

Abstract
Clinical interpretation of germline missense variants represents a major challenge, including those in the TP53 Li-Fraumeni syndrome gene. Bioinformatic prediction is a key part of variant classification strategies. We aimed to optimize the performance of the Align-GVGD tool used for p53 missense variant prediction, and compare its performance to other bioinformatic tools (SIFT, PolyPhen-2) and ensemble methods (REVEL, BayesDel). Reference sets of assumed pathogenic and assumed benign variants were defined using functional and/or clinical data. Area under the curve and Matthews correlation coefficient (MCC) values were used as objective functions to select an optimized protein multi-sequence alignment with best performance for Align-GVGD. MCC comparison of tools using binary categories showed optimized Align-GVGD (C15 cut-off) combined with BayesDel (0.16 cut-off), or with REVEL (0.5 cut-off), to have the best overall performance. Further, a semi-quantitative approach using multiple tiers of bioinformatic prediction, validated using an independent set of non-functional and functional variants, supported use of Align-GVGD and BayesDel prediction for different strength of evidence levels in ACMG/AMP rules. We provide rationale for bioinformatic tool selection for TP53 variant classification, and have also computed relevant bioinformatic predictions for every possible p53 missense variant to facilitate their use by the scientific and medical community.
Keywords: Bioinformatics, variant, classification, TP53, ACMG
INTRODUCTION
Germline pathogenic variants in the TP53 gene (MIM# 191170) predispose to a wide range of cancers, collectively known as Li-Fraumeni syndrome (LFS) (MIM# 151623). With the advent of multi-gene panel testing, new TP53 variants are being identified in cancer patients with no or few LFS features. Most of these are rare (<1% minor allele frequency) predicted missense substitution variants that have not been previously reported in LFS families, and for which clinical interpretation is complicated. Given the medical implications of a TP53 pathogenic variant, it is essential to correctly classify these variants as pathogenic or benign. The challenge of interpreting the clinical significance of rare missense variants is not unique to TP53, and many bioinformatic tools have been designed to predict the effect of missense variants on gene/protein function. An advantage of TP53 among other genes is that yeast-based assays have been used to measure the transactivation activity of every possible missense variant (Kato et al., 2003); these data are available in the IARC TP53 Database (Bouaoun et al., 2016).
Bioinformatic predictions can be used to prioritize missense variants for laboratory functional assays, and also directly as a component of variant classification algorithms. Specifically, bioinformatic predictions can be considered as “Supporting” level of evidence for or against pathogenicity according to the ACMG/AMP guidelines for variant interpretation (Richards et al., 2015). There are many bioinformatic tools available for the prediction of pathogenicity of missense variants. Commonly used pathogenicity predictors for generic “exome-wide” analyses include SIFT (http://sift.jcvi.org/) and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/). SIFT uses evolutionary conservation to construct position-specific scoring matrices (PSSMs), and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), combines evolutionary conservation with some protein structure and functional information to do the same. Ensemble methods which combine multiple tools with different algorithms, such as REVEL (Ioannidis et al., 2016) and BayesDel (Feng, 2017), are gaining popularity and are proving to be more accurate.
The Align-GVGD tool (http://agvgd.hci.utah.edu) is aimed at gene-specific analysis and considers physicochemical properties of amino acids in the context of hand-curated protein multiple sequence alignments (pMSAs). This tool is available for a limited number of genes, and is thus not included in ensemble methods. Align-GVGD is available for TP53 variant analysis, for which it has been shown to predict well the transactivation activity of most missense variants (Mathe et al., 2006). There are two existing pMSAs for TP53 available from the Align-GVGD website: Human to Frog (comprising 8 species) and Human to Zebrafish (9 species), which have an average number of substitutions per position of 2.15 and 2.95, respectively. It has been previously suggested that specificity of alignment-based bioinformatic prediction is optimal when using a pMSA with an evolutionary history containing at least three times the substitutions as the number of amino acids in the protein, in order to decrease the likelihood that absolute conservation of an amino acid occurs by chance alone (Greenblatt et al., 2003; Tavtigian, Greenblatt, Lesueur, Byrnes, & Group, 2008).
In this work, we aimed to optimize the performance of the Align-GVGD tool for p53 missense variant prediction of pathogenicity, by improving the quality of the curated p53 pMSA. After comparing the performance of the optimized Align-GVGD with other bioinformatic prediction tools, alone and in combinations, we propose a calibrated ACMG-compliant (Richards et al., 2015; Tavtigian et al., 2018) prediction tool for missense variants in the TP53 gene.
METHODS
This research has been approved by the Human Research Ethics Committee of the QIMR Berghofer Medical Research Institute.
Reference sets of assumed pathogenic and assumed benign p53 missense variants
Reference sets of assumed pathogenic and assumed benign missense variants were selected for subsequent analyses, using strict functional and clinical evidence as detailed in Table 1. For assumed pathogenic variants, we selected variants that were non-functional in each of the eight transactivation assays (Kato et al., 2003) or variants found in tumors with reported dominant-negative effect, none of them present in population databases. For assumed benign variants, we selected variants that were functional in each assay or with an allele frequency of at least 0.03% in population databases, none of them present in the IARC TP53 Germline database (Bouaoun et al., 2016). Since the purpose of the reference sets was to compare different bioinformatic tools, bioinformatic information relating to effect of missense variants was not included as evidence for or against pathogenicity. Notably, 240 variants out of the total 247 assumed pathogenic variants had been seen at least once in the germline or tumor of a proband in the IARC TP53 Database (Bouaoun et al., 2016). For assumed benign variants from population databases, MaxEntScan was used to exclude variants likely to impact mRNA splicing (Vallee et al., 2016). Reference set variants, evidence for assignment to reference groups, and individual bioinformatic predictions, are detailed in Supp. Table S1.
Table 1.
Definition of reference sets of assumed pathogenic and assumed benign p53 missense variants
| Reference sets | |
|---|---|
|
| |
| Assumed pathogenic (n=247) Variants with ≤20% activity in each transactivation assay (Kato et al., 2003) OR Variants found in tumors from the cBioPortal database (Cerami et al., 2012) with reported dominant-negative effect according to the IARC TP53 Database (Bouaoun et al., 2016) AND absent in ExAC-nonTCGA/gnomAD population databases (Lek et al., 2016) |
Assumed benign (n=71) Variants with ≥75% - ≤140% activity in each transactivation assay (Kato et al., 2003) OR Variants with ≥0.03% allele frequency in at least one population in ExAC-nonTCGA/gnomAD population databases (excluding Ashkenazi Jewish) (Lek et al., 2016) AND absent in IARC TP53 Germline database (Bouaoun et al., 2016) |
Validation sets selected on the basis of functional data only
We used data from the systematic transactivation assays (Kato et al., 2003) to define validation datasets enriched for pathogenic or benign variants based on extremes of functional activity. Non-functional variants (enriched pathogenic) were selected based on average activity ≤20%, and functional variants (enriched benign) had average activity between 75% and 140%. After excluding any variant present in our reference sets (Supp. Table S1), the validation sets included 194 non-functional and 1133 functional missense variants (Supp. Table S2).
pMSA construction to optimize the Align-GVGD tool
Protein sequences were obtained from UniProt and aligned using T-Coffee (Wallace, O’Sullivan, Higgins, & Notredame, 2006). Resulting pMSAs were visualized and hand-modified for minor corrections using the Unipro UGENE software (Okonechnikov, Golosova, Fursov, & team, 2012) (Supp. Figure S1). The number of substitutions per position in the pMSAs was calculated using the PHYLIP program protpars (Felsenstein, 1989). Following an iterative process, different species were dropped or added into the existing pMSAs (Human to Frog and Human to Zebrafish) so that new pMSAs were created with an increasing number of substitutions per position. A total of 20 species were considered for inclusion in new pMSAs, because of their use in previous pMSAs for p53 and in pMSAs for other genes, or their similarity with the human p53 sequence. Ordered by phylogenetic distance from human, these species were: Homo sapiens (human), Pan troglodytes (chimpanzee), Pongo abelii (orangutan), [Rhinopithecus bieti (monkey), Macaca mulatta (monkey)], Callithrix jacchus (marmoset), [Rattus norvegicus (rat), Mus musculus (mouse)], [[Orcinus orca (orca), Camelus ferus (camel), Bos taurus (cow)], [Felis catus (cat), Canis lupus familiaris (dog)]], Dasypus novemcinctus (armadillo), Sarcophilus harrisii (Tasmanian devil), [Anolis carolinensis (lizard), Gallus gallus (chicken)], Xenopus laevis (frog), Latimeria chalumnae (coelacanth), and Danio rerio (zebrafish). We specifically excluded from our pMSAs: Loxodonta africana (elephant), as this species has multiple copies of TP53 (Abegglen et al., 2015); Sus scrofa (pig), as the pig genome is known to have undergone changes during domestication affecting DNA damage repair genes (Chen, Baxter, Muir, Groenen, & Schook, 2007); and Ornithorhynchus anatinus (platypus), as the sequence did not have high enough quality. Ideally, we attempted to include one representative species from each phylogenetic node and avoid including two or more similar species in the same pMSA, unless the accuracy of the predictions was shown to be improved. Following rationale previously detailed (Tavtigian et al., 2008), we also attempted to include enough variation to identify pathogenic variants, but without over-predicting i.e. as close as possible to an average of three substitutions/position. A phylogenetic tree with all these species considered based on the NCBI taxonomy, generated with phyloT (Letunic & Bork, 2016), is shown in Supp. Figure S2. Species included in selected examples of pMSAs are shown in Supp. Table S3.
Bioinformatic tools selected for comparison
Bioinformatic tools considered for p53 missense variant prediction of pathogenicity were Align-GVGD (http://agvgd.hci.utah.edu) paired with an optimized pMSA, SIFT (http://sift.jcvi.org/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), and the ensemble methods REVEL (Ioannidis et al., 2016) and BayesDel used without allele frequency (Feng, 2017) (which both include SIFT and Polyphen-2). The bioinformatic predictors were each considered alone, or in independent combinations.
Statistical analyses
In order to compare the performance between existing and new pMSAs for Align-GVGD, two independent statistical analyses were used: (i) receiver operating characteristic (ROC) the area under the curve (AUC) with 95% CI in RStudio (Team, 2015); (ii) the Matthews correlation coefficient (MCC), which takes into account the number of true positives (TP), true negatives (TN), false positives (FP), and false negatives (FN) using the following formula:
The MCC alone was then used for comparing the performance of optimized Align-GVGD with that of other bioinformatic tools, as well as for comparing the correlation of predictions with transactivation activity. When combining independent bioinformatic tools, some variants had no agreement between tools and therefore could not be considered as true/false positives/negatives in the MCC calculations (“discordant variants”). To capture this aspect of performance for combined tools, we noted for each combination the percentage of discordant variants out of 318 total variants in our reference sets, in addition to the proportion of mispredicted variants (combined false-positive and false-negative predictions).
The MCC value was also used for selecting the best cut-offs of pathogenicity for REVEL, BayesDel and optimized Align-GVGD. Qualitative tiers for SIFT (tolerated, damaging) and PolyPhen-2 (benign, possibly damaging, probably damaging) were assigned using the web-based tools available on November 15, 2017. Positive likelihood ratios of pathogenicity (LRs) were calculated by dividing the percentage of assumed pathogenic variants (or non-functional validation variants) by that of assumed benign variants (or functional validation variants). In instances where the number of variants was 0, we assumed %=1.
RESULTS
The overview of the study design is summarized in Figure 1.
Figure 1.
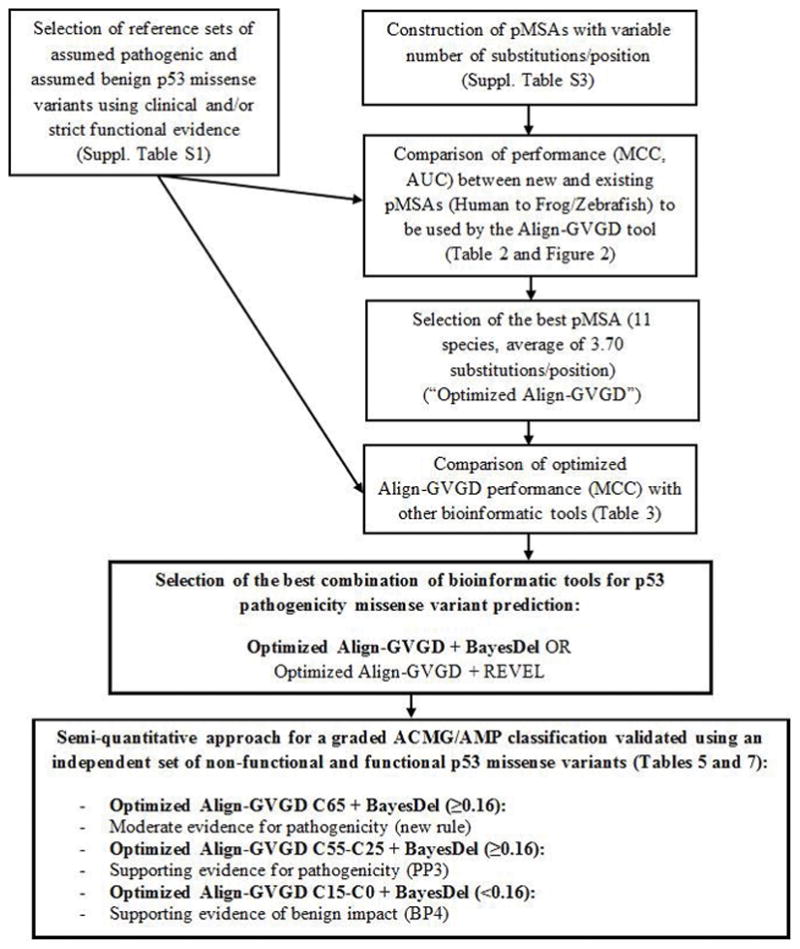
Study design overview
Comparison of performance between existing and new pMSAs for Align-GVGD
Comparing the existing alignments, Human to Zebrafish performed better than Human to Frog. After calculating MCC and AUC values for multiple new pMSAs with an increasing order of average of substitutions/position, a new pMSA (“optimized pMSA/Align-GVGD”) with increased variation was selected as having the best performance, with performance better using a C15 cut-off (MCC=0.622), compared to a C25 cut-off (MCC=0.614). The summary of the performance characteristics of existing and the optimized pMSA are shown in Table 2 and Figure 2. The optimized Align-GVGD-alignment pairing was remarkably better than the existing Human to Frog and slightly better than the existing Human to Zebrafish, and therefore selected for comparison with other bioinformatic tools, as described below.
Table 2.
Summary of performance characteristics of existing pMSAs and the optimized pMSA for Align-GVGD (C15 cut-off)
| Alignment | Species (n) | Average n substitutions/position | AUC (95% CI) | True Positives | True Negatives | False Positives | False Negatives | MCC |
|---|---|---|---|---|---|---|---|---|
| Human to Frog | 8 | 2.15 | 0.862 (0.786, 0.937) | 219 | 53 | 18 | 28 | 0.605 |
| Human to Zebrafish | 9 | 2.95 | 0.869 (0.795, 0.943) | 219 | 54 | 17 | 28 | 0.616 |
| Optimized pMSA | 11 | 3.70 | 0.907 (0.881, 0.934) | 220 | 54 | 17 | 27 | 0.622 |
Figure 2.
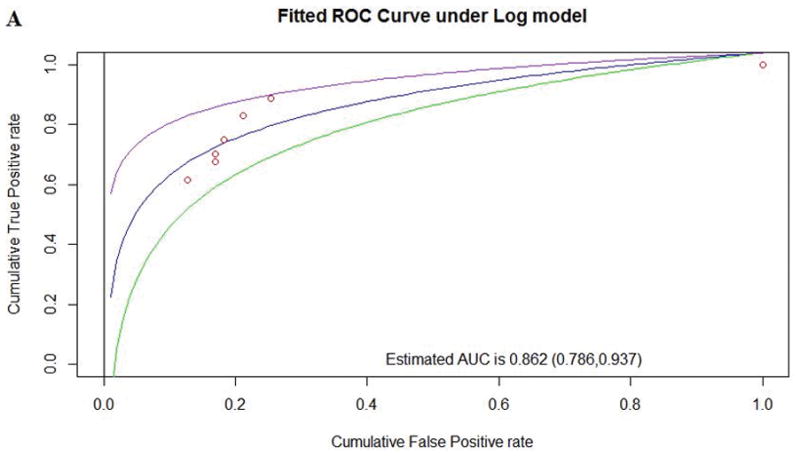
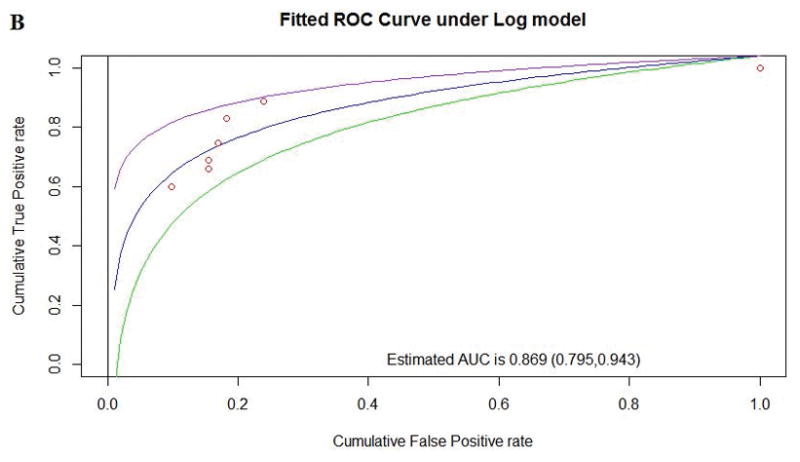
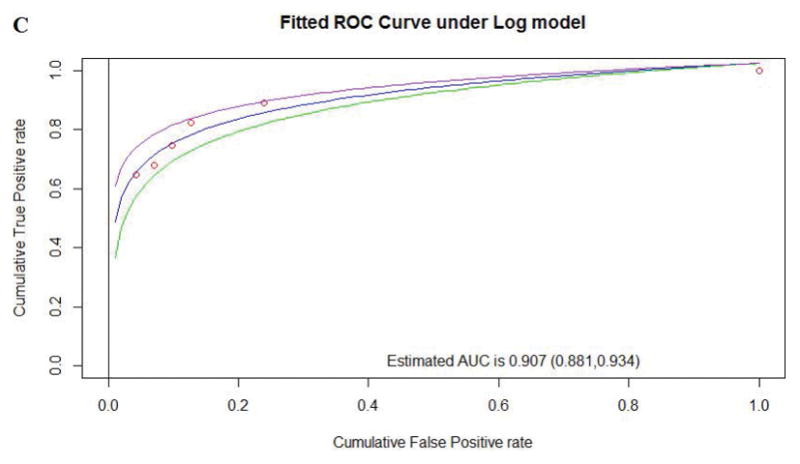
Comparison of performance between pMSAs for Align-GVGD according to the AUC for existing Human to Frog alignment (A), existing Human to Zebrafish alignment (B), and the optimized pMSA
Comparison of performance of optimized Align-GVGD and other bioinformatic tools
We first tested performance (MCC) using different continuous cut-offs for the ensemble method BayesDel, for which the best-performing cut-off was 0.16. For REVEL, we selected 0.5 as the best performing cut-off, as proposed in the original article (Ioannidis et al., 2016), and also observed to have the best performance with our reference sets (data not shown). MCC scores were then calculated for bioinformatic predictions derived using tools individually, or for combinations of independent tools (Table 3). Table 3 also shows the percentage of variants which are mispredicted (false positive or false negative prediction), or discordantly predicted by combinations of tools.
Table 3.
Comparison of performance between bioinformatic tools according to MCC
| Bioinformatic tool or combination of tools | True Positives | True Negatives | False Positives | False Negatives | MCC | % of total variants mispredicted (FN+FP) | % of total variants discordant between tools |
|---|---|---|---|---|---|---|---|
| PolyPhen-2 | 222 | 44 | 27 | 25 | 0.524 | 16.35 | NA |
| SIFT | 231 | 42 | 29 | 16 | 0.568 | 14.15 | NA |
| REVEL (≥0.5) | 237 | 42 | 29 | 10 | 0.620 | 12.26 | NA |
| SIFT + PolyPhen-2 | 218 | 35 | 20 | 12 | 0.621 | 10.06 | 10.38 |
| Optimized Align-GVGD (≥C15) | 220 | 54 | 17 | 27 | 0.622 | 13.84 | NA |
| Optimized Align-GVGD (≥C15) + PolyPhen-2 | 211 | 41 | 14 | 16 | 0.666 | 9.43 | 11.32 |
| Optimized Align-GVGD (≥C15) + SIFT + PolyPhen-2 | 208 | 34 | 13 | 12 | 0.675 | 7.86 | 16.04 |
| BayesDel (≥0.16) | 218 | 60 | 11 | 29 | 0.675 | 12.58 | NA |
| Optimized Align-GVGD (≥C15) + SIFT | 217 | 40 | 15 | 13 | 0.680 | 8.81 | 10.38 |
| Optimized Align-GVGD (≥C15) + REVEL (≥0.5) | 219 | 42 | 16 | 9 | 0.719 | 7.86 | 10.06 |
| Optimized Align-GVGD (≥C15) + BayesDel (≥0.16) | 209 | 49 | 6 | 18 | 0.756 | 7.55 | 11.32 |
Align-GVGD paired with the optimized pMSA showed better performance (MCC=0.622) than SIFT, Polyphen-2 (individually and combined), and the ensemble method REVEL. BayesDel showed better performance (MCC=0.675) than any other individual tool or the combination of optimized Align-GVGD with SIFT and PolyPhen-2. The improved performance of Align-GVGD and BayesDel over other tools appears to be due to an increase in the proportion of true negatives, and concomitant decrease in proportion of false positives, over all other individual tools.
When combining tools, MCC scores improved compared to those for the component tools alone. The proportion of total variants for which predictions were false, or excluded due to discordance between tools, ranged from 12% to 24%. The combination of optimized Align-GVGD and BayesDel gave the best MCC score (MCC=0.756), with a total false/discordant prediction rate of 18.87%. The second best combined MCC score (0.719) was for optimized Align-GVGD and REVEL (total false/discordant prediction rate 17.92%). Considering the two best combinations of tools, optimized Align-GVGD with BayesDel had a much lower false/discordant prediction rate of benign variants (31%) compared to Align-GVGD with REVEL (41%), whereas the false/discordant prediction rate for pathogenic variants was 15% and 11%, respectively.
From binary to graded classifications for the ACMG/AMP guidelines
In general, if there are enough data to evaluate a multi-category classifier, the result can be superior to a binary classifier because the multi-category classifier loses less information than the binary classifier, and edge-effect inaccuracies should be less severe. For BRCA1 and BRCA2, Align-GVGD is used as a calibrated multi-category missense variant prediction tool (Tavtigian et al., 2009), and for the MMR genes a linear combination of MAPP and PolyPhen-2 is used as a calibrated continuous value missense variant prediction tool (Thompson et al., 2013). Moreover, a Bayesian re-analysis of the ACMG/AMP variant classification guidelines (Richards et al., 2015) allows for calibration into a multi-category tool; specifically, under a reasonable set of assumptions, the ACMG category “Strong” corresponds to 350≥ Odds Pathogenicity >18.7, “Moderate” corresponds to 18.7≥ Odds Pathogenicity >4.3, and “Supporting” corresponds to 4.3≥ Odds Pathogenicity >2.08 (Tavtigian et al., 2018). From this Bayesian re-analysis, the benign categories simply have inverse Odds Pathogenicity intervals to the pathogenic categories, while the interval of 2.08≥ Odds Pathogenicity ≥1/2.08 does not provide evidence.
Within this framework, using our reference sets of assumed pathogenic and assumed benign variants, we estimated positive LRs for a similar series of Align-GVGD categories to those used for evaluation of key domain missense variants in BRCA1 and BRCA2 (Table 4).
Table 4.
Proportion of pathogenic and benign variants falling in each Align-GVGD class (optimized pMSA) and corresponding estimated LR
| Align-GVGD class (optimized pMSA) | Assumed pathogenic (n) | % | Assumed benign (n) | % | Positive LR (95% CI) |
|---|---|---|---|---|---|
| C65 | 146 | 59.11 | 0 | NA | 59.11 |
| C55+C25 | 57 | 23.08 | 9 | 12.68 | 1.82 (0.95, 3.49) |
| C15 | 17 | 6.88 | 8 | 11.27 | 0.61 (0.28, 1.36) |
| C0 | 27 | 10.94 | 54 | 76.06 | 0.14 (0.10, 0.21) |
| Total | 247 | 71 |
Notably, these categories produced an ordered series of positive LRs from C65 (strongest evidence for pathogenicity) to C0 (strongest evidence of benign impact). When a variant was predicted pathogenic with C65, the LR was appreciably higher (59.11, consistent with ACMG “Strong” criterion in favor of pathogenicity) than for C55-C25 (LR=1.82, very close to ACMG “Supporting” criterion in favor of pathogenicity). The C15 class alone would not provide any evidence for or against pathogenicity (LR=0.61, 95% CI 0.28–1.36), and the C0 class would be consistent with the ACMG “Moderate” criterion against pathogenicity (LR=0.14). While it is clear that more data are needed to generate reliable LRs for use in a quantitative classification model, the observed trend suggested that introducing graded bioinformatic predictions would improve variant classification using a qualitative system.
Given our results demonstrating improved prediction using combinations of bioinformatic tools, we then assessed if predicted positive LRs would be more linear in distribution by adding BayesDel to the Align-GVGD scores and, in that case, apply the results to the ACMG/AMP guidelines for variant classification (Richards et al., 2015) (Table 5).
Table 5.
Semi-quantitative approach for TP53 variant bioinformatic prediction using Align-GVGD class (optimized pMSA) and BayesDel score
| Category | Assumed pathogenic (n) | % | Assumed benign (n) | % | Positive LR (95% CI) | Proposed ACMG/AMP rule |
|---|---|---|---|---|---|---|
| Optimized Align-GVGD C65 + BayesDel ≥0.16 | 146 | 59.11 | 0 | NA | 59.11 | Moderate evidence of pathogenicity (new rule) |
| Optimized Align-GVGD C65 + BayesDel <0.16 | 0 | NA | 0 | NA | NA | No data |
| Optimized Align-GVGD C55-C25 + BayesDel ≥0.16 | 50 | 20.24 | 3 | 4.22 | 4.79 (1.54, 14.90) | Supporting evidence of pathogenicity (PP3) |
| Optimized Align-GVGD C55-C25 + BayesDel <0.16 | 7 | 2.83 | 5 | 8.45 | 0.33 (0.12, 0.97) | No evidence |
| Optimized Align-GVGD C15 + BayesDel ≥0.16 | 13 | 5.26 | 3 | 4.22 | 1.25 (0.37, 4.25) | No evidence |
| Optimized Align-GVGD C15 + BayesDel <0.16 | 4 | 1.62 | 5 | 7.04 | 0.23 (0.06, 0.83) | Supporting evidence of benign impact (BP4) |
| Optimized Align-GVGD C0 + BayesDel ≥0.16 | 10 | 4.05 | 5 | 7.04 | 0.57 (0.20, 1.63) | No evidence |
| Optimized Align-GVGD C0 + BayesDel <0.16 | 18 | 7.29 | 49 | 69.01 | 0.11 (0.07, 0.17) | Supporting evidence of benign impact (BP4) |
| Total | 247 | 71 |
There were four notable observations: BayesDel had no impact on the optimized Align-GVGD C65 class; a combined C55-C25 class split into two - one with higher evidence of pathogenicity and another with little evidence in either direction; BayesDel had a clear impact on the Align-GVGD C15 class which split into two – one with no or little evidence of pathogenicity and another with higher evidence of benign impact; and the C0 class also split into two - one with higher evidence of benign impact and another with little evidence in either direction.
Further, the LR of the optimized Align-GVGD C65 + BayesDel ≥0.16 category, which included more than 50% of the assumed pathogenic missense variants, could qualify as ACMG/AMP strong evidence of pathogenicity, and the optimized Align-GVGD C55-C25 + BayesDel ≥0.16 class could qualify as moderate evidence of pathogenicity. Similarly, the optimized Align-GVGD C15-C0 + BayesDel <0.16 could qualify as moderate evidence of benign impact. However, acknowledging that the criteria used to define our reference set variants are not universally accepted as standard criteria for variant classification, and also wanting to avoid over-estimating evidence at the median edges of the categories defined here, it would seem appropriate to downgrade the strength of each of these categories by one step. Therefore, we propose optimized Align-GVGD C65 + BayesDel ≥0.16 as moderate evidence of pathogenicity, optimized Align-GVGD C55-C25 + BayesDel ≥0.16 as supporting evidence of pathogenicity, and optimized Align-GVGD C15-C0 + BayesDel <0.16 as supporting evidence of benign effect, as long as there is no evidence of spliceogenicity from MaxEntScan (Vallee et al., 2016).
Validation of the proposed binary and graded Align-GVGD + BayesDel prediction schemes for TP53 variant classification
For the analyses through to Table 5, strict criteria (defined in Table 1) were used to define our reference sets of assumed pathogenic and benign missense variants. For a validation analysis, we used an independent dataset of 194 non-functional (enriched pathogenic) and 1133 functional (enriched benign) missense variants characterized using transactivation assays (Kato et al., 2003), which excluded the variants present in our first reference sets. We recognize that transactivation activity does not equal variant pathogenicity, which implies that the validation set could have a higher false classification rate than the assumed pathogenic and assumed benign reference sets. Using this independent dataset of functional variants, we reassessed the performance of the optimized pMSA for Align-GVGD in comparison to the existing alignments, as well as that of the two best binary bioinformatic prediction schemes for these missense variants (Table 6).
Table 6.
Correlation of binary bioinformatic predictions with p53 transactivation activity according to MCC
| Bioinformatic tools | TP | TN | FP | FN | MCC |
|---|---|---|---|---|---|
| Human to Frog (Align-GVGD ≥C15) | 164 | 818 | 315 | 30 | 0.417 |
| Human to Zebrafish (Align-GVGD ≥C15) | 159 | 871 | 262 | 35 | 0.447 |
| Optimized pMSA (Align-GVGD ≥C15) | 160 | 887 | 246 | 34 | 0.466 |
| Optimized Align-GVGD (≥C15) + REVEL (≥0.5) | 158 | 586 | 210 | 11 | 0.525 |
| Optimized Align-GVGD (≥C15) + BayesDel (≥0.16) | 156 | 819 | 137 | 14 | 0.632 |
As expected, an optimized Align-GVGD correlated marginally better to p53 transactivation activity than existing alignments, and this correlation improved when adding REVEL or BayesDel. In general, the predictions of the two best combinations of tools (Table 3) correlated relatively well with the transactivation activity according to the MCC values (0.5–0.6), with the combination of the optimized Align-GVGD with BayesDel performing better than with REVEL (0.629 vs 0.526). Overall, ~81% of the 194 non-functional p53 missense variants would be predicted pathogenic according to both the optimized Align-GVGD with BayesDel and with REVEL. In contrast, 72.29% of the 1201 functional p53 missense variants would be predicted benign using optimized Align-GVGD with BayesDel, as opposed to only 51.72% for optimized Align-GVGD with REVEL. Using these independent sets of non-functional and functional missense variants, we then re-tested the graded categories defined in Table 5 above (Table 7).
Table 7.
Validation of semi-quantitative approach for TP53 variant bioinformatic prediction using Align-GVGD class (optimized pMSA) and BayesDel score, using an independent set of non-functional and functional variants
| Category | Non-functional (n) | % | Functional (n) | % | Positive LR (95% CI) | LR consistent with proposed ACMG/AMP rule (Table 5) |
|---|---|---|---|---|---|---|
| Optimized Align-GVGD C65 + BayesDel ≥0.16 | 94 | 48.45 | 49 | 4.32 | 11.20 (8.22, 15.27) | Yes - Moderate evidence of pathogenicity (new rule):18.7≥ Odds Path >4.3 |
| Optimized Align-GVGD C55-C25 + BayesDel ≥0.16 | 51 | 26.29 | 61 | 5.38 | 4.88 (3.48, 6.86) | Yes - Supporting evidence of pathogenicity (PP3): 4.3≥ Odds Path >2.08 |
| Optimized Align-GVGD C15 + BayesDel ≥0.16 | 11 | 5.67 | 27 | 2.38 | 2.38 (1.20, 4.72) | No - Not consistent enough with initial results to justify PP3 |
| Optimized Align-GVGD C15 + BayesDel <0.16 | 1 | 0.51 | 42 | 3.71 | 0.14 (0.02, 1) | Yes - Supporting evidence of benign impact (BP4): 0.23< Odds Path <0.48 |
| Optimized Align-GVGD C0 + BayesDel <0.16 | 14 | 7.22 | 819 | 72.29 | 0.10 (0.06, 0.17) | Yes - Supporting evidence of benign impact (BP4): 0.23< Odds Path <0.48 |
The positive LRs estimated with the functional validation sets supported the ACMG/AMP rules for specific bioinformatic tiers, as proposed based on analysis of our first reference sets in Table 5 above. In particular, the LR of the optimized Align-GVGD C65 + BayesDel ≥0.16 category, which included almost 50% of the assumed pathogenic missense variants, would fall into the ACMG/AMP moderate evidence of pathogenicity category, and even the lower bound of its 95% confidence interval falls within that category. Similarly, the optimized Align-GVGD C55-C25 + BayesDel ≥0.16 class would qualify as moderate evidence of pathogenicity, though we still prefer downgrading it to supporting evidence. The optimized Align-GVGD C15-C0 + BayesDel <0.16 would qualify as moderate evidence of benign impact, though we also prefer downgrading it to supporting evidence – with the continued requirement that there is no evidence of spliceogenicity from MaxEntScan (Vallee et al., 2016). On the other hand, evidence from the Align-GVGD C15 + BayesDel ≥0.16 was inconsistent between the two analyses and therefore not strong enough to place in a pathogenicity category.
CONCLUSION
Align-GVGD is a popular bioinformatic tool used for the prediction of the effect of missense variants in the TP53 gene, and is currently paired with two curated, publicly available, TP53 pMSAs (Human to Frog and Human to Zebrafish) created in 2005 for variant analyses. Taking advantage of the fact that many more full-length vertebrate p53 sequences are currently available, we revisited the existing pMSAs. We considered relatively divergent species not previously included (lizard, coelacanth, etc), and dropped most species that were in the same evolutionary node (dog vs cow, mouse vs rat, etc.) relative to humans. We attempted to increase the average number of substitutions per position, and in so doing have increased prediction accuracy as shown by both MCC and AUC values.
The optimized pMSA for Align-GVGD also outperformed the SIFT and PolyPhen-2 bioinformatic tools commonly used for missense variant predictions, and the REVEL composite prediction tool promoted for use by the ClinGen consortium for variant classification using the ACMG/AMP variant classification guidelines (Ghosh, Oak, & Plon, 2017). This improvement in the overall prediction was mainly due to the ability of Align-GVGD to better identify benign variants, reducing false positives, with little improvement in the identification of pathogenic variants. The performance of SIFT and PolyPhen-2 could also potentially be improved by specifically modifying the pMSA used by these tools, but this was not an aim of this study. When combining independent bioinformatic tools, the best performance was for the optimized Align-GVGD together with BayesDel (0.16 cut-off) or with REVEL (0.5 cut-off), which reduced the proportion of false positives and false negatives, albeit at some expense to the true negative and true positive rate. When comparing these two combinations, optimized Align-GVGD with BayesDel showed a lower proportion of pathogenic and benign variants whose prediction would be missed due to discordance between both tools (Table 3), and also correlated better with p53 transactivation activity (Table 6). We openly acknowledge that the criteria used to define our reference set variants are not universally accepted as standard criteria for variant classification, with an obvious but necessary exclusion of computational evidence for classification of variants in these reference sets. However, this is unlikely to have impacted on results since all tools were compared using predictions for exactly the same reference set variants. Further, only 7 of 247 presumed pathogenic variants with abrogated transactivation activity and also absent from reference control datasets had not yet been reported in the IARC germline or tumor datasets (Bouaoun et al., 2016), and exclusion of these variants did not affect overall results.
The ACMG/AMP guidelines (Richards et al., 2015) relating to use of bioinformatic evidence for variant interpretation state “Multiple lines of computational evidence support a deleterious effect on the gene or gene product” as Supporting evidence for pathogenicity (PP3), and “Multiple lines of computational evidence suggest no impact on gene or gene product” as Supporting evidence of benign impact (BP4), but these guidelines do not specify which computational evidence to use, generically or for specific genes. Further, great variability in predictions by different algorithms has been observed, particularly for benign variants (Ghosh et al., 2017). Based on the comparisons conducted in this study (Table 3), we suggest that the combination of the newly optimized Align-GVGD plus ensemble tool BayesDel (0.16 cut-off), or optimized Align-GVGD plus ensemble tool REVEL (0.5 cut-off), should be considered as the bioinformatic tools to be used for the pathogenicity prediction of p53 missense variants in the context of ACMG/AMP and other qualitative classification schemes. Between these two combinations, optimized Align-GVGD with BayesDel showed the best overall performance.
Based on our analysis of bioinformatic predictions as graded categories (Tables 5 and 7), we also suggest upgrading PP3 ACMG/AMP rule to moderate when the variant has a high Align-GVGD class (C65) in addition to a BayesDel score ≥0.16, whereas the evidence level will be supporting of pathogenicity with Align-GVGD scores of C55-C25 and a BayesDel score ≥0.16. In addition, in instances where a variant has conflicting BayesDel and Align-GVGD predictions, the evidence will be supporting of benign impact if it has a C15 class and BayesDel score <0.16 (and, obviously, if the variant has C0 class). These proposed multi-tiered ACMG/AMP classifiers were supported by our original reference sets of assumed pathogenic and assumed benign missense variants, as well as an independent validation dataset of non-functional and functional missense variants selected using transactivation assay data (Kato et al., 2003). There was not enough evidence to predict a variant to be pathogenic or benign when it has Align-GVGD C15 + BayesDel score ≥0.16 from analysis of our reference sets, although the results from the functional replication set (LR 2.38) suggest that further analysis of larger datasets would be useful to assess if this bioinformatic tier could provide supporting evidence towards pathogenicity. It is important to note, as recommended by the ACMG/AMP guidelines (Richards et al., 2015), that variant classifications should be generated from a combination of different sources of evidence (clinical, functional, population, etc) and not from bioinformatic predictions alone. We note also that our analyses have not included consideration of variant effect (at the nucleotide level) on mRNA splicing, and recommend that all presumed missense substitution variants, and also silent substitution variants, are assessed for potential effect on mRNA transcript profile.
Pathogenicity predictions with REVEL, BayesDel and optimized Align-GVGD have been computed for every possible missense variant (Supp. Table S4) and will be included in the public IARC TP53 Database website to facilitate their use by the scientific and medical community.
Supplementary Material
Acknowledgments
FUNDING
We acknowledge useful discussions with the ClinGen TP53 Expert Panel about adaptation of the ACMG/AMP guidelines for TP53 variant classification, and give particular thanks to the group coordinator Kristy Lee. C.F is supported by a University of Queensland (UQ) International Scholarship from the UQ School of Medicine. A.B.S is supported by an NHMRC Senior Research Fellowship (ID1061779). S.V.T received funding from a US NIH NCI R01CA121245.
Footnotes
CONFLICT OF INTEREST
Tina Pesaran is a paid employee of Ambry Genetics. All other authors have declared no conflicts of interest.
References
- Abegglen LM, Caulin AF, Chan A, Lee K, Robinson R, Campbell MS, … Schiffman JD. Potential Mechanisms for Cancer Resistance in Elephants and Comparative Cellular Response to DNA Damage in Humans. JAMA. 2015;314(17):1850–1860. doi: 10.1001/jama.2015.13134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouaoun L, Sonkin D, Ardin M, Hollstein M, Byrnes G, Zavadil J, Olivier M. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum Mutat. 2016;37(9):865–876. doi: 10.1002/humu.23035. [DOI] [PubMed] [Google Scholar]
- Chen K, Baxter T, Muir WM, Groenen MA, Schook LB. Genetic resources, genome mapping and evolutionary genomics of the pig (Sus scrofa) Int J Biol Sci. 2007;3(3):153–165. doi: 10.7150/ijbs.3.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. PHYLIP - Phylogeny Inference Package (Version 3.2) Cladistics. 1989;5:164–166. citeulike-article-id:2344765. [Google Scholar]
- Feng BJ. PERCH: A Unified Framework for Disease Gene Prioritization. Hum Mutat. 2017;38(3):243–251. doi: 10.1002/humu.23158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh R, Oak N, Plon SE. Evaluation of in silico algorithms for use with ACMG/AMP clinical variant interpretation guidelines. Genome Biol. 2017;18(1):225. doi: 10.1186/s13059-017-1353-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenblatt MS, Beaudet JG, Gump JR, Godin KS, Trombley L, Koh J, Bond JP. Detailed computational study of p53 and p16: using evolutionary sequence analysis and disease-associated mutations to predict the functional consequences of allelic variants. Oncogene. 2003;22(8):1150–1163. doi: 10.1038/sj.onc.1206101. [DOI] [PubMed] [Google Scholar]
- Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, … Sieh W. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am J Hum Genet. 2016;99(4):877–885. doi: 10.1016/j.ajhg.2016.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato S, Han SY, Liu W, Otsuka K, Shibata H, Kanamaru R, Ishioka C. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc Natl Acad Sci U S A. 2003;100(14):8424–8429. doi: 10.1073/pnas.1431692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;44(W1):W242–245. doi: 10.1093/nar/gkw290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathe E, Olivier M, Kato S, Ishioka C, Hainaut P, Tavtigian SV. Computational approaches for predicting the biological effect of p53 missense mutations: a comparison of three sequence analysis based methods. Nucleic Acids Res. 2006;34(5):1317–1325. doi: 10.1093/nar/gkj518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okonechnikov K, Golosova O, Fursov M U team. Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics. 2012;28(8):1166–1167. doi: 10.1093/bioinformatics/bts091. [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J … Committee, A. L. Q. A. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavtigian SV, Greenblatt MS, Harrison SM, Nussbaum RL, Prabhu SA, Boucher KM, Biesecker LG. Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genet Med. 2018 doi: 10.1038/gim.2017.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavtigian SV, Greenblatt MS, Lesueur F, Byrnes GB, Group IUGVW. In silico analysis of missense substitutions using sequence-alignment based methods. Hum Mutat. 2008;29(11):1327–1336. doi: 10.1002/humu.20892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavtigian SV, Oefner PJ, Babikyan D, Hartmann A, Healey S, Le Calvez-Kelm F, … Chenevix-Trench G. Rare, evolutionarily unlikely missense substitutions in ATM confer increased risk of breast cancer. Am J Hum Genet. 2009;85(4):427–446. doi: 10.1016/j.ajhg.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team, R. RStudio: Integrated Development for R. 2015. [Google Scholar]
- Thompson BA, Greenblatt MS, Vallee MP, Herkert JC, Tessereau C, Young EL, … Tavtigian SV. Calibration of multiple in silico tools for predicting pathogenicity of mismatch repair gene missense substitutions. Hum Mutat. 2013;34(1):255–265. doi: 10.1002/humu.22214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallee MP, Di Sera TL, Nix DA, Paquette AM, Parsons MT, Bell R, … Tavtigian SV. Adding In Silico Assessment of Potential Splice Aberration to the Integrated Evaluation of BRCA Gene Unclassified Variants. Hum Mutat. 2016;37(7):627–639. doi: 10.1002/humu.22973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace IM, O’Sullivan O, Higgins DG, Notredame C. M-Coffee: combining multiple sequence alignment methods with T-Coffee. Nucleic Acids Res. 2006;34(6):1692–1699. doi: 10.1093/nar/gkl091. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.