

Abstract
Caspases belong to a diverse clan of proteolytic enzymes known as clan CD with highly disparate functions in cell signaling. The caspase members of this clan are only found in animals, and most of them orchestrate the demise of cells by the highly distinct regulated cell death phenotypes known as apoptosis and pyroptosis. This review looks at the mechanistic distinctions between the activity and activation mechanisms of mammalian caspases compared to other members of clan CD. We also compare and contrast the role of different caspase family members that program anti-inflammatory and pro-inflammatory cell death pathways.
Keywords: Apoptosis, pyroptosis, regulated cell death, protease, gasdermin
1. INTRODUCTION
1.1 General preamble on caspases
Caspases are proteases that elicit and propagate signaling events resulting in cellular death: apoptotsis implemented by apoptotic caspases and pyroptosis performed by inflammatory caspases (Fig 1). Their name derives from the common use of a Cys side chain acting as nucleophile during peptide bond hydrolysis, and a rare primary specificity for cleaving after Asp – they are cysteine-dependent aspartate-specific proteases. The ancestor of caspases is ancient, and its descendants (officially peptidase clan CD) are found widespread across all life kingdoms [1], but the distinctive Asp specificity is unique to metazoans [2]. In vertebrates the apoptotic caspases can be further subdivided into initiator (caspases-8, −9 and −10) and effector (caspases-3, −6 and −7) caspases [3]. The apoptotic and inflammatory caspases have moderately well defined biochemical and biological mechanisms, but confusion reigns regarding the role of caspase-2. Caspase-2 has been reported to serve many roles within and outside apoptotic networks, and recent data suggests its participation in cell cycle regulation [4]. Readers interested in the many (sometimes conflicting) conflicting functions of caspase-2 are directed to the following reviews [4–9]. Caspase-14, generally not considered an apoptotic or inflammatory caspase, seems to have a highly specialized, though mechanistically obscure, role in keratinization of the skin [10].
Figure 1. Peptidase clan CD dendrogram illustrating evolutionary relationships of human caspases and their homologs.
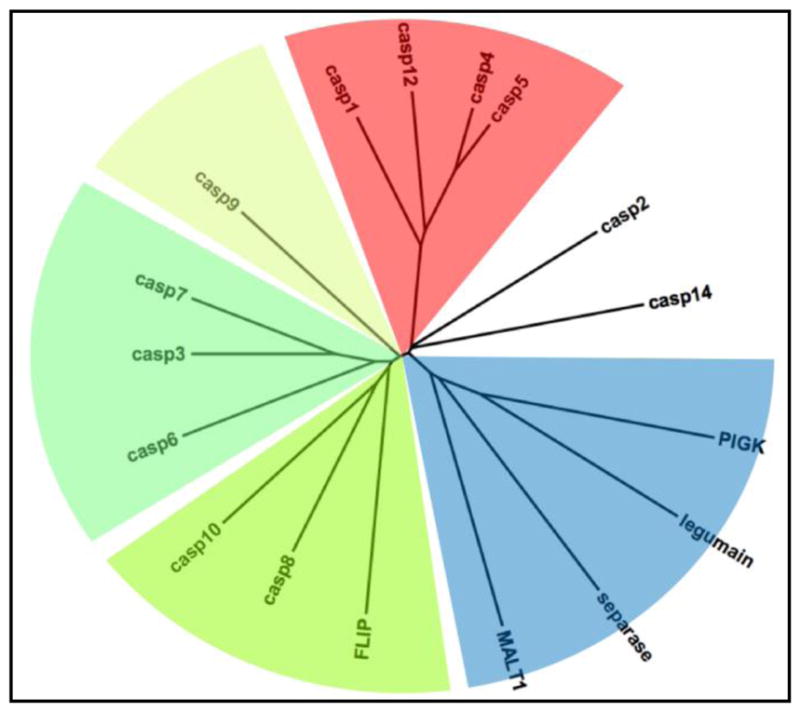
Apoptotic caspases are featured in green hues, inflammatory caspases in red, and the remaining four human members of peptidase clan CD are in blue. Caspases-2 and −14 are uncolored and unassigned, as they have primary roles unassociated with apoptosis or inflammation. Pseudoproteases, FLIP and caspase-12, are proteins with the characteristic fold but mutations in their catalytic machinery render them proteolytically incompetent.
With the exception of caspase-1 - enriched in monocytes/macrophages, and caspase-14 - restricted to keratinocytes [11], caspases are widely expressed. Caspases are obligate cytosolic/nucleoplasmic proteins. Their sequences encode no export or import signals and, although they have been occasionally reported to be associated with mitochondria, these appear to have been artifactual [12]. Caspases begin life as single chain zymogens (enzymes awaiting activation) consisting of an N-terminal domain followed by a catalytic domain. The N-terminal domain is variable, can encode recruitment and activation signals, and defines the type of mechanism that caspases use. The C-terminal protease catalytic unit is a single domain, but often split into two chains by proteolytic cleavage during maturation. The catalytic dyad residues Cys and His reside in the large chain while the substrate recognition groove is formed primarily through residues from the small chain (Fig 2).
Figure 2. Domain structures of human caspases.
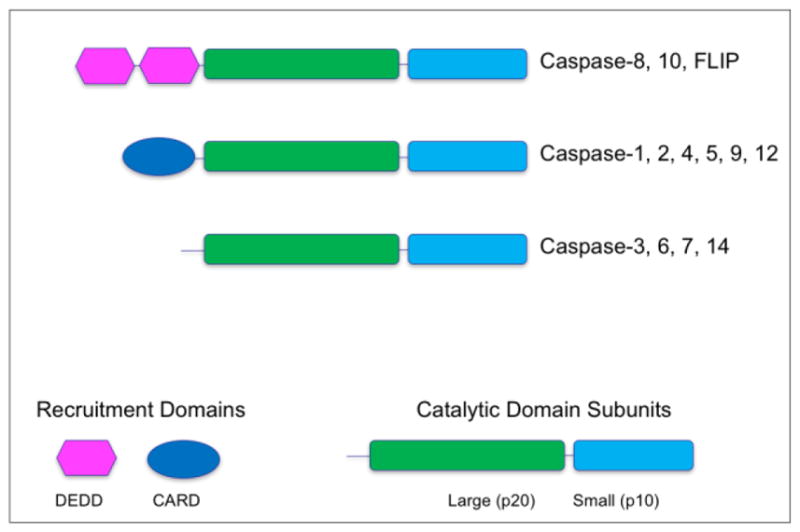
Apical caspases contain domains that serve to recruit the zymogens to activation complexes through homotypic protein interactions – DED (Death Effector Domain) and CARD (Caspase Recruitment Domain). Effector caspases lack these recruitment domains, and are activated by cleavage within the catalytic domain by apical caspases. Cleavage within the catalytic domain is obligatory for effector caspase activation, but dispensable for apical caspase activation.
2. ACTIVATION MECHANISMS OF APOPTOTIC CASPASES
Caspases are restrained as inactive zymogens awaiting appropriate activation signals. The zymogens of apoptotic effector caspases 3 and 7 are obligate dimers and their activity is held in check by a linker separating the large and small chains. Proteolytic processing of the linker allows assembly of the catalytic site through rearrangement of characteristic mobile loops. Auto-proteolysis resulting in removal of the N-terminal pro domain or cleavage of the inter-chain linker sometimes follows activation. Although this has no impact on the inherent proteolytic activity [13, 14] cleavage of the linker enhances dimer stability, and contributes to other downstream regulatory events [3, 15]. There is essentially no dispute about this activation mechanism.
The zymogens of apoptotic initiator caspases are inert monomers, and there is little dispute about their activation mechanism (Fig 3). The most parsimonious model for apical caspase activation, sometimes known as the induced proximity model [16, 17], has been widely tested and holds that apical caspases require dimerization for activation, and that dimerization is the fundamental activating event. Initiator caspases are recruited by adaptor molecules to oligomeric activation platforms following an apoptotic signal. The induced proximity model postulates that local increase in concentration drive proximity-induced dimerization and therefore activation [15]. Much of the biochemical and structural work on activation of apical caspases has focused on caspases-8 and 9, but the same concept is thought to hold true for the activation of pro-inflammatory caspases, as we describe below. In cultured cells treated to undergo apoptosis, apoptotic caspases drive a characteristic morphology that includes membrane blebbing, chromosomal DNA fragmentation, packaging of cell constituents into “apoptotic bodies” and eventually cell death. In vivo few of these morphologies can be observed because apoptotic cell fragments are rapidly cleared by macrophages [18, 19], but in the nematode C. elegans the entire process of cell death and disposal may be visualized. Apoptosis is an immunologically silent cell demise, indeed it may be anti-inflammatory, and therefore complex signaling networks activated by apoptotic caspases are required to dismantle and package cells for removal [19–21].
Figure 3. Distinct activation mechanisms of apical and effector caspases.
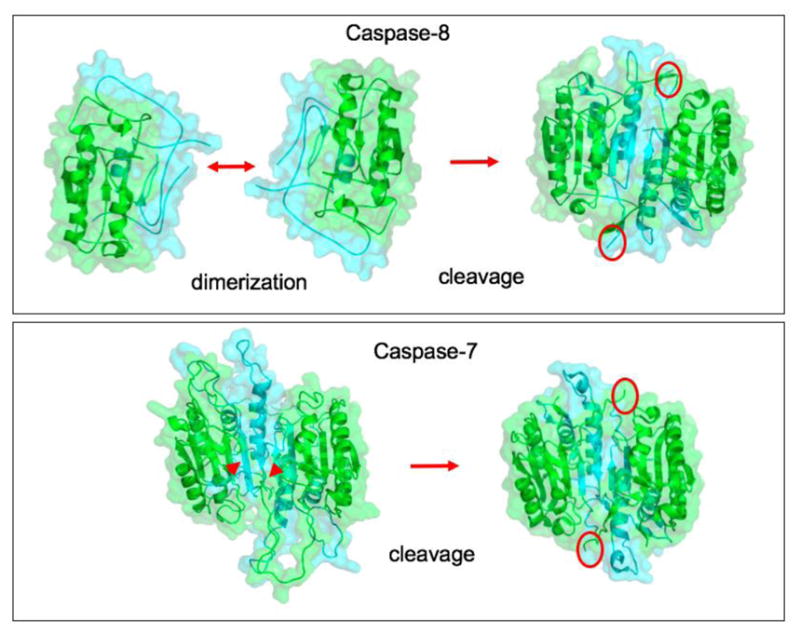
Apical caspase-8 and effector caspase-7 shown as examples, based on the crystal structures reported in PDB codes 2K7Z, 3KJQ, 1GQF, 1F1J. Apical caspases undergo proximity-induced dimerization within activation complexes. Sometimes they also undergo cleavage within the catalytic domain to yield short segments of secondary structure that stabilize the resulting dimer (red circles). Cleavage of the pre-formed zymogen dimer of caspase-7 (red arrowheads) results in activation with an analogous further stabilization of the dimer (red circles).
3. PLATFORMS FOR APICAL CASPASE ACTIVATION
The role of the apical apoptotic caspases is to activate downstream effector caspases, particularly caspases-3 and 7. The apical caspases are the point at which a cellular death signal is converted to proteolytic activity, and this is achieved by remarkably coordinated clustering mechanisms that lead to apical caspase dimerization. Although apical apoptotic caspases respond to very different signals, and are activated in distinct activation platforms, the nature of such activation has striking similarities.
3.1 The DISC
The DISC (Death Inducing Signaling Complex) is a transmembrane assembly that acts as a conduit for extracellular death signals emanating from engagement of certain death receptors of the TNF family. The cytosolic face of the complex contains the adapter protein FADD, which acts as a bridge between the apical caspase and the C-terminal of the transmembrane death receptor. The key components of the recruitment are the DEDs of FADD and caspases-8, 10 and/or FLIP. Current models suggest that the caspase-8 and FLIP form elongated chains connected by their DEDs [22]. Importantly, only the long transcript of FLIP (FLIPL) can encode a caspase activator because the short transcript (FLIPS) has a stop codon following the DEDs, and encodes no catalytic domain homology unit. Thus, FLIPL activates caspase-8 by heterodimerization, on the platform of DISC chains, to generate a single active site. Indeed, in models of DISC formation, FLIPL is a better activator of caspase-8 than is caspase-8 itself – heterodimerization is favored over homodimerization [23, 24]. Because FLIP is an NFkB-regulated gene its concentration in cells is variable and this, accompanied by transcript splicing events to generate differential amounts of FLIPL or FLIPS, would have a major impact on the role of FLIP in activating apical caspases. Some rodents (mice and rats) express only one apical caspase of the extrinsic pathway – caspase-8, but other rodents (guinea pig and squirrel), and primates possess genes for both caspase-8 and caspase-10. Although there has always been debate about the function of caspase-10, current thinking is that it participates in NFkB activation by re-wiring the DISC towards cell survival outcomes [25], and in apoptosis in cell lines deprived caspase-8 [26].
3.2 The Apoptosome
Apaf-1 (Apoptotic Protease Activating Factor-1) is a AAA-ATPase protein that, upon release of cytochrome-C from mitochondria, undergoes polymerization in an ATP or dATP dependent mechanism [15, 27]. Unlike the DISC, and as we will see later the Inflammasome, the Apaf-1 polymers seem to be self-limiting and form a donut-shaped ring of 7 or 8 monomers [28]. At the center of the donut are the CARDs of Apaf-1 which recruit caspase-9 via its CARD to form the apoptosome. Induced proximity of caspase-9 monomers results in their dimerization and generation of a proteolytic signal [29]. Upon activation caspase-9 undergoes auto-cleavage between the large and small subunits of the catalytic domain with two important results. Cleavage unmasks a neo-epitope on the small subunit that permits binding of its cognate inhibitor XIAP to terminate activity, while at the same time promotes dissociation from the apoptosome with subsequent monomerization. Thus the apoptosome acts as a proteolytic-based molecular timer to generate a pulse of apoptotic signal [30]. The utility of the timer, while mechanistically-intriguing, awaits explanation from a cell survival/cell death perspective.
4. INFLAMMATORY CASPASES AND PYROPTOSIS
The innate immune response of macrophages to bacteria and viruses is initiated by a highly conserved group of receptors known as pattern recognition receptors (PRRs) that recognize pathogen-associated molecular patterns (PAMPs) derived from pathogens. There are several PRR families, such as Toll-like receptors (TLRs), c-type lectin receptors and scavenging receptors localized on the outer cell surface or endosomal membrane, retinoic acid inducible gene-I-like receptors, absent in melanoma 2 (AIM2)-like receptors (ALRs) and nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) located in cytoplasm [31]. Some of these receptors signal to NFkB to induce cytokine expression, and some signal to inflammasomes – activators of inflammatory caspases [32].
Activation of inflammatory caspases results in a lytic cell death known as pyroptosis. The term ‘pyroptosis’, in analogy to apoptosis but substituting the first part of the word with pyro, relating to fire or fever, was first proposed to describe a pro-inflammatory programed cell death and distinguish it from apoptosis [33, 34]. While apoptosis is generally considered to be non-lytic and quietly removes unwanted cells, pyroptosis is a lytic and plasma membrane disrupting form of cell death, removes pathogen infected cells and prompts the recruitment of monocytes to site of injury through the release of inflammatory cytokines [35]. Uncontrolled or excessive pyroptosis can lead to widespread cell death, resulting in tissue damage, organ failure and lethal septic shock [36]. The secretion of inflammatory cytokines has been studied for several years, but not until recently was Gasdermin D (GSDMD) discovered to be a substrate of inflammatory caspases that is responsible for membrane pore formation [37, 38]. Cleavage of GSDMD produces an N-terminal fragment that forms pores in the plasma membrane and leads to the rapid lysis of cells – reviewed in [39], thus explaining the old question of how cytokines of the IL1 family – devoid of secretion signals – could exit a cell following an inflammatory stimulus. The exit of these cytokines is coupled to cell lysis. Caspase-1 also cleaves and activates the precursors of cytokines proIL-1b and proIL18, primary pro-inflammatory cytokines, thus the processing of these cytokines is intimately linked to cell lysis driven by GSDMD cleavage. However, there exists somewhat of a controversy in that cytokine exit has been reported to be dissociated from cell death [40, 41], although no mechanisms have been proposed. Perhaps the controversy results from the definition of cell death versus membrane permeabilization, and this seems important to rationalize.
4.1 Inflammatory Caspases-1, −4, −5, −12
Interleukin-1b (IL-1b) converting enzyme (ICE), now known as caspase-1, was discovered as the enzyme responsible for processing inactive proIL-1b to active IL-1b in macrophages [42–44] and confirmed by caspase-1 knock-out mice resistant to the lethal effect of endotoxins [45, 46]. ProIL-18, also known as interferon-γ-inducing factor, is also processed and activated by caspase-1. Caspase-1 sponsors inflammation by effecting the maturation of pro-interleukins. The caspase-1 subfamily, generally referred to as inflammatory caspases, includes human caspase-4, −5 and −12 showing the closest sequence identity to caspase-1, the first caspase to be discovered [3, 42]. Genes of inflammatory caspases are tightly linked forming an inflammatory cluster [34, 47, 48]. Sequence analysis of human caspase-4 and −5 suggest these enzymes originated from gene duplication of mouse caspase-11. Therefore, caspase-11 is the murine ortholog of human caspases-4 and-5 [49].
The current consensus is that caspase-12 is an inactive homolog, based on conflicting results in the literature [50–52] and the substitution of critical residues that surround the active site in mouse, rat and human caspase-12. In humans, a single nucleotide polymorphism at amino-acid position 125 in caspase-12 results in the synthesis of either a truncated or a full-length enzyme. The full-length allele was identified only in populations of African descent (about 20% of them) [53]. However, some species caspase-12, including African elephant, cat and dog, seems to contain sequences compatible with activity. Thus, it is possible that caspase-12 may serve as an active caspase in its own right, or an activator of caspases-1 or 11, depending on the species.
5. ACTIVATION MECHANISM OF INFLAMMATORY CASPASES
In principle, inflammatory caspases are synthesized as inactive zymogens, just like initiator apoptotic caspases. Structurally, the inflammatory caspases have an N-terminal CARD followed by a catalytic domain with an architecture very similar to the apoptotic initiator caspase-9 (Figure 2). In innate immune cells they are considered as monomers awaiting recruitment to their cognate activation platforms, known as inflammasomes, to be dimerized and become active enzymes, and thus initially it was thought that inflammasomes may have the same architecture as the apoptosome [54]. However, more recent studies have provided for alternative models, spurred on by the finding that macrophages seem to possess a single caspase activation focus per cell [55].
5.1 Canonical Inflammasomes
Pyroptosis relies on proteolytic activity of inflammatory to undergo canonical (caspase-1-mediated) or non-canonical (caspase-4/11 or caspase-5-mediated) pathways [56]. The canonical inflammasome activation is dependent on the adapter protein ASC (apoptosis associated speck-like protein containing a CARD) and caspase-1. The formation of canonical inflammasomes varies in degree to the insult or threat the cell faces, ultimately all induce inflammatory responses by processing caspase-1. Canonical inflammasomes are protein complexes of either NOD-like receptors (NLRs) or pyrin domain (PYD)- containing non-NLRs assembling apoptosis-associated speck-like protein containing a C-terminal CARD (ASC) and pro-caspase-1 [31, 57]. Some of the most well studied canonical inflammasomes include the NLR family, which includes NLRC4, NLRP1, NLRP3 and AIM2, all differentially stimulated by different ligands. The activation mechanism has been proposed to result in the ACS-dependent polymerization of either chains [58] or large spherical assemblies [55] of inflammasome components, forming a much larger activation platform than is seen for caspase-9. The hypothesis has been put forward that this single focus results from rounds of polymerization that, unlike the caspase-9 apoptosome where polymerization is self-limiting, results in huge macromolecular assemblies of filamentous inflammasome components [59]. The subsequent activation of caspase-1 remains unknown, but is probably analogous to apoptotic apical caspases, with the inflammasome assemblies acting as platforms for proximity-induced dimerization.
5.2 Non-Canonical Inflammasomes
Recent studies suggest that caspase-4, −5 and −11 may participate in a non-canonical activation process that does not require inflammasome components [32, 56]. Mouse caspase-11 and human orthologous caspases-4 and −5 are components of the non-canonical inflammasome, which do not require adaptor molecule, ASC. Caspase-11 was the first enzyme discovered to participate in a non-canonical or non-caspase-1 mediated inflammasome [56] as a response to gram-negative bacteria. This then led to the discovery of caspase-11’s proposed role as direct sensor of intracellular LPS from gram-negative bacteria during macrophage-mediated inflammatory responses [60, 61]. Recombinant studies using caspase-4, −5 and −11 propose the formation of a multimer in the presence of LPS and monomers in the absence of LPS [60].
6. ACTIVATION MECHANISMS OF OTHER CLAN CD PROTEASES
Clan CD is an ancient group of proteases with representatives in every biotic kingdom. MALT1, also known as paracaspase [62], is the closest relative to the caspases in terms of its activation mechanism, though its biological function and primary specificity are totally different. Like caspases it is an obligate dimer, and like apical caspases it is activated by dimerization [63, 64]. Other clan CD members are distinct in their activation mechanisms, sampling most of the possible activation mechanism of proteases [1]. Legumain is a secreted and/or lysosomal protease involved in peptide degradation and maturation, and is activated by a pH switch that results in autolytic removal of a pro-domain that blocks access of substrates to the active site [65]. Separase is required for sister chromatid separation during anaphase, and is activated by proteasomal degradation of its cognate inhibitor, securin [66, 67]. PIGK (Glycosylphosphatidylinositol:protein transamidase) removes a C-terminal peptide from target proteins before they attach glycosylphosphatidylinositol anchors in the endoplasmic reticulum, but the mechanism of PIGK activation remains unclear, although it may be constitutively active because of its mechanism of action in the endoplasmic reticulum.
7. CONTEMPORARY AND FUTURE DIRECTIONS
The requirement of two inflammatory caspases for the different inflammatory pathways is still not understood, however it has been proposed that the non-canonical pathway abrogates the need for the priming step of the canonical pathway. Thus, canonical inflammasome activation is dependent on two sequential signals in macrophages- 1) priming and 2) triggering. Priming is the process in which PAMPs bind to PRR on macrophage cell surfaces recognize stimulants such as LPS, resulting in upregulation of NF-kB pathway and production of inflammasome components. Triggering is the signal necessary for full activation of inflammasomes. The direct recognition of LPS rapidly oligomerizes caspases-4, −5 and −11 resulting in generation of inflammatory proteolytic signals without the need for NFkB-mediated gene transcription. Although inflammatory caspases fall neatly into the apoptotic initiator caspase description, the activation mechanism of these enzymes has not been well explored. Biochemical studies targeting the dimerization, auto-processing and catalytic sites should be able to shed light on their activation mechanism. Another important aspect to further probe is the involvement of LPS in caspase activation. Are there other proteins or complexes that aid caspase-4 −5 and −11 in the direct sensing of LPS that so far have remain hidden? Most studies seem to suggest that caspases 1, 4 and 5 are biochemically synonymous. Is this correct, or are there differences in the substrate repertoire of inflammatory caspases?
The significance of caspase-12 as a potential inhibitor to inflammatory caspases is a very intriguing field. FLIPL is a non-catalytic enzyme that heterodimerizes with apoptotic caspase-8. This heterodimer is thought to alter the targets of caspase-8 activity leading it away from apoptosis and towards blocking necroptosis. Could it be possible that caspase-12 acts as a modifier to inflammatory caspases in order to alter their specificity in an analogous manner? From this perspective, could caspase-10 also function as a modulator of caspase-8 activity by heterodimerization, given the findings that caspase-10 can apparently drive NFkB activation [25]?
The pyroptotic mediator GSDMD is not only cleaved by inflammatory caspases, but also by caspase-3. However, the caspase-3 cleavage inactivates the pyroptotic potential of the protein, with the consequence that apoptosis seems to prevent pyroptosis [68]. This may be a mechanism to ensure that infected macrophages that evaded pyroptosis are eliminated, and so an interesting question becomes what subtype of macrophages allow pathogens and viruses to utilize them without eliciting pyroptosis or apoptosis. Are there alterations in expression or mutations in GSDMD in pathologies associated with inflammatory diseases? Links between the different regulated cell death pathways may exist. Thus DFNA5 (deafness, autosomal dominant 5 – a Gasdermin family member) has been suggested to switch apoptosis to pyroptosis via cleavage by caspase-3 [69]. At first glance this may seem to be a strange outcome, since one would expect an inflammatory-silent form of cell death (apoptosis) to absolutely reject an inflammatory one. The pyroptosis-inducing outcome of caspase-3 activating DFNA5 seems at odds with the pyroptosis-defeating outcome of caspase-3 inactivating GSDMD, and it seems that highly cell-specific events must be invoked to explain these observations.
Table 1.
Human clan CD proteases, their biological functions, primary specificity, and activation mechanisms.
| Clan CD member | Biological Role | Primary specificity | Activation mechanism |
|---|---|---|---|
| Caspase-1, 4, 5 | Inflammatory caspases | Asp | No consensus – probably dimerization |
| Caspase-12 | Putative pseudocaspase - inflammation | - | |
| Caspase-9 | Intrinsic pathway apical caspase | Asp | Dimerization |
| Caspase-8, 10 | Extrinsic pathway apical caspases | Asp | Dimerization |
| FLIP | Pseudocaspase – extrinsic pathway | - | |
| Caspase-3, 6, 7 | Apoptotic effector (executioner) caspases | Asp | Intra-domain cleavage |
| Caspase-2 | “Multifunctional” caspase | Asp | No consensus |
| Caspase-14 | Keratinocyte differentiation | Asp | Intra-domain cleavage |
| MALT1 | B, T-cell activation via NFkB | Arg | Dimerization |
| Legumain | Antigen presentation, lysosomal protein degradation | Asn | Removal of N-domain |
| Separase | Sister chromatid separation during anaphase | Arg | Removal of blocking inhibitor |
| PIGK | C-terminal processing of proteins destined for GPI anchor addition | Asn/Ser* | Unknown |
Acknowledgments
This work was supported by the National Institutes of Health [grant number GM099040].
Abbreviations
- PRRs
pattern recognition receptors
- PAMPs
pathogen-associated molecular patterns
- TLRs
Toll-like receptors
- AIM2
absent in melanoma 2
- ALRs
AIM2-like receptors
- NOD
nucleotide-binding oligomerization domain
- NLRs
NOD-like receptors
- PYD
pyrin domain
- CARD
caspase recruit domain
- DED
death effector domain
- ASC
apoptosis-associated speck like protein containing a C-terminal CARD
- GSDMD
gasderminD
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.McLuskey K, Mottram JC. Comparative structural analysis of the caspase family with other clan CD cysteine peptidases. Biochem J. 2015;466:219–32. doi: 10.1042/BJ20141324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Salvesen GS, Hempel A, Coll NS. Protease Signaling in Animal and Plant Regulated Cell Death. FEBS J. 2015 doi: 10.1111/febs.13616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fuentes-Prior P, Salvesen GS. The protein structures that shape caspase activity, specificity, activation and inhibition. Biochem J. 2004;384:201–32. doi: 10.1042/BJ20041142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fava LL, Bock FJ, Geley S, Villunger A. Caspase-2 at a glance. J Cell Sci. 2012;125:5911–5. doi: 10.1242/jcs.115105. [DOI] [PubMed] [Google Scholar]
- 5.Troy CM, Shelanski ML. Caspase-2 redux. Cell Death Differ. 2003;10:101–7. doi: 10.1038/sj.cdd.4401175. [DOI] [PubMed] [Google Scholar]
- 6.Troy CM, Ribe EM. Caspase-2: vestigial remnant or master regulator? Sci Signal. 2008;1:pe42. doi: 10.1126/scisignal.138pe42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bouchier-Hayes L, Green DR. Caspase-2: the orphan caspase. Cell Death Differ. 2012;19:51–7. doi: 10.1038/cdd.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Puccini J, Dorstyn L, Kumar S. Caspase-2 as a tumour suppressor. Cell Death Differ. 2013;20:1133–9. doi: 10.1038/cdd.2013.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olsson M, Forsberg J, Zhivotovsky B. Caspase-2: the reinvented enzyme. Oncogene. 2015;34:1877–82. doi: 10.1038/onc.2014.139. [DOI] [PubMed] [Google Scholar]
- 10.Denecker G, Ovaere P, Vandenabeele P, Declercq W. Caspase-14 reveals its secrets. J Cell Biol. 2008;180:451–8. doi: 10.1083/jcb.200709098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Denecker G, Hoste E, Gilbert B, Hochepied T, Ovaere P, Lippens S, et al. Caspase-14 protects against epidermal UVB photodamage and water loss. Nat Cell Biol. 2007;9:666–74. doi: 10.1038/ncb1597. [DOI] [PubMed] [Google Scholar]
- 12.van Loo G, Saelens X, Matthijssens F, Schotte P, Beyaert R, Declercq W, et al. Caspases are not localized in mitochondria during life or death. Cell Death Differ. 2002;9:1207–11. doi: 10.1038/sj.cdd.4401101. [DOI] [PubMed] [Google Scholar]
- 13.Stennicke HR, Jurgensmeier JM, Shin H, Deveraux Q, Wolf BB, Yang X, et al. Pro-caspase-3 is a major physiologic target of caspase-8. J Biol Chem. 1998;273:27084–90. doi: 10.1074/jbc.273.42.27084. [DOI] [PubMed] [Google Scholar]
- 14.Stennicke HR, Salvesen GS. Biochemical characteristics of caspases-3, −6, −7, and −8. J Biol Chem. 1997;272:25719–23. doi: 10.1074/jbc.272.41.25719. [DOI] [PubMed] [Google Scholar]
- 15.Riedl SJ, Salvesen GS. The apoptosome: signalling platform of cell death. Nat Rev Mol Cell Biol. 2007;8:405–13. doi: 10.1038/nrm2153. [DOI] [PubMed] [Google Scholar]
- 16.Salvesen GS, Dixit VM. Caspase activation: The induced-proximity model. Proc Natl Acad Sci U S A. 1999;96:10964–7. doi: 10.1073/pnas.96.20.10964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boatright KM, Renatus M, Scott FL, Sperandio S, Shin H, Pedersen IM, et al. A unified model for apical caspase activation. Mol Cell. 2003;11:529–41. doi: 10.1016/s1097-2765(03)00051-0. [DOI] [PubMed] [Google Scholar]
- 18.Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, et al. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature. 2010;467:863–7. doi: 10.1038/nature09413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hochreiter-Hufford A, Ravichandran KS. Clearing the dead: apoptotic cell sensing, recognition, engulfment, and digestion. Cold Spring Harbor perspectives in biology. 2013;5:a008748. doi: 10.1101/cshperspect.a008748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henson PM, Bratton DL. Antiinflammatory effects of apoptotic cells. J Clin Invest. 2013;123:2773–4. doi: 10.1172/JCI69344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martin SJ, Henry CM, Cullen SP. A perspective on mammalian caspases as positive and negative regulators of inflammation. Mol Cell. 2012;46:387–97. doi: 10.1016/j.molcel.2012.04.026. [DOI] [PubMed] [Google Scholar]
- 22.Dickens LS, Boyd RS, Jukes-Jones R, Hughes MA, Robinson GL, Fairall L, et al. A death effector domain chain DISC model reveals a crucial role for caspase-8 chain assembly in mediating apoptotic cell death. Mol Cell. 2012;47:291–305. doi: 10.1016/j.molcel.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boatright KM, Deis C, Denault JB, Sutherlin DP, Salvesen GS. Activation of caspases-8 and −10 by FLIP(L) Biochem J. 2004;382:651–7. doi: 10.1042/BJ20040809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hughes MA, Powley IR, Jukes-Jones R, Horn S, Feoktistova M, Fairall L, et al. Co-operative and Hierarchical Binding of c-FLIP and Caspase-8: A Unified Model Defines How c-FLIP Isoforms Differentially Control Cell Fate. Mol Cell. 2016;61:834–49. doi: 10.1016/j.molcel.2016.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Horn S, Hughes MA, Schilling R, Sticht C, Tenev T, Ploesser M, et al. Caspase-10 Negatively Regulates Caspase-8-Mediated Cell Death, Switching the Response to CD95L in Favor of NF-kappaB Activation and Cell Survival. Cell reports. 2017;19:785–97. doi: 10.1016/j.celrep.2017.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tanzer MC, Khan N, Rickard JA, Etemadi N, Lalaoui N, Spall SK, et al. Combination of IAP antagonist and IFNgamma activates novel caspase-10- and RIPK1-dependent cell death pathways. Cell Death Differ. 2017;24:481–91. doi: 10.1038/cdd.2016.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a Human Protein Homologous to C. elegans CED-4, Participates in Cytochrome c-Dependent Activation of Caspase-3. Cell. 1997;90:405–13. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- 28.Yu X, Wang L, Acehan D, Wang X, Akey CW. Three-dimensional Structure of a Double Apoptosome Formed by the Drosophila Apaf-1 Related Killer. J Mol Biol. 2006;355:577–89. doi: 10.1016/j.jmb.2005.10.040. [DOI] [PubMed] [Google Scholar]
- 29.Pop C, Timmer J, Sperandio S, Salvesen GS. The apoptosome activates caspase-9 by dimerization. Mol Cell. 2006;22:269–75. doi: 10.1016/j.molcel.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 30.Malladi S, Challa-Malladi M, Fearnhead HO, Bratton SB. The Apaf-1*procaspase-9 apoptosome complex functions as a proteolytic-based molecular timer. Embo J. 2009;28:1916–25. doi: 10.1038/emboj.2009.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yi YS. Caspase-11 non-canonical inflammasome: a critical sensor of intracellular lipopolysaccharide in macrophage-mediated inflammatory responses. Immunology. 2017;152:207–17. doi: 10.1111/imm.12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–26. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 33.Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol. 2001;9:113–4. doi: 10.1016/s0966-842x(00)01936-3. [DOI] [PubMed] [Google Scholar]
- 34.Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun. 2005;73:1907–16. doi: 10.1128/IAI.73.4.1907-1916.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7:99–109. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aglietti RA, Dueber EC. Recent Insights into the Molecular Mechanisms Underlying Pyroptosis and Gasdermin Family Functions. Trends Immunol. 2017;38:261–71. doi: 10.1016/j.it.2017.01.003. [DOI] [PubMed] [Google Scholar]
- 37.Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–71. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- 38.Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–8. doi: 10.1038/nature18629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi J, Gao W, Shao F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem Sci. 2017;42:245–54. doi: 10.1016/j.tibs.2016.10.004. [DOI] [PubMed] [Google Scholar]
- 40.Conos SA, Lawlor KE, Vaux DL, Vince JE, Lindqvist LM. Cell death is not essential for caspase-1-mediated interleukin-1beta activation and secretion. Cell Death Differ. 2016;23:1827–38. doi: 10.1038/cdd.2016.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen KW, Gross CJ, Sotomayor FV, Stacey KJ, Tschopp J, Sweet MJ, et al. The neutrophil NLRC4 inflammasome selectively promotes IL-1beta maturation without pyroptosis during acute Salmonella challenge. Cell reports. 2014;8:570–82. doi: 10.1016/j.celrep.2014.06.028. [DOI] [PubMed] [Google Scholar]
- 42.Black RA, Kronheim SR, Merriam JE, March CJ, Hopp TP. A pre-aspartate-specific protease from human leukocytes that cleaves pro-interleukin-1b. J Biol Chem. 1989;264:5323–6. [PubMed] [Google Scholar]
- 43.Kostura MJ, Tocci MJ, Limjuco G, Chin J, Cameron P, Hillman AG, et al. Identification of a monocyte specific pre-interleukin 1b convertase activity. Proc Natl Acad Sci USA. 1989;86:5227–31. doi: 10.1073/pnas.86.14.5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, et al. A novel heterodimeric cysteine protease is required for interleukin-1beta processing in monocytes. Nature. 1992;356:768–74. doi: 10.1038/356768a0. [DOI] [PubMed] [Google Scholar]
- 45.Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MSS, et al. Altered cytokine export and apoptosis in mice deficient in interleukin-1-beta converting enzyme. Science. 1995;267:2000–3. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- 46.Li P, Allen H, Bannerjee S, Franklin S, Herzog L, Johnston C, et al. Mice deficient in IL-1b-converting enzyme are defective in production of mature IL-1b and resistant to endotoxic shock. Cell. 1995;80:401–11. doi: 10.1016/0092-8674(95)90490-5. [DOI] [PubMed] [Google Scholar]
- 47.Lamkanfi M, Dixit VM. The inflammasomes. PLoS Pathog. 2009;5:e1000510. doi: 10.1371/journal.ppat.1000510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martinon F, Tschopp J. Inflammatory caspases and inflammasomes: master switches of inflammation. Cell Death Differ. 2007;14:10–22. doi: 10.1038/sj.cdd.4402038. [DOI] [PubMed] [Google Scholar]
- 49.Wang S, Miura M, Jung Y-K, Zhu H, Yuan J. Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell. 1998;92:501–9. doi: 10.1016/s0092-8674(00)80943-5. [DOI] [PubMed] [Google Scholar]
- 50.Kalai M, Lamkanfi M, Denecker G, Boogmans M, Lippens S, Meeus A, et al. Regulation of the expression and processing of caspase-12. J Cell Biol. 2003;162:457–67. doi: 10.1083/jcb.200303157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saleh M, Vaillancourt JP, Graham RK, Huyck M, Srinivasula SM, Alnemri ES, et al. Differential modulation of endotoxin responsiveness by human caspase-12 polymorphisms. Nature. 2004;429:75–9. doi: 10.1038/nature02451. [DOI] [PubMed] [Google Scholar]
- 52.Jimenez Fernandez D, Lamkanfi M. Inflammatory caspases: key regulators of inflammation and cell death. Biol Chem. 2015;396:193–203. doi: 10.1515/hsz-2014-0253. [DOI] [PubMed] [Google Scholar]
- 53.Fischer H, Koenig U, Eckhart L, Tschachler E. Human caspase 12 has acquired deleterious mutations. Biochem Biophys Res Commun. 2002;293:722–6. doi: 10.1016/S0006-291X(02)00289-9. [DOI] [PubMed] [Google Scholar]
- 54.Faustin B, Lartigue L, Bruey JM, Luciano F, Sergienko E, Bailly-Maitre B, et al. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell. 2007;25:713–24. doi: 10.1016/j.molcel.2007.01.032. [DOI] [PubMed] [Google Scholar]
- 55.Man SM, Hopkins LJ, Nugent E, Cox S, Gluck IM, Tourlomousis P, et al. Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc Natl Acad Sci U S A. 2014;111:7403–8. doi: 10.1073/pnas.1402911111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–21. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 57.Zamboni DS, Lima-Junior DS. Inflammasomes in host response to protozoan parasites. Immunol Rev. 2015;265:156–71. doi: 10.1111/imr.12291. [DOI] [PubMed] [Google Scholar]
- 58.Zhang L, Chen S, Ruan J, Wu J, Tong AB, Yin Q, et al. Cryo-EM structure of the activated NAIP2-NLRC4 inflammasome reveals nucleated polymerization. Science. 2015;350:404–9. doi: 10.1126/science.aac5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hauenstein AV, Zhang L, Wu H. The hierarchical structural architecture of inflammasomes, supramolecular inflammatory machines. Curr Opin Struct Biol. 2015;31:75–83. doi: 10.1016/j.sbi.2015.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–5. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 61.Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science. 2013;341:1250–3. doi: 10.1126/science.1240988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Uren AG, O’Rourke K, Aravind LA, Pisbarro MT, Seshagiri S, Koonin EV, et al. Identification of paracaspases and metacaspases: two ancient fmilies of caspase-like proteins, one of which plays a key role in MALT lymphoma. Mol Cell. 2000;6:961–7. doi: 10.1016/s1097-2765(00)00094-0. [DOI] [PubMed] [Google Scholar]
- 63.Hachmann J, Snipas SJ, van Raam BJ, Cancino EM, Houlihan EJ, Poreba M, et al. Mechanism and specificity of the human paracaspase MALT1. Biochem J. 2012;443:287–95. doi: 10.1042/BJ20120035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wiesmann C, Leder L, Blank J, Bernardi A, Melkko S, Decock A, et al. Structural determinants of MALT1 protease activity. J Mol Biol. 2012;419:4–21. doi: 10.1016/j.jmb.2012.02.018. [DOI] [PubMed] [Google Scholar]
- 65.Dall E, Brandstetter H. Mechanistic and structural studies on legumain explain its zymogenicity, distinct activation pathways, and regulation. Proc Natl Acad Sci U S A. 2013;110:10940–5. doi: 10.1073/pnas.1300686110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Waizenegger I, Gimenez-Abian J, Wernic D, Peters J. Regulation of human separase by securin binding and autocleavage. Curr Biol. 2002;12:1368. doi: 10.1016/s0960-9822(02)01073-4. [DOI] [PubMed] [Google Scholar]
- 67.Lin Z, Luo X, Yu H. Structural basis of cohesin cleavage by separase. Nature. 2016;532:131–4. doi: 10.1038/nature17402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Taabazuing CY, Okondo MC, Bachovchin DA. Pyroptosis and Apoptosis Pathways Engage in Bidirectional Crosstalk in Monocytes and Macrophages. Cell Chem Biol. 2017;24:507–14. e4. doi: 10.1016/j.chembiol.2017.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547:99–103. doi: 10.1038/nature22393. [DOI] [PubMed] [Google Scholar]