

Abstract
We describe progressive spastic paraparesis in two male siblings and the daughter of one of these individuals. Onset of disease occurred within the first decade, with stiffness and gait difficulties. Brisk deep tendon reflexes and extensor plantar responses were present, in the absence of intellectual disability or dermatological manifestations. Cerebral imaging identified intracranial calcification in all symptomatic family members. A marked upregulation of interferon stimulated gene (ISG) transcripts was recorded in all three affected individuals and in two clinically unaffected relatives. A heterozygous IFIH1 c.2544T>G missense variant (p.Asp848Glu) segregated with interferon status. Although not highly conserved (Supporting figure 1; CADD score 10.08 versus MSC-CADD score of 19.33) and predicted as benign by in silico algorithms, this variant is not present on publically available databases of control alleles, and expression of the D848E construct in HEK293T cells indicated that it confers a gain-of-function. This report illustrates, for the first time, the occurrence of autosomal dominant spastic paraplegia with intracranial calcifications due to an IFIH1-related type 1 interferonopathy.
Keywords: IFIH1, interferonopathy, spastic paraparesis, brain calcification
Introduction
Hereditary spastic paraparesis (HSPs) are Mendelian neurodegenerative diseases characterized by progressive spasticity and hyperreflexia of the lower limbs (Esteves et al., 2014). At least 76 loci and 58 corresponding genes linked to HSPs have been identified so far (Parodi et al., 2017). Autosomal-dominant HSPs have been described due to mutations in 19 SPG genes (Klebe et al., 2015). Brain calcification has been reported in subjects with CYP2U1 mutations (Tesson et al., 2012), but this imaging feature is otherwise rarely described in the context of HSP. The molecular basis of a number of familial cases of HSP remains undefined.
Aicardi-Goutières syndrome (AGS) is a severe childhood onset degenerative encephalopathy associating brain calcification, white matter disease, cerebral atrophy and psychomotor retardation (Aicardi and Goutières, 1984). Seven genes have so far been described which, when mutated, can cause an AGS phenotype: TREX1 (AGS1) (Crow et al., 2006a), RNASEH2A (AGS3), RNASEH2B (AGS2), RNASEH2C (AGS4) (Crow et al., 2006b), SAMHD1 (AGS5) (Rice et al., 2009), ADAR1 (AGS6) (Rice et al., 2012) and IFIH1 (MIM# 606951) (AGS7) (Rice et al., 2014). AGS is most frequently transmitted as an autosomal recessive trait, although cases of inherited and de novo autosomal dominant occurrence are also documented. An underlying activation of the interferon signalling pathway is considered as a direct pathogenic driver, although definitive proof of this in the human context is yet to be determined (Crow and Manel, 2015). Assessment of interferon status has proven to be a very reliable disease marker. This method gives an interferon score comparing the expression of six interferon-stimulated genes using quantitative PCR in peripheral blood in patients and controls (Rice et al., 2013; Rodero et al., 2017). Using this screening assay, a remarkably broad spectrum of associated neurologic phenotypes has been revealed to be associated with mutations in AGS1-7, with isolated spastic paraparesis reported in a limited number of patients with mutations in ADAR1, IFIH1 and RNASEH2B (Crow et al., 2014, 2015).
Here we describe a Portuguese family segregating an autosomal dominant HSP phenotype with intracranial calcification due to a gain-of-function mutation in IFIH1 and demonstrating clinical non-penetrance in certain individuals.
Clinical histories
This Portuguese family includes three clinically affected individuals: a 14 year old girl, her father and her paternal uncle. The proband developed walking difficulties with moderate spasticity of the lower limbs from 3 years of age. On examination aged 10 years, she was able to stand on one foot, had brisk deep tendon reflexes, extensor plantar responses and a stiff gait. Her growth was delayed, with a height at -3SD. Growth velocity deceleration was noticed from the age of 10 years, contrasting with normal weight (3rd percentile) and OFC (-1.5 SD). She was proficient at school with good social integration. At age 14 years she is unable to walk on tip-toes and unipodal balance is unstable. Muscle strength is normal. Her brain CT-scan is remarkable for extensive calcifications in the deep white matter of the frontal lobes and at the white-gray junction (Figure 1G-I). Her twin brother is asymptomatic, with unremarkable clinical examination including normal deep tendon reflexes. Her twin brother and her father have height in the normal range.
Figure 1.
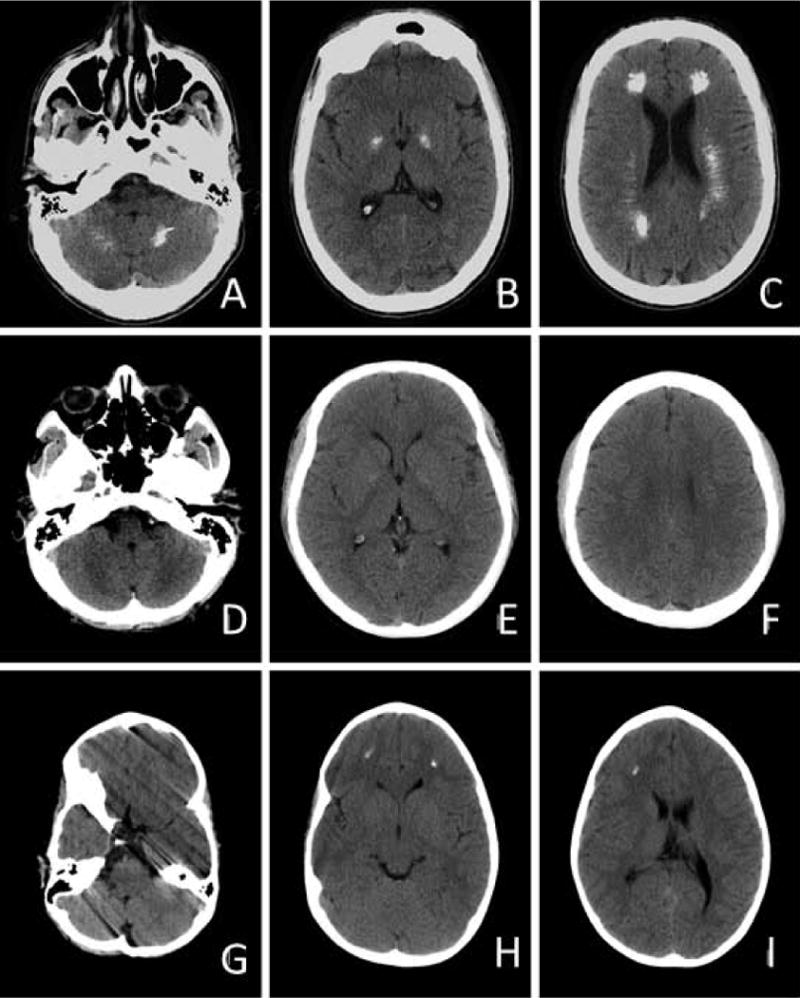
The father of the proband reported onset of walking difficulties from 2 years of age, associated with an increased frequency of falls. A slowly progressive course was noted, still allowing him to run when aged 24 years. When evaluated at 41 years of age he had severe spastic weakness, MRC grade 4, with marked hyperreflexia and bilateral Achilles clonus. Dysarthria and occasional dysphagia were also noted. He has normal higher intellectual functions. Brain CT performed at age 24 years revealed extensive calcifications in the deep white matter (centrum semi-ovale), involving frontal and temporal regions. Punctate deposits were also present in the posterior fossa, globi pallidi and dentate nuclei (Figure 1A-C).
The maternal uncle experienced progressive lower limb motor impairment since the age of 13 years. On examination, aged 38 years, an MRC grade 4 spastic weakness with marked hyperreflexia and bilateral Achilles clonus was noted. Cognition was considered as normal. His brain CT-scan indicates subtle changes similar to those of his niece (Figure 1D-F). His height is in the normal range.
The paternal grandparents of the proband are asymptomatic. Cerebral CT of the grandfather revealed likely non-pathological falx calcifications.
Methods
Written, informed consent for DNA analysis was obtained from patients and parents for the children. The study was approved by our local ethics committee.
Interferon signature
The expression of six interferon-stimulated genes (ISGs), namely IFI27 (Hs01086370_m1), IFI44L (Hs00199115_m1), IFIT1 (Hs00356631_g1), ISG15 (Hs00192713_m1), RSAD2 (Hs01057264_m1), and SIGLEC1 (Hs00988063_m1) was measured by quantitative PCR according to a previously reported method (Rice et al., 2014). The median fold change, when compared with the median of 29 healthy controls, was used to create an interferon score for each patient. Scores higher than the mean of the controls plus two SD (>2·466) were designated as positive.
Sanger sequencing of IFIH1
DNA was extracted from whole blood samples using standard methods, and sequencing performed using primers designed to amplify the coding exons of IFIH1. Purified PCR amplification products were sequenced using BigDye Terminator chemistry and an ABI 3130 DNA sequencer. Mutation description is based on the reference cDNA sequence NM_022168.3, with nucleotide numbering beginning from the first A in the initiating ATG codon. Variants were assessed using the in silico programs SIFT (http://sift.jcvi.org) and Polyphen2 (http://genetics.bwh.harvard.edu/pph2/), and population allele frequencies obtained from the gnomAD (http://gnomad.broadinstitute.org) database.
Interferon reporter assay
The pFLAG-CMV4 plasmid encoding IFIH1 has been described elsewhere (Rice et al., 2014). The indicated mutations were introduced using KAPA HiFi DNA polymerase. Human embryonic kidney (HEK) 293T cells (American Type Culture Collection, ATCC) were maintained in 48-well plates in DMEM (Cellgro) supplemented with 10% heat-inactivated FCS and 1% L-glutamine. Cells were tested for mycoplasma. At ~80% confluence, cells were cotransfected with pFLAG-CMV4 plasmids encoding wild-type or mutant IFIH1 (10 ng), IFN-β promoter–driven firefly luciferase reporter plasmid (100 ng) and a constitutively expressed Renilla luciferase reporter plasmid (pRL-CMV; 10 ng) using Lipofectamine 2000 (Life Technologies) according to the manufacturer’s protocol. Medium was changed 6 hours after transfection, and cells were subsequently stimulated with poly I:C (0.5 μg/ml; Invivogen) using Lipofectamine 2000. Cells were lysed 16 hours after stimulation, and IFN-β promoter activity was measured using the Dual-Luciferase Reporter assay (Promega) and a Synergy2 plate reader (BioTek). Firefly luciferase activity was normalized against Renilla luciferase activity. Each experiment (n = 4) was performed in triplicate and data are presented as mean ± SEM. Statistical significance was determined by two tailed, unpaired Student’s t test with * and ** indicating p values <0.05 and <0.01 respectively. Comparable expression of construct proteins was verified by western blotting (data not shown).
Results
Based on the lower limb spasticity with early onset, slow progression and the presence of brain calcifications, an initial diagnosis of Fahr syndrome was considered in the proband and her father. Standard karyotypes were 46,XX and 46,XY and array CGH (Agilent 180K) was normal. SPG4 and SPG3 genes, encoding spastin and atlastin were sequenced without identifying any likely pathogenic variant. Subsequently, the possibility of an interferonopathy was considered. Quantitative RT-PCR (qPCR) demonstrated persistent and robust upregulation of ISG expression in whole blood in the proband, her affected father and her affected uncle, with scores ranging from 10 to 53 (NR<2.44). Furthermore, the proband’s asymptomatic twin brother and her unaffected paternal grandfather also demonstrated significant upregulation of interferon signalling (Figure 2A).
Figure 2.
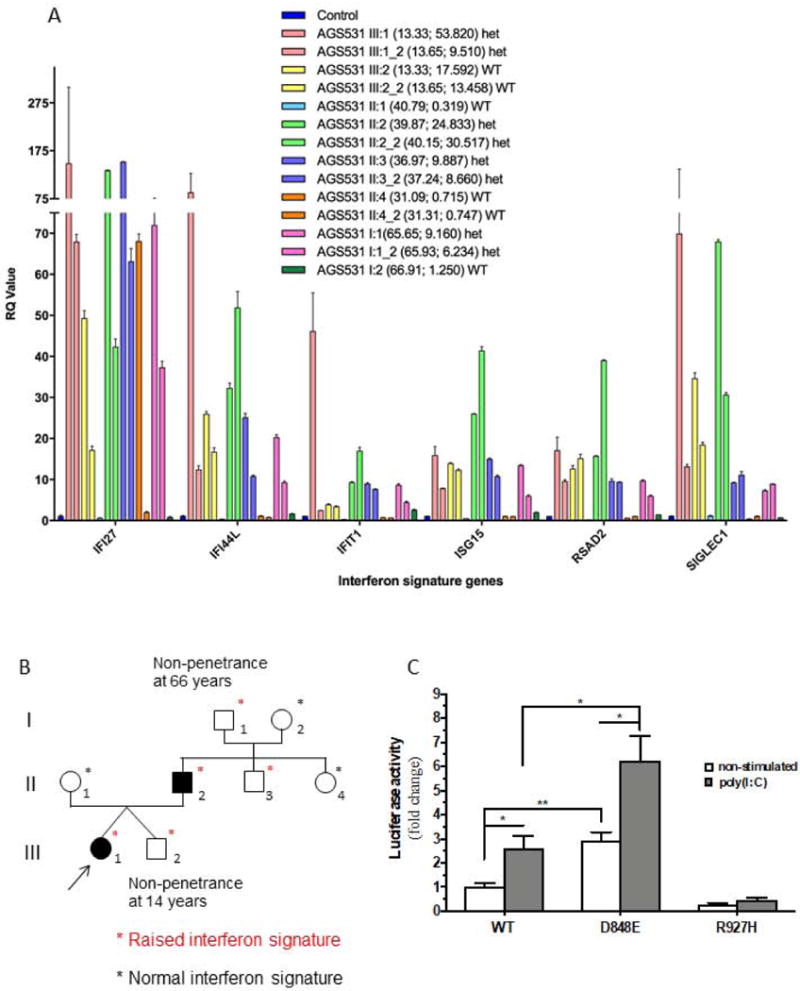
Sequencing of IFH1 identified a heterozygous c.2544T>G sequence alteration resulting in a missense p.Asp848Glu change in every family member displaying abnormal ISG expression (Figure 2B). This variant is predicted as benign according to in silico analyses, and has not been reported in other patients. However, the variant is not present in 240 000 control alleles recorded in the gnomAD cohort. Furthermore, interferon β (IFN-β) reporter stimulatory activity of wild-type and variant IFIH1 in HEK293T cells indicated that the p.Asp848Glu confers a gain-of-function, comparable to that recorded with other known IFIH1 mutations (Figure 2C) (Rice et al., 2014). The aspartate at position 848 is not highly conserved (Supporting figure 1), the p.Asp848Glu substitution is predicted as benign according to in silico analyses (CADD score 10.08 versus MSC-CADD score of 19.33), and this variant has not been reported in other patients.
Discussion
IFIH1 gain-of-function mutations were first reported in 2014 in patients with a variety of neuro-immunological phenotypes including AGS (Rice et al., 2014). Subsequently, IFIH1 mutations were also identified in patients with non-neurological phenotypes, including complex dermatological and skeletal anomalies in the context of Singleton-Merten syndrome (Bursztejn et al., 2015; Rutsch et al., 2015; Buers et al., 2017; De Carvalho et al., 2017), manifesting dental dysplasia, glaucoma, psoriasis, aortic calcification and skeletal abnormalities. Patients with IFIH1 mutations have been previously described to demonstrate lower limb spasticity (Oda et al., 2014; Rice et al., 2014; Bursztejn et al., 2015; Hacohen et al., 2015; Marguet et al., 2016; Buers et al., 2017; Pettersson et al., 2017). Isolated spastic paraplegia was observed in the 31 year old father of a girl presenting with a complex phenotype encompassing spasticity and a lupus-like illness (Rice et al., 2014; Hacohen et al., 2015). Of note, apparently isolated spastic paraplegia was reported in three patients belonging to two unrelated families from North Africa with mutations in RNASEH2B, and in a singleton of European ancestry harbouring a de novo ADAR1 mutation (Crow et al., 2014), both being AGS-associated genes.
In addition to variable clinical expression already described in the type I interferonopathy context, our family underlines the possibility of discordant penetrance between relatives, including twins inheriting the same mutation from their affected father, one of them being clinically asymptomatic despite a robust interferon signature. The follow-up of this brother will determine if he eventually develops a phenotype. Such a contrast between non-identical twins points to the effect of environmental triggers or genetic modifiers.
Although the IFIH1 variant that we describe was predicted as benign according to SIFT and Polyphen softwares, functional studies with dosage of IFN-β receptor activity were in favour of pathogenicity, raising issues about the interpretation of IFIH1 variants based solely on prediction algorithms.
Summarising, our observations suggest that an IFIH1-related interferonopathy should be taken into consideration in the differential diagnosis of autosomal dominant spastic paraparesis, either in the presence of normal neuroimaging or in association with the presence of intracranial calcification.
Supplementary Material
Acknowledgments
Y.J.C. acknowledges funding from the European Research Council (GA 309449: Fellowship to Y.J.C.) and a state subsidy managed by the National Research Agency (ANR, France) under the “Investments for the Future” (ANR-10-IAHU-01). L.V.E. acknowledges funding from FWO-Vlaanderen. H. S. acknowledges the NIH grant R01AI111784.
References
- Aicardi J, Goutières F. A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann Neurol. 1984;15:49–54. doi: 10.1002/ana.410150109. [DOI] [PubMed] [Google Scholar]
- Buers I, Rice GI, Crow YJ, Rutsch F. MDA5-Associated Neuroinflammation and the Singleton-Merten Syndrome: Two Faces of the Same Type I Interferonopathy Spectrum. J Interferon Cytokine Res Off J Int Soc Interferon Cytokine Res. 2017;37:214–219. doi: 10.1089/jir.2017.0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bursztejn A-C, Briggs TA, del Toro Duany Y, Anderson BH, O’Sullivan J, Williams SG, Bodemer C, Fraitag S, Gebhard F, Leheup B, Lemelle I, Oojageer A, et al. Unusual cutaneous features associated with a heterozygous gain-of-function mutation in IFIH1: overlap between Aicardi-Goutières and Singleton-Merten syndromes. Br J Dermatol. 2015;173:1505–1513. doi: 10.1111/bjd.14073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ, Chase DS, Schmidt JL, Szynkiewicz M, Forte GMA, Gornall HL, Oojageer A, Anderson B, Pizzino A, Helman G, Abdel-Hamid MS, Abdel-Salam GM, et al. Characterization of Human Disease Phenotypes Associated with Mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet A. 2015;0:296–312. doi: 10.1002/ajmg.a.36887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, Black DN, van Bokhoven H, Brunner HG, Hamel BC, Corry PC, Cowan FM, et al. Mutations in the gene encoding the 3’-5’ DNA exonuclease TREX1 cause Aicardi-Goutières syndrome at the AGS1 locus. Nat Genet. 2006a;38:917–920. doi: 10.1038/ng1845. [DOI] [PubMed] [Google Scholar]
- Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E, Ali M, Semple C, Aicardi J, Babul-Hirji R, Baumann C, Baxter P, et al. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutières syndrome and mimic congenital viral brain infection. Nat Genet. 2006b;38:910–916. doi: 10.1038/ng1842. [DOI] [PubMed] [Google Scholar]
- Crow YJ, Manel N. Aicardi-Goutières syndrome and the type I interferonopathies. Nat Rev Immunol. 2015;15:429–440. doi: 10.1038/nri3850. [DOI] [PubMed] [Google Scholar]
- Crow YJ, Zaki MS, Abdel-Hamid MS, Abdel-Salam G, Boespflug-Tanguy O, Cordeiro NJV, Gleeson JG, Gowrinathan NR, Laugel V, Renaldo F, Rodriguez D, Livingston JH, et al. Mutations in ADAR1, IFIH1, and RNASEH2B presenting as spastic paraplegia. Neuropediatrics. 2014;45:386–393. doi: 10.1055/s-0034-1389161. [DOI] [PubMed] [Google Scholar]
- de Carvalho LM, Ngoumou G, Park JW, Ehmke N, Deigendesch N, Kitabayashi N, Melki I, Souza FFL, Tzschach A, Nogueira-Barbosa MH, Ferriani V, Louzada-Junior P, et al. Musculoskeletal disease in MDA5-related type I interferonopathy - a Mendelian mimic of Jaccoud’s arthropathy. Arthritis Rheumatol. 2017;69:2081–2091. doi: 10.1002/art.40179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteves T, Durr A, Mundwiller E, Loureiro JL, Boutry M, Gonzalez MA, Gauthier J, El-Hachimi KH, Depienne C, Muriel M-P, Acosta Lebrigio RF, Gaussen M, et al. Loss of association of REEP2 with membranes leads to hereditary spastic paraplegia. Am J Hum Genet. 2014;94:268–277. doi: 10.1016/j.ajhg.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacohen Y, Zuberi S, Vincent A, Crow YJ, Cordeiro N. Neuromyelitis optica in a child with Aicardi-Goutières syndrome. Neurology. 2015;85:381–383. doi: 10.1212/WNL.0000000000001792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebe S, Stevanin G, Depienne C. Clinical and genetic heterogeneity in hereditary spastic paraplegias: from SPG1 to SPG72 and still counting. Rev Neurol (Paris) 2015;171:505–530. doi: 10.1016/j.neurol.2015.02.017. [DOI] [PubMed] [Google Scholar]
- Marguet F, Laquerrière A, Goldenberg A, Guerrot A-M, Quenez O, Flahaut P, Vanhulle C, Dumant-Forest C, Charbonnier F, Vezain M, Bekri S, Tournier I, et al. Clinical and pathologic features of Aicardi-Goutières syndrome due to an IFIH1 mutation: A pediatric case report. Am J Med Genet A. 2016;170A:1317–1324. doi: 10.1002/ajmg.a.37577. [DOI] [PubMed] [Google Scholar]
- Oda H, Nakagawa K, Abe J, Awaya T, Funabiki M, Hijikata A, Nishikomori R, Funatsuka M, Ohshima Y, Sugawara Y, Yasumi T, Kato H, et al. Aicardi-Goutières syndrome is caused by IFIH1 mutations. Am J Hum Genet. 2014;95:121–125. doi: 10.1016/j.ajhg.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parodi L, Fenu S, Stevanin G, Durr A. Hereditary spastic paraplegia: More than an upper motor neuron disease. Rev Neurol (Paris) 2017;173:352–360. doi: 10.1016/j.neurol.2017.03.034. [DOI] [PubMed] [Google Scholar]
- Pettersson M, Bergendal B, Norderyd J, Nilsson D, Anderlid B-M, Nordgren A, Lindstrand A. Further evidence for specific IFIH1 mutation as a cause of Singleton-Merten syndrome with phenotypic heterogeneity. Am J Med Genet A. 2017;173:1396–1399. doi: 10.1002/ajmg.a.38214. [DOI] [PubMed] [Google Scholar]
- Rice GI, Bond J, Asipu A, Brunette RL, Manfield IW, Carr IM, Fuller JC, Jackson RM, Lamb T, Briggs TA, Ali M, Gornall H, et al. Mutations involved in Aicardi-Goutières syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet. 2009;41:829–832. doi: 10.1038/ng.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, Del Toro Duany Y, Jenkinson EM, Forte GM, Anderson BH, Ariaudo G, Bader-Meunier B, Baildam EM, Battini R, Beresford MW, Casarano M, Chouchane M, et al. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat Genet. 2014;46:503–509. doi: 10.1038/ng.2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, Forte GMA, Szynkiewicz M, Chase DS, Aeby A, Abdel-Hamid MS, Ackroyd S, Allcock R, Bailey KM, Balottin U, Barnerias C, Bernard G, et al. Assessment of interferon-related biomarkers in Aicardi-Goutières syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: a case-control study. Lancet Neurol. 2013;12:1159–1169. doi: 10.1016/S1474-4422(13)70258-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, Kasher PR, Forte GMA, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, Jenkinson EM, Bacino CA, et al. Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat Genet. 2012;44:1243–1248. doi: 10.1038/ng.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodero MP, Decalf J, Bondet V, Hunt D, Rice GI, Werneke S, McGlasson SL, Alyanakian M-A, Bader-Meunier B, Barnerias C, Bellon N, Belot A, et al. Detection of interferon alpha protein reveals differential levels and cellular sources in disease. J Exp Med. 2017;214:1547–1555. doi: 10.1084/jem.20161451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutsch F, MacDougall M, Lu C, Buers I, Mamaeva O, Nitschke Y, Rice GI, Erlandsen H, Kehl HG, Thiele H, Nürnberg P, Höhne W, et al. A specific IFIH1 gain-of-function mutation causes Singleton-Merten syndrome. Am J Hum Genet. 2015;96:275–282. doi: 10.1016/j.ajhg.2014.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesson C, Nawara M, Salih MAM, Rossignol R, Zaki MS, Al Balwi M, Schule R, Mignot C, Obre E, Bouhouche A, Santorelli FM, Durand CM, et al. Alteration of fatty-acid-metabolizing enzymes affects mitochondrial form and function in hereditary spastic paraplegia. Am J Hum Genet. 2012;91:1051–1064. doi: 10.1016/j.ajhg.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.