

Abstract
Deleterious variants in dihydropyrimidine dehydrogenase (DPD, DPYD gene) can be highly predictive of clinical toxicity to the widely prescribed chemotherapeutic 5-fluorouracil (5-FU). However, there is very limited data pertaining to the functional consequences of the >450 reported non-synonymous DPYD variants. We developed a DPYD-specific variant classifier (DPYD-Varifier) using machine learning and in vitro functional data for 156 missense DPYD variants. The developed model showed 85% accuracy and outperformed other in silico prediction tools. An examination of feature importance within the model provided additional insight into functional aspects of the DPD protein relevant to 5-FU toxicity. In the absence of clinical data for un-studied variants, prediction tools like DPYD-Varifier have great potential to individualize medicine and improve the clinical decision-making process.
Keywords: 5-Fluorouracil, Dihydropyrimidine Dehydrogenase, DPD Deficiency, Single Nucleotide, Polymorphism, SNPs, Pharmacogenetics, Random Forest (RF), Prediction
Introduction
5-Fluorouracil (5-FU) is commonly used to treat a wide variety of cancers, including colorectal and breast cancer. Although clinically effective, severe and potentially life-threatening toxicity remains a major limitation. Dihydropyrimidine dehydrogenase (DPD, encoded by the DPYD gene) is the initial and rate-limiting enzyme of 5-FU catabolism and, as such, is a critical determinant of 5-FU toxicity risk. While a few DPYD alleles have been characterized as risk variants, the molecular basis for most toxicity cases remains unknown. There is now strong evidence from a number of large clinical studies and meta-analyses that two missense DPYD variants (encoding p.I560S1 and p.D949V2) and a splice-site variant (c.1905+1G>A, also referred to as *2A3,4) act as valid predictors of 5-FU toxicity risk. More recently, a fourth variant, rs75017182 and the linked haplotype termed HapB3, has been shown to be associated with 5-FU toxicity5,6 by moderately reducing the expression of canonical DPYD through alternative splicing.7
In addition to the aforementioned four well-characterized variants, large-scale sequencing efforts such as the 1000 Genomes Project8 and the NIH Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project (ESP)9 have greatly expanded our knowledge of the genetic diversity of variants in the coding region for DPYD. There are currently more than 450 missense DPYD variants reported in NCBI dbSNP.10 Notably, very few of these variants have been studied functionally and/or for clinical association with 5-FU toxicity. Our lab has recently developed and utilized recombinant cellular model systems to examine the DPD enzyme activity of missense DPYD variants.11-13 Data from these studies suggest that approximately 41% of DPYD variants reduce DPD enzyme activity and, therefore, likely contribute to increased risk of 5-FU toxicity.11 Notably, these missense variants are more prevalent in non-Caucasian populations, which have been historically underrepresented in clinical studies.11,12 With continued progress in next-generation sequencing technologies, the number of variants with potential clinical relevance to 5-FU toxicity is expected to increase.
Given the rarity and unequal racial distribution of most DPYD variants, it is not feasible to conduct statistically powered clinical studies or association analyses. Therefore, in vitro and computational methods provide attractive alternatives to identify toxicity-associated variants. The primary goal of the present study was to develop a highly accurate in silico classifier to predict the functional impact of DPYD variants on DPD activity. To accomplish this, we measured the in vitro enzyme activity of 69 DPYD variants, which was combined with previous data from 87 variants.11-14 The developed multi-parameter gene-specific classifier, which we named DPYD-Varifier (DPYD-specific variant classifier) showed improved accuracy over existing in silico tools. In addition to acting as a classifier, the model also provided key insight into the features that contribute to DPD function.
Results
Enzyme activity of DPYD variants
We previously reported the functional effects of 87 DPYD variants.11-14 To generate data from an adequate number of variants on which to build a prediction model, we tested the relative DPD activity of all additional 69 variants that were reported in the 1000 Genomes Project Phase 3 Browser8 and the Exome Sequencing Project9 at the time of this study using previously described methods.11,13 As such, all variants from these two sources were tested. A 30% reduction in activity was chosen as a threshold for the binary classification of variants as “deleterious.” This threshold was chosen because all variants with enzyme activities below 70% showed significant differences (p<0.05) from wildtype DPD.
Of the 69 variants tested, 25 (36%) showed a 30% or greater reduction in activity compared to wildtype DPD (Figure 1A and Supplementary Table S1). Of the 25 deleterious variants, 9 exhibited DPD activities that were less than 15% of wildtype activity (K63E, R208Q, I281T, A549P, M680T, G795R, T737I, I948N, and H978Q). Eight variants showed modest reductions that were 15–35% of wildtype activity (M77V, C87F, Q156R, I560N, T573I, R692W, G767V, and S994T). Eight additional variants had moderate to mild reductions in DPD activity that were 35–70% of wildtype activity (T132S, V586A, G603E, K616Q, S644N, G676E, V714A, and A777D). The activities of the remaining 44 variants were greater than 70% of wildtype DPD activity, and these variants were, therefore, considered neutral even if p-values were less than 0.05. To confirm that the measurements of DPD activity were not specific to the cell type used to produce protein, the results for selected variants (*2A, p.Y186C, p.I560S, p.D949V, and p.H978Q) and wildtype DPD were measured in a second cell line. DPD activities were similar for variants regardless of whether they were expressed in HEK293T cells or HCT116 cells (r2 = 0.98, p = 1.20×10-4; Figure 1B).
Figure 1. In vitro characterization of DPYD missense variants.
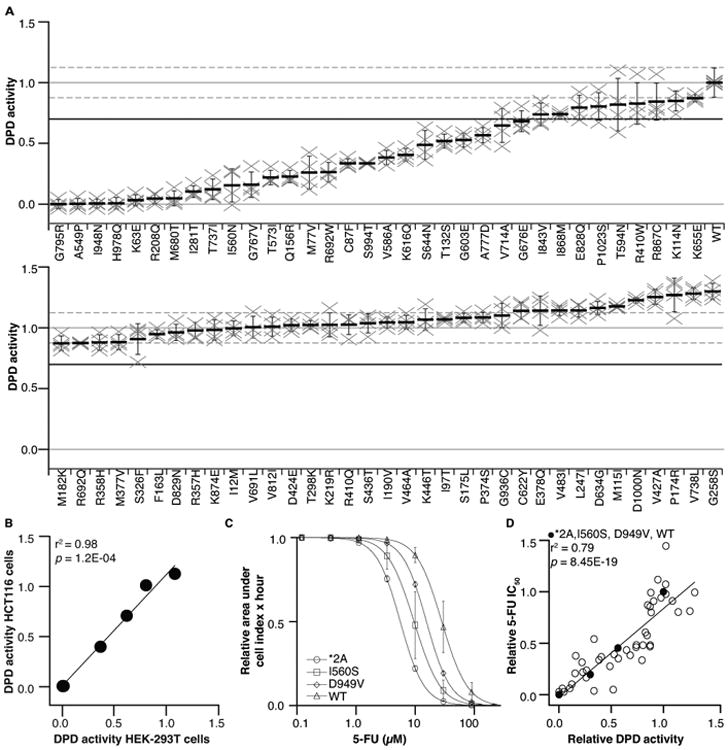
A. In vitro DPD activity was measured for 69 DPYD variants. Each “×” represents an independent biological replicate that is the mean of three technical replicates. The mean of four biological replicates is presented as a horizontal bar ± SD. The mean relative activity for wildtype DPD is presented as a horizontal solid gray line (crossing vertical axis at 1.0); the mean SD range for wildtype DPD is presented as dashed gray lines. The 70% threshold separating deleterious and neutral variants is presented as a solid black line. B. DPD activity of variants expressed in HEK-293T and HCT116 cells was positively correlated. C. 5-FU dose response was measured for cells expressing the indicated variants. Data points represent the mean ± SD from three independent experiments. D. In vitro DPD activity correlated with in vivo 5-FU sensitivity.
Cellular sensitivity to 5-FU conferred by DPYD variants
To confirm that changes in in vitro DPD activity translated to altered cellular sensitivity to 5-FU, we determined the IC50 for 5-FU in cells expressing DPYD variants. All 25 deleterious variants were tested, along with 22 randomly selected neutral variants and the 5-FU toxicity-associated variants *2A, p.I560S, and p.D949V. Dose-response curves for *2A, p.I560S, p.D949V, and wildtype DPD showed a gradient that mirrored the results observed for DPD activity in vitro (Figure 1C). A strong positive correlation between in vitro DPD activity and 5-FU IC50 was observed (r2 = 0.79, p = 8.45×10-19; Figure 1D).
Performance of existing in silico prediction tools
The accuracy of eight in silico tools (PROVEAN15, SIFT15, PolyPhen-216, FATHMM17, PhD-SNP18, SNP&GO19, UMD-Predictor20, and MutantAssessor21) for predicting the function of 156 DPYD variants characterized in the present and previous studies11-14 was assessed. PhD-SNP and PolyPhen-2 were the most accurate algorithms; however, all were <71% accurate when default score thresholds were used (Figure 2A, gray bars, and Supplementary Table S2). Additionally, the Matthews correlation coefficient (MCC), which is insensitive to unequal sample sizes in deleterious and neutral groups22, was computed at each method's default score threshold (Supplementary Table S2). Based on MCC, PROVEAN and PhD-SNP performed better than other tools (Supplementary Table S2).
Figure 2. Sensitivity, specificity, and accuracy of online missense mutation functional prediction tools.
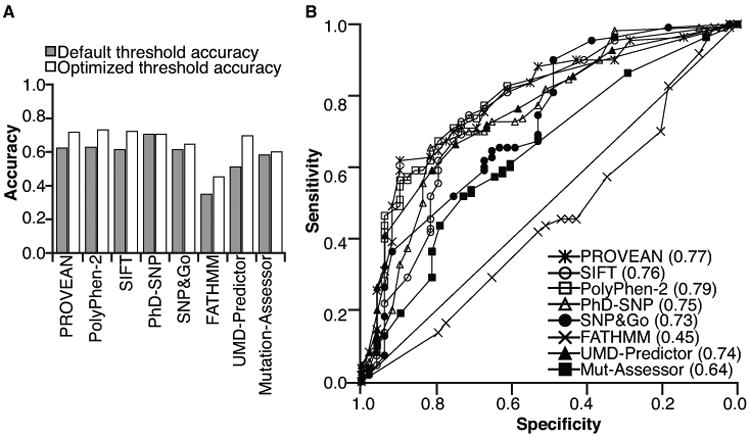
A. Accuracies of the indicated prediction tools were determined using default settings (gray bars) and using thresholds that were adjusted to balance sensitivity and specificity (white bars). B. ROC curves were generated for each tool. The AUC for each tool is presented in parenthesis.
To assess if any of the tools were biased toward sensitivity or specificity at the expense of the other parameter, we determined the area under the receiver operating characteristic (ROC) curve (AUC) for each tool (Figure 2B). Discrepancies were noted between AUC and accuracy estimates prompting us to identify the optimized threshold where sensitivity and specificity intersect for each tool (Supplementary Figure S1). Except for PhD-SNP, which utilized the optimized threshold by default, balancing sensitivity and specificity improved the accuracy of the tools (Figure 2A, compare gray and white bars). Overall, PolyPhen-2 (AUC = 0.79, accuracy = 0.72, MCC = 0.42), SIFT (AUC = 0.76, accuracy = 0.73, MCC = 0.43), and PROVEAN (AUC = 0.77, accuracy = 0.72, MCC = 0.41) performed the best (Supplementary Table S2).
Developing a DPYD-specific variant classifier (DPYD-Varifier)
Because existing in silico prediction tools were developed using data from multiple proteins, we reasoned that a model with a narrow focus on a single gene/protein would improve accuracy. To build our classifier, we trained a random forest model using in vitro DPD activity data from the 156 DPYD variants described above. Fifteen features of the DPD protein, including amino acid coordinates, 3D structural information, and changes in amino acid properties23-25, were used to split the random forest nodes (Figure 3 and Supplementary Table S3). Stratified sampling was used to avoid bias caused by unequal sample sizes in the deleterious and neutral groups. Grid searching was used to optimize the training parameters described below. The total number of trees was set to 200. The number of variables randomly sampled as candidates at each split was set to 1, and the node size was set to 5. In the developed model, the most important features (indicated by mean decrease in Gini coefficients) were the amino acid conservation score and the distances to the substrate-, iron sulfur cluster 1 (FeS-I)-, flavin mononucleotide (FMN)-, and flavin adenine dinucleotide (FAD)-binding domains (Figure 4A).
Figure 3. Overview of DPYD-Varifier.
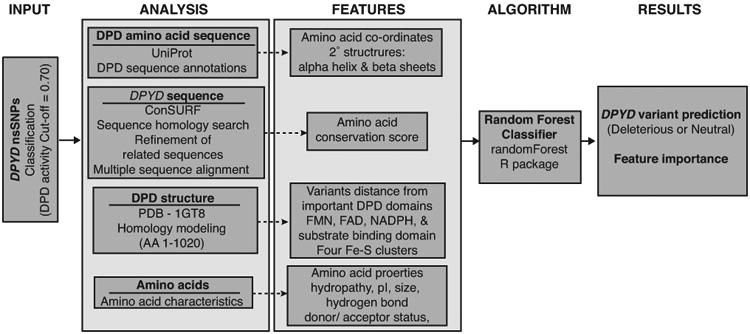
DPYD-Varifier was trained using relative DPD activity data obtained from in vitro experiments. DPYD and DPD sequence analysis was performed to obtain different features built on the classifier, including conservation scores, protein secondary structures, distance from domains, and amino acid biochemical properties. A random forest algorithm was utilized to obtain variant predictions and to assess feature importance to the model.
Figure 4. Performance of the DPYD-specific classifier.
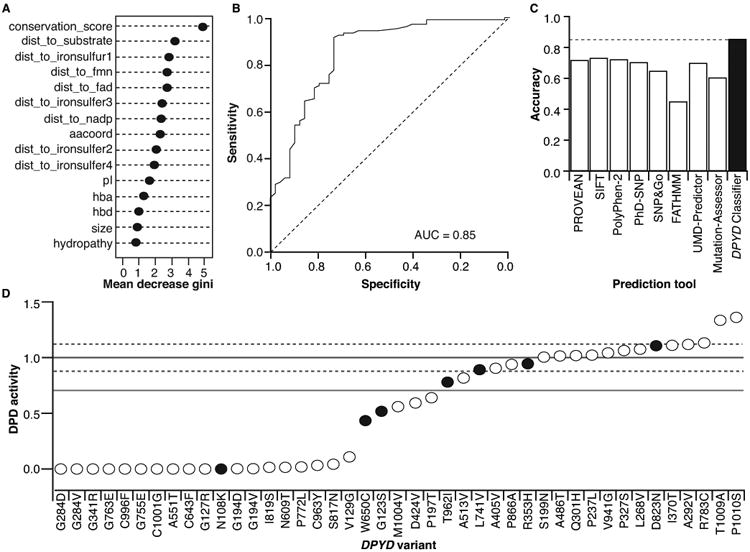
A. The importance of the 15 features utilized in the DPYD classifier is presented as the mean decrease in Gini coefficient. B. An ROC curve was generated for DPYD-Varifier predictions. C. The accuracy of DPYD-Varifier was assessed relative to other prediction tools. D. DPD activity was measured for 43 variants in an independent validation set. The activity of variants that matched DPYD-Varifier prediction is presented as open circles; the activity of variants that did not match the prediction is presented as closed black circles. The mean relative activity and SD for wildtype DPD are presented as solid and dashed gray lines, respectively. The 70% threshold separating deleterious and neutral variants is presented as a solid black line.
To measure the performance of the classifier algorithm, we used a leave-one-out cross-validation approach. To compare this classifier to the other prediction tools, we measured performance as the AUC (Figure 4B), the percent of accurate predictions (Figure 4C), and MCC (Supplementary Table S2). DPYD-Varifier was 85% accurate by both AUC (Figure 4B) and percent accurate predictions (Figure 4C), which was higher than any other tool tested. The MCC value for DPYD-Varifier (0.65) was also higher than other tools (Supplementary Table S2). To determine if DPYD-Varifier lacked any significant features inherent in the three other in silico models that performed the best (see Figure 2A), we constructed three additional classifiers using the PROVEAN, SIFT, and PolyPhen-2 scores as a 16th feature. The 16th feature ranked as one of the top two features in all three models (Supplementary Figure S2A–C). However, the additional features had minimal effect on AUC measures, further suggesting that DPYD-Varifier contains all of the relevant features that are inherent to the other three models (Supplementary Figure S2D).
Independent validation of the novel classifier
DPYD-Varifier was used to predict the functional effects of all additional 295 germline variants reported in dbSNP.10 Within this dataset, 30% of variants (N = 89) were predicted to be deleterious (Supplementary Table S4). As an independent assessment of accuracy for DPYD-Varifier, we randomly selected 25 deleterious and 18 neutral variants from this group for further analysis (Figure 4D). When the predictions were compared to in vitro enzyme activity, the classifier demonstrated 84% accuracy (36 of 43) and MCC value of 0.67, which was higher than that of any of the other in silico tools that were tested. The accuracy observed was similar to the 85% observed using leave-one-out validation (Figure 4C).
Structural modeling of variants
To begin to gain insight into the mechanisms through which deleterious variants affect DPD function, we mapped the locations of variants using a consensus model of human DPD based on the pig protein (93% homology11). Deleterious variants were enriched at conserved residues (p = 9.5×10-17; Figure 5A) and in close proximity to the substrate-binding site (p = 2.2×10-11), the FeS-I-binding site (p = 4.9×10-7), and the FMN-binding site (p = 2.8×10-10; Figure 5B). Interestingly, deleterious changes tended to be located farther from the FAD-binding site (p = 0.049; Figure 5B). However, after adjusting for substrate proximity using a linear model, deleterious amino acids were significantly closer to the FAD-binding site (mean distances in Ångströms, Å: deleterious amino acid = 33.7Å and neutral amino acid = 40.5Å; p = 0.014).
Figure 5. Structural modeling of DPYD variants.

Mean conservation scores (A) and mean 3-dimensional distances of variants from geometric center of substrate (Subs.)-binding, FeS-I-binding, FMN-binding, and FAD-binding sites are presented for deleterious (Del) and neutral (Neu) variants (B). C. The five domains of modeled human DPD are color-coded: Domain I – cyan, Domain II – orange, Domain III – purple, Domain IV – blue, and Domain V – green. UAA, uracil-4-acetic acid (uracil analog used to model the closed/open state in PDB); FMN, flavin mononucleotide; FeS1-4, iron sulfur clusters 1-4; FAD, flavin adenine dinucleotide; and NADPH are labeled. Amino acids #636–671 and amino acids #736–737, which directly interact with uracil/5-FU, are shown in white. The location of variants within 15Å of uracil/5-FU (D), FeS-I (E), FMN (F), and FAD (G) are presented on the DPD 3-dimensional structure and are color coded based on DPYD-Varifier predictions and in vitro data. Red, true deleterious variants (predicted deleterious, deleterious in vitro); magenta, true neutral variants (predicted neutral, neutral in vitro); yellow, false deleterious (predicted deleterious, neutral in vitro); green, false neutral (predicted neutral, deleterious in vitro).
The DPD protein consists of five major domains (Figure 5C), which have been detailed previously.26,27 To identify amino acid changes that might interfere with binding to substrate, FeS-I, FMN, and FAD, we examined variants within 15Å of the geometric center of those sites. Deleterious variants were generally enriched near these locations. Eight deleterious and one neutral variant were located within 15Å of the substrate-coordinating site (Figure 5D). Two deleterious variants (p.R592W and p.R592Q) were located at the same residue 14.3Å from the substrate site. Four of the six variants near the FeS-I site were deleterious (Figure 5E), and nine of 15 variants near the FMN-interacting site impaired DPD activity (Figure 5F). Of the FMN-proximal variants that affected the same amino acids, both p.I560S and p.I560N were deleterious, whereas neither p.V732G nor p.V732I affected DPD activity. Three deleterious and three neutral variants were detected near the FAD-binding site (Figure 5G).
Allele frequencies of deficient variants
DPYD-Varifier permitted us, for the first time, to estimate the overall frequency of deleterious variants in various racial/geographic groups (Figure 6A–C). Notably, the well-studied variants *2A, p.I560S, and p.D949V were most prevalent in European and European American populations, but they were generally absent in East Asian populations. Other deleterious variants (both those identified in in vitro studies and using DPYD-Varifier) were generally more prevalent as a group in African, East Asian, and South Asian populations (Figure 6A–C). Individual deleterious variants identified by DPYD-Varifier, for which in vitro data are not available, ranged in allele frequency from 0.000015 to 0.000728 within ExAC populations (Figure 6C).
Figure 6. Population frequencies of deleterious variants.
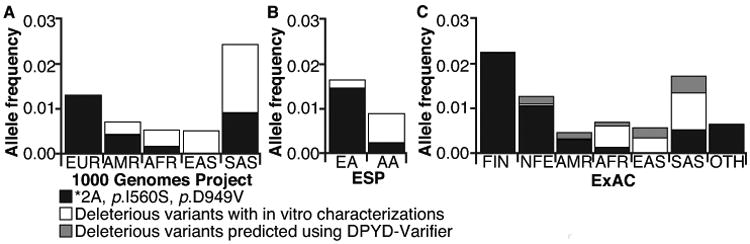
A. Allele frequency data for deleterious DPYD variants among individuals of European (EUR), American (AMR), African (AFR), East Asian (EAS), and South Asian (SAS) ancestry were obtained from the 1000 Genomes Project. B. Data from individuals of European American (EA) and African American (AA) ancestry were obtained from NHLBI ESP. C. Data from individuals of European Finnish (FIN), non-Finnish European (NFE), Latino (AMR), African (AFR), East Asian (EAS), South Asian (SAS), and other (OTH) ancestry were obtained from the ExAC database. Cumulative allele frequencies for the three 5-FU toxicity-associated variants (*2A, p.I560S, and p.D949V) are presented in black. Cumulative frequencies of additional variants established as deleterious based on in vitro functional data are presented in white. Cumulative frequencies of variants classified as deleterious by DPYD-Varifier, but not tested in vitro, are presented in gray.
Discussion
Variants in DPYD are generally accepted to have the potential to greatly increase the risk of severe and possibly deadly toxicity to 5-FU. Identifying the risk variants among the >450 non-synonymous SNPs that have been reported in the DPYD gene10 is of the utmost importance for identifying at-risk individuals and potentially adjusting 5-FU dose. As with most pharmacological risk variants, this task is further complicated by the lack of any outward indicators of toxicity risk until after the drug is administered. In the current study, we developed a DPYD- specific classifier (DPYD-Varifier) to predict the function of any missense DPYD variant. This model was built using in vitro DPD activity data for 156 variants. Overall, DPYD-Varifier was more accurate (85%) than other existing in silico prediction tools. Using this model, we were able to estimate the combined allele frequencies for all reported deleterious DPYD variants.
Four deleterious DPYD variants, *2A, p.I560S, p.D949V, and rs75017182, have been reproducibly associated with reduced DPD function and increased 5-FU toxicity in clinical and laboratory studies.28-32 However, the expanding release of genome- and exome-level data has made it clear that the genetic diversity within the DPYD gene is far greater than previously appreciated. The clinical implications of nearly all of these newly reported variants are unknown. The primary goal of this study was to develop a highly accurate model for classifying DPYD variants. Existing in silico prediction tools rely upon data from public sources, such as ClinVar33 and Human Gene Mutation Database (HGMD)34, which contain a paucity of information that is specific to DPYD. Therefore, we chose to develop a classifier that included features specific to DPYD and was based on in vitro data obtained from a reproducible isogenic system. It has been previously established that features pertaining to a particular protein, such as biochemical properties and structural characteristics, are diluted as a consequence of adding additional proteins into prediction models.35 Therefore, accuracy is expected to be higher in a protein-specific tool35,36, which is consistent with the improved accuracy of DPYD-Varifier (Figure 2B, 4B–C). The predicted phenotypes obtained from DPYD-Varifier have the potential to be of clinical benefit for carriers of rare deleterious variants (i.e., see Supplementary Table S4). It should be noted that the threshold for establishing deleterious/neutral status for variants was determined using in vitro data and that the clinical validity of that threshold cannot be rationally tested. Therefore, additional testing in the clinical setting (e.g., ex vivo DPD activity or post-treatment therapeutic dose monitoring) may be considered to further identify optimal therapeutic dosing for carriers of deleterious variants.
The location of altered amino acids within the protein structure can be used to better understand the mechanisms through which specific variants affect protein function. For instance, the deleterious variant p.D949V is located near the cofactor-binding region, whereas the neutral p.C29R variant is unlikely to affect cofactor or substrate interactions as previously discussed.37 As expected, our data demonstrate that deleterious variants are enriched at conserved amino acid residues and are generally located near important binding/coordinating sites within DPD (e.g., substrate, FeS-I, FMN; Figure 5). Additionally, our data show that p.K63E, one of the few variants that has been previously reported relative to the protein structure, is located 12.3Å from the FeS-II site. This proximity could indicate that p.K63E may interfere with electron shuttling between FeS sites, as has been previously suggested.38 Unexpectedly, our data indicated that deleterious variants are more likely to be distal from the FAD-binding site (Figure 5B). However, it was noted that the FAD- and substrate-binding sites are located at opposite sides of the 3-dimensional protein structure (Figure 5C). Therefore, we hypothesized that the strong contribution of substrate-binding proximity was masking the potential contribution of FAD-site proximity in our analyses. Consistent with this hypothesis, when the distance to substrate was treated as a covariate, deleterious variants were significantly closer to the FAD-binding site than neutral variants (p = 0.014). Similar observations were noted for variants located in proximity to the NAPDH-binding site (data not shown).
Because distances to substrate-/cofactor-binding sites were highly important features in DPYD-Varifier, we were concerned that the model might be biased toward classifying all variants in proximity to these sites as deleterious. This, however, does not appear to be the case. Of the 36 variants that were tested within 15Å of these sites, only 6 were misclassified. This 17% error rate is similar to the 15% error rate for the algorithm overall. In terms of important features, there were no significant differences in conservation scores and distances to substrate- or cofactor-binding sites between correctly and incorrectly predicted variants. There also does not appear to be a bias toward false-deleterious predictions since three false-deleterious (p.V738L, p.N151D, and p.T768K) and three false-neutral (p.M77V, p.C87F, and p.I281T) predictions were observed (Figure 5D–G). The performance of DPYD-Varifier was similar for predicting variants that were not in close proximity to these important sites, with an error rate of 13.6%. These findings suggest that, while substrate and cofactor binding distances are important to the prediction algorithm, additional variables contribute to the performance of the overall model.
Variants that are relevant to the pharmacology of a wide variety of compounds are being identified and characterized at a steadily increasing rate. Perhaps most importantly, the classifier model that we developed has further elucidated the importance of population stratification as it relates to the carrier frequencies of pharmacologically relevant variants in different racial/geographic populations. As such, our data highlight the fact that comparatively little is known regarding risk variants outside of Caucasian haplotypes, which represent the predominant study cohorts used for most clinical studies of 5-FU toxicity. In conclusion, the integration of relevant and accurate data from computational and laboratory models into the precision medicine decision-making process has the potential to improve patient care in situations where decisive clinical data is unavailable.
Materials and Methods
Cells
HEK293T/c17 (culture CRL-11268) and HCT116 (culture CCL-247) cells were obtained from the American Type Culture Collection (ATCC). Cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM; Corning Inc., Corning, NY) supplemented with 10% fetal bovine serum (FBS; MilliporeSigma, Billerica, MA), 100 IU mL-1 penicillin (Corning), and 1% L-Glutamate (Corning) in a humidified 37°C incubator with 5% CO2 atmosphere. Cells were cultured for no more than 20 total passages or 2 months, whichever occurred first. Culture identity and health were monitored by microscopy.13 Cultures were periodically monitored for mycoplasma contamination by Hoechst staining (Sigma-Aldrich, St. Louis, MO).
Vector construction
Expression vectors were constructed as previously described.11,13 Briefly, variants were introduced into a DPYD expression plasmid using PCR-based site-directed mutagenesis using primers reported in Supplementary Table S5. Plasmid identity was confirmed by restriction digestion and sequencing using previously reported primers.13 All primers were obtained from IDT (Coralville, IA), and sequencing was performed by the Mayo Clinic Cancer Center Gene Analysis Shared Resource in the Mayo Clinic Advanced Genomics Technology Center.
DPD enzyme activity measurement
The activity of recombinant DPD was measured as previously detailed.11 Transfections were performed using 1 μg of plasmid per well of a 6-well plate using X-tremeGENE HP (Roche, Indianapolis, IN) according to the manufacturer's instructions. Three individual wells were transfected in parallel for each variant (considered technical replicates). Each set of technical replicates was independently tested at least four times (considered biological replicates). Transfections for wildtype DPYD and a coding sequence mimic of the catalytically inactive variant *2A were performed in parallel for each experiment. Forty-eight hours after transfection, cell lysates were prepared, and total protein concentration (BioRad Protein Assay; BioRad, Hercules, CA) and protein expression (Bio-Dot SF; BioRad, Hercules, CA) were quantified.
Cell lysates were incubated with NADPH (200 μmol L-1; Sigma-Aldrich) and 5-[14C]-FU (8.23 μM L-1; Moravek Biochemicals, Brea, CA) at 37°C for 30 minutes with agitation in a reaction buffer consisting of 35 mmol L-1 potassium phosphate buffer at pH 7.4 supplemented with 2.5 mmol L-1 MgCl2, 0.035% 2-mercaptoethanol, and complete EDTA-free protease inhibitor cocktail (Roche). 5-[14C]-FU and [4C]-Dihydro-FU (DHFU) were separated by high-performance liquid chromatography (HPLC; Dionex/Thermo Fisher Scientific, Waltham, MA) using a reverse-phase C18 column (Grace, Columbia, MD) connected to a Radiomatic 625TR flow scintillation analyzer (PerkinElmer, Waltham, MA). 5-FU and DHFU peak area integration was performed using Chromeleon 7.2 (Thermo Scientific, Waltham, MA). DPD enzyme activity values were normalized to DPD protein expression for each variant relative to controls that were transfected and prepared in parallel, as previously described.11
Cellular sensitivity to 5-FU
Cell viability was monitored using the xCELLigence MP Real-Time Cell Analysis (RTCA) system (Acea Biosciences, San Diego, CA) as previously described.13 Background impedance values for RTCA E-View plates (Acea Biosciences) were acquired using complete DMEM before plating cells. Cells were seeded at a concentration of 7,000 cells per well. Twenty-four hours after plating, 0.1 μg of expression vector was transfected using X–tremeGENE HP (Roche). Impedance values (expressed as cell index, CI, units) were recorded every 15 minutes. Twenty-four hours after transfection, cells were treated with 5-FU at various concentrations. CI measurements were acquired for an additional 6 days. For each experiment, wildtype DPYD and *2A mimic controls were tested in parallel. Average of results from at least two independent experiments, consisting of three technical replicates, were used in this manuscript.
CI values were normalized to the value that was recorded immediately following the application of drug to cells. The area under the curve for the normalized CI was determined between the treatment time point and the time point at which the untreated CI reached its maximum CI as previously described.13 Four Parameter Logistic (4PL) nonlinear regression analysis was used to determine IC50 concentrations (JMP, SAS Institute Inc., Cary, NC). Linear regression was utilized to determine the correlation between DPD activity and 5-FU IC50.
In silico functional prediction tools
The performance of eight different prediction tools, including PROVEAN15, SIFT15, PolyPhen-216, FATHMM17, PhD-SNP18, SNP&GO19, MutantAssessor21, and UMD-Predictor20, was assessed using 156 DPYD variants. The HumDiv-trained PolyPhen-2 algorithm, which has been suggested to be more accurate for predicting the effects of rare variants16, was used instead of the HumVar-trained model. Prediction scores were dichotomized based on the default threshold for each tool, and the prediction accuracy was determined by comparing individual variant predictions to in vitro results. The balanced measurement of qualitative outcomes prediction was also calculated as the MCC, which generates scores that range from -1 to 1. MCC is one of the elective metrics in the US FDA-led MicroArray Quality Control (MAQC-II) initiative, which compares and validates predictive models in individualized medicine.39 To further optimize the predictive ability of each tool, a new prediction score threshold was selected that balanced sensitivity and specificity. Accuracy and MCC were determined at the optimized thresholds. Additionally, ROC curves were generated to compare the performance of each tool using the AUC.
Development of a novel computational classification model for DPYD variants
The DPYD-specific computational model was developed using the Random Forest R package v.4.6-12 (Random Forest Classification algorithm).40 A total of 156 DPYD variants with predetermined in vitro classifications and fifteen different features were used to train the model. The two-dimensional DPD amino acid sequence alignment was utilized to obtain amino acid coordinates and map secondary structures. Conservation scores for each amino acid were determined using ConSurf (ConSeq method) as described.41-43 A homology-based model for the human DPD structure was generated in Chimera44 using the crystal structure for pig DPD (PDB ID 1GT8)26. The homology structure was used to measure distances between amino acids and 3D geometric center of substrate-, FeS-, FAD-, FMN-, or NADP-binding/coordinating regions. Changes in amino acid properties (i.e., hydropathy, isoelectric point, size, and hydrogen bond donor or acceptor status) were also included as features in the random forest model.23-25 To minimize this potential bias from unequal sample sizes for neutral (n = 107) and deleterious (n = 49) variants, stratified sampling was used to generate bootstrap data for the training set. Grid search was used to determine the optimal values of parameters such as total number of trees, the number of features to use at each split, and the minimal number of samples a node can have. Gini index (measured as mean decrease in Gini coefficient), which is a measure of node purity or how well a given feature splits a given dataset, was used to evaluate feature importance. Leave-one-out cross validation was performed to estimate DPYD-Varifier AUC and accuracy.
DPYD-Varifier was also validated using an independent set of variants. Briefly, DPYD-Varifier was used to predict the function for all DPYD variants reported in dbSNP with no known functional data available at the time. Then, 25 deleterious and 25 neutral variants were randomly chosen and site directed mutagenesis performed to generate the requisite expression plasmids. The plasmid sequences for all 25 deleterious variants were confirmed, and 18 of 25 neutral variants were confirmed, following one attempt at mutagenesis. These 43 variants were used for the validation study. We did not further explore the reasons why sequencing failed for 7 neutral variant expression plasmids, nor did we re-attempt construction.
Structural modeling
A homology-directed model of human DPD (NP_000101.2) was generated using the crystal structure of substrate-bound porcine DPD (PDB ID 1GT8)27 using Chimera Modeler (UCSF Chimera version 1.8)44.
Allele frequency determination
Allele frequencies were obtained from the 1000 Genomes Project8, NHLBI ESP9, and ExAc45 databases. Details about specific populations can be found on the projects' websites.8,9,45
Statistics
General statistical analyses, including Pearson correlation, two-tailed t-test analyses, and linear regression model were performed using R v.3.3.1. The probabilistic classifier for the random forest algorithm was performed using the Random Forest R package (v.4.6-12). The AUC was computed using PRISM v.7.0a (GraphPad Software Inc.). Data transformations and analyses of IC50 were performed using JMP v.9.0.3 (SAS Institute Inc.).
Supplementary Material
Figure S1. Optimization of prediction score threshold. New optimized thresholds were established where sensitivity and specificity were balanced for PROVEAN (A), SIFT (B), PolyPhen-2 (C), PhD-SNP (D), SNP&GO (E), FATHMM (F), UMD-Predictor (G), and MutationAssessor (H). Closed circles, specificity; open circles, sensitivity; gray vertical lines, default score thresholds; black vertical lines, optimized score thresholds.
Figure S2. DPYD-Varifier performance with scores from existing prediction tools. Mean decreases in Gini coefficients are presented for models incorporating scores from PROVEAN (A), SIFT (B), and PolyPhen-2 (C) as 16th features in DPYD-Varifier. D. ROC curves for the three models are presented. The AUC for each model is shown in parentheses.
Table S1. In vitro DPD activity and 5-FU IC50 values for DPYD non-synonymous SNPs identified from NCBI dbSNP, 1000 Genomes Project, and Exome Sequencing Projects
Table S2. Comparison of different in silico tools and DPYD-Varifier for prediction of DPYD variants in the leave-one-out validation set
Table S3. Complete list of features considered for use in DPYD-Varifier
Table S4. Binary predictions for novel variants identified in 1000 Genomes database using DPYD-Varifier
Table S5. List of primers used for cloning DPYD variants
Study Highlights.
What is the current knowledge on the topic?
Adverse responses to chemotherapy remain a significant problem. Deleterious variants in the dihydropyrimidine dehydrogenase gene (DPYD) have been associated with increased risk for 5-fluorouracil (5-FU)-associated toxicity. There is very limited clinical data for most of the 400+ non-synonymous variants that have been reported in DPYD.
What question did this study address?
This study evaluated the performance of a novel in silico classifier DPYD-Varifier that was developed using protein specific data and the in vitro enzyme activity data from 156 reported DPYD variants.
What does this study add to our knowledge?
DPYD-Varifier exhibited improved accuracy over existing tools for identifying deleterious DPYD variants that are likely to impair 5-FU catabolism and thus contribute to an increased risk of 5-FU toxicity. By applying this model to large-scale sequence databases, we report the estimated carrier frequencies for deleterious DPYD variants in different racial/geographic populations.
How might this change clinical pharmacology or translational science?
For patients carrying DPYD variants that lack clinical annotations, DPYD-Varifier can be used to integrate genotype information to guide individualized 5-FU dosing.
Acknowledgments
The authors thank Kelly Bouchonville, Ph.D., for proofreading and technical editing of the manuscript. We thank the Mayo Clinic Cancer Center for supporting shared resources used in generating data for this manuscript (NIH CA 15083).
Funding: This study is funded in part by NIH P30 CA15083 grant.
Footnotes
Conflict of Interest: The authors declared no competing interests for this work.
Author Contributions: S.S., C.Z., H.L., S.M.O., and R.B.D. wrote the manuscript; S.S., S.M.O., and R.B.D. designed the research; S.S., C.Z., C.R.J., and Q.N. performed the research; S.S., C.Z., and S.M.O. analyzed the data.
References
- 1.Johnson MR, Wang K, Diasio RB. Profound dihydropyrimidine dehydrogenase deficiency resulting from a novel compound heterozygote genotype. Clin Cancer Res. 2002;8:768–774. [PubMed] [Google Scholar]
- 2.van Kuilenburg AB, et al. Clinical implications of dihydropyrimidine dehydrogenase (DPD) deficiency in patients with severe 5-fluorouracil-associated toxicity: identification of new mutations in the DPD gene. Clin Cancer Res. 2000;6:4705–4712. [PubMed] [Google Scholar]
- 3.Johnson MR, Diasio RB. Importance of dihydropyrimidine dehydrogenase (DPD) deficiency in patients exhibiting toxicity following treatment with 5-fluorouracil. Advances in Enzyme Regulation. 2001;41:151–157. doi: 10.1016/s0065-2571(00)00011-x. [DOI] [PubMed] [Google Scholar]
- 4.Ridge SA, et al. Dihydropyrimidine dehydrogenase pharmacogenetics in patients with colorectal cancer. Br J Cancer. 1998;77:497–500. doi: 10.1038/bjc.1998.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amstutz U, Farese S, Aebi S, Largiader CR. Dihydropyrimidine dehydrogenase gene variation and severe 5-fluorouracil toxicity: a haplotype assessment. Pharmacogenomics. 2009;10:931–944. doi: 10.2217/pgs.09.28. [DOI] [PubMed] [Google Scholar]
- 6.van Kuilenburg AB, et al. Intragenic deletions and a deep intronic mutation affecting pre-mRNA splicing in the dihydropyrimidine dehydrogenase gene as novel mechanisms causing 5-fluorouracil toxicity. Human genetics. 2010;128:529–538. doi: 10.1007/s00439-010-0879-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nie Q, et al. Quantitative contribution of rs75017182 to dihydropyrimidine dehydrogenase mRNA splicing and enzyme activity. Clinical pharmacology and therapeutics. 2017;102:662–670. doi: 10.1002/cpt.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.The 1000 Genomes Browser. Vol. 2016 Hinxton, Cambridgeshire (UK): The European Bioinformatics Institute (EBI); 1000 Genomes release 13. [Google Scholar]
- 9.Exome Variant Server. Vol. 2015. Seattle (WA): NHLBI GO Exome Sequencing Project (ESP); Seattle, WA: EVS data release ESP6500SI-V2. [Google Scholar]
- 10.Database of Single Nucleotide Polymorphisms (dbSNP) Vol. 2016. Bethesda (MD): National Center for Biotechnology Information, National Library of Medicine; Bethesda, MD: dbSNP Build ID: 138. [Google Scholar]
- 11.Offer SM, et al. Comparative functional analysis of DPYD variants of potential clinical relevance to dihydropyrimidine dehydrogenase activity. Cancer research. 2014;74:2545–2554. doi: 10.1158/0008-5472.CAN-13-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Offer SM, et al. A DPYD Variant (Y186C) in Individuals of African Ancestry Is Associated With Reduced DPD Enzyme Activity. Clin Pharmacol Ther. 2013;94:158–166. doi: 10.1038/clpt.2013.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Offer SM, Wegner NJ, Fossum C, Wang K, Diasio RB. Phenotypic profiling of DPYD variations relevant to 5-fluorouracil sensitivity using real-time cellular analysis and in vitro measurement of enzyme activity. Cancer Res. 2013;73:1958–1968. doi: 10.1158/0008-5472.CAN-12-3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elraiyah T, et al. Novel Deleterious Dihydropyrimidine Dehydrogenase Variants May Contribute to 5-Fluorouracil Sensitivity in an East African Population. Clinical Pharmacology & Therapeutics. 2016:n/a–n/a. doi: 10.1002/cpt.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31:2745–2747. doi: 10.1093/bioinformatics/btv195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Current protocols in human genetics / editorial board, Jonathan L Haines … [et al] 2013;Chapter 7:Unit7 20. doi: 10.1002/0471142905.hg0720s76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shihab HA, et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat. 2013;34:57–65. doi: 10.1002/humu.22225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Capriotti E, Altman RB. Improving the prediction of disease-related variants using protein three-dimensional structure. BMC Bioinformatics. 2011;12 Suppl 4:S3. doi: 10.1186/1471-2105-12-S4-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thusberg J, Olatubosun A, Vihinen M. Performance of mutation pathogenicity prediction methods on missense variants. Hum Mutat. 2011;32:358–368. doi: 10.1002/humu.21445. [DOI] [PubMed] [Google Scholar]
- 20.Salgado D, et al. UMD-Predictor: A High-Throughput Sequencing Compliant System for Pathogenicity Prediction of any Human cDNA Substitution. Hum Mutat. 2016;37:439–446. doi: 10.1002/humu.22965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39:e118. doi: 10.1093/nar/gkr407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baldi P, Brunak S, Chauvin Y, Andersen CA, Nielsen H. Assessing the accuracy of prediction algorithms for classification: an overview. Bioinformatics. 2000;16:412–424. doi: 10.1093/bioinformatics/16.5.412. [DOI] [PubMed] [Google Scholar]
- 23.3.0, C.H.P.V. Amino Acids with pKa. 2000;2017 [Google Scholar]
- 24.Foulquier E. In: Amino acids. Ginestoux C, editor. 2001. IMGT®, the international ImMunoGeneTics information system®. [Google Scholar]
- 25.UniProt. UniProtKB - Q12882 (DPYD_HUMAN) Vol. 2017. UniProtKB: 2017. [Google Scholar]
- 26.Dobritzsch D, Ricagno S, Schneider G, Schnackerz KD, Lindqvist Y. Crystal structure of the productive ternary complex of dihydropyrimidine dehydrogenase with NADPH and 5-iodouracil. Implications for mechanism of inhibition and electron transfer. J Biol Chem. 2002;277:13155–13166. doi: 10.1074/jbc.M111877200. [DOI] [PubMed] [Google Scholar]
- 27.Dobritzsch D, Schneider G, Schnackerz KD, Lindqvist Y. Crystal structure of dihydropyrimidine dehydrogenase, a major determinant of the pharmacokinetics of the anti-cancer drug 5-fluorouracil. EMBO J. 2001;20:650–660. doi: 10.1093/emboj/20.4.650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boige V, et al. DPYD Genotyping to Predict Adverse Events Following Treatment With Flourouracil-Based Adjuvant Chemotherapy in Patients With Stage III Colon Cancer: A Secondary Analysis of the PETACC-8 Randomized Clinical Trial. JAMA Oncol. 2016 doi: 10.1001/jamaoncol.2015.5392. [DOI] [PubMed] [Google Scholar]
- 29.Li Q, et al. Influence of DPYD Genetic Polymorphisms on 5-Fluorouracil Toxicities in Patients with Colorectal Cancer: A Meta-Analysis. Gastroenterol Res Pract. 2014;2014:827989. doi: 10.1155/2014/827989. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Meulendijks D, et al. Clinical relevance of DPYD variants c.1679T>G; c.1236G>A/HapB3, and c.1601G>A as predictors of severe fluoropyrimidine-associated toxicity: a systematic review and meta-analysis of individual patient data. The Lancet Oncology. 2015;16:1639–1650. doi: 10.1016/S1470-2045(15)00286-7. [DOI] [PubMed] [Google Scholar]
- 31.Lee AM, et al. DPYD Variants as Predictors of 5-fluorouracil Toxicity in Adjuvant Colon Cancer Treatment (NCCTG N0147) Journal of the National Cancer Institute. 2014;106 doi: 10.1093/jnci/dju298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee AM, et al. Association between DPYD c.1129-5923 C>G/hapB3 and severe toxicity to 5-fluorouracil-based chemotherapy in stage III colon cancer patients: NCCTG N0147 (Alliance) Pharmacogenet Genomics. 2016;26:133–137. doi: 10.1097/FPC.0000000000000197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Landrum MJ, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44:D862–868. doi: 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stenson PD, et al. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat. 2003;21:577–581. doi: 10.1002/humu.10212. [DOI] [PubMed] [Google Scholar]
- 35.Crockett DK, et al. Utility of gene-specific algorithms for predicting pathogenicity of uncertain gene variants. J Am Med Inform Assoc. 2012;19:207–211. doi: 10.1136/amiajnl-2011-000309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jordan DM, et al. Development and validation of a computational method for assessment of missense variants in hypertrophic cardiomyopathy. Am J Hum Genet. 2011;88:183–192. doi: 10.1016/j.ajhg.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Kuilenburg AB. Dihydropyrimidine dehydrogenase and the efficacy and toxicity of 5-fluorouracil. Eur J Cancer. 2004;40:939–950. doi: 10.1016/j.ejca.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 38.Kleibl Z, et al. Influence of dihydropyrimidine dehydrogenase gene (DPYD) coding sequence variants on the development of fluoropyrimidine-related toxicity in patients with high-grade toxicity and patients with excellent tolerance of fluoropyrimidine-based chemotherapy. Neoplasma. 2009;56:303–316. doi: 10.4149/neo_2009_04_303. [DOI] [PubMed] [Google Scholar]
- 39.Boughorbel S, Jarray F, El-Anbari M. Optimal classifier for imbalanced data using Matthews Correlation Coefficient metric. PLoS One. 2017;12:e0177678. doi: 10.1371/journal.pone.0177678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brieman Leo C, Adele Liaw, Andy Matthew Weiner. Breiman and Cutler's Random Forests for Classification and Regression. 2015 [Google Scholar]
- 41.Berezin C, et al. ConSeq: the identification of functionally and structurally important residues in protein sequences. Bioinformatics. 2004;20:1322–1324. doi: 10.1093/bioinformatics/bth070. [DOI] [PubMed] [Google Scholar]
- 42.Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N. ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010;38:W529–533. doi: 10.1093/nar/gkq399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ashkenazy H, et al. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016;44:W344–350. doi: 10.1093/nar/gkw408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pettersen EF, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 45.Karczewski KJ, et al. The ExAC browser: displaying reference data information from over 60 000 exomes. Nucleic Acids Res. 2017;45:D840–D845. doi: 10.1093/nar/gkw971. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Optimization of prediction score threshold. New optimized thresholds were established where sensitivity and specificity were balanced for PROVEAN (A), SIFT (B), PolyPhen-2 (C), PhD-SNP (D), SNP&GO (E), FATHMM (F), UMD-Predictor (G), and MutationAssessor (H). Closed circles, specificity; open circles, sensitivity; gray vertical lines, default score thresholds; black vertical lines, optimized score thresholds.
Figure S2. DPYD-Varifier performance with scores from existing prediction tools. Mean decreases in Gini coefficients are presented for models incorporating scores from PROVEAN (A), SIFT (B), and PolyPhen-2 (C) as 16th features in DPYD-Varifier. D. ROC curves for the three models are presented. The AUC for each model is shown in parentheses.
Table S1. In vitro DPD activity and 5-FU IC50 values for DPYD non-synonymous SNPs identified from NCBI dbSNP, 1000 Genomes Project, and Exome Sequencing Projects
Table S2. Comparison of different in silico tools and DPYD-Varifier for prediction of DPYD variants in the leave-one-out validation set
Table S3. Complete list of features considered for use in DPYD-Varifier
Table S4. Binary predictions for novel variants identified in 1000 Genomes database using DPYD-Varifier
Table S5. List of primers used for cloning DPYD variants