

Abstract
Background
Patients with late‐life depression may be at the preclinical stage of dementia. However, the neurodegenerative processes in late‐life depression are poorly understood. This study aimed to investigate the distribution patterns of amyloid pathology and neurodegeneration in a depressive population without dementia.
Methods
The study recruited 63 middle‐aged and elderly patients with major depressive disorder (MDD) and 22 control subjects. The MDD patients were further subdivided into those with mild cognitive impairment (MCI) (n = 24) and non‐MCI (n = 39) patients. We used the global standardized uptake value ratio of 18F‐florbetapir (AV‐45/Amyvid) positron emission tomography imaging as a biomarker of cerebral amyloidosis and the hippocampal volume as a biomarker for neurodegeneration. Cutoff points of brain amyloid positivity and hippocampal atrophy were determined using independent data obtained from clinically diagnosed Alzheimer's disease (AD) patients in a previous study.
Results
Most of the control subjects (81.8%) were biomarker‐negative, in contrast to the MCI MDD patients (37.5%). A relatively high proportion of the MCI MDD patients (12.5%) exhibited both amyloid positivity and hippocampal atrophy as compared to the control subjects (4.5%) and non‐MCI patients (5.1%). However, a considerable proportion of the MCI MDD patients (29.2%) were categorized into the group with hippocampal atrophy alone, and negative amyloid deposition, as compared to the control subjects (0%) and non‐MCI patients (5.1%).
Conclusions
This study highlights the expected heterogeneity of the processes of neurodegeneration in MDD patients. The diverse neurodegenerative processes may have important etiologic and therapeutic implications regarding neurodegenerative pathophysiology in late‐life depression.
Keywords: 18F‐florbetapir (AV‐45/Amyvid), Alzheimer's disease, amyloid, dementia, hippocampal atrophy, major depressive disorder, mild cognitive impairment
1. INTRODUCTION
Several meta‐analyses (Diniz, Butters, Albert, Dew, & Reynolds, 2013; Jorm, 2001; Ownby et al., 2006) have consistently suggested that a history of depression approximately doubles an individual's risk of developing dementia later in life, including Alzheimer's disease (AD) and non‐AD dementia. One pilot postmortem study (Rapp et al., 2006) showed that AD patients with a lifetime history of major depression have more pronounced amyloid plaque and neurofibrillary tangle, as compared to AD patients without a history of depression. Our previous studies (Wu et al., 2013, 2016) indicated increased cerebral amyloid accumulation as measured by 18F‐florbetapir uptake in specific brain regions of nondemented patients with lifetime major depression relative to comparison subjects. These findings point toward the possibility that patients with lifetime major depression might be at an early preclinical stage of the disease in which the criteria for dementia or even mild cognitive impairment (MCI) have not yet been reached.
Insight accumulated over the years regarding dynamic change in biomarkers of AD pathology has led to the establishment of new research and diagnostic criteria (Jack et al., 2009, 2010). These developments provide guidance on the early detection of underlying AD pathology and early prediction of neurocognitive degeneration. A new series of criteria was recently developed by the task force of the National Institute on Aging and the Alzheimer Association (NIA‐AA), mainly for research purposes, which made specific assumptions about dynamic relationships among AD biomarkers in an ordered manner (Albert et al., 2011; Jack et al., 2012; McKhann et al., 2011). Amyloid biomarkers as assessed by positron emission tomography (PET) imaging of amyloid or cerebrospinal fluid (CSF) amyloid‐β can be detected to be abnormal as early as 20 years before significant clinical symptoms appear. Neurodegenerative biomarkers such as CSF tau, 18F‐fluorodeoxyglucose (18F‐FDG)‐PET, and hippocampal volume as assessed by magnetic resonance imaging (MRI) become abnormal later and are then followed by significant clinical symptoms of cognitive impairment (Sperling et al., 2011). Biomarkers can be classed into two categories: those of an underlying amyloid pathology (CSF amyloid‐β or amyloid PET) and those of neurodegenerative features (hippocampal atrophy on MRI, CSF tau, and hypometabolism on 18F‐FDG‐PET).
Several researchers have reported that up to 50% of depressed elderly subjects meet the criteria for clinical diagnosis of MCI, despite differences in methodology and the definition of cognitive impairment (Bhalla et al., 2006; Lee et al., 2007; Yeh et al., 2011). This rate is far higher than the prevalence of MCI reported in the general population, which ranges from 3% to 19% (Gauthier et al., 2006). This implies that some neurodegenerative processes might underlie the high prevalence of MCI among elderly depressed patients. Whereas patients with late‐life depression represent an etiologically heterogeneous group (i.e., different age at onset, differing severity and episodes, differing medical comorbidities), it is not surprising that cognitive impairment in late‐life depression should involve different ongoing mechanisms. However, the patterns of the neurodegenerative processes underlying cognitive impairment in elderly depressed patients are poorly understood (Jellinger, 2013).
The recently published NIA‐AA criteria mentioned above might provide new insight and framework to explore the patterns of neurodegenerative processes in elderly depressed patients, and may allow them to be categorized into different biomarker‐based groups. In the present study, we focused on a population of nondemented patients with major depression and aimed to apply the two categories of biomarker proposed in the NIA‐AA criteria to investigate the distribution patterns of amyloid pathology and abnormal neurodegeneration in a depressed population.
2. METHODS
2.1. Subjects and protocol
The subjects enrolled in the present study were recruited from a longitudinal clinical cohort study launched in 2011, which was performed to investigate cerebral amyloid deposition in nondemented patients with major depressive disorder (MDD). The patients were recruited consecutively from geriatric psychiatric outpatients at Chang Gung Medical Center from August 2011 to July 2015. The control subjects were recruited through public advertisements during the same period. Every MDD patient was assessed for the presence of lifetime DSM‐IV major depressive episodes by clinical interview, and medical information was obtained from medical records and attending physicians. Control subjects were confirmed as having a lifetime absence of psychiatric illness. All subjects were aged >50 years, and functioned well in activities of daily living; they did not have clinically significant medical or neurological diseases, and had not abused alcohol or other substances within the past 1 year at the time of study enrollment. None of the subjects met the NINCDS‐ADRDA criteria for probable AD or the DSM‐IV criteria for dementia. All eligible subjects underwent 18F‐florbetapir PET study, brain MRI, and cognitive assessment. The patients’ Apolipoprotein E (ApoE) genotype was also classified by polymerase chain reaction, and vascular risk factors as defined by the Framingham stroke risk score were identified, as were clinical characteristics of lifetime major depression. Written informed consent was obtained from all subjects, and the study protocol was approved by the Institutional Review Boards of the Ministry of Health and Welfare and Chang Gung Medical Center.
2.2. Non‐MCI and MCI MDD patients
Cognitive assessment in the present study was performed as previously described (Wu et al., 2016) and included global screening using the Mini‐Mental Status Examination (MMSE) and the Clinical Dementia Rating (CDR), as well as assessment of domain‐specific measures using a comprehensive battery of neuropsychological tests. The neuropsychological tests were used to both confirm the cognitive normality of the control subjects and to divide the MDD patients into MCI and non‐MCI groups. The battery of tests included the Wechsler Adult Intelligence Scale—Third Edition (WAIS‐III) digit symbol (Wechsler, 1997) and Trail‐making A tests (Reitan & Wolfson, 1985) for information‐processing speed assessment; the Controlled Oral Word Association (COWA) (Albert, 1973) Frontal Assessment Battery (FAB) (Dubois, Slachevsky, Litvan, & Pillon, 2000), Trail‐making B (Benton, Hamsher, & Sivan, 1989), and WAIS‐III‐similarity tests (Wechsler, 1997) to assess executive function; the 12‐item, six‐trial selective reminding test (SRT) (Buschke & Fuld, 1974), the total number of words learned in six trials, and delayed recall following a 15‐min delay to assess memory; the WAIS‐III‐language test (Wechsler, 1997) to assess language; and the WAIS‐III‐digit span test (Wechsler, 1997) to assess attention.
Individual original scores were transformed into standardized z‐scores, which were generated using regression‐based norms and adjusted for age and educational level according to independent normative data for Taiwan (Yeh et al., 2011). MCI was defined in MDD patients who exhibited impairment in at least one of the cognitive domains, as shown by a score of 1.5 SD below the age‐ and education level‐adjusted norm (Petersen, 2004; Petersen et al., 2001). The CDR had to be only 0 or 0.5 for all subjects. We used the CDR Sum of Boxes (CDR‐SB) method to characterize cognitive and functional performance.
2.3. Image acquisition
The radiosynthesis of 18F‐florbetapir (Yao et al., 2010) and amyloid PET data acquisition (Lin et al., 2010) followed the same procedures as previously carried out by our group. Each 18F‐florbetapir PET scan at 50–60 min postinjection was obtained using a Biograph mCT PET/CT System (Siemens Medical Solutions, Malvern, PA, USA) with 378 ± 18 MBq of 18F‐florbetapir. T1‐weighted MRI images were obtained for all subjects using a 3T Siemens Magnetom TIM Trio scanner (Siemens Medical Solutions).
2.4. Image analysis
All PET image data were processed and analyzed using PMOD image analysis software (version 3.3; PMOD Technologies Ltd, Zurich, Switzerland) (Hsiao et al., 2013). Seven volumes of interest (VOIs), the frontal, anterior cingulate, posterior cingulate, precuneus, parietal, occipital, and temporal areas, were selected (Hsiao et al., 2013), and the regional standardized uptake value ratio (SUVR) using the whole cerebellum as the reference region was calculated. Moreover, the average SUVR from 7 cerebral cortical VOIs was computed as the global cortical SUVR for further analysis.
FreeSurfer image analysis software (version 5.3.0; https://surfer.nmr.mgh.harvard.edu/) was used to measure the hippocampal and intracranial volumes. To reduce intersubject variability, hippocampal volumes were corrected for the intracranial volume (ICV). A normalization method based on linear regression between the VOI and ICV was applied (Voevodskaya et al., 2014) in order to obtain the adjusted hippocampal volume (HVa) as follows:
where HV was the raw hippocampal volume and ICV indicated the intracranial volume for each subject. For correction, β was the slope of the regression line between the ICV and hippocampal volume of the controls, and ICVmean was the average ICV of the control group.
2.5. Imaging biomarker cutoff points
As the imaging biomarkers were all continuous measures in the present study, every biomarker based on the NIA‐AA criteria was required to be designated normal or abnormal (Sperling et al., 2011). Thus, cutoff points needed to be selected to dichotomize biomarkers in order to divide the subjects into normal or abnormal groups. As FDG‐PET and CSF data were not available in our study, we employed the global 18F‐florbetapir SUVR obtained by PET and the HVa as measured by MRI as cerebral amyloidosis and neurodegenerative biomarkers, respectively, to categorize MDD patients in accordance with the NIA‐AA criteria.
The results of a previous study published by our group (Huang et al., 2013), which included 12 clinically diagnosed AD patients and 11 cognitively normal controls who had undergone the same 18F‐florbetapir PET and MRI analyses, were used to set imaging biomarker cutoff points. The threshold for global cortical amyloid positivity was constructed by the ROC method, as previously described (Huang et al., 2013). The cerebral amyloid‐positive cutoff point was 1.178, with a sensitivity of 92% and a specificity of 91%. The same ROC method was applied to determine the cutoff point for hippocampal atrophy: the HVa cutoff point was 6,879 mm3, with a sensitivity of 88% and a specificity of 100%.
2.6. Statistical analysis
Data were expressed as means ± SD or an absolute number with a proportion for descriptive statistics. Group comparisons between the controls, non‐MCI and MCI MDD patients and across the four biomarker groups were made using nonparametric Kruskal–Wallis tests with Dunn's multiple comparison post hoc analysis for continuous variables and χ2 tests for categorical data. A p value of 0.05 was defined as the threshold of statistical significance in each test.
3. RESULTS
The study recruited 63 nondemented MDD patients and 22 control subjects. Twenty‐four (38.1%) MDD patients met the clinical criteria for MCI at the time of imaging study. Table 1 presents the demographic, clinical and imaging characteristics of the control subjects, non‐MCI and MCI MDD patients. The MCI MDD patients had a significantly lower educational duration and lower MMSE scores, more depressive symptoms and impaired function according to the CDR‐SB score in comparison with the other groups; they also had more lifetime major depressive episodes than the non‐MCI MDD patients. In terms of imaging characteristics, the MCI MDD subjects had the lowest HVa among the three groups (p < 0.001); they also had a higher global 18F‐florbetapir SUVR than the other two groups, but this was not statistically significant (p = 0.131). All subjects in each group were further categorized into one of four types based on the presence or absence of amyloid deposition and neurodegenerative features as measured by the global 18F‐florbetapir SUVR and the MRI HVa, respectively. The biomarker cutoff points indicating normal and abnormal status as described above were used. The results of subject categorization using the four imaging biomarkers in the control, non‐MCI and MCI MDD subjects are shown in Table 1 and Figure 1. As expected, most of the control subjects (81.8%) were biomarker‐negative, in contrast to the MCI MDD patients (37.5%). A substantially higher proportion of the MCI MDD patients (29.2%) were categorized into the group with hippocampal atrophy alone as compared to the control subjects (0%) and non‐MCI patients (5.1%). A relatively higher proportion of the MCI MDD patients (12.5%) had both amyloid positivity and hippocampal atrophy as compared to the control subjects (4.5%) and non‐MCI patients (5.1%).
Table 1.
Demographic, clinical, and imaging characteristics of the control subjects, non‐MCI, and MCI MDD patients
| Characteristic | Controls n = 22 | Non‐MCI MDD n = 39 | MCI MDD n = 24 | p‐Value |
|---|---|---|---|---|
| Age (years) | ||||
| Mean ± SD | 66.7 ± 6.9 | 65.1 ± 6.5 | 66.9 ± 5.5 | 0.393 |
| Female gender, n (%) | 13 (59.1) | 28 (71.8) | 19 (79.2) | 0.320 |
| Education (years) | ||||
| Mean ± SD | 10.8 ± 4.1 | 8.5 ± 4.2*a | 7.0 ± 3.8**a | 0.006 |
| HAM‐D | ||||
| Mean ± SD | 2.0 ± 1.4 | 6.9 ± 6.4***a | 9.0 ± 4.8***a , *b | <0.001 |
| MMSE | ||||
| Mean ± SD | 27.6 ± 1.8 | 25.7 ± 2.3**a | 23.6 ± 2.9***a , **b | <0.001 |
| CDR‐SB | ||||
| Mean ± SD | 0.0 ± 0.0 | 0.3 ± 0.4*a | 1.2 ± 0.7***a , ***b | <0.001 |
| ApoE4, n (%) | 4 (18.2) | 9 (23.1) | 5 (20.8) | 0.903 |
| FSRS | ||||
| Mean ± SD | 7.4 ± 3.0 | 8.7 ± 4.6 | 8.8 ± 3.6 | 0.460 |
| 18F‐florbetapir SUVRs | ||||
| Mean ± SD | 1.1 ± 0.1 | 1.1 ± 0.1 | 1.2 ± 0.1 | 0.131 |
| 18F‐florbetapir SUVRs >1.178, n (%) | 4 (18.2) | 10 (25.6) | 8 (33.3) | 0.503 |
| HVa | ||||
| Mean ± SD | 8,091.5 ± 817.2 | 7,900.2 ± 760.3 | 7,061.7 ± 1,108.0***a , **b | <0.001 |
| HVa < 6,879 mm3, n (%) | 1 (4.5%) | 4 (10.3%) | 10 (41.7%)**a , **b | 0.002 |
| Biomarker group, n (%) | ||||
| All biomarkers negative | 18 (81.8) | 27 (69.2) | 9 (37.5)**a ,*b | 0.005 |
| Amyloid‐positive only | 3 (13.6) | 8 (20.5) | 5 (20.8) | 0.824 |
| Hippocampal atrophy only | 0 (0) | 2 (5.1) | 7 (29.2) *a , *b | 0.003 |
| Amyloid‐positive + hippocampal atrophy | 1 (4.5) | 2 (5.1) | 3 (12.5) | 0.550 |
| Age at onset (years) | ||||
| Mean ± SD | 54.3 ± 12.6 | 57.6 ± 8.2 | 0.381 | |
| Duration since onset of depression (years) | ||||
| Mean ± SD | 10.8 ± 10.8 | 9.4 ± 5.3 | 0.392 | |
| Number of depressive episodes | ||||
| Mean ± SD | 1.7 ± 1.0 | 2.5 ± 1.3**b | 0.007 | |
| Late‐onset MDD, n (%) | 16 (41.0) | 8 (33.3) | 0.541 | |
ApoE 4: Apolipoprotein E ε4 carrier; CDR‐SB: Clinical Dementia Rating–Sum of Boxes; FSRS: Framingham stroke risk score; HAM‐D: 17‐item Hamilton Depression Rating Scale; HVa: adjusted hippocampal volume; MCI: mild cognitive impairment; MDD: major depressive disorder; MMSE: Mini‐Mental Status Examination; SUVR: standardized uptake value ratio.
aSignificant difference compared with control subjects: *p < 0.05, **p < 0.01, ***p < 0.001. bSignificant difference compared with non‐MCI MDD patients: *p < 0.05, **p < 0.01, ***p < 0.001.
Figure 1.
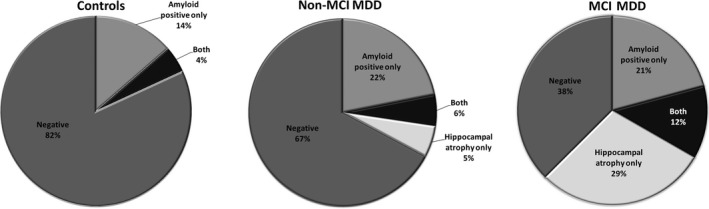
Biomarker distributions within each group. Each group from control subjects, non‐MCI, and MCI MDD patients was divided into the four imaging biomarker types including amyloid positive/negative and/or hippocampal atrophy positive/negative. MCI: mild cognitive impairment; MDD: major depressive disorder
Comparisons of the four biomarker groups in the non‐MCI and MCI MDD patients are presented in Tables 2 and 3, respectively. The highest global 18F‐florbetapir SUVRs were consistently observed in the subjects categorized into the group with both amyloid positivity and hippocampal atrophy, followed by the subjects categorized into the group with amyloid positivity only, regardless of the presence of MCI (p < 0.001) or not (p = 0.002). The smallest HVas were similarly observed in the subjects categorized into the group with hippocampal atrophy alone, followed by those categorized into the group with both amyloid positivity and hippocampal atrophy (non‐MCI, p = 0.013; MCI MDD, p = 0.001). The HVa of the subjects with amyloid positivity only was similar to that of the biomarker‐negative group. A trend of a higher percentage of ApoE4 carriers in the subjects with both amyloid positivity and hippocampal atrophy was observed, although this was not significant across the four biomarker groups in the non‐MCI patients.
Table 2.
Demographic, clinical, and imaging characteristics of the non‐MCI MDD patients categorized by imaging biomarker group
| Characteristic | Biomarkers negative, n = 27 | Amyloid only, n = 8 | Hippocampal atrophy only, n = 2 | Amyloid + hippocampal atrophy, n = 2 | p‐Value |
|---|---|---|---|---|---|
| Age (years) | |||||
| Mean ± SD | 63.9 ± 5.6 | 69.6 ± 7.1*a | 69.0 ± 2.8 | 58.5 ± 10.6 | 0.041 |
| Female gender, n (%) | 18 (66.7) | 6 (75.0) | 2 (100) | 2 (100) | 0.517 |
| Education (years) | |||||
| Mean ± SD | 9.1 ± 4.3 | 6.6 ± 3.9 | 6.0 ± 0.0 | 11.0 ± 5.7 | 0.258 |
| HAM‐D | |||||
| Mean ± SD | 7.8 ± 7.3 | 4.9 ± 3.2 | 5.5 ± 4.9 | 4.5 ± 3.5 | 0.865 |
| MMSE | |||||
| Mean ± SD | 25.7 ± 2.6 | 25.9 ± 1.0 | 25.5 ± 2.1 | 25.5 ± 2.1 | 0.935 |
| CDR‐SB | |||||
| Mean ± SD | 0.2 ± 0.4 | 0.2 ± 0.3 | 0.5 ± 0.7 | 0.8 ± 0.4 | 0.232 |
| ApoE4, n (%) | 6 (22.2) | 2 (25.0) | 0 (0) | 1 (50.0) | 0.799 |
| FSRS | |||||
| Mean ± SD | 7.6 ± 3.9 | 12.0 ± 5.0 | 11.0 ± 1.4 | 7.5 ± 9.2 | 0.169 |
| 18F‐florbetapir SUVRs | |||||
| Mean ± SD | 1.1 ± 0.1 | 1.2 ± 0.0***a | 1.2 ± 0.0 | 1.3 ± 0.1*a | <0.001 |
| HVa | |||||
| Mean ± SD | 8,072.1 ± 672.8 | 7,932.7 ± 665.7 | 6,547.9 ± 467.6*a , *b | 6,802.2 ± 57.6*a , *b | 0.013 |
| Age at onset (years) | |||||
| Mean ± SD | 51.7 ± 12.1 | 63.1 ± 11.8 | 56.5 ± 10.6 | 52.0 ± 17.0 | 0.101 |
| Duration since onset of depression (years) | |||||
| Mean ± SD | 12.2 ± 11.5 | 6.5 ± 9.5 | 12.5 ± 7.8 | 6.5 ± 6.4 | 0.389 |
| Number of depressive episodes | |||||
| Mean ± SD | 1.8 ± 1.1 | 1.4 ± 0.5 | 1.5 ± 0.7 | 2.5 ± 2.1 | 0.768 |
| Late‐onset MDD, n (%) | 8 (29.6) | 6 (75.0) | 1 (50.0) | 1 (50.0) | 0.071 |
| Cognitive domain z‐scores, mean ± SD | |||||
| Executive function | 0.0 ± 0.5 | 0.3 ± 0.6 | 0.8 ± 1.2 | 0.0 ± 0.5 | 0.327 |
| Memory | −0.2 ± 0.7 | 0.1 ± 1.0 | −0.2 ± 0.5 | −0.6 ± 0.4 | 0.670 |
| Processing speed | −0.4 ± 0.7 | −0.6 ± 0.7 | −0.1 ± 0.6 | −1.0 ± 0.6 | 0.387 |
| Language | 1.4 ± 0.8 | 0.8 ± 0.7 | 1.9 ± 1.4 | 1.2 ± 0.7 | 0.329 |
| Attention | 0.5 ± 0.7 | 0.1 ± 0.9 | 2.0 ± 0.8 | 0.1 ± 0.7 | 0.072 |
ApoE 4: Apolipoprotein E ε4 carrier; CDR‐SB: Clinical Dementia Rating–Sum of Boxes; FSRS: Framingham stroke risk score; HAM‐D: 17‐item Hamilton Depression Rating Scale; HVa: adjusted hippocampal volume; MCI: mild cognitive impairment; MDD: major depressive disorder; MMSE: Mini‐Mental Status Examination; SUVR: standardized uptake value ratio.
aSignificant difference compared with biomarker‐negative subjects: *p < 0.05, **p < 0.01, ***p < 0.001. bSignificant difference compared with amyloid‐positive only subjects: *p < 0.05, **p < 0.01, ***p < 0.001.
Table 3.
Demographic, clinical, and imaging characteristics of the MCI MDD patients categorized by imaging biomarker group
| Characteristic | Biomarkers negative, n = 9 | Amyloid only, n = 5 | Hippocampal atrophy only, n = 7 | Amyloid + hippocampal atrophy, n = 3 | p‐Value |
|---|---|---|---|---|---|
| Age (years) | |||||
| Mean ± SD | 64.0 ± 2.7 | 63.4 ± 3.4 | 69.1 ± 3.6*a , *b | 76.0 ± 6.9*a , *b | 0.007 |
| Female gender, n (%) | 7 (77.8) | 4 (80.0) | 7 (100) | 1 (33.3) | 0.119 |
| Education (years) | |||||
| Mean ± SD | 8.1 ± 4.7 | 7.8 ± 2.7 | 5.4 ± 3.8 | 6.0 ± 0.0 | 0.419 |
| HAM‐D | |||||
| Mean ± SD | 9.1 ± 6.6 | 7.8 ± 4.7 | 9.9 ± 3.0 | 8.7 ± 4.2 | 0.794 |
| MMSE | |||||
| Mean ± SD | 24.2 ± 3.1 | 21.4 ± 3.4 | 24.4 ± 2.2 | 23.7 ± 2.5 | 0.358 |
| CDR‐SB | |||||
| Mean ± SD | 0.8 ± 0.8 | 1.5 ± 0.8 | 1.5 ± 0.3 | 1.0 ± 0.5 | 0.077 |
| ApoE4, n (%) | 3 (33.3) | 0 (0.0) | 0 (0.0) | 2 (66.7)*b , *c | 0.050 |
| FSRS | |||||
| Mean ± SD | 8.1 ± 3.4 | 7.0 ± 3.5 | 9.1 ± 2.9 | 12.7 ± 3.8 | 0.203 |
| Mean ± SD | 10.4 ± 4.3 | 8.9 ± 1.8 | 8.3 ± 2.0 | 9.0 ± 1.6 | 0.721 |
| 18F‐florbetapir SUVRs | |||||
| Mean ± SD | 1.1 ± 0.1 | 1.3 ± 0.2**a | 1.1 ± 0.0**b | 1.4 ± 0.1*a , **c | 0.002 |
| HVa | |||||
| Mean ± SD | 7,698.3 ± 801.5 | 7,922.9 ± 567.0 | 5,931.1 ± 795.1**a , **b | 6,354.9 ± 330.8*a , *b | 0.001 |
| Age at onset (years) | |||||
| Mean ± SD | 54.1 ± 4.1 | 55.4 ± 5.8 | 59.1 ± 9.6 | 67.0 ± 12.0 | 0.213 |
| Duration since onset of depression (years) | |||||
| Mean ± SD | 10.0 ± 3.3 | 8.0 ± 4.5 | 10.0 ± 7.6 | 9.0 ± 6.9 | 0.918 |
| Number of depressive episodes | |||||
| Mean ± SD | 2.9 ± 1.4 | 2.2 ± 0.8 | 2.7 ± 1.8 | 1.7 ± 0.6 | 0.535 |
| Late‐onset MDD, n (%) | 1 (11.1) | 1 (20.0) | 4 (57.1) | 2 (66.7) | 0.148 |
| Cognitive domain z‐scores, mean ± SD | |||||
| Executive function | −0.8 ± 1.0 | −1.1 ± 0.9 | −0.8 ± 0.5 | −0.1 ± 0.7 | 0.424 |
| Memory | −1.4 ± 1.1 | −1.8 ± 1.4 | −1.2 ± 0.8 | −1.3 ± 0.7 | 0.757 |
| Processing speed | −1.2 ± 0.4 | −1.4 ± 0.3 | −1.3 ± 1.4 | −0.5 ± 1.1 | 0.602 |
| Language | 0.2 ± 0.6 | 0.6 ± 1.2 | 0.6 ± 0.6 | 1.6 ± 1.0 | 0.174 |
| Attention | −0.5 ± 1.0 | −0.2 ± 1.0 | 0.1 ± 0.9 | −0.3 ± 0.7 | 0.724 |
ApoE 4: Apolipoprotein E ε4 carrier; CDR‐SB: Clinical Dementia Rating–Sum of Boxes; FSRS: Framingham stroke risk score; HAM‐D: 17‐item Hamilton Depression Rating Scale; HVa: adjusted hippocampal volume; MCI: mild cognitive impairment; MDD: major depressive disorder; MMSE: Mini‐Mental Status Examination; SUVR: standardized uptake value ratio.
aSignificant difference compared with biomarker‐negative subjects: *p < 0.05, **p < 0.01, ***p < 0.001. bSignificant difference compared with amyloid‐positive only subjects: *p < 0.05, **p < 0.01, ***p < 0.001. cSignificant difference compared with subjects with hippocampal atrophy only: *p < 0.05, **p < 0.01, ***p < 0.001.
4. DISCUSSION
The present study was a preliminary study that employed a conceptual model of biomarkers based on the newly published NIA‐AA criteria for AD pathology to examine the distributions of MDD patients in four imaging biomarker groups categorized by the presence or absence of cerebral amyloidosis and hippocampal atrophy. We found that the MCI MDD patients had significantly higher amyloid deposition and greater hippocampal atrophy, followed by the non‐MCI MDD patients, as compared to the control subjects.
Our amyloid‐positive cutoff point was in accordance with that of Fleisher et al. (2011) which was determined from antemortem PET study and postmortem pathology data. Their pathology‐based threshold measured using the same 18F‐florbetapir PET as employed in this study was 1.17, which was similar to our result of 1.178. Our cutoff point for the adjusted hippocampal volume was comparable to the volumetric measurement of the bilateral hippocampus by Wang, Lirng, Lin, Chang, and Liu (2006) for individuals with MCI and AD derived from a prospective study in Taiwan. These results provided the rationale for data analyses in this study.
Cerebral amyloid burden and hippocampal atrophy as assessed separately from the 18F‐florbetapir SUVR and the HVa from MRI were significantly different among the three groups, in the following order of abnormality: MCI MDD > non‐MCI MDD > control subjects. The percentages of amyloid positivity, hippocampal atrophy and both were highest in the MCI MDD patients, intermediate in the non‐MCI MDD patients, and lowest in the control subjects. These results supported the conceptual model of AD pathophysiology proposed by the NIA‐AA criteria (Sperling et al., 2011). Of note, the MCI MDD patients who were amyloid‐positive only or, in particular, those with both amyloid positivity and hippocampal atrophy, might be at high risk of progression of MCI to AD dementia in the future, which is in accordance with the hypothesized AD model. Among the four imaging biomarker groups, the subjects in the group with amyloid positivity plus hippocampal atrophy had the highest 18F‐florbetapir SUVR, followed by the subjects in the amyloid‐positive only group. This finding also strengthened the core concept of the proposed AD model of the NIA‐AA criteria, indicating neurodegenerative progression from amyloid positivity first to amyloid positivity plus neurodegeneration.
In particular, an important finding of the present study was the high percentage of MCI MDD patients who were amyloid‐negative but had hippocampal atrophy. This finding clearly provided information that conflicted with the biomarker model of AD proposed by Jack et al. (2010, 2013) in which amyloid deposition becomes apparent first, and precedes other neurodegenerative biomarkers such as hippocampal atrophy or hypometabolism according to FDG‐PET. In addition, the proposed model also implied that by the time of development of symptomatic cognitive impairment with MCI, both amyloid positivity and neurodegeneration should be present (Heister, Brewer, Magda, Blennow, & McEvoy, 2011). This finding deserves further attention. As the proposed model of Jack et al. (2010, 2013) was developed on the basis of the amyloid cascade of AD, and predicts disease progression to AD, the MCI MDD patients with hippocampal atrophy alone in the present study might be subjected to an ongoing neurodegenerative pathway that is completely distinct from the process of AD degeneration. This observation was also made in another study (Petersen et al., 2013; Prestia et al., 2013), in which some MCI patients did not fit the Jack et al. model and were designated “suspected non‐AD pathway”(sNAP) subjects (Jack et al., 2012; Petersen et al., 2013) although these patients were not of a depressive population. Thus, our study indicates the frequency of sNAP patients in different groups is 29.2% of MCI MDD (hippocampal atrophy alone but cerebral amyloid negative), 5.1% of non‐MCI MDD, and 0% of control subjects, respectively. The present study provided the evidence of the heterogeneity of neurodegeneration in MDD patients. In particular, the results of this study implied large proportion of MCI MDD patients with sNAP might enter the neurodegenerative process of non‐AD types of dementia. Taken together, our results provided partial support for the recent NIA‐AA criteria for an AD model, but also suggested that underlying factors other than the amyloid cascade of AD pathology can drive neurocognitive degeneration in MDD patients.
Several studies identified a reduced hippocampal volume in MDD patients, which has been reported to be a consequence of repeated episodes of major depression (Hickie et al., 2005; Sheline, 2003; Sheline et al., 2003; Videbech & Ravnkilde, 2004). The mechanism behind the reduced hippocampal volume remains unclear. It has been well‐documented that hypothalamic–pituitary–adrenal (HPA) axis dysfunction might lead to hypercortisolism (Arborelius, Owens, Plotsky, & Nemeroff, 1999; Checkley, 1996), which is toxic to the hippocampus and further results in hippocampal shrinkage (McEwen, 2000; Sapolsky, 2000). However, it is not known whether the hippocampal atrophy observed in MDD patients might lead to changes in dementia status in later life, nor which types of dementia may be affected (Videbech & Ravnkilde, 2004).
4.1. Limitations
Some issues and limitations need to be raised. The hippocampal volume was selected as the neurodegenerative biomarker in the present study because it has been well‐studied as a validated MRI measure, and is also one of the neurodegenerative biomarkers included in the newly published NIA‐AA criteria. One limitation of this study was that only the neurodegenerative biomarker of hippocampal volume was used, and no FDG‐PET imaging or other CSF biomarkers were employed. Thus, the distribution rates might have differed if other biomarkers such as FDG‐PET and CSF biomarkers had been included. In addition, although the newly published NIA‐AA criteria provide a conceptual framework, several operational issues remain to be resolved, including standardization methods for biomarker measures, and consensus in the definitions of cutoff points for biomarkers (Jack et al., 2012). Thus, a population‐based means of defining abnormality was unavailable in this study, and some subjects at the margins of the biomarker cutoff points would inevitably have been classified into incorrect biomarker groups. Together, these operational issues limited and hampered mutual comparison of data obtained from different studies. However, in attempting to implement the NIA‐AA criteria, we performed a preliminary study in a MDD population that could be used as a basis for further exploration.
One additional limitation was the small sample size influenced the distribution of subjects into the different biomarker groups. The small sample size also meant that the MCI MDD patients could not be further subdivided into subgroups according to different domains of cognitive deficit (e.g., amnestic or nonamnestic MCI). Of note, this study was a clinical‐based study; thus, the control and MDD subjects differed from samples from the community or those in population‐based research. Our results cannot be generalized to the general population. Future long‐term studies with large sample sizes employing more neurodegenerative biomarkers are needed in order to examine in depth the neurodegenerative processes in elderly depressed patients.
5. CONCLUSION
This study highlights the expected heterogeneity of the processes of neurodegeneration in MDD patients. Some of the MCI MDD patients had entered the neurodegenerative process and were evident in the prodromal stage of AD dementia. In particular, other MCI MDD patients who were amyloid‐negative but had abnormal hippocampal atrophy might represent prodromal stages of other non‐AD types of dementia.
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest.
AUTHOR CONTRIBUTIONS
KY Wu, KJ Lin, and IT Hsiao designed the study. KY Wu, KJ Lin, IT Hsiao, CH Chen, and CY Liu acquired the data. KY Wu, KJ Lin, CS Chen, SY Huang, TC Yen, and IT Hsiao analyzed the data. KY Wu, KJ Lin, TC Yen, and IT Hsiao wrote the article. All authors revised and approved the article for publication.
ROLE OF THE FUNDER/SPONSOR
The funding sources had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and the decision to submit the manuscript for publication.
ACKNOWLEDGMENTS
We thank Avid Radiopharmaceuticals Inc. (Philadelphia, PA, USA) for providing the precursor for the preparation of 18F‐florbetapir. This study was carried out with financial support from the National Science Council and the Ministry of Science and Technology, Taiwan (MOST 105‐2314‐B‐182A‐061‐, MOST 106‐2314‐B‐182‐017‐MY3, MOST 104‐2314‐B‐182A‐034‐, NSC 101‐2314‐B‐182‐054‐MY2, NSC 100‐2314‐B‐182‐041), and grants from the Research Fund of Chang Gung Memorial Hospital (CMRPG3F1031, CMRPG3E2052, CMRPG3E2051, CMRPD1C0383, CMRPG3D1802, BMRP 488, and CMRPD1E0303). This study was also supported by the Ministry of Health and Welfare, Taiwan (MOHW105‐TDU‐B‐212‐113020, MOHW106‐TDU‐B‐212‐113005). We also thank the Center for Advanced Molecular Imaging and Translation, Chang Gung Memorial Hospital, Linkou, for technical support.
Wu K‐Y, Lin K‐J, Chen C‐H, et al. Diversity of neurodegenerative pathophysiology in nondemented patients with major depressive disorder: Evidence of cerebral amyloidosis and hippocampal atrophy. Brain Behav. 2018;8:e01016 10.1002/brb3.1016
REFERENCES
- Albert, M. L. (1973). A simple test of visual neglect. Neurology, 23, 658–664. 10.1212/WNL.23.6.658 [DOI] [PubMed] [Google Scholar]
- Albert, M. S. , DeKosky, S. T. , Dickson, D. , Dubois, B. , Feldman, H. H. , Fox, N. C. , … Petersen, R. C. (2011). The diagnosis of mild cognitive impairment due to Alzheimer's disease: Recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimer's & Dementia, 7(3), 270–279. 10.1016/j.jalz.2011.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arborelius, L. , Owens, M. , Plotsky, P. , & Nemeroff, C. (1999). The role of corticotropin‐releasing factor in depression and anxiety disorders. Journal of Endocrinology, 160(1), 1–12. 10.1677/joe.0.1600001 [DOI] [PubMed] [Google Scholar]
- Benton, A. , Hamsher, K. , & Sivan, A. (1989). Multilingual aphasia examination. Iowa City, IA: AJA Associates, Inc. [Google Scholar]
- Bhalla, R. K. , Butters, M. A. , Mulsant, B. H. , Begley, A. E. , Zmuda, M. D. , Schoderbek, B. , … Becker, J. T. (2006). Persistence of neuropsychologic deficits in the remitted state of late‐life depression. American Journal of Geriatric Psychiatry, 14(5), 419–427. 10.1097/01.JGP.0000203130.45421.69 [DOI] [PubMed] [Google Scholar]
- Buschke, H. , & Fuld, P. A. (1974). Evaluating storage, retention, and retrieval in disordered memory and learning. Neurology, 24(11), 1019–1025. 10.1212/WNL.24.11.1019 [DOI] [PubMed] [Google Scholar]
- Checkley, S. (1996). The neuroendocrinology of depression and chronic stress. British Medical Bulletin, 52(3), 597–617. 10.1093/oxfordjournals.bmb.a011570 [DOI] [PubMed] [Google Scholar]
- Diniz, B. S. , Butters, M. A. , Albert, S. M. , Dew, M. A. , & Reynolds, C. F. (2013). Late‐life depression and risk of vascular dementia and Alzheimer's disease: Systematic review and meta‐analysis of community‐based cohort studies. The British Journal of Psychiatry, 202(5), 329–335. 10.1192/bjp.bp.112.118307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois, B. , Slachevsky, A. , Litvan, I. , & Pillon, B. (2000). The FAB A frontal assessment battery at bedside. Neurology, 55(11), 1621–1626. 10.1212/WNL.55.11.1621 [DOI] [PubMed] [Google Scholar]
- Fleisher, A. S. , Chen, K. , Liu, X. , Roontiva, A. , Thiyyagura, P. , Ayutyanont, N. , … Pontecorvo, M. J. (2011). Using positron emission tomography and florbetapir F 18 to image cortical amyloid in patients with mild cognitive impairment or dementia due to Alzheimer disease. Archives of Neurology, 68(11), 1404 10.1001/archneurol.2011.150 [DOI] [PubMed] [Google Scholar]
- Gauthier, S. , Reisberg, B. , Zaudig, M. , Petersen, R. C. , Ritchie, K. , Broich, K. , … Chertkow, H. (2006). Mild cognitive impairment. The Lancet, 367(9518), 1262–1270. 10.1016/S0140-6736(06)68542-5 [DOI] [PubMed] [Google Scholar]
- Heister, D. , Brewer, J. B. , Magda, S. , Blennow, K. , McEvoy, L. K. ; Alzheimer's Disease Neuroimaging Initiative . (2011). Predicting MCI outcome with clinically available MRI and CSF biomarkers. Neurology, 77(17), 1619–1628. 10.1212/WNL.0b013e3182343314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickie, I. , Naismith, S. , Ward, P. , Turner, K. , Scott, E. , Mitchell, P. , … Parker, G. (2005). Reduced hippocampal volumes and memory loss in patients with early‐and late‐onset depression. The British Journal of Psychiatry, 186(3), 197 10.1192/bjp.186.3.197 [DOI] [PubMed] [Google Scholar]
- Hsiao, T. , Huang, C.‐C. , Hsieh, C.‐J. , Wey, S.‐P. , Kung, M.‐P. , Yen, T.‐C. , & Lin, K.‐J. (2013). Perfusion‐like template and standardized normalization‐based brain image analysis using 18F‐florbetapir (AV‐45/Amyvid) PET. European Journal of Nuclear Medicine and Molecular Imaging, 40(6), 908–920. 10.1007/s00259-013-2350-x [DOI] [PubMed] [Google Scholar]
- Huang, K.‐L. , Lin, K.‐J. , Hsiao, T. , Kuo, H.‐C. , Hsu, W.‐C. , Chuang, W.‐L. , … Wai, Y.‐Y. (2013). Regional amyloid deposition in amnestic mild cognitive impairment and Alzheimer's disease evaluated by [18F] AV‐45 positron emission tomography in Chinese population. PLoS ONE, 8(3), e58974 10.1371/journal.pone.0058974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack, C. R. , Knopman, D. S. , Jagust, W. J. , Petersen, R. C. , Weiner, M. W. , Aisen, P. S. , … Weigand, S. D. (2013). Tracking pathophysiological processes in Alzheimer's disease: An updated hypothetical model of dynamic biomarkers. The Lancet Neurology, 12(2), 207–216. 10.1016/S1474-4422(12)70291-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack, C. R. , Knopman, D. S. , Jagust, W. J. , Shaw, L. M. , Aisen, P. S. , Weiner, M. W. , … Trojanowski, J. Q. (2010). Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. The Lancet Neurology, 9(1), 119–128. 10.1016/S1474-4422(09)70299-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack, C. R. , Knopman, D. S. , Weigand, S. D. , Wiste, H. J. , Vemuri, P. , Lowe, V. , … Ivnik, R. J. (2012). An operational approach to National Institute on Aging–Alzheimer's Association criteria for preclinical Alzheimer disease. Annals of Neurology, 71(6), 765–775. 10.1002/ana.22628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack, C. R. , Lowe, V. J. , Weigand, S. D. , Wiste, H. J. , Senjem, M. L. , Knopman, D. S. , … Kemp, B. J. (2009). Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer's disease: Implications for sequence of pathological events in Alzheimer's disease. Brain, 1355–1365. 10.1093/brain/awp062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger, K. A. (2013). Organic bases of late‐life depression: A critical update. Journal of Neural Transmission, 120(7), 1109–1125. 10.1007/s00702-012-0945-1 [DOI] [PubMed] [Google Scholar]
- Jorm, A. F. (2001). History of depression as a risk factor for dementia: An updated review. Australian & New Zealand Journal of Psychiatry, 35(6), 776–781. 10.1046/j.1440-1614.2001.00967.x [DOI] [PubMed] [Google Scholar]
- Lee, J. S. , Potter, G. G. , Wagner, H. R. , Welsh‐Bohmer, K. A. , Steffens, D. C. , Lee, J. S. , … Steffens, D. C. (2007). Persistent mild cognitive impairment in geriatric depression. International Psychogeriatrics, 19(1), 125–135. 10.1017/S1041610206003607 [DOI] [PubMed] [Google Scholar]
- Lin, K.‐J. , Hsu, W.‐C. , Hsiao, I.‐T. , Wey, S.‐P. , Jin, L.‐W. , Skovronsky, D. , … Yao, C. H. (2010). Whole‐body biodistribution and brain PET imaging with [18F] AV‐45, a novel amyloid imaging agent—A pilot study. Nuclear Medicine and Biology, 37(4), 497. [DOI] [PubMed] [Google Scholar]
- McEwen, B. S. (2000). Effects of adverse experiences for brain structure and function. Biological Psychiatry, 48(8), 721–731. 10.1016/S0006-3223(00)00964-1 [DOI] [PubMed] [Google Scholar]
- McKhann, G. M. , Knopman, D. S. , Chertkow, H. , Hyman, B. T. , Jack, Jr, C. R. , Kawas, C. H. , … Mayeux, R. (2011). The diagnosis of dementia due to Alzheimer's disease: Recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimer's & Dementia, 7(3), 263–269. 10.1016/j.jalz.2011.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ownby, R. L. , Crocco, E. , Acevedo, A. , John, V. , Loewenstein, D. , Ownby, R. L. , … Loewenstein, D. (2006). Depression and risk for Alzheimer disease: Systematic review, meta‐analysis, and metaregression analysis. Archives of General Psychiatry, 63(5), 530–538. 10.1001/archpsyc.63.5.530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen, R. C. (2004). Mild cognitive impairment as a diagnostic entity. Journal of Internal Medicine, 256(3), 183–194. 10.1111/j.1365-2796.2004.01388.x [DOI] [PubMed] [Google Scholar]
- Petersen, R. C. , Aisen, P. , Boeve, B. F. , Geda, Y. E. , Ivnik, R. J. , Knopman, D. S. , … Rocca, W. A. (2013). Mild cognitive impairment due to Alzheimer disease in the community. Annals of Neurology, 74(2), 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen, R. C. , Doody, R. , Kurz, A. , Mohs, R. C. , Morris, J. C. , Rabins, P. V. , … Winblad, B. (2001). Current concepts in mild cognitive impairment. Archives of Neurology, 58(12), 1985 10.1001/archneur.58.12.1985 [DOI] [PubMed] [Google Scholar]
- Prestia, A. , Caroli, A. , van der Flier, W. M. , Ossenkoppele, R. , Van Berckel, B. , Barkhof, F. , … Schöll, M. (2013). Prediction of dementia in MCI patients based on core diagnostic markers for Alzheimer disease. Neurology, 80(11), 1048–1056. 10.1212/WNL.0b013e3182872830 [DOI] [PubMed] [Google Scholar]
- Rapp, M. A. , Schnaider‐Beeri, M. , Grossman, H. T. , Sano, M. , Perl, D. P. , Purohit, D. P. , … Haroutunian, V. (2006). Increased hippocampal plaques and tangles in patients with Alzheimer disease with a lifetime history of major depression. Archives of General Psychiatry, 63(2), 161–167. 10.1001/archpsyc.63.2.161 [DOI] [PubMed] [Google Scholar]
- Reitan, R. M. , & Wolfson, D. (1985). The Halstead–Reitan neuropsychological test battery: Theory and clinical interpretation (Vol. 4). Mesa, AZ: Reitan Neuropsychology. [Google Scholar]
- Sapolsky, R. M. (2000). The possibility of neurotoxicity in the hippocampus in major depression: A primer on neuron death. Biological Psychiatry, 48(8), 755–765. 10.1016/S0006-3223(00)00971-9 [DOI] [PubMed] [Google Scholar]
- Sheline, Y. I. (2003). Neuroimaging studies of mood disorder effects on the brain. Biological Psychiatry, 54(3), 338–352. 10.1016/S0006-3223(03)00347-0 [DOI] [PubMed] [Google Scholar]
- Sheline, Y. I. , Gado, M. H. , Kraemer, H. C. , Sheline, Y. I. , Gado, M. H. , & Kraemer, H. C. (2003). Untreated depression and hippocampal volume loss [see comment]. American Journal of Psychiatry, 160(8), 1516–1518. 10.1176/appi.ajp.160.8.1516 [DOI] [PubMed] [Google Scholar]
- Sperling, R. A. , Aisen, P. S. , Beckett, L. A. , Bennett, D. A. , Craft, S. , Fagan, A. M. , … Montine, T. J. (2011). Toward defining the preclinical stages of Alzheimer's disease: Recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimer's & Dementia, 7(3), 280–292. 10.1016/j.jalz.2011.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Videbech, P. , & Ravnkilde, B. (2004). Hippocampal volume and depression: A meta‐analysis of MRI studies. American Journal of Psychiatry, 161(11), 1957–1966. 10.1176/appi.ajp.161.11.1957 [DOI] [PubMed] [Google Scholar]
- Voevodskaya, O. , Simmons, A. , Nordenskjöld, R. , Kullberg, J. , Ahlström, H. , Lind, L. , … Alzheimer's Disease Neuroimaging Initiative . (2014). The effects of intracranial volume adjustment approaches on multiple regional MRI volumes in healthy aging and Alzheimer's disease. Frontiers in Aging Neuroscience, 6, 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, P. , Lirng, J. , Lin, K. , Chang, F. , & Liu, H. (2006). Prediction of Alzheimer's disease in mild cognitive impairment: A prospective study in Taiwan. Neurobiology of Aging, 27(12), 1797–1806. 10.1016/j.neurobiolaging.2005.10.002 [DOI] [PubMed] [Google Scholar]
- Wechsler, D. (1997). WAIS‐III, wechsler adult intelligence scale: Administration and scoring manual. San Antonio, TX: Psychological Corporation. [Google Scholar]
- Wu, K.‐Y. , Hsiao, T. , Chen, C.‐S. , Chen, C.‐H. , Hsieh, C.‐J. , Wai, Y.‐Y. , … Lin, K. J. (2013). Increased brain amyloid deposition in patients with a lifetime history of major depression: Evidenced on 18F‐florbetapir (AV‐45/Amyvid) positron emission tomography. European Journal of Nuclear Medicine and Molecular Imaging, 41, 714–722. [DOI] [PubMed] [Google Scholar]
- Wu, K.‐Y. , Liu, C.‐Y. , Chen, C.‐S. , Chen, C.‐H. , Hsiao, T. , Hsieh, C.‐J. , … Lin, K.‐J. (2016). Beta‐amyloid deposition and cognitive function in patients with major depressive disorder with different subtypes of mild cognitive impairment: 18F‐florbetapir (AV‐45/Amyvid) PET study. European Journal of Nuclear Medicine and Molecular Imaging, 43, 1067–1076. 10.1007/s00259-015-3291-3 [DOI] [PubMed] [Google Scholar]
- Yao, C.‐H. , Lin, K.‐J. , Weng, C.‐C. , Hsiao, I.‐T. , Ting, Y.‐S. , Yen, T.‐C. , … Wey, S.‐P. (2010). GMP‐compliant automated synthesis of [18F] AV‐45 (Florbetapir F 18) for imaging β‐amyloid plaques in human brain. Applied Radiation and Isotopes, 68(12), 2293–2297. 10.1016/j.apradiso.2010.07.001 [DOI] [PubMed] [Google Scholar]
- Yeh, Y.‐C. , Tsang, H.‐Y. , Lin, P.‐Y. , Kuo, Y.‐T. , Yen, C.‐F. , Chen, C.‐C. , … Chen, C.‐S. (2011). Subtypes of mild cognitive impairment among the elderly with major depressive disorder in remission. American Journal of Geriatric Psychiatry, 19(11), 923 10.1097/JGP.0b013e318202clc6 [DOI] [PubMed] [Google Scholar]