

Abstract
There is increasing evidence that statin treatment can be beneficial in certain cancer patients. To determine if these benefits are a direct result of the cholesterol-lowering effects of statins or a result of secondary, protein transcription effects, the impacts of pravastatin and a cholesterol sequestrating agent methyl-β-cyclodextrin (MβCD) on mRNA expression in the breast cancer cell MDA-MB-231 and the lung carcinoma cell Calu-1 have been compared by microarray techniques. The effects of these agents on cholesterol-rich rafts and caveolae, which have significance in cancer signaling, have also been examined. Both treatments caused a general downregulation of not only signal transduction including cancer pathway proteins, but also apoptosis and chemokine pathways, with statins impacting 35 genes by twofold or greater in MDA-MB-231 and >300 genes in Calu-1. These manifold dysregulations could also explain the various side effects reportedly caused by statins. MβCD produced far fewer statistical events than pravastatin in the breast cancer line but many more in the lung cell line. Pravastatin increased expression of CAV1 but caveolae density decreased and overall raft density was unaffected. MβCD also caused an increase in CAV1 expression and reduced the prevalence of both rafts and caveolae. It is proposed that sequestration of cholesterol from the membrane by MβCD is not equivalent to blockade of the cholesterol pathway and causes different effects on microdomain-mediated signal transduction dependant on the cell line. The profound effects of statins on mRNA expression can be explained by the failure of caveolin-1 to properly complex with cholesterol in an altered sterol environment, with caveolae acting as the main loci for signaling directed towards those transcription processes unaffected by MβCD. Targeted inhibition of the postmevalonate pathway could offer an opportunity to specifically reduce caveolae-based signaling in cancer cells. The observed impact of pravastatin on gene expression may explain the pleiotropic effects of statins when they are used as adjuvants in chemotherapy and suggests impact on gene expression as a possible cause of side effects from statin use.
Key words: Cancer, Gene expression, Pravastatin, Rafts, Caveolae
INTRODUCTION
Statins inhibit the in vivo expression of inflammatory cytokines (27), C-reactive protein (CRP) (28,41), interleukins, tumor necrosis factor (TNF), and matrix metalloproteins (MMPs) (3). These inflammatory mediators are involved in many different diseases (10) including coronary disease (25), type 2 diabetes (23,43), and Alzheimer’s disease (20). The possible clinical benefits of statin use outside the normal lipid-lowering applications have been reported and these pleiotropic effects have attracted considerable interest (1). The use of statins to control cancer has also been explored (7,22), with some studies showing beneficial use in prostate cancer recurrence after surgery (31) and radiation therapy (14), colorectal cancer (2,17), and ovarian cancer (9). Others, notably cancers in the lung and bladder (39), do not respond (6). Some researchers have postulated that statin use could even promote tumor growth (13,29) through upregulation of proteins involved in angiogenesis (11), although the evidence is by no means conclusive (40).
Several putative models have been proposed for the proapoptotic and antimetastatic effects of statins, including the direct downregulation of specific genes such as survivin in prostate cancer cells (19) and in breast cancer cells, and transcription factor activation of c-Jun, part of the mitogen activated protein kinase group that induce apoptosis and inhibit growth (21). In contrast, two mechanisms that are closely linked to the mevalonate pathway inhibited by statins are i) reduction in geranylgeranylpyrophosphate and farnesylpyrophosphate that cause isoprenylation and activation of RhoA, Ras, and other pro-oncogenic proteins (12,40) and ii) reduction of caveolin-1 and cholesterol-dependent endocytosis leading to noncanonical signaling and tumor development in colon cells (16), presumably through reduction of the complexing of Cav-1 with cholesterol to form caveolae in a reduced cholesterol environment. Direct downregulation of CAV1 expression is another route to fewer caveolae and generally reduced signal transduction. It is possible that more than one mechanism is involved in the manifold effects of statins on cancer progression.
Statins are known to affect gene expression in calcium regulatory (e.g., SERCA3) and membrane repair systems and this has been postulated as a cause of statin-associated peripheral myopathy (30). These “extensive” changes in protein turnover have also been found in nonmyopathic patients receiving statins (8). Although the in vivo effects of statins on gene expression have been studied in aortic cells (26) and carotid explants (34) there have been few, if any, attempts to measure genome-wide mRNA changes following exposure to statins in human cancer cells despite the plethora of data suggesting involvement by statins in cancer pathways. It is difficult to discriminate between the cholesterol-related effects of the statin and other effects the molecule may have on the proteome.
The objective of this study was to investigate the impact on gene expression by microarray-based techniques using the ER-negative and p53-mutant human cell line MDA-MB-231 as a model invasive breast cancer and the human lung carcinoma line Calu-1 as an example of an aggressive lung cancer. Much of the epidemiological work linking statin use with reduced morbidity and mortality has alluded to possible antimetastatic effects. For this reason these two highly invasive cell lines were tested to examine if genes associated with metastasis were affected. Pravastatin was chosen as a model HMG-CoA reductase inhibitor. To examine if the observed effects on gene expression were caused by a reduction of cholesterol per se, methyl-β-cyclodextrin (MβCD), a cyclic oligomer of glucose that is able to entrap cholesterol in its hydrophobic core and specifically sequester the sterol from the membrane (35), was used for comparison because it mimics only the ultimate cholesterol-lowering effects of the statin.
Rafts and caveolae are morphologically and chemically distinct platforms that rely on high concentrations of cholesterol and, once assembled, serve as platforms for multiple signaling systems. To determine if these cholesterol-rich domains were disrupted by the treatments, flotillin was used as a general indicator of overall raft density and caveolin-1 as a specific marker of caveolae. Both were assayed using immunofluorescence techniques.
MATERIALS AND METHODS
Sources
MDA-MB-231 and Calu-1 cells were obtained from Cell Lines Service (Eppenheim, Germany). Explorer protein microarrays were purchased from Full Moon Biosciences Inc. (USA). Illumina Human HT12_V4_0_R2_15002873_B human expression microarray was purchased from Gen-Probe Ltd (UK). RNeasy Maxi Kit was purchased from Qiagen Ltd. All other reagents were sourced from Sigma Aldrich Ltd (UK) except where noted.
Treatments
The final concentrations of pravastatin and MβCD in culture flasks were 8.0 μM and 0.00085% (w/v), respectively, dissolved in DMEM plus 10% v/v serum. The dose of pravastatin was chosen because low millimolar serum levels are attainable in vivo at high doses of other statins (lovastatin) (27). Treatment exposure was for 24 h beginning after cells reached 80% confluence. The same treatment regime was used in the antibody assays at a range of doses was used.
All treatments and controls were conducted in quadruplicate and the microarray was performed using these four biological replicates. There were no technical replicates. Cells were treated in 174-ml culture flasks (Nunc) containing 40 ml of DMEM with 10% (v/v) FBS per treatment. Negative control flasks contained only the FBS-supplemented media. Treatments were 24 h and treatment start time was 24 h after subculture. Incubation was at 37°C with 5.0% CO2. Cells from each treatment were harvested with 0.5 g/L porcine trypsin w/v and 0.2 g/L w/v EDTA in Dulbecco phosphate buffer and immediately spun down to a cell pellet. The cells were then resuspended in PBS containing 0.1% of Sigma Protease inhibitor cocktail and then recentrifuged. The resultant cell pellet was then stored in LN2 prior to RNA or protein extraction.
mRNA Expression Profiling
Array analysis was performed in accordance with the manufacturer’s guidance. Each treatment was conducted in quadruplicate and there were no technical replicates in this study.
Protein Assay
Treatments and controls were prepared as above. Each Explorer array slide has 656 protein probes in duplicate. Mean spot intensities and coefficient of variations were recorded to provide standard errors.
Immunofluorescence Assays
A conjugate of fluorescein isothiocyanate (Ex495 and Em525) and anti-Flotillin antibody was prepared using the Sigma Fluorotag kit and affinity isolated anti-Flotillin-1 produced in rabbit. The second antibody used in the experiment was anti-Caveolin-1 that was purchased preconjugated to the cyanine dye Cy3 (Ex550 nm and Em570nm). Phosphate buffer solution (1.25 ml) was added to 1 mg of lyophilized protein and the light-protected tube vortexed for 1 min. It was used without further preparation. Both antibodies were used at 1 μg/ml in 96 × 100 μl plates (Sterilin). Readings were taken using a Biochrom 480 fluorescence plate reader after 1-h exposure posttreatment followed by three gentle washes with phosphate buffer, pH 7.4.
Statistical Treatment
Raw array data were assessed for quality, and outliers removed. The remaining arrays were normalized and array features annotated. A threshold for significance was adjusted to p < 0.01 and significant loci in each comparison were assessed for functional enrichment of KEGG pathways, and GO terms, based on their annotation information. The p-values were adjusted using Benjamini & Hochberg method for multiple testing, to 0.001 for the comparison of significant array features.
Normalization of the 47,319 features across all arrays was achieved using robust spline normalization after data were subjected to a variance stabilizing transformation. Raw data were transformed using a variance stabilizing transformation (VST) method prior to normalization across all arrays using the robust spline normalization (RSN) method. Expression measures (summarized intensities) are in log base 2.
Probesets on the array may have been annotated as being a member of a KEGG pathway (www.kegg.jp). Significant genes (adjusted p < 0.01) from each comparison were analyzed for enrichment of KEGG pathway membership using a hypergeometric test. Enrichment (p < 0.05) was assessed for upregulated and downregulated genes separately. Significant genes (adjusted p < 0.01) from each comparison were analyzed for enrichment of GO terms across all three GO ontologies using a hypergeometric test. Enrichment (p < 0.001) was assessed for upregulated and downregulated genes separately.
RESULTS
Pravastatin Treatment Relative to Negative Control
One hundred and one array features were statistically significant at p < 0.01 (27 upregulated, 74 downregulated). Within the significant features, ANGPTL2, COL5A1, COPS2, DST, FOS, GAS2L1, GPR56, GPRC5C, ID1, and ID2 were upregulated. Within the significant features, ABCA1, ADM, ANGPTL4, C10orf10, C13orf15, C15orf48, C7orf68, CCL20, CCL26, and CDCP1 were downregulated.
The predominant (number of p < 0.01 features are in parenthesis) upregulated pathways include those associated with TGF-β signaling (3), focal adhesion (2), and ECM-receptor interactions (2).
The predominant downregulated pathways include those associated with cytokine–cytokine receptor interactions (9), chemokine signaling (5), NOD-like receptor signaling (5), cancer (4), and MAPK signaling (3). In terms of observed fold change (independent of statistical threshold), no features exhibited greater than twofold upregulation, while 24 features exhibited greater than twofold downregulation. Fold changes ranged from twofold up to 10.4-fold down.
Twenty KEGG pathways were statistically enriched. Members of ECM–receptor interaction pathways were among those enriched in upregulated loci. Members of cytokine–cytokine receptor interaction and NOD-like receptor signaling pathways were among those enriched in downregulated loci. CAV1 was upregulated by 10%, FLOT1 by 1%, and overall gene expression was downregulated by 0.55% compared to the control group. In Calu-1 cells, 5,107 array features were statistically significant (2,535 upregulated, 2,572 downregulated). The predominant upregulated pathways include metabolic pathways (154), pathways in cancer (37), endocytosis (35), insulin signaling pathway (29), MAPK signaling pathway (28), lysosome (26), and cytokine–cytokine receptor interaction (22). The predominant downregulated pathways include metabolic pathways (126) and pathways in cancer (57).
In terms of observed fold change (independent of statistical threshold), 219 features exhibited greater than twofold upregulation, while 174 features exhibited greater than twofold downregulation. Fold changes ranged from 11.1-fold up to 5.1-fold down. Within the biggest change loci, NUPR1, GDF15, TRIB3, RNF165, DDIT3, ASNS, DDIT4, PDE5A, CTH, and PCK2 were upregulated. Within the biggest change loci, TXNIP, MMP3, STC1, CTGF, GLIPR1, NPPB, BMPER, EDN1, MAP2K3, and CYP24A1 were downregulated. FLOT1 was upregulated with a log2 fold change of 0.51 (p = 0.0024). CAV1 was also upregulated but this was not statistically significant.
Fifty-nine KEGG pathways and 408 GO terms were statistically enriched. Members of steroid biosynthesis were enriched in upregulated loci. Members of spliceosome, cell cycle, and DNA replication pathways were among those enriched in downregulated loci.
MβCD Treatment Relative to Negative Control
There were 79 statistically significant (3 upregulated, 76 downregulated) array features. Within the significant features, MARCH4, NQO1, and SNX6 were upregulated and ABCA1, ADAM8, ADM, AGR2, ANGPTL4, C10orf10, C13orf15, C15orf48, C7orf68, and CCL20 were downregulated. The predominant downregulated pathways include those associated with cytokine–cytokine receptor interactions (9), cancer (6), chemokine signaling (5), NOD-like receptor signaling (5), MAPK signaling (3), Toll-like receptor signaling (3), type 1 diabetes mellitus (3), bladder cancer (3), metabolism(2), apoptosis (2), and VEGF signaling (2).
In terms of observed fold change (independent of statistical threshold), no features exhibited greater than twofold upregulation, while 13 features exhibited greater than twofold downregulation. Fold changes ranged from 1.6-fold up to 3.2-fold down. Within the biggest change loci, THBS1, AMY1C, FTLP2, NQO1, ID3, FLNC, CAV1, RPL35, SLC7A5, and PSMC1 were upregulated. Cytokine–cytokine receptor interaction and NOD-like receptor signaling pathways were downregulated loci. CAV1 was upregulated by 25% and the entire treatment caused a global increase in gene expression of 8.64% compared to the control group. FLOT1 was not significantly affected (increasing by 1.9%).
In Calu-1 cells, 6,868 array features were statistically significant (3,417 upregulated, 3,451 downregulated). The predominant upregulated pathways include metabolic pathways (173), pathways in cancer (50), endocytosis (39), MAPK signaling pathway (37), insulin signaling pathway (34), lysosome (30), and cytokine–cytokine receptor interaction (29). The predominant downregulated pathways include metabolic pathways (174), pathways in cancer (73), spliceosome (61), and cell cycle (53). In terms of observed fold change (independent of statistical threshold), 393 features exhibited greater than twofold upregulation, while 349 features exhibited greater than twofold downregulation. Fold changes ranged from 16.4-fold up to 8.1-fold down. Within the biggest change loci, GDF15, NUPR1, TRIB3, DDIT3, RNF165, ASNS, PDE5A, CTH, DDIT4, and FBXO32 were upregulated. Within the biggest change loci, CTGF, STC1, NPPB, BMPER, CYP24A1, GLIPR1, TXNIP, MMP3, EDN1, and MARCH4 were downregulated. FLOT1 was upregulated by log2 fold change of 0.56 (p = 0.00069) but CAV1 was not affected. Sixty-six KEGG pathways were statistically enriched. Members of steroid biosynthesis were among those enriched in upregulated loci. Members of spliceosome, cell cycle, and DNA replication pathways were among those enriched in downregulated loci.
Five hundred GO terms were statistically enriched with members annotated with intracellular, intracellular part, and membrane-bound organelle GO terms among those upregulated loci. Members annotated with organelle part, intracellular organelle part, and organelle GO terms were among those enriched in downregulated loci.
Pravastatin Relative to MβCD
There were 27 statistically significant array features (16 upregulated, 11 downregulated). Within the significant features, ANGPTL2, GAS2L1, GPR56, GPRC5C, ID1, ID2, IGFBP6, ITGB4, MALL, and MXD4 were upregulated while ABCA1, CRY1, FST, IGFBP3, IL11, LOX, MMP1, PTGER4, and PTGS2 were downregulated. The predominant upregulated pathways include TGF-β signaling (2). The predominant downregulated pathways include pathways in cancer (2). In terms of observed fold change (independent of statistical threshold), no features exhibited greater than twofold upregulation, while three features exhibited greater than twofold downregulation. Fold changes ranged from 1.8-fold up to 3.3-fold down. Six KEGG pathways and 21 gene ontology terms were statistically enriched. Genes annotated with regulation of localization, negative regulation of transport, and negative regulation of hormone secretion gene ontology terms were among those enriched in downregulated loci.
In Calu-1, 382 array features were statistically significant (210 upregulated, 172 downregulated). The predominant upregulated pathways include metabolic pathways (8), cytokine–cytokine receptor interaction (7), and protein processing in endoplasmic reticulum (6). The predominant downregulated pathways include metabolic pathways (17), cell cycle (8), pathways in cancer (7), and DNA replication (6). Twenty-three KEGG pathways were statistically enriched with members of prion diseases, protein processing in endoplasmic reticulum, and NA pathways among those upregulated loci. Members of DNA replication, cell cycle, and pancreatic cancer pathways were among those enriched in downregulated loci.
One hundred and thirteen GO terms were statistically enriched. Members annotated with cellular response to stress, response to stress, and cellular response to stimulus GO terms were among those enriched in upregulated loci. Members annotated with cell division, DNA replication, and organelle fission GO terms were among those enriched in downregulated loci.
DISCUSSION
The response in mRNA expression caused by the two agents suggests some, but not total, commonality in mechanism. The volcano plots of mRNA events in MB231 cells reveal that in the statin treatment there is a bias towards downregulation (Fig. 1). MβCD versus statin (Fig. 2) shows that both treatments cause similar responses, with some additional downregulations caused by the statin. In Calu-1 cells the response is very different with a symmetrical distribution of statistical events in both treatments (Fig. 3) and some additional upregulations caused by the statin. In these two cell lines both the statin and MβCD showed considerable crossover in impact on ABCA1, ADM, ANGPTL4, C10orf10, C13orf15, C15orf48, C7orf68, and CCL20 and this suggests a similar mode of action. Significantly, ABCA1 is a cholesterol efflux regulator (33), while C13orf15 and ANGPTL4 control the cell cycle (15,18,36) and increase tumor growth, respectively. A direct DNA suppression of these latter genes by both agents seems unlikely so an indirect cholesterol raft-mediated mechanism is an intriguing possibility. The results suggest that removal of cholesterol by either statin or MβCD causes changes in gene expression unfavorable to cancer development, with both treatments causing cancer pathways specifically to be downregulated.
Figure 1.
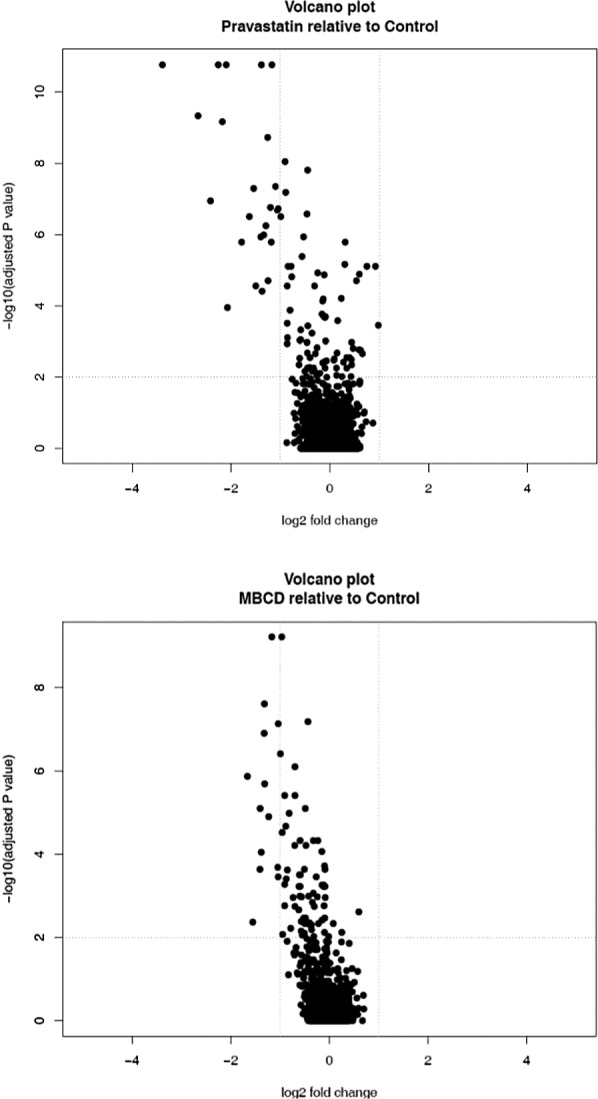
Pravastatin causes many more and greater intensity downregulations compared to MβCD. The different response in terms of gene expression could be a result of the type of membrane domain affected by the two treatments.
Figure 2.
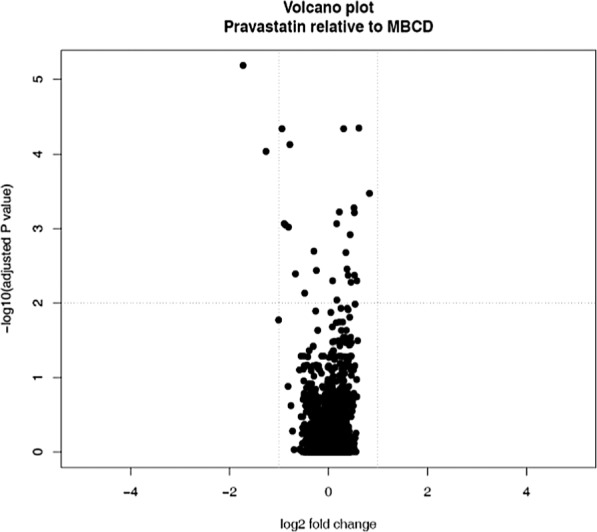
Volcano plot of pravastatin relative to MβCD. The treatments do not have equivalent effects on mRNA expression.
Figure 3.
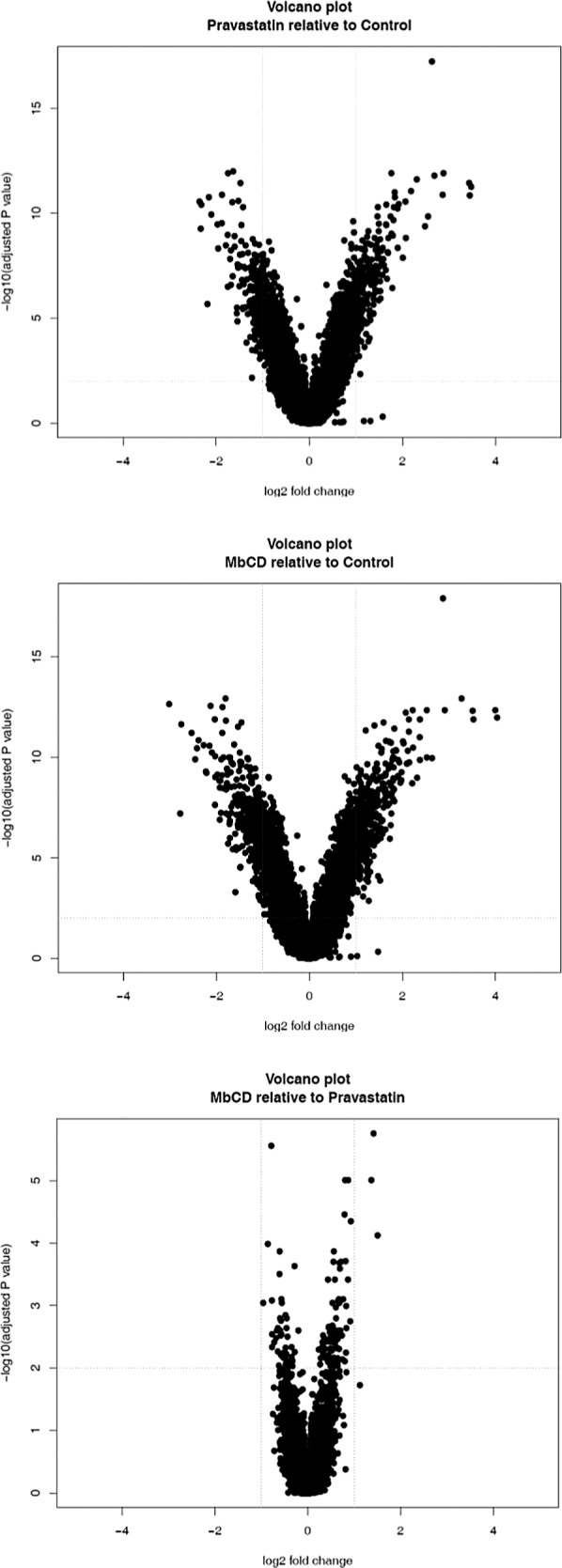
Volcano plots of fold changes and significance in Calu-1 lung cells.
However, apoptotic pathways are also downregulated. In MDA-MB-231 cells MβCD produced fewer events at p < 0.001 (regardless of impact) than pravastatin, with many fewer genes upregulated. At the doses used in this research MβCD invoked downregulation of the proteins involved in cholesterol synthesis to a greater extent than pravastatin, suggesting that normal lipid homeostasis is secondary to cholesterol raft-based signal transduction directed at the genome. In Calu-1 MβCD had a drastic effect on gene regulation with 742 features exhibiting a greater than twofold change. This suggests that Calu-1 is more than 20 times as susceptible to the effects of MβCD as the breast cancer cell line but the genes most affected are upregulated and are not related to cancer pathways. The global gene events caused by the treatments are given in Table 1. Further analysis of the data set reveals that many of these features are highly significant but have a low fold change value and are therefore unlikely to affect cell health. In MDA-MB231 cells pravastatin downregulated four cancer pathways and increased CAV1 expression by 10% and FLOT1 by 1% (p < 0.05). Overall gene expression was reduced by 0.55%. In Calu-1 cells 37 cancer pathways were upregulated and 57 were downregulated. MβCD treatment, in contrast, caused six cancer pathways in MDA-MB231 cells to be downregulated and CAV1 expression to be upregulated by 25%. Overall gene expression in this experiment was increased by 8.64% (p < 0.05). However, in Calu-1 cells 37 cancer pathways were upregulated and 57 were downregulated by statin treatment and mirrored the result of MβCD treatment (50 upregulated and 73 downregulated). When the two treatments in MDA-MB-231 cells are compared (Table 2) two genes specifically related to cancer are significantly affected by statin but not MβCD: PTGS2 (log FC −1.73; p = 6.50E-06) and IL-8 (log FC −1.01; p = 0.017). In Calu-1 cells seven cancer-related genes have low p-values but the fold change is minimal. These include IL-6, CCND1, and SMAD3. The data from the MβCD relative to pravastatin treatments suggests that removing cholesterol from the bilayer is not biologically equivalent to inhibition of the mevalonate pathway.
TABLE 1.
CHANGES TO ARRAY FEATURES BY TREATMENT WITH PRAVASTATIN AND MβCD
| Cell Type | Comparison | Significant Array Features at p < 0.001 (Corrected Using Benjamini and Hochberg Method for Multiple Testing) | Features Greater Than Twofold Change |
|---|---|---|---|
| MDA-MB-231 | Pravastatin relative to control | 101 | 35 |
| MDA-MB-231 | MβCD relative to control | 79 | 34 |
| MDA-MB-231 | Pravastatin relative to MβCD | 27 | 8 |
| Calu-1 | Pravastatin relative to control | 2,013 | 393 |
| Calu-1 | MβCD relative to control | 3,149 | 742 |
| Calu-1 | Pravastatin relative to MβCD | 30 | 4 |
TABLE 2.
MOST SIGNIFICANTLY AFFECTED GENES
| Treatment | Cell Type | Gene Identifier | Log Fold Change | Adjusted Significance |
|---|---|---|---|---|
| Pravastatin–control | MDA-MB-231 | PTGS2* | −2.10 | 1.62E-11 |
| MβCD–control | MDA-MB-231 | Lipocalin-2† | −0.96 | 6.19E-10 |
| Pravastatin–MβCD | MDA-MB-231 | PTGS2 | −1.73 | 6.50E-06 |
| Pravastatin–control | Calu-1 | ASNS‡ | 2.63 | 5.43E-18 |
| MβCD–control | Calu-1 | ASNS | 2.87 | 1.18E-18 |
| Pravastatin–MβCD | Calu-1 | RNU1-5§ | 1.42 | 1.75E-06 |
When the 50 genes with the highest fold change are ranked according to adjusted statistical significance these four genes are most impacted.
Prostaglindin-endoperoxide synthase-2 is a component of the pathways in cancer and small cell lung cancer pathways.
Steroid transport protein.
Asparagine synthetase.
snRNA component of the spliceosome.
MDA-MB-231 was analyzed for differences in the densities of cholesterol-rich rafts and caveolae following treatment with pravastatin and MβCD as determined by immunofluorescence (Figs. 4 and 5). The results indicate that both treatments cause a reduction of available Cav-1 at the membrane, despite the observed upregulation of CAV1, with pravastatin causing a significantly greater reduction. This difference in response to the treatments was not seen in Flot-1 availability, suggesting that pravastatin causes a specific reduction in caveolae but not rafts per se. Caveolae have unique signaling functionality in cancer that can vary by cell type and stage of disease progression (4) and caveolae require cholesterol for their formation. Pravastatin causes depletion of available sterol to perform this function but also causes significant reduction in membrane Cav-1 (as assayed by immunofluorescence) favoring the formation of rafts rather than caveolae with any available cholesterol. This is despite an upregulation in CAV1 gene expression. Indeed, rafts containing other sterol intermediates are likely to be viable signaling platforms whereas caveolae may have a specific requirement for cholesterol so that the Cav-1 protein can oligomerize and coordinate with the other lipids correctly. However, the statin is able to significantly reduce membrane Cav-1 at higher doses. Neither treatment caused any change to the amount of flotillin actually being transcribed by RNA so it seems likely that the changes in membrane flotillin and caveolin are not driven by transcription events but rather they are a downstream result of the reduction of cholesterol. Statins are known to induce COX-2 gene expression in a manner consistent with farnesyl transferase inhibitors, geranylgeranyltransferase inhibition and impairment of G-protein prenylation (5) but this does not explain the breadth of response at the mRNA level. It has been suggested that statins specifically antagonize a set of genes modulated by L-NAME-induced hypertension in vivo (32), but the results presented here reveal that 35 genes are modulated greater than twofold by pravastatin in breast cancer cells and more than 300 are affected greater than twofold in lung cancer cells.
Figure 4.

The effects of pravastatin and MβCD on caveolin-1 protein detection by immunofluorescence assay. MβCD caused a reduction in caveolin-1 concentrations at both doses tested. The statin did not reduce cav-1 at 8 μM but at much higher concentrations did reduce levels of the protein with a clear dose response. *Statistical significant difference p < 0.05 between treatment and control data by ANOVA two-tailed test, n = 3.
Figure 5.
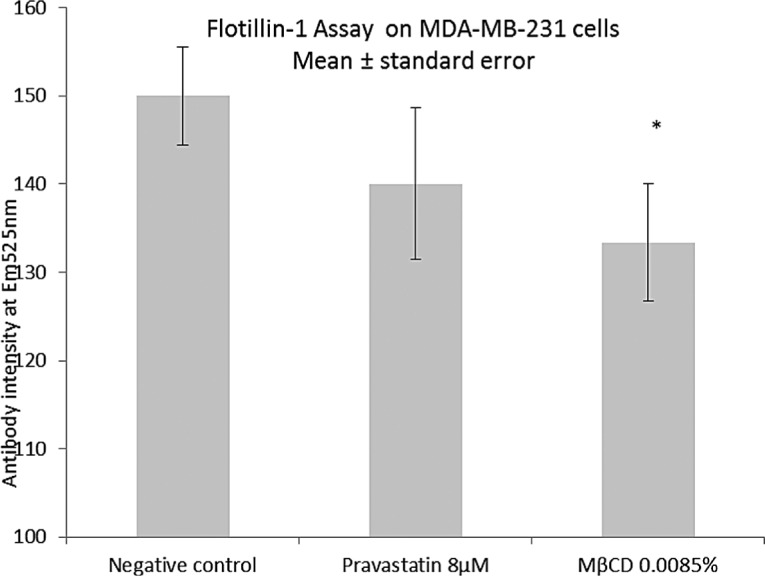
The effect of pravastatin and MβCD on flotillin-1 protein detection by immunofluorescence assay. Pravastatin did not reduce flotillin concentrations, indicating that statins may not reduce prevalence of cholesterol-rich rafts in MB231. MβCD did reduce raft density. *Statistical significant difference p < 0.05 between treatment and control data by ANOVA two-tailed test, n = 3.
Statins are among the most prescribed pharmaceuticals and have undoubted health benefits not limited to lipid-lowering indications. However, membrane repair genes are activated during statin treatment irrespective of clinical myopathy and this could be due to cholesterol-deprived membranes becoming more permeable to ion leakage as the bilayer becomes fluidized without sufficient sterol reenforcement. Calcium leakage is associated with myopathy (8). It has been reported that 5% of patients using statins suffer from toxic muscle damage (38). Common side effects of statin treatment include peripheral myopathy and mood disturbance (42), but multiple others can be expected given the profound alterations that statins cause to both membrane microdomains and gene expression.
While the anti-inflammatory and antioncogenic characteristics of some statin treatments are unexpected but welcome it is possible that rebound effects on gene expression following termination of long-term statin use—similar to those seen in inflammatory response (24)—might result in reduced or reversed biochemical impact in cancer patients benefiting from statin treatment.
The overall effect on cancer of statin treatment may be either deleterious or beneficial depending on the cell type, cancer phenotypes, and tissue environment. Cholesterol is, after all, primarily a structural component of the plasma membrane and it seems reasonable to assume that its effects can be measured by the density of those microdomains that are rich in this sterol—be they predominantly cholesterol–lipid or cholesterol–protein in nature. Many of the anticancer and anti-inflammatory effects noted by other researchers could be explained by a diminution of either raft-or caveolae-based canonical pathways leading to mRNA dysregulation or abortive noncanonical signaling.
ACKNOWLEDGMENTS
D. Garnett’s work has been funded by Brightwater Research LLP.
Footnotes
T. Greenhough declares no potential conflict of interest.
REFERENCES
- 1. Almuti K.; Rimawi R.; Spevack D.; Ostfeld R. J. Effects of statins beyond lipid lowering: Potential for clinical benefits. Int. J. Cardiol. 109:7–15; 2006. [DOI] [PubMed] [Google Scholar]
- 2. Bardou M.; Barjun A. N.; Martel M. Prolonged statin use weakly decreases the risk of colorectal cancer (CRC): A meta-analysis of 21 observational studies totalling more than 1.6 million patients. Gastroenterology 138:S349–S350; 2010. [Google Scholar]
- 3. Bellosta S.; Via D.; Canavesi M.; Pfister P.; Fumagalli R.; Pauletti R.; et al. HMG-CoA reductase inhibitors reduce MMP-9 secretion by macrophages. Arterioscler. Thromb. Vasc. Biol. 18:1671–1678; 1998. [DOI] [PubMed] [Google Scholar]
- 4. Burgermeister E.; Liscovitch M.; Rocken C.; Schmid R.; Ebert M. Caveats of caveolin-1 in cancer progression. Cancer Lett. 268:187–201; 2008. [DOI] [PubMed] [Google Scholar]
- 5. Chen J-C.; Huang K-C.; Wingerd B.; Wu W-T.; Lin W-W. HMG-CoA reductase inhibitors induce COX-2 gene expression in murine macrophages: Role of MAPK cascades and promoter elements for CREB and C/EBPβ. Exp. Cell Res. 301:305–319; 2004. [DOI] [PubMed] [Google Scholar]
- 6. Cheng M-H.; Chiu H-F.; Ho S-C.; Yang C-Y. Statin use and the risk of female lung cancer: A population-based case-control study. Lung Cancer. 75(3):275–279; 2012. [DOI] [PubMed] [Google Scholar]
- 7. Dale K. M.; Coleman C. I.; Henyan N. N.; Kluger J.; White C. M. Statins and cancer risk: A meta-analysis. JAMA 295:74–80; 2006. [DOI] [PubMed] [Google Scholar]
- 8. Draeger A.; Sanchez-Freire V.; Monastyrskaya K.; Hoppeler H.; Mueller M.; Breil F.; Mohaupt M. G.; Babiychuk E. B. Statin therapy and the expression of genes that regulate calcium homeostasis and membrane repair in skeletal muscle. Am. J. Pathol. 177(1):291–299; 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Elmore R. G.; Ioffe Y.; Scoles D. R.; Karlan B. Y.; Li A. J. Impact of statin therapy on survival in epithelial ovarian cancer. Gynecol. Oncol. 111:102–105; 2008. [DOI] [PubMed] [Google Scholar]
- 10. Forrester J. S.; Libby P. L. The inflammation hypothesis and its potential relevance to statin therapy. Am. J. Cardiol. 99:732–738; 2007. [DOI] [PubMed] [Google Scholar]
- 11. Garjani A.; Rezazadeh H.; Maleki-Dizaji N.; Barer J.; Omidi Y. Mevalonate independent effects of atorvastatin on angiogenesis: Relevance to cancer. Biosci. Hypoth. 1:67–69; 2008. [Google Scholar]
- 12. Ghosh-Choudhury N.; Charan Mandal C.; Ghosh-Choudhury N.; Ghosh-Choudhury G. Simvastatin induces depression of PTEN expression via NFκB to inhibit breast cancer growth. Cell. Signal. 22:749–758; 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Goldstein M. R.; Mascitelli L.; Pezzetta F. Might the widespread use of statin drugs explain the increase in prevalence of breast carcinoma in situ? Med. Hypoth. 74:613–621; 2010. [DOI] [PubMed] [Google Scholar]
- 14. Gutt R.; Tonlaar N.; Kunnavakkam R.; Karrison T.; Weichselbaum R. R.; Liauw S. L. Statin use and risk of prostate cancer recurrence in men treated with radiation therapy. J. Clin. Oncol. 28:2653–2659; 2010. [DOI] [PubMed] [Google Scholar]
- 15. Huang W. Y.; Li Z. G.; Rus H.; Wang X.; Jose P. A.; Chen S. Y. RGC-32 mediates transforming growth factor-beta-induced epithelial-mesenchymal transition in human renal proximal tubular cells. J. Biol. Chem. 284(14):9426–9432; 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jacobs R.; Weil N.; Kodach L.; Voorneveld P.; Wildenberg M.; Hommes D.; Hardwick J. Statin treatment of colon cancer cells leads to increased cell surface levels of BMP receptors and a shift from noncanonical to canonical BMP signaling. Gastroenterology 140(5):S482; 2011. [Google Scholar]
- 17. Katz M. S.; Minsky B. D.; Saltz L. B.; Riedel E.; Chessin D. B.; Guillem J. G. Association of statin use with a pathologic complete response to neoadjuvant chemoradiation for rectal cancer. Int. J. Radiat. Oncol. Biol. Phys. 63:1363–1370; 2005. [DOI] [PubMed] [Google Scholar]
- 18. Kim I.; Kim H. G.; Kim H.; Kim H. H.; Park S. K.; Uhm C. S.; Lee Z. H.; Koh G. Y. Hepatic expression, synthesis and secretion of a novel fibrinogen/angiopoietin-related protein that prevents endothelial-cell apoptosis. Biochem. J. 346(3):603–610; 2000. [PMC free article] [PubMed] [Google Scholar]
- 19. Koike H.; Sekine Y.; Furuya Y.; Morikawa Y.; Matsui H.; Shibata Y.; Suzuki K. Statin inhibits the proliferation of human prostate cancer cells via downregulation of the survivin. Eur. Urol. Suppl. 9:273; 2010. [Google Scholar]
- 20. Koistinaho M.; Koistinaho J. Interactions between Alzheimer’s disease and cerebral ischemia—focus on inflammation. Brain Res. Rev. 48:240–250; 2005. [DOI] [PubMed] [Google Scholar]
- 21. Koyuturk M.; Ersoz M.; Altiok N. Simvastatin induces apoptosis in human breast cancer cells: p53 and estragon receptor independent pathway requiring signalling through JNK. Cancer Lett. 250:220–228; 2007. [DOI] [PubMed] [Google Scholar]
- 22. Kuoppala J.; Lamminpaa A.; Pukkala E. Statins and cancer: A systematic review and meta-analysis. Eur. J. Cancer 44:2122–2132; 2008. [DOI] [PubMed] [Google Scholar]
- 23. Lang C. H.; Dobrescu C.; Bagby G. J. Tumour necrosis factor impairs insulin action on peripheral glucose disposal and hepatic glucose output. Endocrinology 130:43–52; 1992. [DOI] [PubMed] [Google Scholar]
- 24. Li J-J.; Li Y-S.; Chen J.; Yang J-Q. Rebound phenomenon of inflammatory response may be a major mechanism responsible for increased cardiovascular events after abrupt cessation of statin therapy. Med. Hypoth. 66:1199–1204; 2006. [DOI] [PubMed] [Google Scholar]
- 25. Libby P. L. Inflammation in atherosclerosis. Nature 420:868–874; 2002. [DOI] [PubMed] [Google Scholar]
- 26. Liu S-L.; Li, Yi-H.; Shi G-Y.; Jiang M-J.; Chang J-H.; Wu H-L. The effect of statin on the aortic gene expression profiling. Int. J. Cardiol. 114:71–77; 2007. [DOI] [PubMed] [Google Scholar]
- 27. Lyngdoh T.; Vollenweider P.; Waeber G.; Marques-Vidal P. Association of statins with inflammatory cytokines: A population-based Colaus study. Athersclerosis 219:253–258; 2011. [DOI] [PubMed] [Google Scholar]
- 28. Marz W.; Winkler K.; Nauck M.; Bohm B. O.; Winkelmann R. Effects of statins on C-reactive protein and interleukin-6 (the Ludwigshafen Risk and Cardiovascular health study). Am. J. Cardiol. 92:305–308; 2003. [DOI] [PubMed] [Google Scholar]
- 29. Mascitelli L.; Pezzetta F.; Goldstein M. R. Statin induced angiogenesis and tumour growth. Eur. J. Int. Med. 20:e159; 2009. [DOI] [PubMed] [Google Scholar]
- 30. Mohaupt M.; Karas R.; Babiychuk E.; Sanchez-Freire V.; Monastyrskaya K.; Lyer L.; et al. Association between statin-associated myophathy and skeletal muscle damage. CMAJ 181:E11–E18; 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mondul A. M.; Han M.; Humphreys E. B. Association of station use with pathological tumour characteristics and prostate cancer recurrence after surgery. Eur. Urol. 60:867–871; 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ndaud S.; Dupuis M.; Brocheriou I.; Haloui M.; Louedec L.; Capron F.; Michel J-B.; Soubrier F. Counter-regulation by atorvastatin of gene modulations induced by L-NAME hypertension is associated with vascular protection. Vasc. Pharmacol. 51:253–261; 2009. [DOI] [PubMed] [Google Scholar]
- 33. Oram J. F.; Vaughan A. M. ABCA1-mediated transport of cellular cholesterol and phospholipids to HDL apolipoproteins. Curr. Opin. Lipidol. 11(3):253–260; 2000. [DOI] [PubMed] [Google Scholar]
- 34. Razuvaev A.; Ekstrand J.; Folkersen L.; Agardh H.; Markus D.; Swedenborg J.; et al. Correlations between clinical variables and gene-expression profiles in carotid plaque instability. Hedin U. Eur. J. Vasc. Endovasc. Surg. 42:722–730; 2011. [DOI] [PubMed] [Google Scholar]
- 35. Rodal S. K.; Skretting G.; Garred O.; Vilhart F.; van Deurs B.; Sandvig K. Extraction of cholesterol with methyl-β-cyclodextrin perturbs formation of clathrincoated endocytic vesicles. Mol. Biol. Cell 10:961–974; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Saigusa K.; Imoto I.; Tanikawa C.; Aoyagi M.; Ohno K.; Nakamura Y.; Inazawa J. RGC32, a novel p53-inducible gene, is located on centrosomes during mitosis and results in G2/M arrest. Oncogene 26(8):1110–1121; 2007. [DOI] [PubMed] [Google Scholar]
- 37. Thibault A.; Samid D.; Tompkins A.; Figg W.; Cooper M.; Hohl R.; Trepel J.; Liang B.; Patronas N.; Venzon D. J.; Reed E.; Myers C. E. Phase I study of lovastatin, an inhibitor of the mevalonate pathway, in patients with cancer. Clin. Cancer Res. 2:483–491; 1992. [PubMed] [Google Scholar]
- 38. Thompson P.; Clarkson P.; Karas R. Statin associated myopathy. JAMA 289:1681–1690; 2003. [DOI] [PubMed] [Google Scholar]
- 39. Tsai H. K.; Katz M. S.; Coen J. J.; Zietman A. L.; Kaufman D. S.; Shipley W. U. Association of statin use with local control in patients treated with selective bladder preservation for muscle-invasive bladder cancer. Int. J. Radiat. Oncol. Biol. Phys. 63(2)334; 2005.16168828 [Google Scholar]
- 40. Tsubaki M.; Yamazoe Y.; Yanae M.; Satou T.; Itoh T.; Kaneko J. Blockade of Ras/MEK/ERK and Ras/PI3K/Akt pathways by statins reduces the expression of bFGF, HGF and TGF-β as angiogenic factors in mouse osteosarcoma. Cytokine 54:100–107; 2011. [DOI] [PubMed] [Google Scholar]
- 41. Voleti B.; Agrawal A. Statins and nitric oxide reduce C-reactive protein production while inflammatory conditions persist. Mol. Immunol. 43:891–896; 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. While A.; Keen L. The effects of statins on mood: A review of the literature. Eur. J. Cardiovasc. Nurs. 11(1):85–96; 2012. [DOI] [PubMed] [Google Scholar]
- 43. Yamagishi S.; Matsui T.; Sato T.; Takeuchi M. Protective role of pravastatin in the pathogenesis of the metabolic syndrome. Med. Hypoth. 66:609–611; 2006. [DOI] [PubMed] [Google Scholar]