

Abstract
The mechanism of action and contribution to pathogenesis of many virulence genes are understood. By contrast, little is known about anti‐virulence genes, which contribute to the start, progression, and outcome of an infection. We now report how an anti‐virulence factor in Salmonella enterica serovar Typhimurium dictates the onset of a genetic program that governs metabolic adaptations and pathogen survival in host tissues. Specifically, we establish that the anti‐virulence protein CigR directly restrains the virulence protein MgtC, thereby hindering intramacrophage survival, inhibition of ATP synthesis, stabilization of cytoplasmic pH, and gene transcription by the master virulence regulator PhoP. We determine that, like MgtC, CigR localizes to the bacterial inner membrane and that its C‐terminal domain is critical for inhibition of MgtC. As in many toxin/anti‐toxin genes implicated in antibiotic tolerance, the mgtC and cigR genes are part of the same mRNA. However, cigR is also transcribed from a constitutive promoter, thereby creating a threshold of CigR protein that the inducible MgtC protein must overcome to initiate a virulence program critical for pathogen persistence in host tissues.
Keywords: anti‐virulence protein, CigR, pathogenesis, Salmonella, virulence protein MgtC
Subject Categories: Microbiology, Virology & Host Pathogen Interaction; Transcription
Introduction
In the context of pathogenesis, the genes of a pathogen can be divided into three distinct groups: virulence genes, which promote virulence; anti‐virulence genes, which decrease virulence; and genes that neither promote nor decrease virulence. Investigations over the last several decades have revealed the mechanism of action and contribution to pathogenesis of an increasing number of virulence genes. By contrast, little is known about how anti‐virulence genes function. Anti‐virulence genes have been identified in a wide variety of pathogens (Cunningham et al, 2001; Shimono et al, 2003; Ionescu et al, 2013). The interplay between virulence genes and anti‐virulence genes is believed to regulate the pathogenicity of pathogens (Brown et al, 2016). Here, we report how an anti‐virulence protein governs the onset of a Salmonella virulence program.
Salmonella enterica serovar Typhimurium is a facultative intracellular pathogen that causes gastroenteritis in humans and a typhoid fever‐like disease in mice (Coburn et al, 2007; Fabrega & Vila, 2013). Salmonella can survive and replicate in acidic vacuoles within host phagocytic cells (Buchmeier & Heffron, 1991; Lee et al, 2013), where it senses low pH (Prost et al, 2007; Choi & Groisman, 2016) and expresses virulence genes via the PhoP/PhoQ two‐component regulatory system (Alpuche Aranda et al, 1992; Guo et al, 1997; Bijlsma & Groisman, 2005; Retamal et al, 2009). Salmonella survival in host phagocytic cells is made possible by the precise timing at which virulence gene products are produced (Jones & Falkow, 1996; Marcus et al, 2000). Indeed, growth within acidic vacuoles requires the coordinated expression of several virulence factors (Eriksson et al, 2003).
The mgtC gene mediates intramacrophage survival and proliferation within host tissues in several intracellular pathogens (Blanc‐Potard & Groisman, 1997; Grabenstein et al, 2006; Rang et al, 2007; Alix & Blanc‐Potard, 2008; Lee & Groisman, 2012; Belon et al, 2014; Pontes et al, 2015). In S. enterica serovar Typhimurium, the MgtC protein confers virulence in two distinct ways. On the one hand, MgtC inhibits Salmonella's own F1Fo ATP synthase (Lee et al, 2013), the machine responsible for the synthesis of the majority of adenosine triphosphate (ATP) in the cell (Senior, 1990). Thus, MgtC enables Salmonella to maintain its cytoplasmic pH near 7 when experiencing a mildly acidic pH inside macrophages (Lee et al, 2013) and to reduce transcription of ribosomal rRNA when cytosolic conditions prevent the assembly of functional ribosomes (Pontes et al, 2016). On the other hand, MgtC prevents degradation of PhoP (Yeom et al, 2017), the master regulator of Salmonella pathogenesis (Groisman et al, 1989; Miller et al, 1989), thereby advancing virulence gene expression. Thus, MgtC's actions commit Salmonella to low cytosolic ATP, slow growth, and expression of genes requiring large amounts of the regulator PhoP.
We now report that Salmonella's commitment to the MgtC‐dependent program requires not only the signals promoting expression of the mgtC gene, but also that MgtC protein amounts supersede those of CigR, an anti‐virulence protein (Kidwai et al, 2013; Yin et al, 2016) that binds MgtC, thereby preventing MgtC from inhibiting the F1Fo ATP synthase. Surprisingly, the cigR and mgtC genes are located on the Salmonella pathogenicity island 3 (SPI‐3; Blanc‐Potard et al, 1999) and are part of the same transcription unit under MgtC‐inducing conditions. However, the cigR gene is also transcribed constitutively and independently of MgtC, setting a threshold of CigR protein that MgtC must overcome to exert its virulence and metabolic functions.
Results
CigR is an inner membrane protein that binds to the inner membrane protein MgtC
It was previously suggested that CigR is an effector protein secreted into host cells (Niemann et al, 2011, 2013). However, this does not appear to be the case because (i) CigR lacks the characteristics of effector proteins (Sato et al, 2011); (ii) CigR secretion was detected only in a Salmonella mutant lacking control of secretion and overexpressing a regulatory protein (Niemann et al, 2011); and (iii) CigR localizes to the inner membrane in wild‐type Salmonella (Fig EV1A), in agreement with transmembrane domain predictions (TMHMM v. 2.0). Given that MgtC is also an inner membrane protein (Lee et al, 2013) and that inactivation of cigR renders Salmonella hypervirulent in mice and inside macrophages (Kidwai et al, 2013; Yin et al, 2016), we wondered whether CigR interacts with MgtC, which would implicate these two proteins in the same virulence pathway.
Figure EV1. CigR is an inner membrane protein that interacts with the MgtC protein.

-
AA Western blot analysis of inner membrane (IM), outer membrane (OM), and secreted fractions (SC) prepared from a strain specifying a chromosomally encoded CigR‐HA protein (JY6). (Please note that the CigR‐HA protein is fully functional.) Bacteria were grown in N‐minimal medium (pH 7.7) with 10 μM MgCl2. NADH oxidase activity [100 × μmol of substrate oxidized (ΔOD340)/min/mg of protein] reflects the purity of the inner membrane preparations.
-
B, CSpots (B) and β‐galactosidase activity (C) of Escherichia coli strain BTH101 harboring plasmids pKT25‐CigR and pUT18/pUT18C‐MgtC. Control strains carry the plasmid vector and pUT18/pUT18C‐PmrB along with pKT25 or pKT25‐CigR. Shown are the mean and SD from three independent experiments.
-
D, ESpots (D) and β‐galactosidase activity (E) of E. coli strain BTH101 harboring plasmids pUT18/pUT18C‐CigR and pKT15‐MgtC. Control strains carry the plasmid vector and pKT25‐PmrB along with pUT18/pUT18C or pUT18/pUT18C‐CigR. Shown are the mean and SD from three independent experiments.
-
FWestern blot analysis of outer membrane (OM), inner membrane (IM), and cytosolic fractions (SC) prepared from wild‐type (14028s) and MgtC‐N92T (EL551) Salmonella strains. NADH oxidase activity [100 × μmol of substrate oxidized (ΔOD340)/min/mg of protein] reflects the purity of the inner membrane preparations.
-
Gβ‐galactosidase activity of E. coli strain BTH101 harboring plasmids pUT18‐MgtC/MgtC‐E84A/MgtC‐N92T and pKT25‐CigR. Control strains carry the pKT25 vector and pKT25‐Zip along with pUT18 or pUT18‐Zip. Shown are the mean and SD from three independent experiments.
-
HWestern blot analysis of outer membrane (OM), inner membrane (IM), and cytosolic fractions (SC) prepared from wild‐type (14028s) Salmonella harboring plasmids pUHE‐cigR‐FLAG, pUHE‐D3cigR‐FLAG, or pUHE‐W133AcigR‐FLAG.
-
Iβ‐galactosidase activity of E. coli strain BTH101 harboring plasmids pUT18‐CigR/CigR‐D1/CigR‐D3/CigR‐W133A and pKT25‐MgtC. Control strains carry the pKT25 vector and pKT25‐Zip along with pUT18 or pUT18‐Zip. Shown are the mean and SD from three independent experiments.
Source data are available online for this figure.
We determined that the CigR and MgtC proteins interact in a specific manner. First, bacterial two‐hybrid analysis demonstrated CigR binding to MgtC but not to PmrB (Fig EV1B–E), an inner membrane protein used as a negative control (Wösten et al, 2000). Second, anti‐HA antibodies pulled down MgtC‐FLAG (see low‐intensity band in center bottom panel of Fig 1A), and anti‐FLAG antibodies pulled down CigR‐HA (Fig 1A), in a strain expressing C‐terminally FLAG‐tagged MgtC and C‐terminally HA‐tagged CigR from their normal chromosomal locations. And third, CigR did not bind to YqjA‐FLAG, an inner membrane protein used as a negative control (Fig 1A). In addition, CigR exhibited decreased binding to an MgtC variant with the N92T substitution (Fig 1B), which is defective in the ability to inhibit the F1Fo ATP synthase (Lee et al, 2013) but retains normal localization to the inner membrane (Fig EV1F). By contrast, CigR interacted with the MgtC variants with the E84A, C99A, or W226A amino acid substitutions (Lee et al, 2013), as it did with the wild‐type MgtC protein (Fig 1B). Furthermore, the anti‐HA antibody did not precipitate MgtC‐FLAG from a strain expressing untagged CigR (Fig 1A), and the anti‐FLAG antibody did not precipitate CigR in a strain expressing untagged MgtC (Fig 1A). In sum, the CigR protein specifically interacts with the MgtC protein in vivo.
Figure 1. The AtpB and CigR proteins compete for binding to MgtC.

- Western blot analysis of crude extracts prepared from JY6, EG16539, YS251, JY92, and JY95 Salmonella strains grown in N‐minimal media pH 7.7 containing 10 μM MgCl2 for 6 h, followed by immunoprecipitation and detection with anti‐HA and anti‐FLAG antibodies, as indicated. Relevant bands are boxed.
- Western blot analysis of crude extracts prepared from wild‐type (14028s), EL549, EL551, EL552, and EL553 Salmonella strains harboring a plasmid expressing CigR‐FLAG grown in N‐minimal media pH 7.7 containing 10 μM MgCl2 and 0.5 mM IPTG for 6 h. These strains express the wild‐type MgtC protein or derivatives with the indicated amino acid substitutions. Immunoprecipitation and detection was carried out with anti‐MgtC and anti‐FLAG antibodies, as indicated. Relevant bands are boxed.
- Dissociation curves for CigR‐HA to MgtC‐FLAG, and AtpB‐HA to MgtC‐FLAG (from Fig EV3G and H; see Materials and Methods). The IC50 corresponds to the concentration at which half of the protein is dissociated from the MgtC‐FLAG protein. Shown are the mean and SD from three independent experiments.
- Western blot analysis of crude extracts from strain EL481 harboring a plasmid expressing the cigR gene following growth in N‐minimal media pH 7.7 containing 10 μM MgCl2 and IPTG (0.01, 0.1, and 1 mM) for 6 h. Immunoprecipitation and detection were carried out with anti‐HA and anti‐FLAG antibodies. Data are representative of three independent experiments, which gave similar results.
Source data are available online for this figure.
CigR and AtpB compete for binding to MgtC
We hypothesized that CigR competes with the F1Fo ATP synthase subunit AtpB for binding to MgtC because the MgtC N92T variant was defective for interaction with AtpB (Lee et al, 2013) and CigR (Figs 1B and EV1G). To test this hypothesis, we performed co‐immunoprecipitation experiments using different amounts of in vitro synthesized MgtC‐FLAG, AtpB‐HA, and CigR‐HA proteins reconstituted into proteoliposomes. As AtpB‐HA amounts increased, binding between CigR and MgtC decreased (Fig EV2A, lower box). Likewise, an increase in CigR‐HA amounts decreased the interaction between MgtC and AtpB (Fig EV2A, upper box). These results argue that CigR and AtpB compete with each other for binding to MgtC.
Figure EV2. CigR competes with AtpB for interaction with MgtC.

- Western blot analysis of proteoliposomes reconstituted with in vitro synthesized F1Fo ATP synthase containing AtpB‐HA, MgtC‐FLAG, and CigR‐HA proteins. At the end of the reconstitution reaction, an aliquot (input) and fractions were immunoprecipitated with either anti‐HA or anti‐FLAG antibodies and analyzed using anti‐HA and anti‐FLAG antibodies. ×5 indicates that five times the amount of the protein is present in the reaction. Relevant bands are boxed.
- Western blot analysis of proteoliposomes reconstituted from in vitro synthesized F1Fo ATP synthase containing AtpB‐HA and CigR‐HA proteins. Proteoliposomes were prepared as described in Materials and Methods. The data are representative of two independent experiments, which gave similar results.
Source data are available online for this figure.
To examine the binding specificity of CigR and AtpB for MgtC, we calculated the half inhibitory concentration (IC50) values of CigR‐HA and AtpB‐HA for MgtC‐FLAG using increasing amounts of competitor AtpB‐HA and CigR‐HA, respectively (Figs 1C and EV3). The IC50 values of CigR‐HA and AtpB‐HA for MgtC‐FLAG are 8.9 and 22.3 μM, respectively (Fig 1C). Thus, MgtC binding to AtpB is stronger than to CigR (Figs 1C and EV3). Additionally, CigR does not appear to bind to AtpB because the intensity of the CigR‐HA band was similar in the presence and absence of AtpB‐FLAG following a pull‐down with anti‐FLAG antibodies (Fig EV2B). Competition between CigR and AtpB was also detected in vivo as expression of the cigR gene from a heterologous promoter reduced the interaction between the AtpB and MgtC proteins (Fig 1D). These experiments demonstrate that CigR binds MgtC directly and that this binding hinders MgtC binding to AtpB. The CigR protein determines whether Salmonella embarks on a pathway that alters normal bacterial physiology.
Figure EV3. CigR and AtpB bind to MgtC with different apparent affinities.

-
A–F(A, F) Western blot analysis of the purified AtpB‐HA, CigR‐HA, and MgtC‐FLAG proteins to calculate apparent binding affinity (A) and to carry out competition experiments (F). Protein amounts were calculated based on known amounts of purified proteins. Proteins were loaded onto the same gel and detected using monoclonal antibodies against the HA and FLAG tags. (B–E) Western blot analysis of the MgtC‐FLAG, CigR‐HA, and AtpB‐HA proteins synthesized using an in vitro transcription/translation system followed by immunoprecipitation and detection with the indicated antibodies. K d values correspond to half of the amount of AtpB‐HA or CigR‐HA proteins that specifically bind to MgtC‐FLAG. (B) Western blot analysis of proteins recovered following incubation of different amounts of AtpB‐HA with a constant amount of MgtC‐FLAG followed by pull‐down with anti‐FLAG antibodies. (C) Affinity of MgtC‐FLAG and AtpB‐HA from panel (B) calculated with purified proteins in panel (A). Shown are the mean and SD from three independent experiments. (D) Western blot analysis of proteins recovered following incubation of different amounts of CigR‐HA and a constant amount of MgtC‐FLAG followed by pull‐down with anti‐FLAG antibodies. (E) Affinity of CigR‐HA and MgtC‐FLAG bands in panel (D) was calculated with purified proteins in panel (A). Shown are the mean and SD from three independent experiments.
-
G, HIn vitro competition between the AtpB‐HA and CigR‐HA proteins for the MgtC‐FLAG protein was determined using a constant amount of MgtC‐FLAG and pull‐down with anti‐FLAG antibodies. The amounts of CigR‐HA and AtpB‐HA were kept constant in (G) and (H), respectively.
Source data are available online for this figure.
The cigR gene decreases survival inside macrophages and increases both ATP levels and intracellular pH, in an mgtC‐dependent manner
If CigR exerts its anti‐virulence function by targeting MgtC, a cigR mutant should exhibit the opposite behavior of an mgtC mutant. As predicted, a strain deleted for the cigR gene achieved four times the number of wild‐type Salmonella inside the macrophage‐like cell line J774A.1 by 20 h post‐infection (Fig 2A). This is in contrast to the mgtC mutant, which survived five times less than the wild‐type strain (Fig 2A; Blanc‐Potard & Groisman, 1997; Rang et al, 2007), albeit not to the low levels as the phoP mutant (Fig 2A). The enhanced intramacrophage survival of the cigR mutant is due to inactivation of cigR (as opposed to the cigR mutation being polar on a downstream gene) because a plasmid harboring the cigR gene restored wild‐type survival, whereas the plasmid vector did not (Fig 2A).
Figure 2. The cigR and mgtC mutants display opposite behaviors.

- Survival inside J774A.1 macrophages of wild‐type (14028s), mgtC (EL4), cigR (JY12), mgtC cigR (JY6), and phoP (MS7953s) Salmonella without/with the indicated plasmids 20 h after infection. The mean and SD from two independent experiments are shown. Please note log10 scale of y‐axis.
- ATP levels in wild‐type (14028s), mgtC (EL4), cigR (JY12), mgtC cigR (JY6), and mgtB Salmonella, and in the cigR mutant harboring the plasmid vector or pcigR. Intracellular ATP levels correspond to picomoles of ATP per ml of cells at a given OD600. The mean and SD from three independent experiments are shown. Bacteria were grown in N‐minimal media pH 7.7 containing 10 μM MgCl2 for 5 h.
- ATP hydrolysis rates in inverted vesicles prepared from wild‐type (14028s), mgtC (EL4), and cigR (JY12) Salmonella. The reaction was initiated by adding ATP and monitored for 5 min. The mean and SD from two independent experiments are shown.
- Intracellular pH of wild type (14028s), mgtC (EL4), and cigR (JY12) Salmonella following bacterial growth in N‐minimal medium pH 7.7 for 1 h and then switched to the same media at pH 4.6 for 1 h. Lines represent the average pH of seven independent replicates.
- Intracellular pH of wild type (14028s), mgtC (EL4), and cigR (JY12) Salmonella when inside macrophages. Bacteria grown in LB medium overnight were used to infect J774A.1 macrophages. pH measurements were carried out 6 h post‐internalization. Numbers represent the average pH of four independent replicates.
- mRNA amounts of the PhoP‐activated pagC and pcgL genes in wild type (14028s), mgtC (EL4), and cigR (JY12) Salmonella. The mean and SD from two independent experiments are shown.
- ATP levels present in wild type (14028s) and mgtC (EL4) Salmonella harboring no added plasmid, the plasmid vector, or plasmids expressing the cigR or mgtC genes. Bacteria were grown in N‐minimal media pH 7.7 containing 10 μM MgCl2 with 200 μM IPTG for 4 h. ATP levels correspond to picomoles of ATP per ml of cells at given OD600. Shown are the mean and SD from three independent experiments.
The cigR mutant exhibited lower ATP levels (Fig 2B) and ATPase activity (Fig 2C) than wild‐type Salmonella, the opposite phenotypes of those displayed by the mgtC mutant (Fig 2B and C; Lee et al, 2013). The plasmid harboring the cigR gene restored wild‐type ATP levels to the cigR mutant, but the vector control did not (Fig 2B). [As previously reported (Lee et al, 2013), a mutant deleted for the mgtB gene, which is co‐transcribed with mgtC (Snavely et al, 1991), retained wild‐type ATP levels (Fig 2B).] Furthermore, the cytoplasmic pH of the cigR mutant was higher than that of wild‐type Salmonella both during growth in defined laboratory media (Fig 2D) and inside macrophages (Fig 2E), unlike the lower pH displayed by the mgtC mutant in both conditions (Fig 2D and E; Lee et al, 2013).
The cigR mutant produced more PhoP‐activated mRNAs than wild‐type Salmonella (Fig 2F). This result reflects that MgtC protects PhoP from degradation (Yeom et al, 2017). Thus, when CigR is absent, the higher amounts of free MgtC further PhoP‐dependent gene transcription (Yeom et al, 2017).
That CigR works primarily (if not exclusively) by inhibiting MgtC is supported by three additional lines of evidence. First, an mgtC cigR double mutant retained the behavior of the mgtC single mutant: It displayed lower intramacrophage survival (Fig 2A) and higher ATP levels (Fig 2B) than wild‐type Salmonella. Second, a plasmid expressing the cigR gene from a heterologous promoter increased ATP levels in wild‐type Salmonella but not in the mgtC mutant (Fig 2G). Taken together with the data presented above (Fig EV1A), these results indicate that the anti‐virulence protein CigR exerts its effects by targeting the virulence protein MgtC. Moreover, they reinforce the notion that the inner membrane protein CigR operates inside Salmonella.
The C‐terminal domain of CigR is required for inhibition of MgtC
A tBLASTN analysis using the deduced amino acid sequence of the S. enterica serovar Typhimurium cigR gene revealed the presence of cigR homologs in several enteric bacterial species (Appendix Fig S1). The C‐terminal region of CigR, which harbors the single predicted transmembrane domain, is much more conserved (82~98% amino acid identity) than the rest of the protein (53~85% amino acid identity for the full‐length CigR; Appendix Fig S1). Therefore, we hypothesized that the C‐terminal region is crucial for CigR function.
To test this hypothesis, we compared the behaviors of wild‐type Salmonella and a strain deleted for the whole cigR open reading frame to those of strains with deletions or nucleotide substitutions in the part of the chromosomal copy of the cigR gene specifying the C‐terminal region of CigR (Appendix Fig S1). We deleted the nucleotide sequence corresponding to three conserved domains (D1, D2, and D3), each four amino acids long (Appendix Fig S1). Because tryptophan residues are generally critical for protein function (Bogan & Thorn, 1998; Rasmussen et al, 2007), we also substituted the nucleotides specifying the single tryptophan at position 133 so that cigR specified an alanine residue at this position.
Bacteria expressing the D3 and W133A CigR variants displayed the enhanced intramacrophage survival (Fig 3A), lower ATP levels during growth in laboratory media (Fig 3B), and higher pH inside macrophages (Fig 3C) that are characteristic of the cigR‐deleted strain. The D3 and W133A CigR proteins localized to the inner membrane like wild‐type CigR (Fig EV1H). By contrast, the D1 and D2 mutants retained a wild‐type behavior (Fig 3A–C). Although bacteria expressing the D3 and W133A CigR variants exhibited the same phenotypes, the corresponding proteins differed in their ability to interact with the MgtC protein in vivo and in vitro. Specifically, the D3 variant interacted with MgtC like wild‐type CigR or the D1 and D2 variants (Fig 3D and E). By contrast, the W133A CigR variant was defective in binding to MgtC (Figs 3D and E, and EV1I). Cumulatively, these data establish that W133 is required for CigR binding to MgtC and that one or more residues substituted in the D3 region are necessary for inhibition of the MgtC protein.
Figure 3. The C‐terminal domain of CigR is required for inhibition of MgtC.

-
A–CBacterial survival inside macrophages (A), ATP levels (B), and intracellular pH (C) of wild‐type Salmonella (14028s) and mutants with deletions or nucleotide substitution in the cigR open reading frame (JY139, JY150, JY151, and JY152). (A) Salmonella survival inside J774A.1 macrophages 20 h after infection. The mean and SD from two independent experiments are shown. Please note log10 scale of y‐axis. (B) ATP levels, corresponding to picomoles of ATP per ml of cells at a given OD600, of the strains listed in (A). Bacteria were grown for 4 h in N‐minimal media pH 7.7 containing 10 μM MgCl2. The mean and SD from three independent experiments are shown. (C) Intracellular pH of the Salmonella strains listed in (A) was measured inside the macrophage‐like cell line J774A.1. Numbers represent the average pH of four independent replicates, which gave similar bacterial colony counts when plated on LB agar plates. Data information: *P < 0.05, **P < 0.01, ***P < 0.001, two‐tailed t‐test with each sample vs. wild type, N.S., not significant.
-
DWestern blot analysis of proteoliposomes reconstituted from in vitro synthesized MgtC‐FLAG and CigR‐HA proteins, followed by immunoprecipitation and detection with antibodies directed to the HA or FLAG epitopes, as indicated. At the end of the reconstitution reaction, an aliquot (input) and fractions immunoprecipitated with antibodies directed to the HA or FLAG epitopes were analyzed. Proteoliposomes were prepared as described in Materials and Methods. The data are representative of two independent experiments, which gave similar results.
-
EWestern blot analysis of crude extracts prepared from wild‐type Salmonella (14028s) harboring a plasmid expressing the CigR‐FLAG protein grown in N‐minimal media pH 7.7 containing 10 μM MgCl2 and IPTG (0.12 mM for wild type, 0.2 mM for D1 and D2, 0.3 mM for D3, and 0.4 mM for W) for 6 h. These strains express the wild‐type CigR‐FLAG protein or derivatives with the indicated amino acid substitution or domain mutations. Immunoprecipitation and detection were carried out with antibodies directed to the MgtC protein and FLAG epitope, as indicated. The data are representative of two independent experiments, which gave similar results.
Source data are available online for this figure.
The cigR gene is transcribed together with and independently of the mgtC gene
Toxins and anti‐toxins are typically encoded by adjacent genes and produced from the same transcript (Gerdes et al, 2005). Although the mgtC and cigR genes are separated by two genes (Fig 4A–C), we hypothesized that they are part of the same polycistronic mRNA because they exhibit comparable expression behavior in a different Salmonella strain (http://bioinf.gen.tcd.ie/cgi-bin/salcom.pl; Kröger et al, 2013). As hypothesized, similar RNA polymerase (RNAP) occupancy of the mgtC and cigR coding regions as well as of the intervening mgtB and mgtR genes was observed during growth in 10 μM Mg2+ (Fig EV4A), a condition promoting mgtCBR expression (Soncini et al, 1996). Moreover, reverse transcriptase PCR (RT–PCR) assays produced mgtC‐cigR, mgtC‐mgtB, mgtB‐cigR, and cigR‐cigR amplicons of the predicted sizes (Fig EV4B–F). These results establish that the mgtCBR operon is longer than previously reported (Alix & Blanc‐Potard, 2008).
Figure 4. Transcription of the cigR gene is partially PhoP‐dependent.

- Diagram of the mgtC‐cigR chromosomal region showing the PhoP‐dependent (green color) and PhoP‐independent (red color) promoters.
- mRNA amounts of the mgtC, mgtB, and cigR genes produced by wild‐type Salmonella (14028s) grown in N‐minimal media pH 7.7 with 10 mM or 10 μM MgCl2 for 4 h. Shown are the mean and SD from three independent experiments.
- mRNA amounts of the mgtC, mgtB, and cigR genes produced by wild‐type (14028s) and phoP mutant (MS7953s) Salmonella following growth in N‐minimal media pH 7.7 with 10 μM MgCl2 for 4 h. mRNA amounts of target genes were normalized to those of the rpoD gene. The mean and SD from two independent experiments are shown.
- Western blot analysis of crude extracts from wild‐type Salmonella (14028s) following growth in N‐minimal media with 10 μM MgCl2 for the indicated times using antibodies directed to the MgtC, CigR, AtpB, or GroEL proteins.
- Number of MgtC, CigR, and AtpB protein molecules present in cell extracts from wild‐type Salmonella (14028s) following growth under non‐inducing conditions (10 mM MgCl2, time zero) and inducing conditions (10 μM MgCl2) at the indicated times. Shown are the mean and SD from three independent experiments.
Source data are available online for this figure.
Figure EV4. The anti‐virulence gene cigR and the virulence gene mgtC are part of the same polycistronic transcript produced during growth in 10 μM Mg2+ for 4 h.

-
ARNA polymerase occupancy of the mgtCBRcigR DNA region in wild‐type Salmonella (14028s) grown in N‐minimal media pH 7.7 with 10 or 50 μM Mg2+ for 4 h. Cross‐linking with formaldehyde, immunoprecipitation, and analysis was performed as described in Materials and Methods. Black color indicates samples after RNAP ChIP‐seq, and gray color indicates input control.
-
BDiagram of the mgtC‐cigR chromosomal region showing the location of the primers used in the reverse transcriptase PCR assay experiment shown below. Numbers above horizontal lines correspond to the size of the predicted amplicons.
-
C–FAgarose gel electrophoresis of reverse transcription–polymerase chain reaction (RT–PCR) products obtained using specific primers for the mgtC‐cigR region. Bacteria were grown in N‐minimal media at pH 7.7 with 10 μM (10), 50 μM (50), or 10 mM (HH) MgCl2 for 4 h. mRNA samples were reverse‐transcribed with primer 15111. (C) Primers 15106/15107 were used for amplification of mgtC_cigR, (D) 15108/15109 for mgtB_cigR, (E) 15106/15112 for mgtC_mgtB, and (F) 15110/15111 for cigR. Chromosomal DNA was used as a positive control (DNA) and samples without RT as negative controls (RNA). Data are representative of two independent experiments, which gave similar results. cDNA samples were prepared as described in Materials and Methods.
Source data are available online for this figure.
Surprisingly, the cigR gene is also transcribed independently of the mgtC gene. That is to say, transcription from the mgtC promoter is strictly dependent on the PhoP protein (Soncini et al, 1996), whose amounts and activity increase during growth in 10 μM Mg2+ and decrease during growth in 10 mM Mg2+ (Soncini et al, 1996). Thus, the mgtC and mgtB mRNA amounts are very low in wild‐type Salmonella grown in 10 mM Mg2+ (Fig 4B) and in a phoP mutant grown in 10 μM Mg2+ (Fig 4C). By contrast, significant amounts of cigR mRNA were produced in these strains and growth conditions (Fig 4B and C).
We identified a PhoP‐independent cigR promoter (Figs 4A and EV5A) which, when mutated, resulted in a strain in which cigR transcription was like mgtC's (i.e., strictly dependent on growth in 10 μM Mg2+; Fig EV5B). These results imply that the CigR protein is present in wild‐type Salmonella before the onset of mgtC‐inducing conditions. Taken together with the genetic (Figs 2 and 3A–C) and biochemical (Figs 1, and 3D and E) data presented above, these results suggest that inhibition of the F1Fo ATP synthase and stabilization of the PhoP protein take place only when MgtC protein amounts exceed those of CigR.
Figure EV5. The CigR protein inhibits the MgtC protein at early times after induction of the PhoP/PhoQ system.
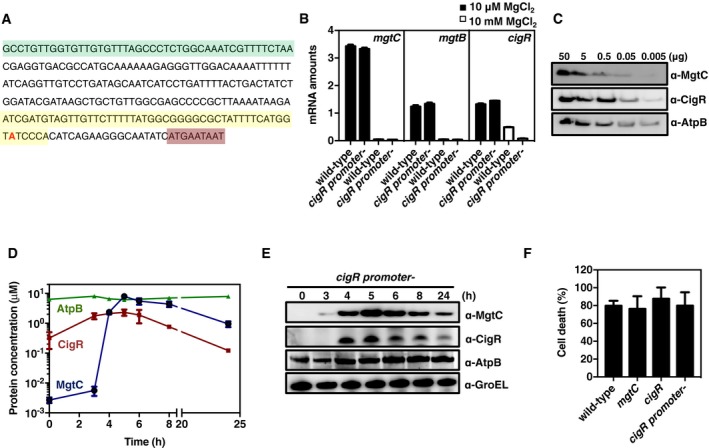
- Nucleotide sequence of the mgtR‐cigR intergenic region showing the 3′ end of the mgtR gene (green color) and the 5′ end of the cigR gene (light brown color). Deleted nucleotides of the cigR promoter mutant are marked in yellow, and the transcription start site for cigR is in red.
- mRNA amounts of the mgtC, mgtB, and cigR genes present in wild‐type (14028s) and cigR promoter mutant (JY372) Salmonella strains following growth in N‐minimal media with 10 mM or 10 μM MgCl2 for 4 h. mRNA amounts of the investigated genes were normalized to those of the rpoD gene. Shown are the mean and SD from three independent experiments.
- Western blot analysis of the indicated purified proteins. Proteins were loaded onto the same gel to calculate protein concentrations and detected using antibodies directed to the MgtC, CigR, or AtpB proteins.
- Protein concentrations of the MgtC, CigR, and AtpB proteins from panel (B) were calculated as described in Materials and Methods. Shown are the mean and SD from three independent experiments.
- Western blot analysis of crude extracts from the cigR promoter mutant (JY372) Salmonella following growth in N‐minimal media with 10 μM MgCl2 for the indicated times. Proteins were detected using antibodies directed to the MgtC, CigR, AtpB, or GroEL proteins.
- The percentage of J774A.1 macrophage death was determined by detecting the amount of lactate dehydrogenase released into the supernatants 20 h after infection with wild‐type (14028s), cigR (JY12), mgtC (EL4), and cigR promoter mutant (JY372) Salmonella. Data correspond to the percentage of host cell death at 20 h after infection. Shown are the mean and SD from four independent experiments.
Source data are available online for this figure.
CigR controls MgtC's virulence function at early times inside macrophages
To test the model presented above, we examined CigR and MgtC protein amounts and MgtC‐dependent phenotypes in a set of isogenic strains at different times following a switch from non‐inducing to mgtC‐inducing conditions. CigR protein amounts were > 20 times higher than MgtC's at early times after wild‐type Salmonella experienced mgtC‐inducing conditions (Figs 4D and E, and EV5C and D). MgtC protein amounts increased dramatically starting at 3 h and exceeded CigR's by 4 h. By contrast, the amounts of the MgtC target AtpB (Lee et al, 2013) did not change over the 24‐h course of the experiment (Fig 4D and E).
The cigR promoter mutant and the mutant lacking the cigR coding region exhibited similar ATP levels at 3 h post‐switch to mgtC‐inducing conditions (Figs 5A and Appendix Fig S2). This was also the case for wild‐type and mgtC Salmonella (Figs 5A and Appendix Fig S2). These results reflect that the mgtC gene is barely expressed at this time (Fig 4E) and that cigR transcription largely originates from the PhoP‐independent promoter (Fig 4A). By 5 h, the ATP levels of the mgtC mutant were much higher than those of the cigR mutant, which, in turn, were lower than those of the wild‐type strain (Fig 5B). The ATP levels of the cigR promoter mutant resembled those of the wild‐type strain (Fig 5B), likely due to the stronger mgtC promoter providing the bulk of cigR transcription at 5 h. Thus, MgtC‐dependent phenotypes are manifested only when MgtC protein amounts exceed CigR's. As expected, the CigR protein was absent from the cigR promoter mutant before the onset of mgtC‐inducing conditions, mimicking the expression of the MgtC protein in this strain (Fig EV5E).
Figure 5. PhoP‐independent CigR expression sets threshold for control of MgtC protein.

-
A, BATP levels in wild type (14028s), mgtC (EL4), cigR (JY12), and cigR promoter mutant (JY372) Salmonella strains following growth in N‐minimal media pH 7.7 containing 10 μM MgCl2 for 3 (A) or 5 h (B). ATP levels correspond to luminescence amounts. The mean and SD from three independent experiments are shown.
-
C, DSurvival inside J774A.1 macrophages of the strains listed in (A, B) at 5 (C) or 20 (D) h after infection. The mean and SD from two independent experiments are shown.
-
EWestern blot analysis of crude extracts from wild type (14028s), cigR (JY12), mgtC (EL4), and cigR promoter mutant (JY372) Salmonella strains inside J774 A.1 macrophages determined at the indicated times after infection.
To test whether initiation of MgtC's virulence program requires that MgtC supersedes the CigR threshold, we investigated the behavior of an mgtC mutant harboring a plasmid that transcribed the mgtC gene from an inducible promoter. In contrast to wild‐type Salmonella, in which the MgtC protein is hardly detectable at early times (i.e., 3 h; Fig 4E), MgtC expression in the engineered strain at early times lowered ATP levels and growth under low Mg2+ conditions (Appendix Fig S3A and B). Furthermore, the mRNA amounts of the PhoP‐activated genes pagC and pgtE increased at early times in the engineered strain (Appendix Fig S3C and D). These results argue that MgtC can overcome the CigR threshold even at early times when MgtC is expressed from a heterologous (as opposed to its normal) promoter. Investigation of this strain at 6 h revealed that the MgtC‐expressing plasmid rescued all four investigated MgtC‐dependent phenotypes (Appendix Fig S3E–G).
We determined that the CigR protein also controls MgtC when Salmonella is inside macrophages. First, the cigR promoter mutant survived slightly better than the wild‐type strain both at 5 and 20 h post‐internalization (Fig 5C). Second, survival of the cigR deletion mutant was significantly higher than that of the cigR promoter mutant at 20 h (Fig 5D). And third, the mgtC mutant was slightly defective at 5 h (Fig 5C) and very defective at 20 h (Fig 5D). These results reflect that at 5 h post‐internalization, the CigR protein is detected in wild‐type and mgtC strains, but not in the cigR promoter mutant (Fig 5E), and that by 20 h, the cigR promoter mutant produces CigR at amounts similar to those of the wild‐type and mgtC strains (Fig 5E). As cigR inactivation did not impact macrophage lysis (Fig EV5F), CigR regulates Salmonella's ability to survive and replicate inside macrophages.
Discussion
Pathogens respond to specific signals by regulating precisely when and where virulence genes are expressed. In addition, the manifestation of a pathogenic behavior may be subject to the action of anti‐virulence genes (Brown et al, 2016). We have now determined that the anti‐virulence protein CigR controls the timing of a virulence program by imposing a threshold for the key virulence factor MgtC (Fig 6). CigR achieves this task by outcompeting MgtC for binding to two virulence factors: the F1Fo ATP synthase (Fig 1D), preventing the MgtC‐dependent decrease in ATP levels (Fig 2B), and the master virulence regulator PhoP, resulting in higher mRNA abundance of two PhoP‐activated MgtC‐dependent genes in the cigR null mutant than in wild‐type Salmonella (Fig 2E). The CigR protein is produced continuously (Fig 4D). Therefore, when an organism first experiences mgtC‐inducing conditions, the high basal levels of CigR are sufficient to bind MgtC and prevent it from executing its virulence program (Fig 5C–E). In other words, CigR sets a threshold for Salmonella's commitment to a pathway that promotes profound physiological changes.
Figure 6. An anti‐virulence protein controls the onset of a Salmonella virulence program.

Under non‐inducing conditions for the master virulence regulator PhoP (gray color), the anti‐virulence protein CigR is expressed independently of PhoP, and the virulence protein MgtC is not expressed (left). At early times under inducing conditions (pink color), PhoP promotes expression of MgtC, which, sequestered by CigR, does not inhibit the F1Fo ATP synthase (ATPase) or protect PhoP from degradation (middle). At late times under inducing conditions (blue color), MgtC amounts supersede CigR amounts, which result in MgtC binding to and inhibition of the ATPase (right). In addition, MgtC protects PhoP from degradation, thereby increasing PhoP amounts and enabling transcription of a subset of PhoP‐activated genes (right).
The MgtC protein is part of a genetic program that, by reducing ATP levels (Lee et al, 2013), lowers ribosome production, protein synthesis, and bacterial growth rate (Pontes et al, 2016), and is anticipated to increase antibiotic tolerance. We have now determined that execution of this program requires not only the signals that promote expression of the mgtC gene, but also that the amounts of the resulting MgtC protein supersede those of the anti‐virulence protein CigR (Fig 6). In this way, CigR functions as a gatekeeper that prevents the accidental entry into a low ATP state when Salmonella experiences mgtC‐inducing signals only transiently (Fig 5A and Appendix Fig S2).
Three negative regulators have been implicated in the control of MgtC amounts and activity: the CigR protein, the peptide MgtR, and the anti‐sense RNA AmgR. MgtR promotes MgtC degradation and is encoded in the same operon as mgtC (Alix & Blanc‐Potard, 2008). AmgR is complementary to the mgtC portion of the mgtCBRcigR polycistronic mRNA (Lee & Groisman, 2010). Whereas mgtR and amgR are transcribed exclusively in a PhoP‐dependent manner, cigR is also transcribed independently of PhoP (Fig 4). Because CigR is present before MgtC, it controls the onset of the MgtC‐dependent activities. This is in contrast to MgtR and AmgR, which, being made after MgtC, control the extent of time MgtC is present.
Our findings illustrate how specific, ordered interactions between virulence and anti‐virulence determinants govern the progression of an infection process. That is to say, once MgtC amounts surpass CigR's, MgtC protects the master virulence regulator PhoP from degradation (Yeom et al, 2017; Fig 6). This protection enables normal transcription of PhoP‐activated genes, including pcgL (Fig 2G), which specifies another anti‐virulence protein, one that destroys a metabolite promoting pathogen growth in host tissues (Mouslim et al, 2002).
The genes specifying the MgtC and CigR proteins are often found in enteric pathogens. By contrast, Yersinia and Mycobacterium have MgtC but not CigR, whereas Citrobacter and Rahnella have CigR but not MgtC. In addition, the mgtC‐cigR operon structure is conserved within Salmonella species whereas enteric bacteria specify mgtC and cigR at different parts of the genome. cigR is a pseudogene in S. enterica serovar Typhi (Parkhill et al, 2001), which may advance the MgtC‐dependent virulence program in this human‐restricted pathogen.
CigR localizes to the Salmonella inner membrane (Fig EV1A), binds to the MgtC protein (Fig 1A), which also localizes to the Salmonella inner membrane, and mediates all investigated behaviors including replication and control of bacterial pH inside macrophages in an mgtC‐dependent manner (Fig 2). Therefore, CigR operates within Salmonella. This is in contrast to CigR's previously proposed site of action inside host mammalian cells (Niemann et al, 2011, 2013), which was based on detection of a reporter based on a fusion protein between adenylate cyclase (CyaA; Sory et al, 1995) and CigR in a Salmonella mutant lacking control of secretion and overexpressing a regulatory protein (Niemann et al, 2011). That the anti‐virulence protein CigR operates within Salmonella is reminiscent of other anti‐virulence gene products, such as PmrA (Choi & Groisman, 2013) and AmgR (Lee & Groisman, 2010), that also function within this pathogen.
Finally, the CigR‐MgtC interaction is reminiscent of that established between anti‐toxins and toxins implicated in antibiotic tolerance (Gerdes et al, 2005). For example, the toxin HipA promotes slow growth and tolerance to antibiotics (Schumacher et al, 2015), and the MgtC protein also promotes slow growth by reducing ATP levels (Pontes et al, 2016), a condition that results in antibiotic tolerance (Conlon et al, 2016; Shan et al, 2017). Moreover, HipB binds to HipA, preventing it from exerting its function (Schumacher et al, 2015), whereas CigR sequesters MgtC with like effects (Figs 1 and 2).
Materials and Methods
Bacterial strains, plasmids, and growth conditions
Bacterial strains and plasmids used in this study are presented in Table 1. All S. enterica serovar Typhimurium strains are derived from strain 14028s (Fields et al, 1986). Phage P22‐mediated transductions were carried out as described (Davis et al, 1980). DNA oligonucleotides used in this study are presented in Table 2. Bacteria were grown at 37°C in Luria‐Bertani broth (LB), N‐minimal media (pH 7.7 or pH 4.8; Snavely et al, 1991) supplemented with 0.1% casamino acids, 38 mM glycerol, and the indicated concentrations of MgCl2. Escherichia coli DH5α was used as the host for preparation of plasmid DNA. Ampicillin was used at 50 μg/ml, chloramphenicol at 25 μg/ml, kanamycin at 50 μg/ml, and tetracycline at 12.5 μg/ml.
Table 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant characteristics | Source |
|---|---|---|
| Escherichia coli | ||
| BTH101 | Host strain used for bacterial two‐hybrid system | Karimova et al (1998) |
| DH5α | Host strain used for generation and propagation of plasmid constructs | Hanahan (1983) |
| Salmonella enterica serovar Typhimurium | ||
| 14028s | Wild type | Fields et al (1986) |
| EG16539 | mgtC‐FLAG | Lee et al (2013) |
| EL4 | ΔmgtC | Lee et al (2013) |
| EL5 | ΔmgtB | Lee et al (2013) |
| EL481 | mgtC‐FLAG/atpB‐HA | Lee et al (2013) |
| EL549 | E84AmgtC | This study |
| EL551 | N92TmgtC | Lee et al (2013) |
| EL552 | C99AmgtC | This study |
| EL553 | W226AmgtC | This study |
| JY2 | cigR‐HA | This study |
| JY6 | ΔmgtC/cigR | This study |
| JY12 | ΔcigR | This study |
| JY92 | mgtC‐FLAG/cigR‐HA | This study |
| JY95 | yqjA‐FLAG/cigR‐HA | This study |
| JY139 | W133AcigR | This study |
| JY150 | D1cigR | This study |
| JY151 | D2cigR | This study |
| JY152 | D3cigR | This study |
| JY372 | cigR promoterless | This study |
| MS7953s | phoP::Tn10 | Fields et al (1989) |
| YS251 | yqjA‐FLAG | Shi et al (2004) |
| Plasmids | ||
| pCP20 | ReppSC101 ts l cI857 FLP AmpR CmR | Datsenko and Wanner (2000) |
| pFPV25.1 | RepColE1 AmpR rpsM::gfpmut3 | Valdivia and Falkow (1996) |
| pKD46 | ReppSC101 ts AmpR ParaBAD‐gbexo | Datsenko and Wanner (2000) |
| pKD3 | RepR6Kg AmpR FRT CmR FRT | Datsenko and Wanner (2000) |
| pKD4 | RepR6Kg AmpR FRT KmR FRT | Datsenko and Wanner (2000) |
| pKT25 | KmR repp15A (pACYC184 derivative) | Battesti and Bouveret (2012) |
| pKT25‐zip | reppSC101KmR T25‐zip | Battesti and Bouveret (2012) |
| pKT25‐cigR | reppSC101KmR T25‐cigR | This study |
| pKT25‐mgtC | reppSC101KmR T25‐mgtC | This study |
| pKT25‐pmrB | reppSC101KmR T25‐pmrB | This study |
| pUHE21‐2lacI q | ReppMB1 lacI q AmpR | Soncini et al (1995) |
| pUHE‐MgtC | ReppMB1 lacI q AmpR Plac‐mgtC | Chamnongpol and Groisman (2002) |
| pUHE‐CigR | ReppMB1 lacI q AmpR Plac‐cigR | This study |
| pUHE‐CigR‐FLAG | ReppMB1 lacI q AmpR Plac‐cigR‐FLAG | This study |
| pUHE‐D1cigR‐FLAG | ReppMB1 lacI q AmpR Plac‐D1cigR‐FLAG | This study |
| pUHE‐D2cigR‐FLAG | ReppMB1 lacI q AmpR Plac‐D2cigR‐FLAG | This study |
| pUHE‐D3cigR‐FLAG | ReppMB1 lacI q AmpR Plac‐D3cigR‐FLAG | This study |
| pUHE‐W133AcigR‐FLAG | ReppMB1 lacI q AmpR Plac‐W133AcigR‐FLAG | This study |
| pUT18 | AmpR reppMB1 (pBlueScriptII derivative) | Battesti and Bouveret (2012) |
| pUT18‐zip | reppMB1 AmpR zip‐UT18 | Battesti and Bouveret (2012) |
| pUT18‐mgtC | reppMB1 AmpR mgtC‐ UT18 | Yeom et al (2017) |
| pUT18‐pmrB | reppMB1 AmpR pmrB‐ UT18 | Yeom et al (2017) |
| pUT18‐cigR | reppMB1 AmpR cigR‐ UT18 | Yeom et al (2017) |
| pUT18‐mgtC‐E84A | reppMB1 AmpR mgtC‐E84A‐UT18 | This study |
| pUT18‐mgtC‐N92T | reppMB1 AmpR mgtC‐N92T‐UT18 | This study |
| pUT18‐cigR‐D1 | reppMB1 AmpR cigR‐D1‐ UT18 | This study |
| pUT18‐cigR‐D3 | reppMB1 AmpR cigR‐D3‐ UT18 | This study |
| pUT18‐cigR‐W133A | reppMB1 AmpR cigR‐W133A‐ UT18 | This study |
Table 2.
Primers used in this study
| No. | Sequence (from 5′ to 3′) |
|---|---|
| 13852 | TAATAATCGCCGTGACCACCGCGGTACTGAGCGCGATCAGTGTAGGCTGGAGCTGCTTC |
| 13853 | AATATCATGAATAATCGTCGTGGTTTAACCGCCGTCCTGTATGAATATCCTCCTTAGT |
| 13861 | GTCACGGCGATTATTAATGGCGTATTTGATTACCCATACGATGTTCCAGATTACGCTTAAGTGTAGGCTGGAGCTGCTTC |
| 13862 | TCCAAACTGGCTGCGCCAATAACGCCTGGTTATGAATATCCTCCTTAGT |
| 12595 | GTCAGGATCCCATGGAGGAACGTATGTTAAT |
| 12596 | GTCATGGTACCCGTTGACTATCAATGCTCCAGT |
| 1388 | AAGGTACCCTGATGCGTTTTCAGC |
| 1389 | CAGGATCCACGGCGTATTACCCGT |
| 16108 | CGCTCTAGAGATGAATAATCGTCGTGGTTTAACCGCCGTCCTGGCGACG |
| 16109 | CGCGGTACCCGATCAAATACGCCATTAATAATCGCCGTGACCAC |
| 14721 | GGGATACCATGAAAATTTAAGACCCACTTTCACA |
| 14722 | AAAATAAGAATCGATGCTAAGCACTTGTCTCCTG |
| 14723 | ACGACGATTATTCATGATATTGCCCTTCTGATGCTTATTTTAAGCGGGGCTCGCCAACAGCAGCTTA |
| 14724 | TAAGCTGCTGTTGGCGAGCCCCGCTTAAAATAAGCATCAGAAGGGCAATATCATGAATAATCGTCGT |
| 14718 | CGCAAGCTTTTACTTGTCATCGTCGTCCTTGTAGTCATCAAATACGCCATTAATAATCGCCGTGACCACCGCG |
| 15088 | CGCAGGATCCATGAATAATCGTCGTGGTTTAACCGCCGTCCTGGCGA |
| 15089 | CGCAAGCTTTTAATCAAATACGCCATTAATAATCGCCGTGACCACCGCG |
| 12718 | CATCATGCGCGCAGGGATGAATGTG |
| 12719 | CACATTCATCCCTGCGCGCATGATG |
| 12722 | GCGGCAACGCTATGGGCTTCGGCGGGCATCGGC |
| 12723 | GCCGATGCCCGCCGAAGCCCATAGCGTTGCCGC |
| 12724 | ATATCACCGCAATTCACGCGAGCATTGATAGTCA |
| 12725 | TGACTATCAATGCTCGCGTGAATTGCGGTGATAT |
| 14057 | GCGAATTAATACGACTCACTATAGGGCTTAAGTATAAGGAGGAAAAAATATGGAGGAACGTATGTTAATGTTTCCTTAT |
| 14058 | AAACCCCTCCGTTTAGAGAGGGGTTATGCTAGTTACTTGTCATCGTCGTCCTTGTAGTCTTGACTATCAATGCTCCAGTGAATTGCGGT |
| 14065 | GCGAATTAATACGACTCACTATAGGGCTTAAGTATAAGGAGGAAAAAATATGAATAATCGTCGTGGTTTAACCGCCGTC |
| 14066 | AAACCCCTCCGTTTAGAGAGGGGTTATGCTAGTTAAGCGTAATCTGGAACATCGTATGGGTAATCAAATACGCCATTAATAATCGCCGTGACCA |
| 14300 | GCGAATTAATACGACTCACTATAGGGCTTAAGTATAAGGAGGAAAAAATATGGCTTCAGAAAATATGACGCCGCAGGAATACATAGG |
| 14301 | AAACCCCTCCGTTTAGAGAGGGGTTATGCTAGTTAAGCGTAATCTGGAACATCGTATGGGTAATGCTCTTCGGACGCCATCGACAGATAG |
| 15106 | CGGCTTAGGGCAGTTCAAAAATGCGCTGGCG |
| 15107 | AGTTGCCTGGCGACAGCATAGCTAATGTCC |
| 15108 | TTTTGCCCGGCATTTCCTTT |
| 15109 | ATTTCGCAGTTGTCGTTGCC |
| 15110 | ATGAATAATCGTCGTGGTTTAACCGCCGTCCTGGCGA |
| 15111 | TTAATCAAATACGCCATTAATAATCGCCGTGACCACCGCG |
| 15112 | ACGCAACACCGTGGCGGTGGTGCGAACC |
| 7530 | CAGCCCGCGCACATTC |
| 7531 | TTGTCTCTGGGATTGGCTTTCT |
| 7763 | TCAGAAAATGATAAGCAGCATAAAAAA |
| 7764 | CCCTGACGATGGCTGTTCA |
| 15171 | ACGTCGAGTCGGACATTAGC |
| 15172 | ATTGTCCCAGCATAGACGCC |
| 4149 | ACCGTGGCACAAATGATGCT |
| 4150 | TCGGCAATCGCCTTATCTG |
| 15208 | GCGAATTAATACGACTCACTATAGGGCTTAAGTATAAGGAGGAAAAAATATGGCTTCAGAAAATATGACGCCGCAGGAATACATAGG |
| 15209 | AAACCCCTCCGTTTAGAGAGGGGTTATGCTAGTTACTTGTCATCGTCGTCCTTGTAGTCATGCTCTTCGGACGCCATCGACAGATAG |
Construction of chromosomal mutants and plasmids
To construct cigR mutant strains, a kan cassette was introduced in the cigR gene as follows: The kan fragment was amplified from plasmid pKD4 using primers 13852/13853, then introduced into wild‐type 14028s and mgtC mutant strains harboring plasmid pKD46 (Datsenko & Wanner, 2000). The resulting strain was kept at 30°C and transformed with plasmid pCP20 to remove the kan cassette (Datsenko & Wanner, 2000).
To construct a strain specifying CigR‐HA, a cat cassette was introduced in the cigR gene as follows: A cat fragment was amplified from plasmid pKD3 using primers 13861/13862, then introduced into wild‐type 14028s, mgtC‐FLAG, and yqjA‐FLAG strains harboring plasmid pKD46. The resulting strains were kept at 30°C and transformed with pCP20 to remove the cat cassette.
To create the cigR promoter mutant strain, we introduced a tetRA cassette in the cigR promoter region as follows: The tetRA fragment was amplified from strain MS7953s using primers 14721/14722. Then, the PCR product was used to electroporate strain 14028s harboring plasmid pKD46. The resulting strain containing the tetRA cassette was kept at 30°C. Then, the tetRA cassette in the cigR promoter region was replaced by annealed oligonucleotides (14723/14724). These oligonucleotides were used to electroporate strain pcigR::tetRA harboring pKD46, and the bacterial suspension was plated on media containing fusaric acid and incubated at 42°C to select against the tetRA genes (Maloy & Nunn, 1981).
Plasmids expressing CigR and CigR derivatives were constructed as follows: The cigR gene was amplified using primers 15088/15089 for cigR and 15088/14718 for cigR‐FLAG and then introduced between the BamHI and HindIII sites of plasmid pUHE21‐2lacI q (Soncini et al, 1995).
Western blot analysis
Cells were grown in N‐minimal medium containing 10 μM or 10 mM Mg2+. Crude extracts were prepared in B‐PER reagent (Pierce) with 100 μg/ml lysozyme and EDTA‐free protease inhibitor (Roche). Samples were loaded onto 4–12% NuPAGE gels (Life Technologies). Then, samples were analyzed by Western blotting using anti‐HA or anti‐FLAG antibodies at 1:2,000 dilution. Mouse anti‐GroEL or anti‐DnaK antibodies (Abcam) at 1:5,000 dilution were used as control. Secondary horseradish peroxidase‐conjugated anti‐rabbit or anti‐mouse antisera (GE healthcare) were used at 1:5,000 dilution. The blots were developed with the Amersham ECL Western Blotting Detection Reagents (GE Healthcare) or SuperSignal West Femto Chemiluminescent system (Pierce).
Pull‐down assay with proteins synthesized using an in vitro transcription/translation system
Proteins were produced using the cell‐free PURExpress in vitro protein synthesis system (NEB) in the presence of 60 μg/ml proteoliposomes at 37°C for 2 h. Proteoliposomes were prepared using soybean L‐α‐phosphatidylcholine (Sigma) in buffer (20 mM Tricine, 20 mM succinic acid, 80 mM NaCl, and 0.6 mM KOH, adjusted to pH 8.0) to a concentration of 32 mg/ml as described (Kuruma et al, 2012). DNA templates for mgtC‐FLAG, cigR‐HA, atpB‐HA, and cigR‐HA domain mutants were used to synthesize the corresponding proteins according to the manufacturer's instructions. DNA templates were made with primers 14057/14058 for mgtC‐FLAG, 14300/14301 for atpB‐HA, and 14065/14066 for cigR‐HA. Each protein was synthesized separately. At the end of the reaction, samples were diluted in TBS (Tris‐buffered saline) buffer 20‐fold. Diluted samples were mixed in 500 μl TBS and incubated at 4°C for 2 h. Then, samples were immunoprecipitated with antibodies directed to the HA or FLAG epitopes at 4°C for 2 h and analyzed by Western blotting using the same antibodies.
In vivo pull‐down assay
The interaction between the MgtC and CigR proteins was investigated using strain JY92, which expresses the FLAG‐tagged mgtC gene and HA‐tagged cigR gene from the normal chromosomal locations. Cells were grown overnight in N‐minimal media containing 10 mM Mg2+. One milliliter of the overnight culture was washed in N‐minimal media without Mg2+ and resuspended in 1 ml of the same media. 1/100 dilution of bacteria was used to inoculate 25 ml of N‐minimal media containing 10 μM Mg2+ and grown for 5 h. Crude extracts were prepared as described above and incubated with anti‐HA magnetic beads (Pierce) or anti‐FLAG magnetic beads (Sigma) at 4°C overnight. After washing the beads, bound proteins were eluted in 100 μl SDS sample buffer and separated on 4–12% SDS–polyacrylamide gel and analyzed by Western blotting using antibodies directed to the HA or FLAG epitopes.
In vitro and in vivo competition assays
CigR‐HA, AtpB‐HA, and MgtC‐FLAG proteins were synthesized using the PURExpress in vitro synthesis system (Promega) per the manufacturer's protocol. To measure the concentrations of the CigR‐HA, AtpB‐HA, and MgtC‐FLAG proteins, known amounts of purified CigR‐HA, AtpB‐HA, and MgtC‐FLAG proteins were run on the same gel and used as standards. Standard curves calculated from purified proteins in the same blots used for calculation of molecular concentrations and binding affinities of the CigR‐HA, AtpB‐HA, and MgtC‐FLAG proteins. All proteins were dissolved in 500 μl TBS and incubated at room temperature for 2 h. Then, samples were pulled down with antibodies directed to the HA or FLAG epitopes at room temperature for 2 h. Proteins were then electrotransferred onto nitrocellulose membrane (iBlot; Life Technologies) following the manufacturer's protocol, detected by immunoblotting using monoclonal antibodies directed to the HA or FLAG epitopes, and a secondary antibody, horseradish peroxidase‐conjugated anti‐mouse IgG fragment (GE). All proteins were visualized by the Supersignal West Femto Chemiluminescent Substrate (Thermo Scientific) and LAS‐4000 (FujiFilm). K m and IC50 were calculated using Prism (Graphpad, ver. 7). The densities of protein bands were determined by quantification using the ImageJ program (NIH, ver. 1.49u). The amounts of CigR‐HA, AtpB‐HA, and MgtC‐FLAG proteins were then calculated from the standard curve derived from serial dilutions of purified protein standards run on the same gel (see Fig EV3A and F). For the in vivo competition assay, bacteria were grown in N‐minimal media pH 7.7 containing 10 μM MgCl2 and IPTG (0.01, 0.1, and 1 mM) for 6 h. Samples were analyzed immunoblotting using antibodies directed to the HA or FLAG epitopes.
Intramacrophage survival assay
The murine‐derived macrophage cell line J774A.1 was cultured in Dulbecco's modified Eagle's medium (DMEM; Life Technologies) supplemented with 10% FBS (Life Technologies) at 37°C under 5% CO2. Confluent monolayers were prepared in 24‐well tissue culture plates. Each well of a 24‐well plate was seeded with 5 × 105 cells suspended in DMEM/10% FBS and incubated at 37°C under 5% CO2 for 20 h. Bacteria were grown in Luria‐Bertani (LB) media at 37°C for 16 h. Bacterial cells were washed two times with PBS, suspended in pre‐warmed DMEM, and then added to the cell monolayer at a multiplicity of infection (MOI) of 10. Following 30‐min incubation, the wells were washed three times with pre‐warmed Dulbecco's phosphate‐buffered saline (DPBS; Life Technologies) to get rid of extracellular bacteria and then incubated with pre‐warmed medium supplemented with 120 μg/ml gentamicin for 1 h to kill the remaining extracellular bacteria. Then, the wells were washed three times with DPBS incubated with pre‐warmed medium supplemented with 10 μg/ml gentamicin. Bacteria were collected at 5 and 20 h post‐infection. Wells were washed two times with DPBS to remove extracellular bacteria. Then, 1 ml DPBS and 0.1% Triton X‐100 was added to the wells, and following a 10‐min incubation, serial dilutions of bacteria were plated on LB agar plates and incubated at 37°C overnight to determine the number of colony forming units.
Measurement of ATP levels
Adenosine triphosphate levels were measured using a microplate reader (Tecan, Infinite M1000 PRO) with modification of a described protocol (Pontes et al, 2015). Briefly, bacteria were grown in N‐minimal media containing 10 mM Mg2+ overnight. One milliliter of the overnight culture was washed three times in N‐minimal medium without Mg2+ and resuspended in 1 ml of the same media. Diluted (1/50) bacteria were inoculated in 1 ml of N‐minimal media containing 10 μM Mg2+ and grown for 4 h. Cells were normalized by OD600 and heated at 70°C for 10 min. Intracellular ATP was measured using the BacTiter‐Glo Microbial Cell Viability Assay Kit (Promega) according to the manufacturer's instructions. ATP levels (picomoles per milliliter of cells at given OD600) were obtained using as reference standards of known concentration.
Measurement of ATP hydrolysis
Inverted vesicles were prepared as described (Suzuki et al, 2007). The ATP hydrolysis reaction was started by adding 1 mM ATP and phosphate release monitored at absorbance 360 nm using EnzChek Phosphate Assay Kit (Life Technologies) according to the manufacturer's instructions. Average of hydrolysis rates in a time period from 1 to 5 min after initiation was calculated and presented as amount of phosphate (μmol) released per min. As a control, ATP was omitted from the reaction.
Measurement of Salmonella pH when inside macrophages
pH was measured with green fluorescent protein as an indicator (Kneen et al, 1998; Olsen et al, 2002; Wilks & Slonczewski, 2007). Bacteria harboring a plasmid containing the gfp gene expressed from a vector promoter (pFPV25.1) were grown in N‐minimal media pH 7.7 containing 10 μM MgCl2 for 5 h. Cells were normalized by OD600 values and resuspended in 150 μl of PBS in a 96‐well black microplate (PerkinElmer). Excitation spectra were measured at 30°C in 500 nm (slit width, 10 nm), using an emission wavelength of 545 nm (slit width, 10 nm) by a microplate reader (Tecan, Infinite M1000 PRO). To measure Salmonella's pH when inside J774A.1 macrophages, cells were seeded in 96‐well black microplates (PerkinElmer) in Dulbecco's modified Eagle's medium (Life Technologies) supplemented with 10% heat‐inactivated fetal bovine serum at a density of 5 × 105 per well 24 h prior to infection with Salmonella harboring plasmid pFPV25.1. Bacteria were grown overnight in LB broth at 37°C, washed with PBS, and used to infect macrophages at a multiplicity of infection of 50:1. At 6 h post‐infection, infected macrophages were resuspended in 150 μl of PBS with 20 mM sodium benzoate for standard curve. A standard curve was determined for green fluorescent protein by measuring fluorescence of samples resuspended in the same buffer at pH 4.8, 5.8, 6.5, 7.5, or 7.7 with addition of 20 mM sodium benzoate, an acid that equilibrates cytoplasmic pH with external pH (Wilks & Slonczewski, 2007). After measuring the spectra, infected macrophages were lysed by adding 0.1% Triton X‐100 and serial dilutions of bacteria were plated on LB agar plates and incubated at 37°C overnight.
Quantitative RT–PCR
To measure mRNA amounts, bacteria were grown in N‐minimal medium containing 10 μM or 10 mM Mg2+ for 4 h. Total RNA was purified by using RNeasy Kit (Qiagen) with on‐column DNase treatment, and cDNA was synthesized by using VILO Super Mix (Life Technologies). Quantification of transcripts was carried out by qRT–PCR using SYBR Green PCR Master Mix (Applied Biosystems) in an ABI 7500 Sequence Detection System (Applied Biosystems). The relative amount of mRNA was determined using a standard curve obtained by PCR with serially diluted genomic DNA, and results were normalized to the amounts of the rpoD gene. The mRNA amounts of the mgtC, cigR, and rpoD genes were measured using the following primer pairs (mgtC, 7530/7531; mgtB, 7763/7764; cigR, 15171/15172; and rpoD, 4149/4150). Data shown are an average from at least two independent experiments.
RNA polymerase ChIP‐sequencing
Wild‐type Salmonella (14028s) was grown overnight in N‐minimal medium (pH 7.7) supplemented with 10 mM MgCl2. Cultures were washed three times with N‐minimal medium without MgCl2 and resuspended in a volume to give the same initial optical density (OD600). Samples were diluted 1:50 in N medium (pH 7.7) containing 10 or 50 μM MgCl2 and grown in a shaking water bath at 250 rpms and 37°C for 4 h (OD600 ≈ 0.34–0.4). ChIP assays were performed as described (Shin & Groisman, 2005; Pontes & Groisman, 2018), with the following modifications. Following formaldehyde cross‐linking, samples were quenched with 220 mM glycine. Immunoprecipitation was performed with MagnaChip Protein A/G Magnetic Beads (Millipore) complexed with a monoclonal antibody directed against RpoC, the β’ subunit of the E. coli RNA polymerase (RNAP; NeoClone). Purified immunoprecipitated DNA samples were submitted for library construction and high‐throughput sequencing at the Yale Center for Genomic Analysis. Assembly of sequenced fragments was performed using CLC Genomics Workbench software.
Reverse transcriptase–PCR
An overnight culture of wild‐type Salmonella was diluted 1:50 into 1 ml of N‐minimal medium (pH 7.7) containing 10 μM, 50 μM, or 10 mM MgCl2 and grown at 37°C for 4 h. Total RNA was isolated using the RNeasy Kit (Qiagen) with on‐column DNase treatment. Reverse transcription was performed with the ThermoScript™ RT–PCR System (Life Technologies) following the manufacturer's instructions. Primers 15106/15107 were used for amplification of mgtC_cigR, 15108/15109 for mgtB_cigR, 15110/15111 for cigR, and 15106/15112 for mgtC_mgtB. PCR was performed as follows: 5 min initial denaturation at 95°C, 27 repetitions of 30 s at 95°C, 0.5~5 min at 57°C, 30 s at 68°C, and a final amplification of 5 min at 68°C. To exclude the possibility of genomic DNA contamination contributing to the measured values, control reactions were performed without reverse transcriptase.
Amounts of Salmonella proteins inside macrophages
The murine‐derived macrophage‐like cell line J774A.1 was cultured in Dulbecco's modified Eagle's medium (DMEM; Life Technologies) supplemented with 10% FBS (Life Technologies) at 37°C under 5% CO2. Confluent monolayers for infection with bacteria were prepared in 12‐well tissue culture plates. Each well was seeded with 106 (12‐well plate) cells suspended in DMEM/10% FBS and incubated at 37°C under 5% CO2 for 5 or 20 h. Bacterial cells were washed two times with DPBS, suspended in pre‐warmed DMEM, and then added to the cell monolayer at a multiplicity of infection (MOI) of 10. After washing with DPBS, cells were lysed with 1 ml/well of cell lysis solution [0.1% (w/v) SDS, 1% (v/v) acidic phenol, and 19% (v/v)] ethanol in double distilled water for 30 min. The cell lysates from two plates were pooled and centrifuged at 5,000 × g for 20 min. Pellets were washed twice with DPBS and resuspended in 100 μl of 100 mM NH4 HCO3, pH 8.4. Recovered Salmonella were subsequently analyzed by Western blot (Shi et al, 2006).
Bacterial two‐hybrid analysis to examine protein–protein interactions
We used the BACTH system (Battesti & Bouveret, 2012) with the following constructs: the mgtC, cigR, pmrB, mgtC derivatives, and domain mutated cigR genes were PCR‐amplified, and the PCR fragments were cloned between the XbaI and KpnI sites of the pUT18/pUT18C vectors and pKT25 to generate genes specifying the corresponding fusion proteins. Recombinant plasmids carrying the mgtC and cigR genes were co‐transformed into strain BTH101. Transformants were plated on LB agar plates containing ampicillin (100 μg/ml) and kanamycin (50 μg/ml) and incubated at 30°C for 24 h. To quantify the interaction between hybrid proteins, bacteria were grown at 30°C overnight as recommended in the BACHT protocol (Battesti & Bouveret, 2012) in LB Amp Kan liquid medium supplemented with 0.5 mM IPTG. All samples were spotted onto LB agar plates or measured β‐galactosidase activity supplemented with ampicillin (100 μg/ml), kanamycin (50 μg/ml), X‐Gal (40 μg/ml), and IPTG (0.5 mM). Values of β‐galactosidase activity are normalized by absorbance (OD595).
Subcellular localization of the MgtC and CigR proteins
Wild‐type (14028s), MgtC‐N92T (EL551), CigR‐HA (JY6), and wild‐type harboring pUHE‐cigR‐FLAG, pUHE‐D3cigR‐FLAG, and pUHE‐W133AcigR‐FLAG were grown in N‐minimal medium, pH 7.7, with 10 mM MgCl2 overnight. Then, bacteria were washed three times with N‐minimal medium, pH 7.7, without MgCl2. Cells were diluted 1:100 in 25 ml of N‐minimal medium, pH 7.7, with 10 μM MgCl2, and grown at 37°C with/without 100 μM IPTG for 5 h. Cells were harvested by centrifugation at 5,000 × g for 10 min at 4°C. Cells were washed once with PBS and resuspended in 4 ml of PBS containing sucrose (20%) and lysozyme (100 μg/ml). The supernatant fraction was filtered using a 0.22‐μm PVDF filter and then concentrated by precipitation with tricloroacetic acid. After 30 min on ice, cells were disrupted by sonication or prench press. Cell debris was removed by centrifugation at 4,000 × g for 15 min, and the whole‐cell lysate was loaded on top of a sucrose gradient made with 4 ml each of 60 and 70% sucrose in a Beckman Ultra‐Clear centrifuge tube followed by centrifugation in an SW40 rotor at 250,000 × g at 4°C for 20 h. Bands between 20 and 60% (upper, reddish band) and between 60 and 70% (lower, white band) sucrose, corresponding to the inner and outer membranes, respectively, were collected and dialyzed against PBS. Protein concentrations were determined by a BCA method, with bovine serum albumin used as a standard protein. NADH oxidase activity was used as a marker for inner membrane purity. Inner and outer membrane preparations (10 μg of protein each) were run in 4–12% NuPAGE gel (Life Technologies), transferred onto a nitrocellulose membrane, and developed with an anti‐HA antibody (Sigma), an anti‐MgtC, an anti‐FLAG (Sigma), an anti‐rabbit immunoglobulin G horseradish peroxidase‐linked antibody (GE Healthcare), and the ECL detection system (Amersham Biosciences).
Determination of MgtC, CigR, and AtpB proteins amounts
An overnight culture of wild‐type (14028s) and cigR promoter mutant (JY372) Salmonella were grown in N‐minimal medium at pH 7.7 with 10 mM MgCl2, washed twice with N‐minimal medium containing no Mg2+, and used to inoculate 50 ml of N‐minimal medium containing 10 μM MgCl2 at a 1:50 dilution. 0.5 ml of each culture was taken at the indicated time points, except for the 2.5 h samples (when 1.5 ml was taken), and 100 μl B‐PER solution was added. Triton X‐100 (final 0.5%; Sigma) was added to the total cell lysates to solubilize the membrane, and the lysates were incubated at room temperature for 30 min. Known amounts of purified MgtC‐FLAG, CigR‐FLAG, and AtpB‐FLAG proteins were run on the same SDS–PAGE and used as standards.
For purification of the MgtC‐FLAG and CigR‐FLAG proteins, overnight cultures of EG16539 or wild‐type Salmonella (14028s) harboring pUHE‐CigR‐FLAG were used to inoculate N‐minimal medium at pH 7.7 with 10 mM MgCl2 media. Cells were grown at 37°C to logarithmic phase (OD600 ≈ 0.5), and the CigR protein was induced by addition of 0.2 mM of IPTG at 30°C for an additional 5 h. Cells were collected, washed once with 10 mM Tris–HCl (pH 8.0), and resuspended in a solution containing 20 mM Tris–HCl (pH 8.0), 20% sucrose, 5 mM M EDTA, 20 mM MgCl2 (final concentration), and 150 μg/ml lysozyme. After a 30‐min incubation at 4°C, cells were centrifuged at 18,000 × g at 4°C for 20 min. Cells were resuspended in 10 mM Tris–HCl (pH 8.0) and 10 mM MgCl2, subjected to sonication, and membranes were collected by centrifugation for 1 h at 25,000 × g at 4°C. Isolated membranes were solubilized in 1× binding buffer [50 mM Tris–HCl, pH 8.0, 150 mM NaCl, 1 mM EDTA, and 0.1% n‐dodecyl α‐D‐maltoside (DM; Sigma)] on ice for 1 h. Solubilized proteins were recovered by centrifugation at 25,000 × g at 4°C for 1 h and applied to FLAG® M Purification Kit (Sigma) per the manufacturer's protocol. Finally, the eluate was exchanged with TKM buffer [10 mM KCl, 10 mM Tris–Cl (pH 7.5), 1 mM MgCl2], followed by TKM buffer containing 50% glycerol, and concentrated using Amicon Ultra‐3 (MW 3,000; Millipore). For purification of the AtpB‐FLAG protein, the AtpB‐FLAG was produced from a DNA template (primers 15208/15209) using the PURExpress system (Promega) per the manufacturer's protocol. Purification steps were the same for the MgtC‐FLAG and CigR‐FLAG proteins.
Proteins were electrotransferred onto a nitrocellulose membrane (iBlot; Life Technologies) following the manufacturer's protocol, detected by immunoblotting using antiserum against MgtC or CigR and the secondary antibody, horseradish peroxidase‐conjugated anti‐rabbit IgG fragment (GE). Both MgtC and CigR proteins were visualized by the Supersignal West Femto Chemiluminescent Substrate (Thermo Scientific) using LAS‐4000 (FujiFilm). The densities of protein bands were determined by quantification with ImageJ program (NIH, ver. 1.49u). The amounts of MgtC, CigR, and AtpB proteins were then calculated from the standard curve derived from a serial dilution of the purified MgtC‐FLAG, CigR‐FLAG, and AtpB‐FLAG protein standards run on the same gel (see Figs 4E and EV5D).
Macrophage cell death assay
The percentage of macrophage death was determined by measuring the release of host cytoplasmic lactate dehydrogenase (LDH). At 20 h after infection, the supernatants were collected, and the release of LDH was quantified by using the Cytotoxicity Detection Kit (Roche). The absorbance at 490 nm was measured by using multidetector (Tecan, Infinite M1000 PRO), and the percentage of host cell death was calculated as [(experimental release – spontaneous release)/(maximum release − spontaneous release)] × 100. The spontaneous release is the amount of LDH released from the cytoplasm of uninfected macrophages, whereas the maximum release is the amount released by total lysis of uninfected macrophages by 2% Triton X‐100.
Author contributions
JY and EAG designed the experiments. JY performed the majority of the experiments. MHP performed the RNA polymerase ChIP‐sequencing experiment. JC performed the in vivo co‐immunoprecipitation experiment. JY and EAG analyzed the data, and JY and EAG discussed the data. JY and EAG wrote the manuscript, and MHP commented on the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Expanded View
Acknowledgements
This research was supported by NIH Grant AI49561 to EAG.
The EMBO Journal (2018) 37: e96977
References
- Alix E, Blanc‐Potard A‐B (2008) Peptide‐assisted degradation of the Salmonella MgtC virulence factor. EMBO J 27: 546–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alpuche Aranda CM, Swanson JA, Loomis WP, Miller SI (1992) Salmonella typhimurium activates virulence gene transcription within acidified macrophage phagosomes. Proc Natl Acad Sci USA 89: 10079–10083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battesti AL, Bouveret E (2012) The bacterial two‐hybrid system based on adenylate cyclase reconstitution in Escherichia coli . Methods 58: 325–334 [DOI] [PubMed] [Google Scholar]
- Belon C, Gannoun‐Zaki L, Lutfalla G, Kremer L, Blanc‐Potard A‐B (2014) Mycobacterium marinum MgtC plays a role in phagocytosis but is dispensable for intracellular multiplication. PLoS One 9: e116052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijlsma JJE, Groisman EA (2005) The PhoP/PhoQ system controls the intramacrophage type three secretion system of Salmonella enterica . Mol Microbiol 57: 85–96 [DOI] [PubMed] [Google Scholar]
- Blanc‐Potard AB, Groisman EA (1997) The Salmonella selC locus contains a pathogenicity island mediating intramacrophage survival. EMBO J 16: 5376–5385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc‐Potard AB, Solomon F, Kayser J, Groisman EA (1999) The SPI‐3 pathogenicity island of Salmonella enterica . J Bacteriol 181: 998–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogan AA, Thorn KS (1998) Anatomy of hot spots in protein interfaces. J Mol Biol 280: 1–9 [DOI] [PubMed] [Google Scholar]
- Brown NA, Urban M, Hammond‐Kosack KE (2016) The trans‐kingdom identification of negative regulators of pathogen hypervirulence. FEMS Microbiol Rev 40: 19–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchmeier NA, Heffron F (1991) Inhibition of macrophage phagosome‐lysosome fusion by Salmonella typhimurium . Infect Immun 59: 2232–2238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamnongpol S, Groisman EA (2002) Mg2+ homeostasis and avoidance of metal toxicity. Mol Microbiol 44: 561–571 [DOI] [PubMed] [Google Scholar]
- Choi J, Groisman EA (2013) The lipopolysaccharide modification regulator PmrA limits Salmonella virulence by repressing the type three‐secretion system Spi/Ssa. Proc Natl Acad Sci USA 110: 9499–9504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Groisman EA (2016) Acidic pH sensing in the bacterial cytoplasm is required for Salmonella virulence. Mol Microbiol 101: 1024–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coburn B, Grassl GA, Finlay BB (2007) Salmonella, the host and disease: a brief review. Immunol Cell Biol 85: 112–118 [DOI] [PubMed] [Google Scholar]
- Conlon BP, Rowe SE, Gandt AB, Nuxoll AS, Donegan NP, Zalis EA, Clair G, Adkins JN, Cheung AL, Lewis K (2016) Persister formation in Staphylococcus aureus is associated with ATP depletion. Nat Microbiol 1: 16051 [DOI] [PubMed] [Google Scholar]
- Cunningham ML, Titus RG, Turco SJ, Beverley SM (2001) Regulation of differentiation to the infective stage of the protozoan parasite Leishmania major by tetrahydrobiopterin. Science 292: 285–287 [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL (2000) One‐step inactivation of chromosomal genes in Escherichia coli K‐12 using PCR products. Proc Natl Acad Sci USA 97: 6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RW, Botstein D, Roth JR (1980) Advanced bacterial genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; [Google Scholar]
- Eriksson S, Lucchini S, Thompson A, Rhen M, Hinton JCD (2003) Unravelling the biology of macrophage infection by gene expression profiling of intracellular Salmonella enterica . Mol Microbiol 47: 103–118 [DOI] [PubMed] [Google Scholar]
- Fabrega A, Vila J (2013) Salmonella enterica Serovar Typhimurium skills to succeed in the host: virulence and regulation. Clin Microbiol Rev 26: 308–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields PI, Groisman EA, Heffron F (1989) A Salmonella locus that controls resistance to microbicidal proteins from phagocytic cells. Science 243: 1059–1062 [DOI] [PubMed] [Google Scholar]
- Fields PI, Swanson RV, Haidaris CG, Heffron F (1986) Mutants of Salmonella typhimurium that cannot survive within the macrophage are avirulent. Proc Natl Acad Sci USA 83: 5189–5193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes K, Christensen SK, Løbner‐Olesen A (2005) Prokaryotic toxin‐antitoxin stress response loci. Nat Rev Microbiol 3: 371–382 [DOI] [PubMed] [Google Scholar]
- Grabenstein JP, Fukuto HS, Palmer LE, Bliska JB (2006) Characterization of phagosome trafficking and identification of PhoP‐regulated genes important for survival of Yersinia pestis in macrophages. Infect Immun 74: 3727–3741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groisman EA, Chiao E, Lipps CJ, Heffron F (1989) Salmonella typhimurium phoP virulence gene is a transcriptional regulator. Proc Natl Acad Sci USA 86: 7077–7081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Lim KB, Gunn JS, Bainbridge B, Darveau RP, Hackett M, Miller SI (1997) Regulation of lipid A modifications by Salmonella typhimurium virulence genes phoP‐phoQ . Science 276: 250–253 [DOI] [PubMed] [Google Scholar]
- Hanahan D (1983) Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166: 557–580 [DOI] [PubMed] [Google Scholar]
- Ionescu M, Baccari C, Da Silva AM, Garcia A, Yokota K, Lindow SE (2013) Diffusible signal factor (DSF) synthase RpfF of Xylella fastidiosa is a multifunction protein also required for response to DSF. J Bacteriol 195: 5273–5284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones BD, Falkow S (1996) Salmonellosis: host immune responses and bacterial virulence determinants. Annu Rev Immunol 14: 533–561 [DOI] [PubMed] [Google Scholar]
- Karimova G, Pidoux J, Ullmann A, Ladant D (1998) A bacterial two‐hybrid system based on a reconstituted signal transduction pathway. Proc Natl Acad Sci USA 95: 5752–5756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidwai AS, Mushamiri I, Niemann GS, Brown RN, Adkins JN, Heffron F (2013) Diverse secreted effectors are required for Salmonella persistence in a mouse infection model. PLoS One 8: e70753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneen M, Farinas J, Li Y, Verkman AS (1998) Green fluorescent protein as a noninvasive intracellular pH indicator. Biophys J 74: 1591–1599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kröger C, Colgan A, Srikumar S, Händler K, Sivasankaran SK, Hammarlöf DL, Canals R, Grissom JE, Conway T, Hokamp K, Hinton JCD (2013) An infection‐relevant transcriptomic compendium for Salmonella enterica Serovar Typhimurium. Cell Host Microbe 14: 683–695 [DOI] [PubMed] [Google Scholar]
- Kuruma Y, Suzuki T, Ono S, Yoshida M, Ueda T (2012) Functional analysis of membranous Fo‐a subunit of F1Fo‐ATP synthase by in vitro protein synthesis. Biochem J 442: 631–638 [DOI] [PubMed] [Google Scholar]
- Lee EJ, Groisman EA (2010) An antisense RNA that governs the expression kinetics of a multifunctional virulence gene. Mol Microbiol 76: 1020–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EJ, Groisman EA (2012) Control of a Salmonella virulence locus by an ATP‐sensing leader messenger RNA. Nature 486: 271–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EJ, Pontes MH, Groisman EA (2013) A bacterial virulence protein promotes pathogenicity by inhibiting the bacterium's own F1Fo ATP synthase. Cell 154: 146–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloy SR, Nunn WD (1981) Selection for loss of tetracycline resistance by Escherichia coli . J Bacteriol 145: 1110–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus SL, Brumell JH, Pfeifer CG, Finlay BB (2000) Salmonella pathogenicity islands: big virulence in small packages. Microbes Infect 2: 145–156 [DOI] [PubMed] [Google Scholar]
- Miller SI, Kukral AM, Mekalanos JJ (1989) A two‐component regulatory system (phoP phoQ) controls Salmonella typhimurium virulence. Proc Natl Acad Sci USA 86: 5054–5058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouslim C, Hilbert F, Huang H, Groisman EA (2002) Conflicting needs for a Salmonella hypervirulence gene in host and non‐host environments. Mol Microbiol 45: 1019–1027 [DOI] [PubMed] [Google Scholar]
- Niemann GS, Brown RN, Gustin JK, Stufkens A, Shaikh‐Kidwai AS, Li J, McDermott JE, Brewer HM, Schepmoes A, Smith RD, Adkins JN, Heffron F (2011) Discovery of novel secreted virulence factors from Salmonella enterica Serovar Typhimurium by proteomic analysis of culture supernatants. Infect Immun 79: 33–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemann GS, Brown RN, Mushamiri IT, Nguyen NT, Taiwo R, Stufkens A, Smith RD, Adkins JN, McDermott JE, Heffron F (2013) RNA type III secretion signals that require Hfq. J Bacteriol 195: 2119–2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen KN, Budde BB, Siegumfeldt H, Rechinger KB, Jakobsen M, Ingmer H (2002) Noninvasive measurement of bacterial intracellular pH on a single‐cell level with green fluorescent protein and fluorescence ratio imaging microscopy. Appl Environ Microbiol 68: 4145–4147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhill J, Dougan G, James KD, Thomson NR, Pickard D, Wain J, Churcher C, Mungall KL, Bentley SD, Holden MT, Sebaihia M, Baker S, Basham D, Brooks K, Chillingworth T, Connerton P, Cronin A, Davis P, Davies RM, Dowd L et al (2001) Complete genome sequence of a multiple drug resistant Salmonella enterica serovar Typhi CT18. Nature 413: 848–852 [DOI] [PubMed] [Google Scholar]
- Pontes MH, Groisman EA (2018) Protein synthesis controls phosphate homeostasis. Genes Dev 32: 79–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontes MH, Lee EJ, Choi J, Groisman EA (2015) Salmonella promotes virulence by repressing cellulose production. Proc Natl Acad Sci USA 112: 5183–5188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontes MH, Yeom J, Groisman EA (2016) Reducing ribosome biosynthesis promotes translation during low Mg2+ stress. Mol Cell 64: 480–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prost LR, Daley ME, Le Sage V, Bader MW, Le Moual H, Klevit RE, Miller SI (2007) Activation of the bacterial sensor kinase PhoQ by acidic pH. Mol Cell 26: 165–174 [DOI] [PubMed] [Google Scholar]
- Rang C, Alix E, Felix C, Heitz A, Tasse L, Blanc‐Potard A‐B (2007) Dual role of the MgtC virulence factor in host and non‐host environments. Mol Microbiol 63: 605–622 [DOI] [PubMed] [Google Scholar]
- Rasmussen A, Rasmussen T, Edwards MD, Schauer D, Schumann U, Miller S, Booth IR (2007) The role of tryptophan residues in the function and stability of the mechanosensitive channel MscS from Escherichia coli . Biochemistry 46: 10899–10908 [DOI] [PubMed] [Google Scholar]
- Retamal P, Castillo‐Ruiz M, Mora GC (2009) Characterization of MgtC, a virulence factor of Salmonella enterica Serovar Typhi. PLoS One 4: e5551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Takaya A, Yamamoto T (2011) Meta‐analytic approach to the accurate prediction of secreted virulence effectors in gram‐negative bacteria. BMC Bioinformatics 12: 442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher MA, Balani P, Min J, Chinnam NB, Hansen S, Vulić M, Lewis K, Brennan RG (2015) HipBA‐promoter structures reveal the basis of heritable multidrug tolerance. Nature 524: 59–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senior AE (1990) The proton‐translocating ATPase of Escherichia coli . Annu Rev Biophys Chem 19: 7–41 [DOI] [PubMed] [Google Scholar]
- Shan Y, Brown Gandt A, Rowe SE, Deisinger JP, Conlon BP, Lewis K (2017) ATP‐dependent persister formation in Escherichia coli . mBio 8: e02267–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Adkins JN, Coleman JR, Schepmoes AA, Dohnkova A, Mottaz HM, Norbeck AD, Purvine SO, Manes NP, Smallwood HS, Wang H, Forbes J, Gros P, Uzzau S, Rodland KD, Heffron F, Smith RD, Squier TC (2006) Proteomic analysis of Salmonella enterica serovar typhimurium isolated from RAW 264.7 macrophages: identification of a novel protein that contributes to the replication of serovar typhimurium inside macrophages. J Biol Chem 281: 29131–29140 [DOI] [PubMed] [Google Scholar]
- Shi Y, Cromie MJ, Hsu F‐F, Turk J, Groisman EA (2004) PhoP‐regulated Salmonella resistance to the antimicrobial peptides magainin 2 and polymyxin B. Mol Microbiol 53: 229–241 [DOI] [PubMed] [Google Scholar]
- Shimono N, Morici L, Casali N, Cantrell S, Sidders B, Ehrt S, Riley LW (2003) Hypervirulent mutant of Mycobacterium tuberculosis resulting from disruption of the mce1 operon. Proc Natl Acad Sci USA 100: 15918–15923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin D, Groisman EA (2005) Signal‐dependent binding of the response regulators PhoP and PmrA to their target promoters in vivo . J Biol Chem 280: 4089–4094 [DOI] [PubMed] [Google Scholar]
- Snavely MD, Miller CG, Maguire ME (1991) The mgtB Mg2+ transport locus of Salmonella typhimurium encodes a P‐type ATPase. J Biol Chem 266: 815–823 [PubMed] [Google Scholar]
- Soncini FC, Garcia Vescovi E, Solomon F, Groisman EA (1996) Molecular basis of the magnesium deprivation response in Salmonella typhimurium: identification of PhoP‐regulated genes. J Bacteriol 178: 5092–5099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soncini FC, Vescovi EG, Groisman EA (1995) Transcriptional autoregulation of the Salmonella typhimurium phoPQ operon. J Bacteriol 177: 4364–4371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sory MP, Boland A, Lambermont I, Cornelis GR (1995) Identification of the YopE and YopH domains required for secretion and internalization into the cytosol of macrophages, using the cyaA gene fusion approach. Proc Natl Acad Sci USA 92: 11998–12002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Ozaki Y, Sone N, Feniouk BA, Yoshida M (2007) The product of uncI gene in F1Fo‐ATP synthase operon plays a chaperone‐like role to assist c‐ring assembly. Proc Natl Acad Sci USA 104: 20776–20781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdivia RH, Falkow S (1996) Bacterial genetics by flow cytometry: rapid isolation of Salmonella typhimurium acid‐inducible promoters by differential fluorescence induction. Mol Microbiol 22: 367–378 [DOI] [PubMed] [Google Scholar]
- Wilks JC, Slonczewski JL (2007) pH of the cytoplasm and periplasm of Escherichia coli: rapid measurement by green fluorescent protein fluorimetry. J Bacteriol 189: 5601–5607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wösten MM, Kox LF, Chamnongpol S, Soncini FC, Groisman EA (2000) A signal transduction system that responds to extracellular iron. Cell 103: 113–125 [DOI] [PubMed] [Google Scholar]
- Yeom J, Wayne KJ, Groisman EA (2017) Sequestration from protease adatpor confers differential stability to N‐end rule substrate. Mol Cell 66: 234–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J, Xia J, Tao M, Xu L, Li Q, Geng S, Jiao X (2016) Construction and characterization of a cigR deletion mutant of Salmonella enterica serovar Pullorum. Avian Pathol 45: 569–575 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Expanded View