao SP, Pethe K, Dick T. Nutrient-starved, Non-replicating Mycobacterium tuberculosis Requires Respiration, ATP Synthase and Isocitrate Lyase for Maintenance of ATP Homeostasis and Viability. Microbiology. 2010;156:81–87. doi: 10.1099/mic.0.033084-0. [DOI] [PubMed] [Google Scholar]
- 36.Ramanujulu PM, Yang TM, Yap SQ, Wong FC, Casey PJ, Wang M, Go ML. Functionalized Indoleamines as Potent, Drug-like Inhibitors of Isoprenylcysteine Carboxyl Methyltransferase (Icmt) Eur J Med Chem. 2013;63:378–386. doi: 10.1016/j.ejmech.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 37.Zimmerman M, Lestner J, Prideaux B, O’Brien P, Dias-Freedman I, Chen C, Dietzold J, Daudelin I, Kaya F, Blanc L, Chen PY, Park S, Salgame P, Sarathy J, Dartois V. Ethambutol Partitioning in Tuberculous Pulmonary Lesions Explains Its Clinical Efficacy. Antimicrob Agents Chemother. 2017;61:e00924–17. doi: 10.1128/AAC.00924-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.