

Abstract
Human parainfluenza virus type 3 (HPIV3), one of the paramyxoviruses, uses its accessory C protein as an antagonist against interferon (IFN)-mediated host innate immunity. We have previously shown that the C protein significantly decreased the IFN-induced phosphorylation of signal transducer and activator of transcription (Stat) 1 and the formation of gamma IFN activation factor (GAF) complex, thus abrogating the antiviral activity of the IFNs against vesicular stomatitis virus (VSV) replication. Here, by mutational analyses we demonstrated that the N-terminal truncation of the C protein (CNΔ25 and CNΔ50) substantially (∼50%) recovers the IFN-induced responses, suggesting the critical role of the N-terminal region of the C protein in IFN signaling. Furthermore, our results indicate that the charged amino acid residues within the N-terminal region of the C protein regulate the antagonistic effect of the C protein on IFN signaling.
Key words: Human parainfluenza virus type 3 (HPIV3), C protein, Interferon (IFN) antagonist
INTRODUCTION
Human parainfluenza virus type 3 (HPIV3) infection causes severe respiratory tract diseases, including croup, bronchiolitis, bronchitis, and pneumonia in infants and young children (10,20), as well as immunocompromised patients (13), especially those undergoing allogeneic hematopoietic stem cells transplantation (2). HPIV3, together with other human parainfluenza virues (HPIV1, HPIV2, and HPIV4), is one of the most common pathogens, second only to the respiratory syncytial virus (RSV), to cause the infectious respiratory tract diseases requiring hospitalization of infants and children.
The virus has a linear nonsegmented, negative strand RNA genome of 15,462 bases. The genome is transcribed into six mRNAs in the infected cells in the order N, P, M, F, HN, and L proteins. The P mRNA encodes the viral phosphoprotein P, an essential cofactor of the viral polymerase protein L for both transcription and replication. The C protein, one of the accessory proteins of HPIV3, is also encoded by the P mRNA through an alternative open reading frame (21). The P mRNA also encodes two other putative accessory proteins D and V by RNA editing, although the V protein has not been detected in the infected cells to date (5). As observed for other para-myxoviruses (4,6,11,26), these accessory proteins of HPIV3 were dispensable for the HPIV3 replication (3). HPIV3 with deletion of C (rC KO PIV3) was able to grow both in cell culture and in the respiratory tracts of animals, but it was significantly attenuated, with 3–4 log lower of the peak titers compared to the wild-type virus (3). Several accessory proteins of paramyxoviruses are shown to counteract the host immune responses by targeting the virus-induced IFN synthesis (1,7,16,17), the IFN signaling pathway (6,14,15,24), or the expression and functions of the IFN-stimulated genes (ISGs) [e.g., the dsRNA-dependent protein kinase (PKR)] (25,26). The C protein of HPIV3 has been reported to inhibit the IFN-induced phosphorylation of signal transducer and activator of transcription (Stat) 1 and the formation of gamma IFN activation factor (GAF) transcriptional complex in the IFN signaling (18), but the mechanism for the inhibition is as yet unknown.
In this study, we established stable cell lines that express the C, CNΔ25, or CNΔ50 proteins of HPIV3, using the lentivirus vector system (18,19) to gain insight into the C protein-mediated inhibition of IFN signaling. The CNΔ25 and CNΔ50 proteins refer to the N-terminal 25 or 50 amino acid deletion mutants of the C protein, respectively (19). We have found that the N-terminal 25 amino acids are required for the IFN antagonistic effect of the C protein of HPIV3 and substitution of the charged amino acids within the N-terminus resulted in less inhibition of the IFN-responsive promoter activation. Our study suggests that the charged amino acids in the N-terminal domain of the C protein regulate the inhibitory activity of the C protein in IFN signaling.
MATERIALS AND METHODS
Cells and Virus
HeLa (S) cells were cultured in DMEM (10% FBS, penicillin/streptomycin). LC-6, CNΔ25, and CNΔ50 cells are HeLa-derived stable cell lines and were cultured in DMEM with blasticidin (18,19). Vesicular stomatitis virus (VSV, Indiana) was grown in CV-1 cells.
IFN Stimulation and Western Blotting
To detect the IFN signaling molecules (e.g., Stat1, p-Stat1, p-Tyk2), cells (HeLa, LC-6, CNΔ25, and CNΔ50, 7 × 106 each) were seeded into dishes (diameter of 100 mm). In the second day, the cells were washed with phosphate-buffered saline (PBS) and cultured in Opti-MEM media containing IFN-α A/D (1000 U/ml) or IFN-γ (1000 U/ml). After 30 min of IFN treatment, cells were washed with PBS and scraped into 100 μl of extraction buffer (10 mM HEPES, pH 7.9, 300 mM NaCl, 0.25% NP-40, 10% glycerol, 1 mM EDTA, 1 mM EGTA, 1 mM Na3VO4, 1 mM DTT, 1 mM PMSF) (24). The cells extracts were quantified by the Bradford assay and used for Western blotting (19). The anti-Stat1 antibody and the HRP-labeled secondary antibody were purchased from Santa Cruz. The anti-p-Stat1 and anti-p-Tyk2 antibodies were ordered from Cell Signaling.
Electrophoretic Mobility Shift Assay (EMSA)
The sequences for GAS probe are 5′-TCGAGCCT GATTTCCCCGAAATGACGGC3′ and TCGAGCC GTCATTTCGGGGAAATCAGGC-3′, human IR element with the GAS core sequence underlined (9). The oligonucleotides were synthesized by Operon and 20 μg/strand of oligonucleotide was annealed in 1× anealing buffer (10 mM Tris-HCl, pH 7.5, 0.1 M NaCl, 1 mM EDTA) by heating at 95°C for 3 min and cooling down to 4°C slowly. EMSA was performed as follows. The double-stranded probe (200 ng) was labeled using Klenow enzyme (6u) at 37°C for 1 h in the presence of 20 μCi of α-[32P]dCTP and labeling mix-dCTP (0.1 mM of dATP, dTTP, and dGTP in 50 mM Tris-HCl, pH 7.5, 50 mM MgCl2, 250 mM NaCl, 25 mM β-mercaptoethanol). α-[32P]-labeled probe was further purified with Quick Spin Column (Roche). In the 20 μl of DNA protein binding reaction, whole cell extract (20 μg) was incubated with α-[32P]-labeled probe (3–4 × 104 cpm), 2 μg of poly(dI-dC) · poly(dI-dC) in 1× binding buffer (100 mM Tris-HCl, pH 7.5, 500 mM NaCl, 50 mM MgCl2, 5 mM DTT, 0.5% NP-40, 5 mM EDTA, pH 8.0, 10% glycerol) for 20 min at room temperature. After adding 10× loading buffer (250 mM Tris-HCl, pH 7.5, 0.2% bromophenol blue, 0.2% xylene cyanol, 40% glycerol), the samples were loaded onto a 5% poly-acrylamide gel and run at 200 V of electronic voltage. The gel was fixed in 7% acetic acid for 5 min, and dried using BioRad drier at 90°C for 1 h. The dried gel was exposed to X-ray film in a −80°C freezer.
Plaque Assay
The virus titer was determined by plaque assay as described previously (19). Briefly, the virus sample was serially diluted in 1 ml of OPTI-MEM. Confluent monolayer of CV1 cells in six-well plates was washed with PBS and incubated with the diluted suspension at 37°C, 5% CO2 for 1.5 h. Then the media were removed and cells were washed with PBS and overlaid with 0.8% methyl cellulose. After 48 h, the methyl cellulose was aspired and cells were stained with 1% crystal violet (in 50% methanol).
Luciferase Assay
The IFN-α/β-responsive plasmid [termed p(9-27)4tkΔ(–39)lucter] and the IFN-γ-responsive plasmid [termed p(GAS)2tkΔ(–39)lucter] were kindly supplied by Dr. Steve Goodbourn. Monolayers of HeLa cells in 24-well plates at 50–70% confluence were transfected with 0.3 μg DNA [0.1 μg of p(9-27)4tkΔ(–39)lucter or p(GAS)2tkΔ(–39)lucter, 0.1 μg of pLR-TK, 0.1 μg of pcDNA3-C or its mutants] and 2 μl lipofectin according to the manufacturer’s instructions. At 48 h posttransfection, the cells were induced with 1000 U/ml of either rHuIFNα A/D or IFN-γ. Four hours later, firefly- and renillar-luciferase activities in cellular lysates were measured by dual luciferase assay (Promega). The ratio of firefly LUC/renilla LUC represents the activation of the IFN-responsive promoter.
RESULTS
IFN-Induced Stat1 Phosphorylation and the Formation of Transcription Complex in the Presence of C Protein or its N-terminal Mutants
We used the C, CNΔ25, or CNΔ50 cell lines to study the effects of the overexpressed proteins in IFN signaling. It is well established that after IFN-α or INF-γ treatment, the Stat1 is phosphorylated, dimerized (Stat1/Stat1 homodimer for IFN-γ and Stat1/ Stat2 heterodimer for IFN-α), and translocated into the cell nucleus, forming the IFN-stimulated gene factor (ISGF) 3 or GAF complex, which transcribes the IFN-activated genes (22,23). Accordingly, we treated cells with IFN-α (1000 U/ml) or IFN-γ (1000 U/ml) for 30 min, then harvested the cell lysates and immunoblotted to detect the IFN-induced p-Stat1. As shown in Figure 1A and B, during the 30 min of IFN induction, the Stat1 synthesis was comparable in the mock, IFN-α-, or IFN-γ-treated HeLa/LC-6/CNΔ25/ CNΔ50 cells. As expected, the IFN-induced p-Stat1 (HeLa + IFN-α/IFN-γ; Fig. 1A, B) was greatly decreased in the LC-6 cells (LC-6 + IFN-α/IFN-γ; Fig. 1A, 1B) (18). Interestingly, phosphorylation of Stat1 was significantly (30–50%) recovered in cells CNΔ25 and CNΔ50 (CNΔ25/CNΔ50 + IFN-α/IFN-γ; Fig. 1A, 1B), suggesting that the IFN signaling was partially restored when the N-terminal 25 or 50 amino acid residues were removed from the C protein.
Figure 1.
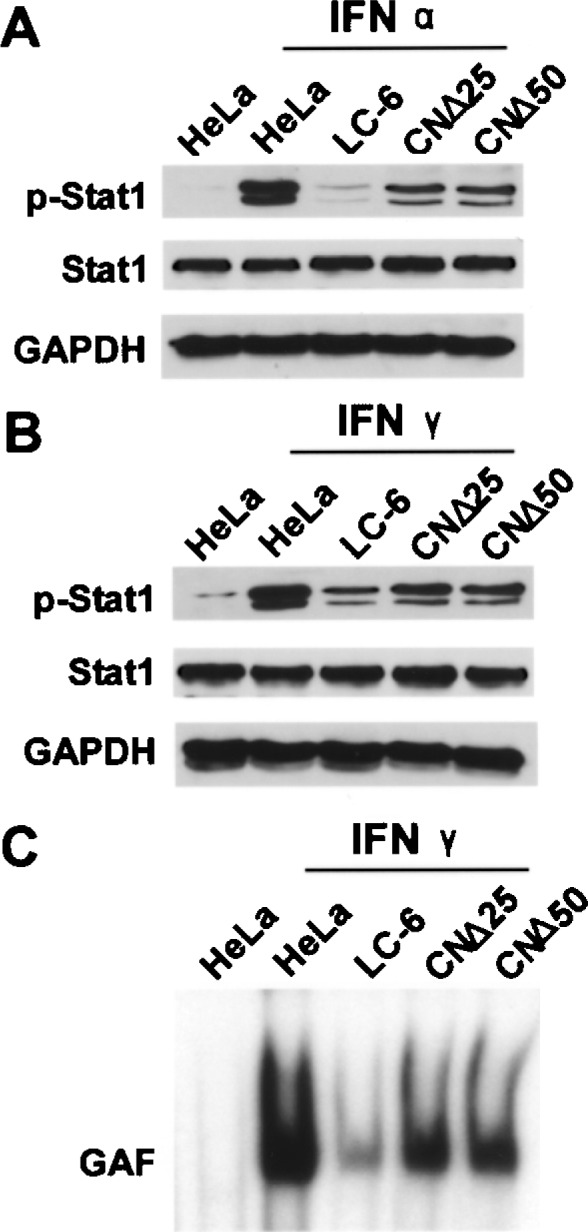
The effects of the C, CNΔ25, and CNΔ50 proteins on the IFN-α or IFN-γ signaling pathway. (A) The HeLa, LC-6, CNΔ25, and CNΔ50 cells were treated with IFN-α (1000 U/ml) for 30 min, then harvested for Western blot analyses using the anti-p-Stat1, anti-Stat1, and anti-GAPDH antibodies, respectively. Nontreated HeLa cells served as a control. GAPDH was used as a loading control. (B) Cells were treated with IFN-γ (1000 U/ml) and harvested for Western blot analyses as described in (A). (C) Cell lysates from (B), were incubated with the 32P-labeled GAS probe for 20 min, and the reaction mixtures were subjected to 5% polyacrylamidine gel to separate the radioactive GAF complex for autoradiography.
The above cell lysates from HeLa and stable cell lines were also used for the electrophoretic mobility shift assay (EMSA) to detect the formation of the GAF transcriptional complex induced by IFN-γ (Fig. 1C). The GAF complex band in the Figure 1C was discernible when 20-fold of cold probes were added, and supershifted in the presence of the anti-Stat1 antibody (data not shown), indicating that the complex was specific. As previously shown (18), the IFN-γ-induced GAF complex, the p-Stat1 homodimer, was dramatically repressed in the LC-6 cells, but clearly augmented to about 50% level in the CNΔ25 or CNΔ50 cells (Fig. 1C), suggesting that the IFN signaling was effectively restored when the N-terminal 25 or 50 amino acid residues were removed.
IFN-Induced Antiviral Activity Against Vesicular Stomatitis Virus (VSV) in the Presence of C Protein or its Mutants
Next, we compared the IFN-induced antiviral state in these stable cell lines. The HeLa, LC-6, CNΔ25, and CNΔ50 cells were pretreated with IFN-α (1000 U/ml) or IFN-γ (1000 U/ml) for 16 h and infected with VSV at an MOI of 0.1. At 24 h postinfection, the cells were stained with crystal violet to test the cell viability, and the corresponding supernatants were harvested for plaque assay. As shown in Figure 2, although all uninfected cells grew well (mock), the infection of VSV caused 50% cell death in HeLa cells, but 100% of LC-6/CNΔ25/CNΔ50 cells were obliterated. Pretreatment with IFN-α or IFN-γ, however, consistently rescued all the HeLa cells and 30–50% of CNΔ25/CNΔ50 cells from VSV infection, but having no effect on LC-6 cells (IFN-α/IFN-γ + VSV). The virus yields were decreased by 2.5–3.5 log in the HeLa cells treated with IFN-α or IFN-γ, similar to the reduction of virus titers in the CNΔ25 and CNΔ50 cells pretreated with IFNs (1–1.5 log). As expected, no inhibitory effect was seen in the LC-6 cells following IFN treatment. Combining the cell viability and virus titer data, it seems that the C protein of HPIV3 abrogated the IFN-induced antiviral state, whereas removal of the N-terminal 25 or 50 amino acid residues partially restored the IFN function. Note that the replication of VSV (+VSV; Fig. 2B), as well as the subsequent cytopathogenicity (+VSV; Fig. 2A), was obviously potentiated in the C protein-expressing cells compared to the HeLa cells, suggesting the role of the C proteins in inhibiting other host innate immune responses (e.g., the virus-induced endogenous IFN synthesis) that are unrelated to the IFN signaling.
Figure 2.

The effects of the C, CNΔ25, and CNΔ50 proteins on the IFN-α- or IFN-γ-induced antiviral state. (A) The HeLa, LC-6, CNΔ25, and CNΔ50 cells were pretreated with IFN-α (1000 U/ml) or IFN-γ (1000 U/ml) for 16 h, then infected with VSV for 24 h. The cells were stained with crystal violet to test the cell viability. (B) The supernatants corresponding to the differently treated cells in (A) were titered by plaque assay. The plaque assay for each sample was performed three times. The average and SD of the viral yields are indicated.
The Inhibition of Upstream Signaling Molecule p-Tyrosine Kinase 2 (p-Tyk2)
It has been previously established that after IFN-α/β is bound to the receptor, the signaling cascade begins with the phosphorylation of Tyk2, which can then cross-phosphorylate Janus kinase 1 (Jak1). Activated Jak1 and Tyk2 are responsible for the sequential phosphorylation of IFN-α/β receptor alpha chain (IFNAR1), Stat2, and Stat1 (22,23). To explore the target of the C protein in the IFN signaling pathway, we followed the appearance of p-Tyk2 using the same cell extracts used in Figure 1. As shown in Figure 3A, p-Tyk2, the first activated molecule in the pathway, was significantly inhibited in LC-6 cells. In contrast, levels of p-Tyk2 in CNΔ25 and CNΔ50 cells were close to that in parent HeLa cells. These results suggest strongly that the C protein negatively regulates the phosphorylation of Tyk2 during IFN-α signaling and its N-terminal domain is required for such inhibition.
Figure 3.

Inhibition of the upstream molecule p-Tyk2 in IFN signaling pathway. (A) The order of activation of molecules in the IFN-α/β signaling pathway. The HeLa, LC-6, CNΔ25, and CNΔ50 cells were treated with IFN-α (1000 U/ml) for 30 min, then harvested for Western blot analyses using the anti-p-Tyk2 antibody. Nontreated HeLa cells served as a control. GAPDH was used as a loading control.
The Role of Charged Amino Acids Within the N-terminal Domain of the C Protein in IFN Signaling Inhibition
We have recently shown that the mutants CNΔ25 and CNΔ50 essentially abrogate transcription of minigenome (19). Further mutational analyses revealed that the charged amino acids within the N-terminal domain regulate the transcriptional inhibitory activity of the C protein. To test the role of charged amino acids in the IFN response, we used the IFN-responsive promoter (ISRE/GAS) luciferase assay to identify the role of residues K3, K6, K12, E16, and R24 within the N-terminal 25aa of the C protein. The charged amino acid residues were substituted with alanine separately, and the HeLa cells were transfected with the IFN-responsive plasmid p(9-27)4tkΔ(–39) lucter or p(GAS)2tkΔ(–39)lucter (27), the internal control pRL-TK plasmid containing the renilla luciferase gene and the plasmid expressing C protein (C, CNΔ25, CNΔ50, CK3, CK6, CK12, CE16, or CR24). At 36 h posttransfection, the cells were treated with IFN-α or IFN-γ for 4 h and harvested for the dual luciferase assay. As shown in Figure 1A, as expected, the C protein abolished the IFN-α-induced activation of the ISRE promoter, represented by the firefly LUC/renilla LUC ratio, whereas the CNΔ25 and CNΔ50 proteins only partially decreased the ISRE activation. When the site-directed C mutants were present, the IFN-induced ISRE activation increased to different extents, particularly the mutant CK3, which lost the inhibitory effect of the C protein, suggesting the critical role of the charged amino acid residues within the N-terminal region of the C protein in inhibiting IFN signaling. Figure 4B shows a similar result for IFN-γ-responsive GAS promoter luciferase assay, providing further evidence for the importance of the charged amino acid residues within 1-25aa of the C protein in antagonizing IFN signaling.
Figure 4.

The effects of the charged amino acid residues within the N-terminal region of the C protein on the activation of the IFN-responsive ISRE and GAS promoters. (A) The IFN-α-responsive ISRE promoter luciferase assay. The HeLa cells were transfected with the firefly luciferase encoding plasmid pISRE, the renilla luciferase encoding plasmid pRL-TK and the C/site-directed C mutant-expressing plasmid. At 36 h posttransfection, the cells were treated with IFN-α for 4 h. The cells were lysed for the dual luciferase assay and the ratio of the firefly LUC/renilla LUC was measured and standardized based on the ratio for the nontreated cells. (B) The IFN-γ-responsive GAS promoter luciferase assay, the same as described for IFN-α in (A).
DISCUSSION
It was previously reported that the N-terminal half of the C protein of the SeV had no effect on the IFN signaling inhibition, and the IFN antagonistic activities of the C, Y1, and Y2 proteins of SeV were comparable (12). In contrast, as shown in the present study, the IFN antagonistic activities of the C, CNΔ25, and CNΔ50 proteins of HPIV3 are different and the CNΔ25 and CNΔ50 proteins lost half of the IFN inhibitory effect of the C protein. Interestingly, the CNΔ25 showed potent ability to inhibit the HPIV3 growth by 5 log, whereas the C protein had no effect on the viral growth (19). Thus, it seems that CNΔ25 and CNΔ50 proteins exert via different pathways to inhibit IFN-mediated host immunity that led to inhibition of viral replication.
To understand the inhibitory mechanism of the C protein of HPIV3 to antagonize IFN signaling, we also tested the IFN-α-induced p-Tyk2, the phosphorylated IFN receptor associated kinases, in the presence of the C protein of HPIV3. Similar to the pStat1 (Fig. 1), we found that the IFN-α-induced p-Tyk2 was decreased in the LC-6 cells, and partially recovered in the CNΔ25 and CNΔ50 cells (Fig. 3). Thus, it seems that the C protein of HPIV3 possibly targets Tyk2 in the IFN-α signaling pathway, directly or indirectly. It was reported that the C protein of SeV physically associated with Stat1 and inhibited the Stat2 and Tyk2 activation (8,14,24). However, we did not detect any interaction between the HPIV3 C protein and Tyk2, or Stat1. Additionally, we found that the IFN-induced activation of JAK1, IFN-γ receptor associated kinase, was similar in all cell lines tested (i.e., HeLa, LC-6, CNΔ25, and CNΔ50 cells) (data not shown), suggesting that JAK1 is not the target of the C protein of HPIV3 in the IFN-γ signaling pathway. Further studies along this line of investigation will provide greater insight to understand the strategies that paramyxovirus apply to combat the host inhnate immune responses.
In summary, we identified the functional domain of the HPIV3 C protein in inhibiting IFN signaling, and found that the N-terminal region of the C protein and the charged amino acid residues within the region are critical for the C protein to antagonize IFN signaling.
ACKNOWLEDGMENTS
We are grateful to Steve Goodbourn (in University of St Andrews) and Richard E. Randall (in University of London) for the IFN-responsive luciferase assay system. This work was supported by the NIH grant to A.K.B. (AI 32037).
REFERENCES
- 1. Childs K.; Stock N.; Ross C.; Andrejeva J.; Hilton L.; Skinner M.; Randall R.; Goodbourn S. Mda-5, but not RIG-I, is a common target for paramyxovirus V proteins. Virology 359(1):190–200; 2007. [DOI] [PubMed] [Google Scholar]
- 2. Cortez K. J.; Erdman D. D.; Peret T. C.; Gill V. J.; Childs R.; Barrett A. J.; Bennett J. E. Outbreak of human parainfluenza virus 3 infections in a hematopoietic stem cell transplant population. J. Infect. Dis. 184(9):1093–1097; 2001. [DOI] [PubMed] [Google Scholar]
- 3. Durbin A. P.; McAuliffe J. M.; Collins P. L.; Murphy B. R. Mutations in the C, D, and V open reading frames of human parainfluenza virus type 3 attenuate replication in rodents and primates. Virology 261(2):319–330; 1999. [DOI] [PubMed] [Google Scholar]
- 4. Escoffier C.; Maine S.; Vincent S.; Muller C. P.; Billeter M.; Gerlier D. Nonstructural C protein is required for efficient measles virus replication in human peripheral blood cells. J. Virol. 73(2):1695–1699; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Galinski M. S.; Troy R. M.; Banerjee A. K. RNA editing in the phosphoprotein gene of the human para-influenza virus type 3. Virology 186(2):543–550; 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gotoh B.; Takeuchi K.; Komatsu T.; Yokoo J.; Kimura Y.; Kurotani A.; Kato A.; Nagai Y. Knockout of the Sendai virus C gene eliminates the viral ability to prevent the interferon-alpha/beta-mediated responses. FEBS Lett. 459(2):205–210; 1999. [DOI] [PubMed] [Google Scholar]
- 7. Gotoh B.; Komatsu T.; Takeuchi K.; Yokoo J. Paramyxovirus strategies for evading the interferon response. Rev. Med. Virol. 2(6):337–357; 2002. [DOI] [PubMed] [Google Scholar]
- 8. Gotoh B.; Takeuchi K.; Komatsu T.; Yokoo J. The STAT2 activation process is a crucial target of Sendai virus C protein for the blockage of alpha interferon signaling. J. Virol. 77(6):3360–3370; 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Haque S. J.; Williams B. R. Identification and characterization of an interferon (IFN)-stimulated response element-IFN-stimulated gene factor 3-independent signaling pathway for IFN-alpha. J. Biol. Chem. 269(30):19523–19529; 1994. [PubMed] [Google Scholar]
- 10. Henderson F. W. Pulmonary infections with respiratory syncytial virus and the parainfluenza viruses. Semin. Respir. Infect. 2(2):112–121; 1987. [PubMed] [Google Scholar]
- 11. Irie T.; Nagata N.; Yoshida T.; Sakaguchi T. Paramyxovirus Sendai virus C proteins are essential for maintenance of negative-sense RNA genome in virus particles. Virology 374(2):495–505; 2008. [DOI] [PubMed] [Google Scholar]
- 12. Kato A.; Ohnishi Y.; Hishiyama M.; Kohase M.; Saito S.; Tashiro M.; Nagai Y. The amino-terminal half of Sendai virus C protein is not responsible for either counteracting the antiviral action of interferons or downregulating viral RNA synthesis. J. Virol. 76(14):7114–7124; 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim Y. J.; Boeckh M.; Englund J. A. Community respiratory virus infections in immunocompromised patients: hematopoietic stem cell and solid organ transplant recipients, and individuals with human immunodeficiency virus infection. Semin. Respir. Crit. Care Med. 28(2):222–242; 2007. [DOI] [PubMed] [Google Scholar]
- 14. Komatsu T.; Takeuchi K.; Yokoo J.; Tanaka Y.; Gotoh B. Sendai virus blocks alpha interferon signaling transducers and activators of transcription. J. Virol. 74(5):2477–2480; 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Komatsu T.; Takeuchi K.; Yokoo J.; Gotoh B. Sendai virus C protein impairs both phosphorylation and dephosphorylation processes of Stat1. FEBS Lett. 511(1–3):139–144; 2002. [DOI] [PubMed] [Google Scholar]
- 16. Komatsu T.; Takeuchi K.; Yokoo J.; Gotoh B. C and V proteins of Sendai virus target signaling pathways leading to IRF-3 activation for the negative regulation of interferon-beta production. Virology 325(1):137–148; 2004. [DOI] [PubMed] [Google Scholar]
- 17. Komatsu T.; Takeuchi K.; Gotoh B. Bovine parainfluenza virus type 3 accessory proteins that suppress beta interferon production. Microbes Infect. 9(8):954–962; 2007. [DOI] [PubMed] [Google Scholar]
- 18. Malur A. G.; Chattopadhyay S.; Maitra R. K.; Banerjee A. K. Inhibition of STAT1 phosphorylation by human parainfluenza virus type 3 C protein. J. Virol. 79(12):7877–7882; 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mao H.; Chattopadhyay S.; Banerjee A. K. N-terminal truncated protein, CNdelta25, of human parainfluenza virus type 3 is a potential inhibitor of viral replication. Virology 394(1):143–148; 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Reed G.; Jewett P. H.; Thompson J.; Tollefson S.; Wright P. F. Epidemiology and clinical impact of parainfluenza virus infections in otherwise healthy infants and young children <5 years old. J. Infect. Dis. 175(4):807–813; 1997. [DOI] [PubMed] [Google Scholar]
- 21. Sanchez A.; Banerjee A. K. Cloning and gene assignment of mRNAs of human parainfluenza virus 3. Virology 147:177–186; 1985. [DOI] [PubMed] [Google Scholar]
- 22. Stark G. R.; Kerr I. M.; Williams B. R.; Silverman R. H.; Schreiber R. D. How cells respond to interferons. Annu. Rev. Biochem. 67:227–264; 1998. [DOI] [PubMed] [Google Scholar]
- 23. Stark G. R. How cells respond to interferons revisited: From early history to current complexity. Cytokine Growth Factor Rev. 18(5–6):419–423; 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Takeuchi K.; Komatsu T.; Yokoo J.; Kato A.; Shioda T.; Nagai Y.; Gotoh B. Sendai virus C protein physically associates with Stat1. Genes Cells 6(6):545–557; 2001. [DOI] [PubMed] [Google Scholar]
- 25. Takeuchi K.; Komatsu T.; Kitagawa Y.; Sada K.; Gotoh B. Sendai virus C protein plays a role in restricting PKR activation by limiting the generation of intracellular double-stranded RNA. J. Virol. 82(20):10102–10110; 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Toth A. M.; Devaux P.; Cattaneo R.; Samuel C. E. Protein kinase PKR mediates the apoptosis induction and growth restriction phenotypes of C protein-deficient measles virus. J. Virol. 83(2):961–968; 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Young D. F.; Didcock L.; Goodbourn S.; Randall R. E. Paramyxoviridae use distinct virus-specific mechanisms to circumvent the interferon response. Virology 269(2):383–390; 2000. [DOI] [PubMed] [Google Scholar]