

Abstract
Family with sequence similarity 20C (Fam20C), the physiological Golgi casein kinase, phosphorylates numerous secreted proteins that are involved in a wide variety of biological processes. However, the role of Fam20C in regulating proteins in the endoplasmic reticulum (ER) lumen is largely unknown. Here, we report that Fam20C interacts with various luminal proteins and that its depletion results in a more reduced ER lumen. We further show that ER oxidoreductin 1α (Ero1α), the pivotal sulfhydryl oxidase that catalyzes disulfide formation in the ER, is phosphorylated by Fam20C in the Golgi apparatus and retrograde‐transported to the ER mediated by ERp44. The phosphorylation of Ser145 greatly enhances Ero1α oxidase activity and is critical for maintaining ER redox homeostasis and promoting oxidative protein folding. Notably, phosphorylation of Ero1α is induced under hypoxia, reductive stress, and secretion‐demanding conditions such as mammalian lactation. Collectively, our findings open a door to uncover how oxidative protein folding is regulated by phosphorylation in the secretory pathway.
Keywords: ER redox, Ero1α, Fam20C, oxidative protein folding, phosphorylation
Subject Categories: Membrane & Intracellular Transport; Post-translational Modifications, Proteolysis & Proteomics
Introduction
Protein phosphorylation regulates almost every aspect of cell life, including the cell cycle, growth, differentiation, migration, and death, due to its simplicity, flexibility, and reversibility (Cohen, 2002). It is estimated that around 30% of human proteins are phosphoproteins. The first phosphoprotein, casein, was reported in 1883 (Hammarsten, 1883) and has been widely used as a model substrate to evaluate protein kinase activity, yet family with sequence similarity 20C (Fam20C), the bona fide kinase catalyzing the phosphorylation of secreted protein casein, was not identified until 2012 (Ishikawa et al, 2012; Tagliabracci et al, 2012). Fam20C is a unique protein kinase that phosphorylates proteins within the S‐x‐E/pS motif, preferentially uses Mn2+ over Mg2+ as a metal ion cofactor and displays insensitivity to the broad‐spectrum protein kinase inhibitor staurosporine (Tagliabracci et al, 2012).
Loss‐of‐function mutations in Fam20C cause a rare and often lethal osteosclerotic bone dysplasia called Raine syndrome (Raine et al, 1989). Indeed, Fam20C has been found to phosphorylate proteins important to biomineralization, including osteopontin, dentin matrix protein‐1, and dentin sialophosphoprotein (Ishikawa et al, 2012; Tagliabracci et al, 2012). Recently, Dixon laboratory reported that Fam20C is the kinase responsible for generating the majority of the secreted phosphoproteome (more than 100 secreted proteins) implicated in a broad‐spectrum of biological processes, such as wound healing, regeneration, and cell migration (Tagliabracci et al, 2015). This implies that Fam20C participates in a wide variety of regulations in human physiology and disease. However, little is known about the role of protein phosphorylation in the endoplasmic reticulum (ER) lumen.
More than 30% of the proteins encoded by the human genome are predicted to enter the secretory pathway (Fass & Thorpe, 2018). Most secreted and membrane proteins (e.g., cytokines, immunoglobulins, and receptors) contain disulfide bonds that stabilize their structures and regulates their functions (Hogg, 2003). The folding of nascent peptides accompanied by disulfide bond formation is called oxidative protein folding. The ER is the central organelle for secreted and membrane protein synthesis, folding, and assembly. The pivotal pathway for catalyzing oxidative protein folding in the ER is composed of sulfhydryl oxidase ER oxidoreductin 1α (Ero1α) and protein disulfide isomerase (PDI) (Wang et al, 2015; Fass & Thorpe, 2018). Ero1α utilizes oxygen as the final electron acceptor for de novo disulfide bond formation and transfers disulfides to PDI, which directly catalyzes disulfide formation in reduced substrates (Mezghrani et al, 2001; Tu & Weissman, 2002; Sevier & Kaiser, 2008). Ero1α is upregulated under hypoxic conditions (May et al, 2005), highly expressed in many cancers (Battle et al, 2013; Kukita et al, 2015), and involved in calcium flux and cell apoptosis (Li et al, 2009; Chin et al, 2011).
The byproduct of Ero1α oxidase activity is hydrogen peroxide (H2O2) (Gross et al, 2006; Wang et al, 2009), which is estimated to account for up to 25% of the cellular reactive oxygen species (ROS) produced during protein synthesis (Tu & Weissman, 2004). Therefore, Ero1α oxidase activity must be tightly controlled to prevent excess ROS accumulation in resting states and it needs to be activated promptly once robust oxidative protein folding capacity is required. The activity of Ero1α is feedback regulated by PDI and its homologues to maintain redox balance in the ER (Sevier et al, 2007; Kim et al, 2012; Zhang et al, 2014). Excessive reduction of PDI in the ER induces Ero1α to undergo regulatory disulfides breakage. Conversely, if the ER is excessively oxidized, the regulatory disulfides will be formed by oxidized PDI to suppress Ero1α activity (Wang et al, 2015; Delaunay‐Moisan et al, 2017). Besides this “regulatory disulfide switch”, whether any other post‐translational modification can regulate Ero1α activity remains unclear.
Here, we report that Fam20C interacts with a wide range of proteins localized within the secretory pathway and that its deletion disrupts the ER redox poise. We further show that Fam20C phosphorylates Ero1α at Ser145 in the Golgi apparatus and that ERp44/KDEL receptor (KDELR) mediates the retrograde transport of phosphorylated Ero1α to the ER. Importantly, phosphorylated Ero1α displays enhanced oxidase activity and promotes oxidative protein folding. Phosphorylation of Ero1α at Ser145 by Fam20C is critical for maintaining ER redox homeostasis, and it is induced under hypoxia, reductive stress, and physiological conditions with increased secretory demands such as mammalian lactation. Our study expands Fam20C's biological roles in the ER lumen and establishes the first link between secretory protein phosphorylation and oxidative protein folding.
Results
Fam20C kinase activity is linked to ER redox homeostasis
To explore the role of secretory kinase Fam20C in the lumen of the secretory pathway, we first identified the Fam20C interactome by co‐immunoprecipitation and mass spectrum (MS) analysis (Fig 1A). We identified a total of 349 proteins localized in the ER and Golgi apparatus that specifically interact with Fam20C (Fig 1B), involved in protein processing in the ER, N‐glycan biosynthesis, fatty acid metabolism etc., as analyzed by DAVID Gene Ontology (Fig 1C). Notably, enzymes responsible for oxidative protein folding were enriched in the Fam20C interactome, including PDI family proteins (PDI, ERp57, P5, ERp44, and TMX3) and the ER sulfhydryl oxidase Ero1α (Dataset EV1). Since ER lumen is the compartment for secretory protein folding and its oxidizing environment is critical for protein disulfide formation, we further studied whether Fam20C could affect ER redox homeostasis.
Figure 1. FAM20C KO triggers a reduced ER.
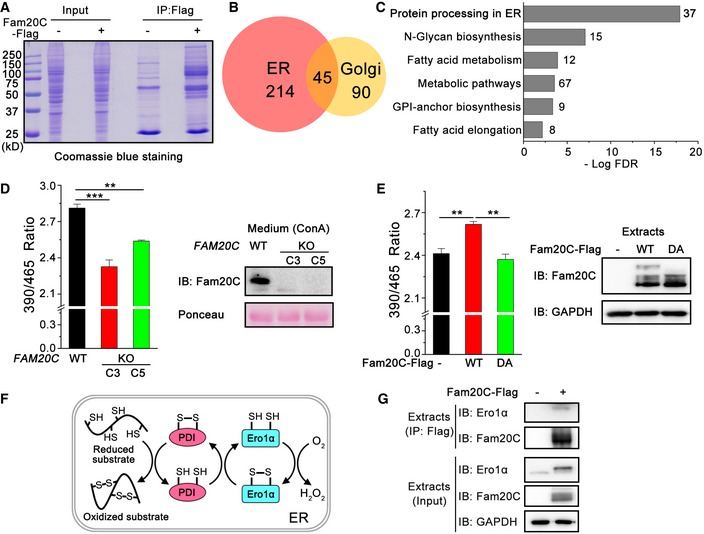
- Coomassie Blue Staining of Flag immunoprecipitates from HeLa cells expressing Fam20C‐Flag for MS analysis.
- Venn diagram of the 349 proteins interacting with Fam20C identified in the ER and Golgi apparatus based on DAVID GO term analysis.
- KEGG pathway mapping of the most enriched biological processes in the Fam20C interactome in the ER and Golgi. The graph represents the top six statistically significant enriched gene clusters ordered by FDR (false discovery rate), with the number of genes in each cluster indicated beside the bars.
- (Left) Fluorescence intensities of superfolded‐roGFP‐iEER in wild‐type (WT) and two clones (C3 and C5) of FAM20C KO HeLa cells at 525 nm were measured with excitation at 390 and 465 nm. The fluorescence ratio at 390/465 nm excitation was calculated. Data are shown as mean ± SEM from five (WT and C3) or three (C5) independent experiments performed in six technical replicates. **P < 0.01, ***P < 0.001 (one‐way ANOVA, the post hoc Tukey's HSD test). (Right) Protein immunoblotting of Concanavalin A (ConA) precipitates from the culture medium of WT and FAM20C KO HeLa cells. Ponceau staining is shown as a loading control.
- (Left) Fluorescence intensities of superfolded‐roGFP‐iEER in FAM20C KO HeLa cells expressing Fam20C WT or its inactive mutant D478A (DA) were measured as in (D). Data are shown as mean ± SEM from four independent experiments performed in six technical replicates. **P < 0.01 (one‐way ANOVA, the post hoc Tukey's HSD test). (Right) Protein immunoblotting of FAM20C KO HeLa cells expressing Fam20C WT or DA.
- Schematic representation of the pivotal pathway constituted by Ero1α and PDI for oxidative protein folding in the mammalian ER.
- Co‐immunoprecipitation of endogenous Ero1α and Flag‐tagged Fam20C in HeLa cells.
Source data are available online for this figure.
Using the CRISPR/Cas9 technology, we obtained two clones of FAM20C knockout (KO) HeLa cells, which harbor frameshift mutations resulting in premature stop codons (Fig EV1A). As a glycosylated protein, Fam20C was detected in Concanavalin A (ConA) precipitates from the conditioned medium of unedited HeLa cells but not of the FAM20C KO cells, by immunoblotting with a polyclonal antibody against the C‐terminal peptide of human Fam20C (Fig EV1B). The depletion of FAM20C showed little effect on thapsigargin (Tg)‐induced unfolded protein response (UPR) signaling, suggesting that FAM20C KO does not trigger broad ER stress (Fig EV1C).
Figure EV1. Generation of FAM20C KO HeLa cells using CRISPR/Cas9 genome editing.
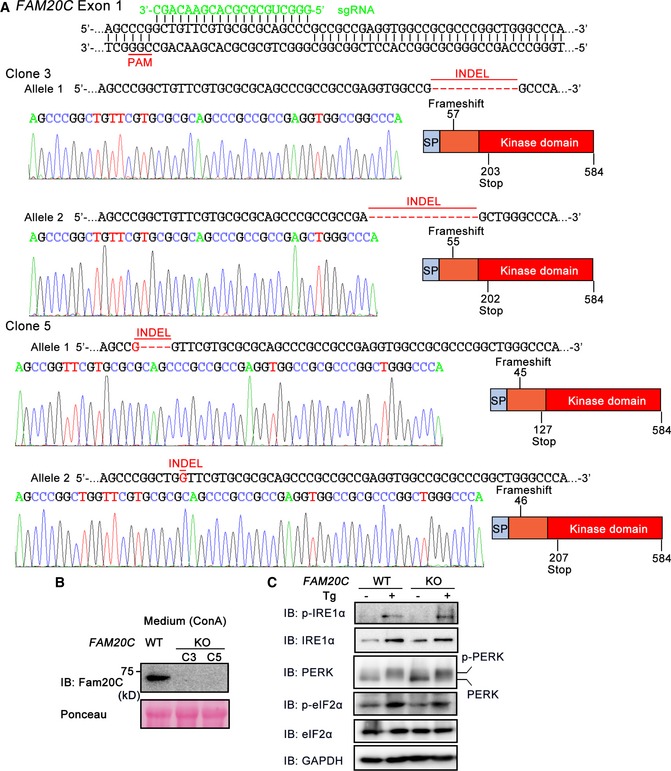
- Schematic representation of the base pairing between the guide RNA (sgRNA) and the targeting locus of exon 1 in the human FAM20C gene. The sequences of the mutated alleles in FAM20C clone 3 (C3) and clone 5 (C5) and representative chromatograms depicting the insertions/deletions (INDELs) are shown. The INDELs were predicted to cause frameshift mutations producing inactive copies of the protein. SP, signal peptide.
- Protein immunoblotting of Concanavalin A‐Sepharose (ConA) precipitates from the culture medium of WT and FAM20C KO HeLa cells. Ponceau staining was used as a loading control.
- FAM20C KO shows little effect on unfolded protein response signaling. Protein immunoblotting of cell extracts from WT and FAM20C KO HeLa cells treated with or without 5 μM Tg for 6 h.
To monitor the ER redox, we made a superfolded ER‐localized redox‐sensitive green fluorescent protein (superfolded‐roGFP‐iEER) referring to previous studies (Birk et al, 2013; Hoseki et al, 2016). The ratio of fluorescence intensity at 390/465 nm excitation of the superfolded‐roGFP‐iEER increased under oxidizing conditions and decreased under reducing conditions (Fig EV2). Interestingly, we observed a more reduced ER in the two clones of FAM20C KO cells (Fig 1D). Replenishment with Fam20C wild‐type (WT) reverted the ER to a state of oxidation, but the catalytically inactive Fam20C D478A (DA) did not (Fig 1E). Among all the ER oxidoreductases identified in the Fam20C interactome, Ero1α is the pivotal oxidase catalyzing de novo disulfide formation, which uses molecular oxygen to oxidize PDI and downstream substrates (Fig 1F). We therefore focused on the interaction between Fam20C and Ero1α. As shown in Fig 1G, endogenous Ero1α was indeed immunoprecipitated with Fam20C. Taken together, these results suggest that the kinase activity of Fam20C is linked to the regulation of ER redox homeostasis.
Figure EV2. Redox response ability of the superfolded‐roGFP‐iEER in HeLa cells.
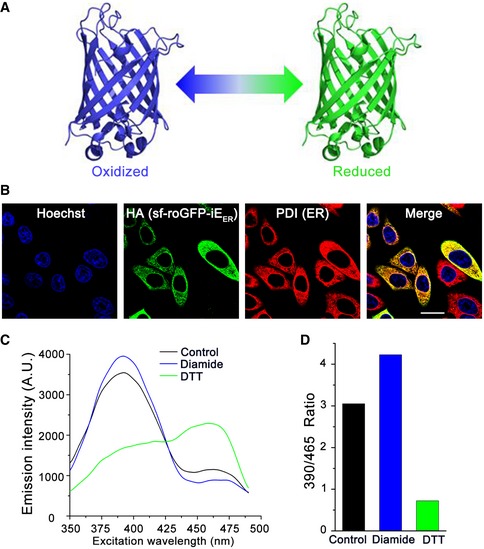
- Schematic representation of superfolded‐roGFP‐iEER exhibiting different fluorescence characteristics under oxidizing (blue) or reducing (green) condition.
- Localization of superfolded‐roGFP‐iEER in HeLa cells analyzed by immunofluorescence. Scale bar = 20 μm.
- Fluorescence excitation spectrum of superfolded‐roGFP‐iEER in HeLa cells untreated or treated with 1 mM diamide or 1 mM DTT.
- The ratios of fluorescence intensities at 525 nm with excitation at 390 and 465 nm were calculated from (C).
Ero1α is phosphorylated at Ser145
To determine whether Ero1α is a phosphoprotein and to identify putative phosphorylation sites, HA‐tagged human Ero1α was expressed in HeLa cells and purified by HA immunoprecipitation (Fig EV3A). Purified Ero1α was digested with trypsin and subjected to liquid chromatography (LC)‐MS/MS. MS analysis yielded 95.7% sequence coverage and identified a single phosphopeptide 137LGAVDESLpSEETQK150 (Fig 2A and B). Especially, the phosphate group‐containing y6 + ion was captured in the MS/MS spectrum (Fig 2A), strongly indicating that Ser145 is a genuine phosphorylation site in Ero1α. Ser145 is C‐terminal to the outer active site‐containing flexible loop (Fig 2C) and highly conserved in higher eukaryotes (Fig 2D). Subsequently, we generated an anti‐phosphorylated Ero1α (p‐Ero1α) antibody specifically recognizing phosphorylated Ser145 of Ero1α (Fig EV3B) to facilitate the study of Ser145 phosphorylation. We could clearly detect p‐Ero1α in HA precipitates from cell extracts and in ConA precipitates from the conditioned medium of HeLa cells overexpressing Ero1α WT, but not Ero1α S145A mutant (Fig 2E). Moreover, when treated with λ‐protein phosphatase, p‐Ero1α signals in the conditioned medium remarkably diminished (Fig 2F). Thus, phosphorylation of Ero1α in cells occurs at Ser145.
Figure EV3. Fam20C is the kinase catalyzing Ero1α phosphorylation.
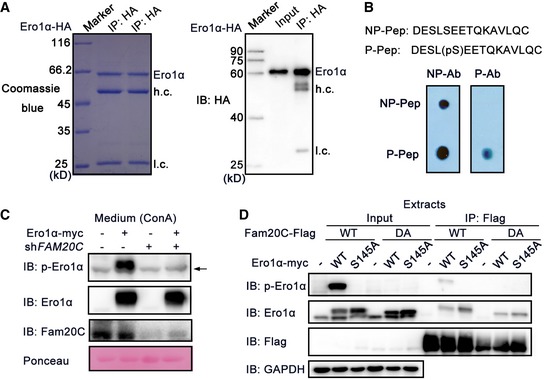
- Coomassie Blue Staining (Left) and protein immunoblotting (Right) of HA immunoprecipitates from HeLa cells expressing Ero1α‐HA. h.c.: heavy chain, l.c.: light chain.
- Non‐phosphopeptide (NP‐Pep) and phosphopeptide (P‐Pep) used for generating non‐phospho‐antibody (NP‐Ab) and phospho‐antibody (P‐Ab). 5 ng NP‐Pep and P‐Pep were spotted on membranes separately, followed by dot blot analysis with NP‐Ab and P‐Ab, respectively.
- Protein immunoblotting of ConA precipitates from the conditioned medium of HeLa cells transfected with Ero1α‐myc and/or shRNA targeting FAM20C. The arrow indicates an unspecific background band.
- Co‐immunoprecipitation of Ero1α and Fam20C in HeLa cells expressing Fam20C‐Flag WT/DA and/or Ero1α‐myc WT/S145A.
Figure 2. Ero1α can be phosphorylated at Ser145.
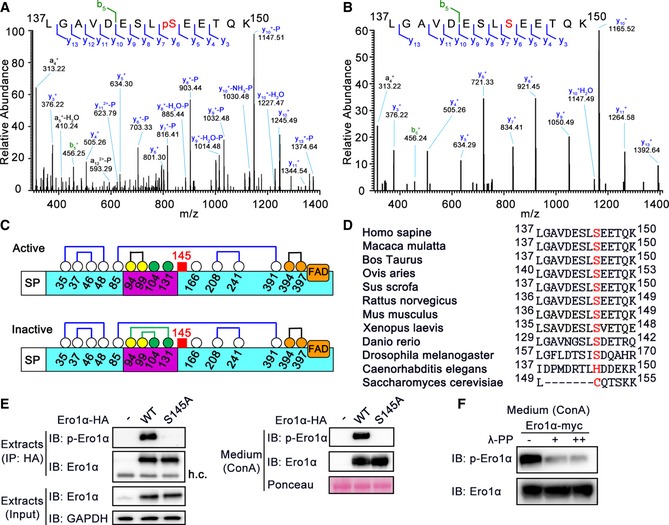
-
A, BRepresentative MS/MS fragmentation spectra of tryptic phosphorylated (A) and non‐phosphorylated (B) peptides (Ero1α Leu137–Lys150) depicting Ero1α Ser145 phosphorylation enriched from Ero1α‐overexpressed HeLa cells.
-
CSchematic representation of human Ero1α active and inactive forms depicting signal peptide (SP), flexible loop (lilac), and cofactor FAD. The cysteines are shown as circles of white, yellow (outer active site), orange (inner active site), or green (regulatory cysteines) with amino acid numbering, disulfides as lines of blue (structural), black (active site), or green (regulatory), and Ser145 as a red square.
-
DAmino acid sequence alignments of Ero1α homologues in several species by BLAST. Residue positions are indicated by numbers counted from the N‐terminus. Human Ero1α Ser145 and its counterparts are shown in red.
-
EDetection of phosphorylated Ero1α (p‐Ero1α) in HeLa cells overexpressing HA‐tagged Ero1α WT or S145A (Left), or in ConA precipitates from the conditioned medium (Right). h.c.: heavy chain.
-
FProtein immunoblotting of ConA precipitates treated with λ‐phosphatase (λ‐PP) from the conditioned medium of HeLa cells overexpressing Ero1α.
Source data are available online for this figure.
Ero1α phosphorylation is catalyzed by Fam20C
Ero1α Ser145 lies within the Fam20C S‐x‐E/pS recognition motif, and we next investigated whether Fam20C phosphorylates Ero1α. An in vitro kinase assay showed that purified Fam20C protein phosphorylated recombinant Ero1α in a time‐dependent manner, as confirmed by the slowly migrating bands and by p‐Ero1α blotting. In contrast, the inactive Fam20C DA mutant failed to incorporate phosphate into Ero1α (Fig 3A). In HeLa and HepG2 cells, overexpressed Fam20C effectively catalyzed phosphorylation of endogenous Ero1α (Fig 3B). When we knocked Fam20C out or down in the HeLa cells, Ero1α was no longer phosphorylated (Figs 3C and EV3C). Moreover, by either immunoblotting (Fig 3D) or immunofluorescence (Fig 3E), we observed p‐Ero1α in cells co‐expressing Ero1α and Fam20C WT but not Fam20C DA. Immunofluorescence analysis also showed that p‐Ero1α and Fam20C displayed subcellular colocalization (Fig 3F), and co‐immunoprecipitation experiments supported the finding that p‐Ero1α and Fam20C interacted physically (Fig EV3D). Convincingly, Fam20C is the bona fide kinase catalyzing Ero1α Ser145 phosphorylation.
Figure 3. Ero1α phosphorylation is catalyzed by Fam20C.
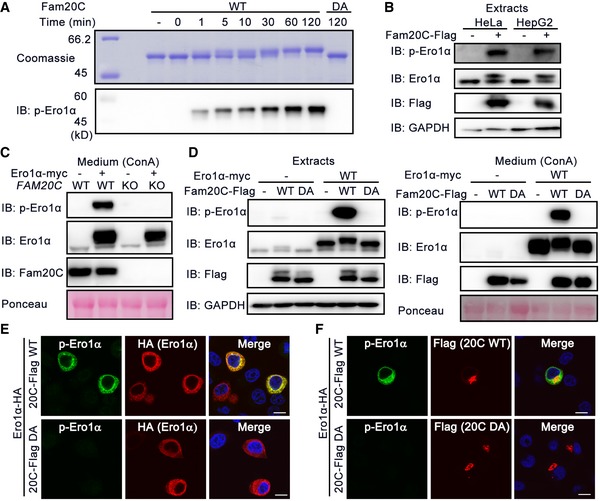
-
ATime‐dependent incorporation of phosphate group into Ero1α catalyzed by recombinant Fam20C WT or DA mutant. Reaction products were analyzed by Coomassie Blue staining and p‐Ero1α immunoblotting.
-
BDetection of endogenous p‐Ero1α in HeLa and HepG2 cells overexpressing Fam20C.
-
CProtein immunoblotting of ConA precipitates from the conditioned medium of WT or FAM20C KO HeLa cells transfected with or without Ero1α‐myc.
-
DProtein immunoblotting of cell extracts (Left) and ConA precipitates from the conditioned medium (Right) of HeLa cells co‐expressing Ero1α‐myc and/or Fam20C‐Flag WT or DA.
-
E, FImmunofluorescence analysis of p‐Ero1α in HepG2 cells co‐expressing Ero1α‐HA and Fam20C‐Flag WT or DA. Scale bars = 10 μm.
Source data are available online for this figure.
Ero1α is phosphorylated by Fam20C in the Golgi and retained in the ER by ERp44
Fam20C‐catalyzed protein phosphorylation is believed to occur in the Golgi lumen or the extracellular space (Tagliabracci et al, 2013). Ero1α is predominantly located in the ER lumen at basal level and can also be secreted into the extracellular space when its expression is greatly elevated (Cabibbo et al, 2000; Anelli et al, 2003). Accordingly, we detected p‐Ero1α in both cell extracts and culture medium (Fig 2E). Thus, a key question is where Ero1α phosphorylation occurs. To address this, we compared HeLa cells co‐expressing Ero1α and Fam20C with co‐cultured cells expressing Ero1α and Fam20C individually. If Ero1α can be phosphorylated in the extracellular space, a p‐Ero1α signal should be observable in the culture medium of the latter group. However, in both the cell extracts and conditioned medium, we detected p‐Ero1α only in cells co‐expressing Ero1α and Fam20C, not in the co‐cultured group. This strongly suggests that Fam20C phosphorylates the secreted Ero1α within cells (Fig 4A).
Figure 4. Ero1α is phosphorylated by Fam20C in the Golgi and retained in the ER by ERp44.
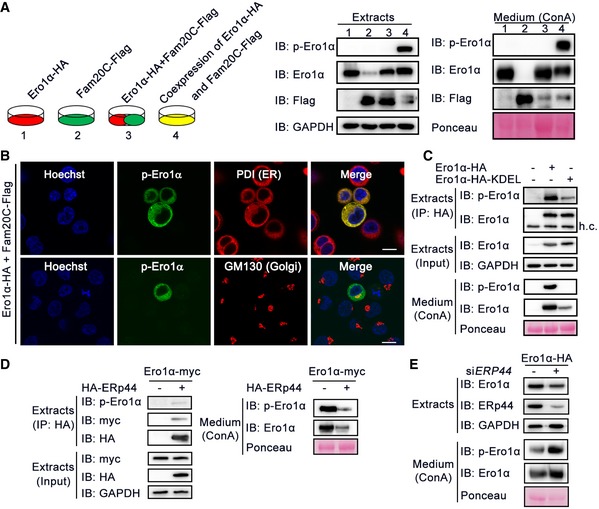
- Protein immunoblotting of cell extracts and ConA precipitates from the conditioned medium of HeLa cells transfected with Ero1α (1) or Fam20C (2) alone, or co‐transfected with Ero1α and Fam20C (4), or co‐cultured cells expressing Ero1α and Fam20C separately (3).
- Immunofluorescence analysis showing the subcellular localization of p‐Ero1α in HepG2 cells co‐expressing Ero1α and Fam20C. PDI and GM130 were used as ER and Golgi markers, respectively. Scale bars = 10 μm.
- Protein immunoblotting of cell extracts and ConA precipitates from the conditioned medium of HeLa cells overexpressing Ero1α‐HA or Ero1α‐HA‐KDEL.
- Co‐immunoprecipitation of ERp44 and Ero1α/p‐Ero1α (Left) and protein immunoblotting of ConA precipitates from the conditioned medium (Right) in HeLa cells expressing Ero1α‐myc alone or with HA‐ERp44.
- Protein immunoblotting of cell extracts and ConA precipitates from the conditioned medium of HeLa cells transfected with Ero1α‐HA and scramble siRNA or siRNA targeting ERP44.
Source data are available online for this figure.
In cells, we observed p‐Ero1α in both the ER and the Golgi lumen by immunofluorescence, using PDI and GM130 as respective markers (Fig 4B), and also by subcellular fractionation (Fig EV4A). To learn whether Ero1α phosphorylation catalyzed by Fam20C occurs in the ER or Golgi apparatus, we engineered a KDEL retrieval sequence to the C‐terminus of Ero1α, which can be sequestrated by cis‐Golgi‐anchored KDELR. This engineering would enhance ER localization of Ero1α and decrease the amount that traverses the late secretory pathway. As expected, Ero1α‐KDEL was largely retained in cells compared to WT Ero1α (Fig 4C). Strikingly, Ero1α‐KDEL was less phosphorylated in cells, indicating that it is in the Golgi cisternae where Fam20C phosphorylates Ero1α and that p‐Ero1α is then retrograde‐transported to the ER (Fig 4C). It is known that the ER retention of Ero1α is accomplished by ERp44, a PDI family chaperone ensuring ER retrieval via KDELR in the early secretory pathway (Anelli et al, 2003; Wang et al, 2008; Vavassori et al, 2013). We propose that relocation of p‐Ero1α to the ER is also achieved by ERp44. Indeed, p‐Ero1α could be co‐immunoprecipitated with ERp44 and the secretion of p‐Ero1α dramatically decreased with ERp44 overexpression (Fig 4D), though ERp44 bound to Ero1α WT, S145A and S145E mutants and retained them within the ER with similar affinities (Fig EV4B and C). When we knocked ERp44 down using siRNA in HeLa cells, a higher amount of p‐Ero1α was released to the culture medium (Fig 4E). Taken together, these results indicate that Ero1α phosphorylation occurs in the Golgi, catalyzed by Fam20C, and that ERp44/KDELR achieves the retention of p‐Ero1α in the ER.
Figure EV4. The ER retention of p‐Ero1α is mediated by ERp44.
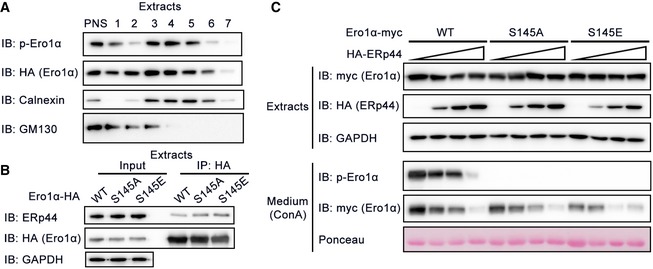
- Subcellular fractionation of HeLa cells. The postnuclear supernatant (PNS) of HeLa cells expressing Ero1α‐HA and Fam20C‐Flag was separated on a 30% Percoll gradient and the fractions were collected and subjected to protein immunoblotting. Calnexin, ER marker; GM130, Golgi marker.
- Co‐immunoprecipitation of Ero1α and endogenous ERp44 in HeLa cells expressing Ero1α‐HA WT/S145A/S145E.
- Protein immunoblotting of cell extracts and ConA precipitates from the conditioned medium of HeLa cells overexpressing Ero1α‐myc WT/S145A/S145E and increasing amounts of HA‐ERp44.
p‐Ero1α displays higher sulfhydryl oxidase activity
Protein phosphorylation is a common way to induce changes in protein conformation and activity. Based on its crystal structure, Ero1α Ser145 is located at the C‐terminus of the outer active site (Cys94‐Cys99)‐containing flexible loop, the movement of which is critical for shuttling electrons from PDI to Ero1α inner active site (Cys394‐Cys397) (Fig 5A). Since Ero1α uses oxygen to catalyze de novo disulfide bond formation in the electron transfer pathways (Fig 5B), we monitored the oxygen consumption rate via an oxygen electrode to study the effect of phosphorylation on Ero1α activity. The Ero1α phosphorylation mimic S145E displayed twofold higher oxidase activity than that of Ero1α WT in the presence of either the small molecule reducing agent dithiothreitol (DTT) (Fig 5C) or the physiological substrate PDI reduced by glutathione (GSH) (Fig 5D). Mutation of the two non‐catalytic cysteines, Cys104 and Cys131, disrupts the regulatory disulfides in Ero1α and results in a constitutively active oxidase (Appenzeller‐Herzog et al, 2008; Baker et al, 2008). Notably, Ero1α C104A/C131A/S145E, a hyperactive Ero1α phosphorylation mimic, showed further increased activity (Fig 5C and D), suggesting that Ser145 phosphorylation is a novel activity controller independent of the known “regulatory disulfide switch” in Ero1α. To further confirm the regulatory role of Ser145 phosphorylation, we prepared genuine phosphorylated Ero1α using an in vitro kinase assay. Consistent with the results obtained from the phosphorylation mimics, both p‐Ero1α WT and p‐Ero1α C104A/C131A exhibited twice the activity of Ero1α WT and C104A/C131A (Fig 5E and F).
Figure 5. Phosphorylation of Ero1α increases its sulfhydryl oxidase activity in vitro .
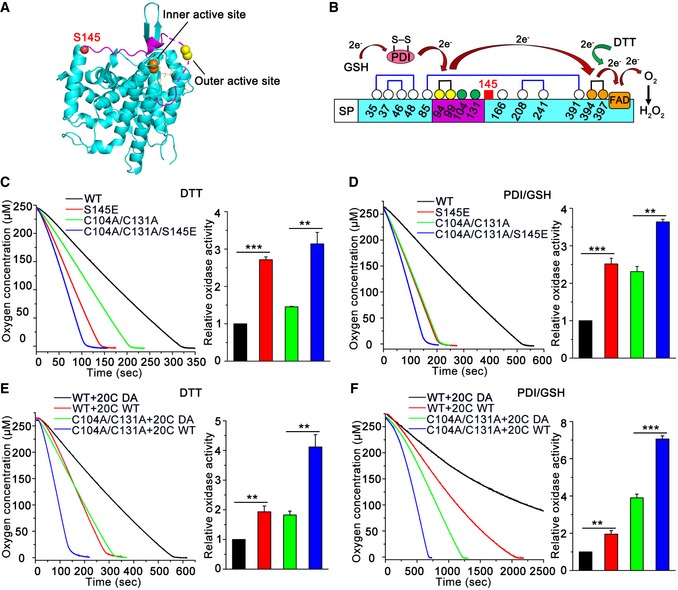
-
ARibbon representation of the active form of human Ero1α structure (PDB: 3AHQ). Outer active site (yellow ball), inner active site (orange ball), cofactor FAD (orange stick), and Ser145 (red ball) are indicated. The outer active site‐containing loop is shown in lilac and the missing region by the dashed line.
-
BSchematic representation of the electron transfer pathway between the active sites of Ero1α and its physiological substrate PDI/GSH or small molecule reducing agent DTT.
-
C–FOxygen consumption catalyzed by Ero1α phosphorylation mimics (C, D) or Fam20C‐treated Ero1α (E, F), on a background of Ero1α WT or hyperactive C104A/C131A, was monitored in the presence of DTT (C, E) or PDI and GSH (D, F). The relative oxidase activity of Ero1α was calculated from the slope of the linear phase of oxygen decrease. Data are shown as mean ± SEM from three independent experiments. **P < 0.01, ***P < 0.001 (two‐tailed, Student's t‐test).
Acceleration of PDI oxidation caused by Ser145 phosphorylation still depended on the presence of the Ero1α outer active site (Fig EV5A), implying that phosphorylation does not alter the electron transfer pathway. Moreover, the steady redox state (Fig EV5B) and reduced PDI‐mediated activation (Fig EV5C) of p‐Ero1α mimics showed no difference from those of Ero1α, suggesting that phosphorylation does not affect allosteric activation of Ero1α. As Ser145 is located between the flexible loop region and an adjacent α‐helix (Fig 5A), we investigated whether its phosphorylation results in a conformational change in Ero1α. Although the overall secondary structure of Ero1α S145E was similar to that of Ero1α WT as shown by the circular dichroism spectrum (Fig EV5D), we observed an increase in intrinsic tryptophan fluorescence in Ero1α S145E (Fig EV5E), implying that microenvironment changes occur around the aromatic residues. We also noticed from the crystal structure of Ero1α that there is a hydrogen bond between the O atom of the Ser145 hydroxyl group and the N atom of the Thr148 main chain, and this hydrogen bond would be disfavored due to the presence of a phosphate group on the side chain of Ser145 (Fig EV5F). The conformational change induced by phosphorylation may increase the flexibility of the outer active site‐containing flexible loop and contribute to the higher oxidase activity of p‐Ero1α.
Figure EV5. Biophysical and biochemical characterization of Ero1α phosphorylation mimics.
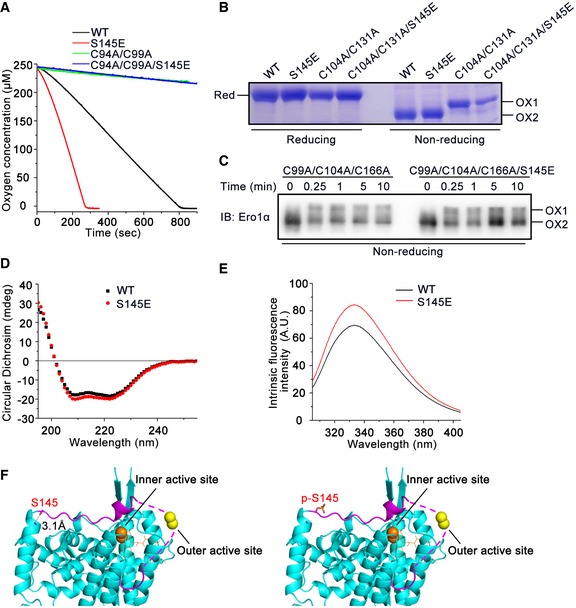
- Oxygen consumption catalyzed by Ero1α WT, inactive Ero1α C94A/C99A, and corresponding phosphorylation mimics was monitored in the presence of PDI and GSH.
- The redox states of recombinant Ero1α WT, hyperactive Ero1α C104A/C131A, and corresponding phosphorylation mimics were determined under reducing and non‐reducing conditions. The active form (OX1), inactive form (OX2), and fully reduced form (Red) are indicated.
- Recombinant oxidized Ero1α C99A/C104A/C166A and Ero1α C99A/C104A/C166A/S145E were mixed with reduced PDI, and aliquots were taken at indicated times for analysis by non‐reducing SDS–PAGE and Ero1α blotting.
- Far UV circular dichroism spectrum of recombinant Ero1α WT and S145E.
- Intrinsic fluorescence spectrum of recombinant Ero1α WT and S145E.
- Ribbon representation of non‐phosphorylated Ero1α (Left) and Ser145‐phosphorylated Ero1α (Right) based on the structure of active human Ero1α (PDB: 3AHQ). The outer active site‐containing loop is shown in lilac, and the missing region in dashed line. Outer active site (yellow ball), inner active site (orange ball), cofactor FAD (orange stick), and the side chains of Ser145 and p‐Ser145 (stick) are indicated. The distance between O atom of Ser145 side chain and N atom of Thr148 main chain is shown.
Phosphorylation of Ero1α Ser145 plays a critical role in ER redox homeostasis
To investigate whether phosphorylation of Ero1α Ser145 by Fam20C is linked to ER redox regulation, we first generated ERO1A KO HeLa cells by means of CRISPR/Cas9 genome editing (Fig EV6). Using the superfolded‐roGFP‐iEER probe, we observed a dramatically reduced ER redox environment in the two clones of ERO1A KO cells (Fig 6A), underlining the importance of Ero1α in maintaining ER redox homeostasis. The redox state in the ER was partially recovered by the replenishment of Ero1α WT and further restored by Ero1α S145E, indicating that p‐Ero1α is also more active in cells (Fig 6B). Importantly, the expression of Fam20C in ERO1A KO cells transfected with Ero1α WT resulted in the phosphorylation of the majority of Ero1α and generated a more oxidized ER (Fig 6C). However, the expression of Fam20C in ERO1A KO cells or in Ero1α S145A‐transfected ERO1A KO cells exhibited little effect on the ER redox state (Fig 6C). Therefore, we conclude that Fam20C participates in ER redox regulation through the phosphorylation of Ero1α Ser145.
Figure EV6. Generation of ERO1A KO HeLa cells using CRISPR/Cas9 genome editing.
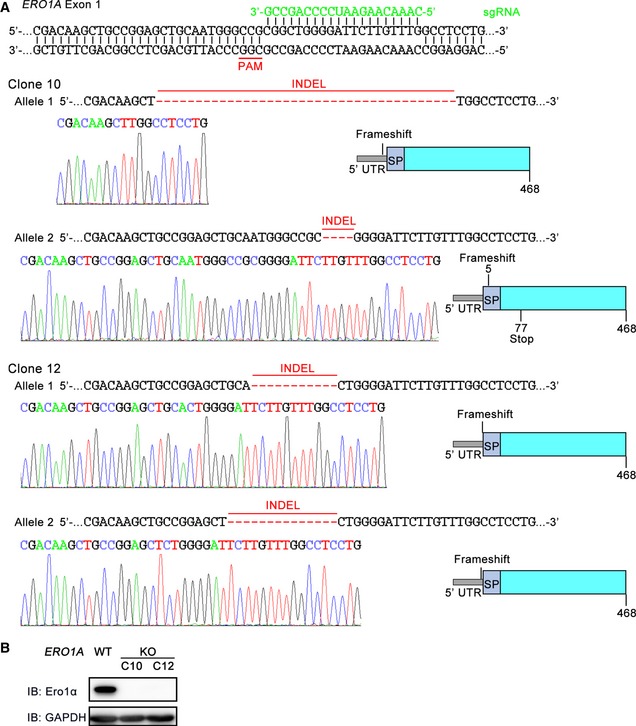
- Schematic representation of the base pairing between the sgRNA and the targeting locus of exon 1 in the human ERO1A gene. The sequences of the mutated alleles in ERO1A clone 10 (C10) and clone 12 (C12) and representative chromatograms depicting the INDELs are shown. The INDELs were predicted to cause frameshift mutations producing inactive copies or terminating translation of the protein. SP, signal peptide.
- Protein immunoblotting of cell extracts from WT and ERO1A KO HeLa cells.
Figure 6. Phosphorylation of Ero1α Ser145 plays a critical role in ER redox homeostasis.
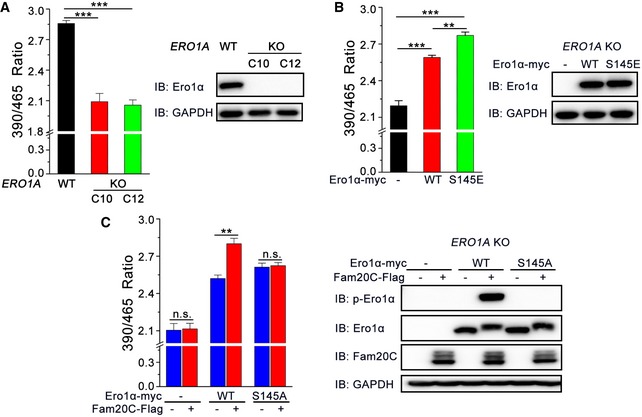
- (Left) The fluorescence intensities of superfolded‐roGFP‐iEER in WT and two clones (C10 and C12) of ERO1A KO HeLa cells were measured as in Fig 1D. (Right) Aliquots of cells in the left panel were analyzed by immunoblotting.
- (Left) The fluorescence intensities of superfolded‐roGFP‐iEER in ERO1A KO HeLa cells expressing Ero1α‐myc WT or S145E were measured as in Fig 1D. (Right) Aliquots of cells in the left panel were analyzed by immunoblotting.
- (Left) The fluorescence intensities of superfolded‐roGFP‐iEER in ERO1A KO HeLa cells expressing Ero1α‐myc WT or S145A and/or Fam20C‐Flag were measured as in Fig 1D. (Right) Aliquots of cells in the left panel were analyzed by immunoblotting.
Phosphorylation of Ero1α promotes oxidative protein folding
We next explored whether Ero1α phosphorylation promotes oxidative protein folding in cells by monitoring the folding of immunoglobulin J‐chain, a specific substrate of Ero1α (Mezghrani et al, 2001). As shown in Fig 7A, when HeLa transfectants expressing Myc‐tagged J‐chain (JcM) were briefly exposed to DTT, most JcM migrated as reduced monomers. Upon removal of DTT, those monomers progressively disappeared, and in turn, oxidized monomers and dimers emerged. Compared to cells co‐expressing an inactive Ero1α mutant C99A/C104A, oxidized JcM monomers and dimers formed more rapidly during the chase phase in cells co‐expressing Ero1α WT, and more high‐molecular‐weight (HMW) species were observed. In accordance with the in vitro results, the Ero1α phosphorylation mimic S145E significantly accelerated the J‐chain folding process in cells compared to Ero1α WT, as quantified by the disappearance of reduced monomers (Fig 7B). Furthermore, the transition of reduced JcM into oxidized species was also faster in HeLa cells overexpressing Fam20C WT than in cells expressing Fam20C DA (Fig 7C and D), probably due to the elevated level of endogenous p‐Ero1α. In contrast, in the ERO1A KO cells, the re‐oxidation of JcM was completely repressed, and ectopic expression of either Fam20C WT or DA had little effect on JcM re‐oxidation (Fig 7C and D). Altogether, these results suggest that phosphorylation of Ero1α Ser145 by Fam20C does promote oxidative protein folding in cells.
Figure 7. Phosphorylation of Ero1α promotes oxidative protein folding in cells.
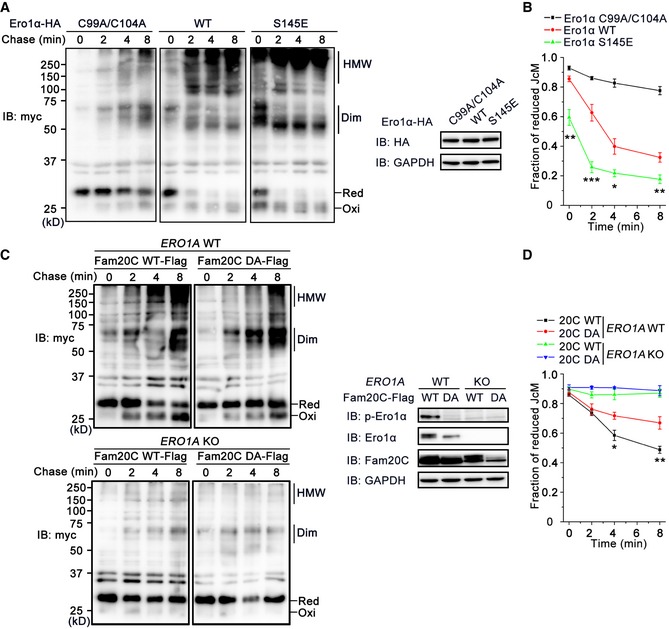
- (Left) HeLa transfectants expressing myc‐tagged JcM with Ero1α‐HA C99A/C104A, WT, or S145E were pulsed with DTT, washed, and chased at indicated time points by non‐reducing myc blotting. The mobility of reduced JcM monomers (Red), oxidized monomers (Oxi), homodimers (Dim), and high‐molecular‐weight (HMW) species is indicated. (Right) Aliquots from cell lysates in the left panel were resolved in reducing conditions and analyzed by immunoblotting.
- The fraction of reduced JcM (Red/[Red + Oxi]) in (A) was quantified by densitometry. Data are shown as mean ± SEM from five independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001, between Ero1α WT and S145E (two‐tailed Student's t‐test).
- (Left) JcM re‐oxidation in WT and ERO1A KO HeLa cells overexpressing Fam20C‐Flag WT or DA was monitored as in (A). (Right) Aliquots from cell lysates in the left were resolved in reducing conditions and analyzed by immunoblotting.
- The fraction of reduced JcM in (C) was quantified by densitometry. Data are shown as mean ± SEM from four independent experiments. *P < 0.05, **P < 0.01, between Fam20C‐Flag WT and DA in WT HeLa cells (two‐tailed Student's t‐test).
Source data are available online for this figure.
Phosphorylation of Ero1α is induced during mammalian lactation, hypoxia, and reductive stress
To determine whether Fam20C can phosphorylate Ero1α in physiological processes, we first focused on mammalian lactation, during which abundant secretory proteins are produced. Figure 8A shows that mRNA levels of FAM20C and its activator FAM20A, which encodes a pseudokinase that forms a functional complex with Fam20C to enhance secretory protein phosphorylation, were dramatically elevated in mammary glands of lactating mice compared to those of virgin mice, consistent with the previous report (Cui et al, 2015). Interestingly, ERO1A and PDIA1 mRNAs were also greatly upregulated in lactating mammary glands. Consistent with their mRNA levels, Ero1α and Fam20C proteins were more abundant in lactating mouse mammary glands (Fig 8B). Notably, we could clearly detect a much stronger p‐Ero1α signal and a significantly higher ratio of p‐Ero1α/Ero1α in the mammary glands of lactating mice than those of virgin mice (Fig 8B and C). Altogether, these results demonstrate that Fam20C phosphorylates Ero1α in the mammary glands of lactating animals.
Figure 8. p‐Ero1α is induced during mammalian lactation, hypoxia, and reductive stress.
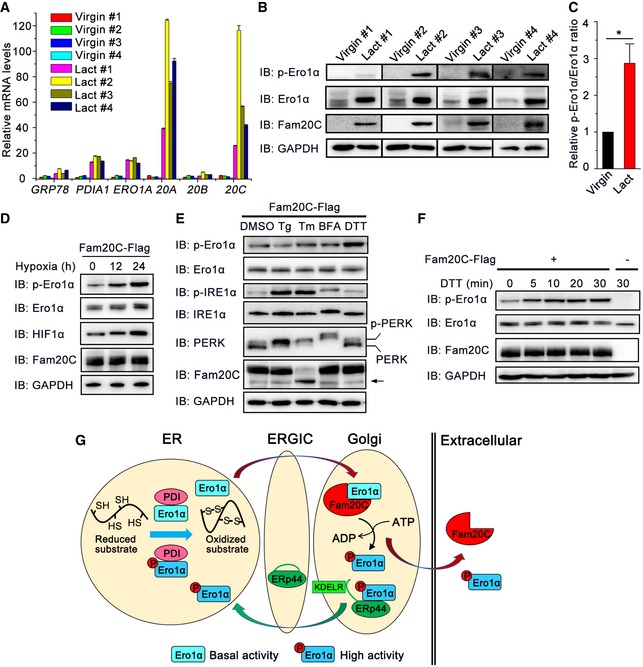
- mRNA levels of several ER folding catalysts and Fam20 family members in the mouse mammary gland. The mammary glands of four virgin and four lactating (Lact) mice were isolated, and their mRNA levels were determined by RT–qPCR and normalized to the mean value of the virgin group. Data are represented as mean ± SEM performed in six technical replicates.
- Protein immunoblotting of extracts from the whole mammary glands of virgin and lactating mice.
- Quantification of relative p‐Ero1α/Ero1α ratio in (B). Data are shown as mean ± SEM of four groups. *P < 0.05 (two‐tailed, Student's t‐test).
- Protein immunoblotting of HeLa cells expressing Fam20C‐Flag following exposure to hypoxia (0.1% oxygen) for the indicated times.
- Protein immunoblotting of HeLa cells expressing Fam20C‐Flag treated with 5 μM thapsigargin (Tg), 5 μg/ml tunicamycin (Tm), 5 μg/ml Brefeldin A (BFA), or 200 μM DTT for 6 h. The arrow indicates an unglycosylated form of Fam20C.
- Protein immunoblotting of HeLa cells transfected with or without Fam20C‐Flag treated with 200 μM DTT for the indicated times.
- A working model for Fam20C tuning ER redox homeostasis and oxidative protein folding. Ero1α and PDI catalyze protein disulfide formation in the ER lumen. During mammalian lactation, hypoxia, and reductive stress, non‐phosphorylated Ero1α with basal activity is transported to the Golgi apparatus and phosphorylated by Fam20C at Ser145. Phosphorylated Ero1α is sequestrated by ERp44, a chaperone primarily localized in the ER‐Golgi intermediate compartment (ERGIC), and relocated to the ER mediated by KDEL receptor (KDELR). Phosphorylated Ero1α displays higher oxidase activity to promote disulfide bond formation and maintain ER redox homeostasis.
Source data are available online for this figure.
It has been known that hypoxic induction of Ero1α is the key adaptive response to improve protein secretion under hypoxia (May et al, 2005). Notably, when Fam20C‐expressing HeLa cells were cultured in a hypoxic chamber with 0.1% oxygen concentration, phosphorylation of Ero1α was dramatically induced along with an increase in total Ero1α and hypoxia‐inducible factor 1 (HIF‐1α) (Fig 8D). We also investigated whether phosphorylation of Ero1α could be induced during ER stress or reductive stress by pharmacological modulation. When Fam20C‐expressing cells were treated with known ER stress inducers, including Tg (an inhibitor of sarco/endoplasmic reticulum Ca2+‐ATPase (SERCA) calcium pump), tunicamycin (Tm; an inhibitor of protein glycosylation), and Brefeldin A (BFA; an inhibitor of ER‐Golgi traffic), both p‐Ero1α and total Ero1α level were unchanged up to 6 h, though IRE1α and PERK UPR branches were activated (Fig 8E). Interestingly, when cells were treated with 200 μM DTT, a concentration sufficient to reduce protein disulfides but not trigger UPR, p‐Ero1α was rapidly and strongly induced whereas total Ero1α protein level did not change (Fig 8E and F). These results suggest that Fam20C‐catalyzed phosphorylation of Ero1α occurs as a post‐translational regulatory mechanism to counteract cellular reductive stress.
Discussion
In this study, we report Fam20C, the Golgi‐localized secretory kinase, has a previously unidentified function of fine‐tuning ER redox homeostasis and oxidative protein folding. Ero1α, an ER sulfhydryl oxidase, can be transported to the Golgi apparatus and effectively phosphorylated by Fam20C at Ser145. Phosphorylation of Ero1α largely enhances its oxidase activity. Similar to non‐phosphorylated Ero1α, phosphorylated Ero1α is relocated to the ER lumen, also mediated by the ERp44/KDELR system, to promote disulfide bond formation and maintain ER redox homeostasis (Fig 8G). This model also provides an answer to the long‐unsolved question of why Ero1α functions in the ER but does not contain an ER retention motif. Indeed, engineered Ero1α‐KDEL protein is more resistant to phosphorylation by Fam20C (Fig 4C). Although it is possible that Fam20C is active during its transit route across the ER, we propose that a rather unique milieu in the Golgi is appropriate for Fam20C activity. Besides Ero1α and the recently reported sarcoplasmic reticulum histidine‐rich calcium‐binding protein (HRC) (Pollak et al, 2017), we identify several ER/Golgi‐localized enzymes and chaperones that also interact with Fam20C (Fig 1 and Dataset EV1). Future studies are required to determine whether these proteins can be phosphorylated by Fam20C and can therefore participate in certain biological processes.
Professional secretory cells and tissues have extremely high demands for oxidative protein folding in the ER. For example, lactating mammary glands produce milk, which contains a large amount of disulfide‐rich proteins (e.g., immunoglobulins, α‐lactalbumin, and serum albumin). Indeed, the expression of both Fam20C and Ero1α is dramatically elevated in lactating mouse mammary glands. Notably, we have detected strong p‐Ero1α signals in the mammary glands of lactating mice, suggesting that Ero1α phosphorylation by Fam20C plays an important role in lactation. Interestingly, we find that p‐Ero1α is not induced under ER stress caused by calcium depletion or aberrant glycosylation; instead, it is rapidly induced once the protein disulfides in the ER are disrupted by the reducing agent DTT. Since oxygen is the ultimate source of oxidizing power for disulfide formation, it is also reasonable that p‐Ero1α is induced under hypoxic condition to increase the bioavailability of oxygen. Previous studies showed that exposure of cells to reductive stress or hypoxia leads to the relocation of Ero1α from the early secretory pathway to the late secretory pathway (Gilady et al, 2010), which is in line with our conclusion that Ero1α is phosphorylated by Fam20C in the Golgi. Altogether, our study highlights the importance of phosphorylation of Ero1α by Fam20C as a novel regulatory mechanism to counteract cellular reductive stress at post‐translational level. Whether Ero1α phosphorylation is also enhanced in other secretory cells, such as plasma cells secreting immunoglobulins and β‐cells secreting insulin, and is important for efficient oxidative protein folding in those cells are still open questions.
Ero1 oxidase catalyzes one disulfide bond formation and produces one molecule of H2O2 as a byproduct; therefore, its activity should be tightly controlled to prevent excess ROS production (Gross et al, 2006; Wang et al, 2009, 2011). Ero1 activity can be allosterically controlled by “regulatory disulfides”, which were first studied in yeast (Gross et al, 2004; Sevier et al, 2007; Vitu et al, 2010; Kim et al, 2012) and later dissected in mammals (Appenzeller‐Herzog et al, 2008; Baker et al, 2008; Inaba et al, 2010; Wang et al, 2011; Zhang et al, 2014). Interestingly, Ser145 in Ero1α is highly conserved in multicellular organisms but missing in yeast Ero1p (Fig 2D), and Fam20C homologue is not present in yeast, implying that Ero1p is lack of phosphorylation regulation. Instead, yeast Ero1p harbors Cys150 in the same position, and disruption of the Cys150‐Cys295 allosteric disulfide is known to enhance the movement of outer active site‐containing loop and increase Ero1p activity (Sevier et al, 2007), though physiological reduction in the Cys150‐Cys295 disulfide requires extremely reducing condition (Niu et al, 2016). The homology between Cys150 in Ero1p and Ser145 in Ero1α further suggests that modification of Ser145 is likely to cause a similar conformational change and enzymatic enhancement. Besides previous reports on the regulatory disulfides of Ero1α, our observation that Ero1α can be phosphor‐regulated via Ser145 adds an additional layer to the regulation of Ero1α activity. These findings suggest that human Ero1α has undergone extensive evolution to optimize the folding efficiency of large, complicated disulfide‐containing secretory proteomes and to simultaneously minimize the risks of overoxidation in the ER. Whether Ero1α possesses other phosphorylation sites besides Ser145, the manner by which these phosphorylation sites orchestrate to regulate ER redox, and the identity of the protein phosphatase for dephosphorylating Ero1α remain open questions.
Apart from the introduction of disulfide bonds, Ero1α has also been reported to regulate ER Ca2+ influx and efflux. A fraction of Ero1α is localized in mitochondrial‐associated ER membranes (Gilady et al, 2010; Ramming & Appenzeller‐Herzog, 2012). Ero1α can inhibit SERCA calcium pump activity (Marino et al, 2015) and activate inositol 1,4,5‐triphosphate receptor 1 (IP3R1) to induce Ca2+ release into the mitochondria and trigger apoptosis (Li et al, 2009; Anelli et al, 2012), doing both in a redox‐dependent manner. Thus, it would be interesting to investigate whether p‐Ero1α with high oxidase activity participates in the regulation of ER calcium homeostasis. Although Ero1α predominantly localizes and functions in the ER lumen under normal conditions, it can be secreted into the extracellular space at an upregulated level of expression (Cabibbo et al, 2000; Anelli et al, 2003). However, the role of secreted Ero1α is largely unknown. In this study, we observe that p‐Ero1α is present in both cell extracts and culture medium (Figs 2E and 3C and D), implying that secreted p‐Ero1α with high activity also functions to regulate extracellular redox homeostasis.
Ero1α is recognized as a cancer‐associated oxidase because its expression is significantly elevated in many cancers such as breast (Kutomi et al, 2013), gastrointestinal (Battle et al, 2013), and pancreatic (Kukita et al, 2015) cancers. Moreover, Ero1α enhances the expression of major histocompatibility complex class I antigen (Kukita et al, 2015), vascular endothelial growth factor (Tanaka et al, 2016), and programmed death‐ligand 1 (Tanaka et al, 2017) via oxidative folding to promote tumor growth. Notably, Fam20C‐dependent phosphorylation of secreted proteins is necessary for the proper adhesion, migration, and invasion of cancer cells (Tagliabracci et al, 2015). Therefore, future efforts to validate whether the p‐Ero1α levels are elevated in cancers and are correlated with a poor prognosis will greatly benefit the use of p‐Ero1α as a potential biomarker and a novel therapeutic target for various types of cancer.
Materials and Methods
Generation of constructs
The pGEX‐6P‐1 plasmids encoding human Ero1α and pQE30‐PDI were laboratory products (Zhang et al, 2014; Niu et al, 2016). The mutation of Ero1α S145E was created using the Fast Mutagenesis System (TransGen). The expression of pcDNA3.1‐Ero1α‐myc, pcDNA3.1‐Ero1α‐HA, and/or pcDNA3.1‐HA‐ERp44 in mammalian cells was as previously described (Wang et al, 2014; Yang et al, 2016). pcDNA3.1‐Ero1α‐HA‐KDEL was generated by insertion of a KDEL motif between HA and the termination codon. pcDNA3.1‐Ero1α S145A and S145E with myc or HA tag were produced using the Fast Mutagenesis System (TransGen). pCCF‐Fam20C‐Flag and pCCF‐Fam20C D478A‐Flag were previously described (Tagliabracci et al, 2012). pcDNA3.1‐roGFP‐iEER was a kind gift from Dr. Christian Appenzeller‐Herzog (University of Basel), and S30R, T39N, N105T, and I171V mutations were induced to generate the superfolded‐roGFP‐iEER as described elsewhere (Hoseki et al, 2016). All constructs were verified by DNA sequencing (Invitrogen).
Protein expression and purification
Recombinant full‐length human Ero1α proteins and PDI were expressed in Escherichia coli BL21 (DE3) cells and purified as before (Wang et al, 2009). Maltose‐binding protein (MBP) tagged human Fam20C protein (141–578) was expressed in Hi5 insect cells and purified from the conditioned medium, and the MBP tag was removed using gel filtration chromatography following tobacco etch virus protease digestion (Xiao et al, 2013). Proteins were quantified using the Bradford method with bovine serum albumin as a standard or spectrophotometrically at 280 nm and stored as aliquots at −80°C.
In vitro kinase assay
The reactions were initiated by adding ATP to a final concentration of 2 mM into buffer A (HEPES–NaOH, pH 7.0, 10 mM MnCl2) containing 1 mg/ml recombinant Ero1α and 20 μg/ml purified Fam20C or Fam20C D478A protein at 30°C, and were terminated at different times by adding 20 mM EDTA. Proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE) and visualized by Coomassie Brilliant Blue staining and immunoblotting.
Oxygen consumption assay
Oxygen consumption was monitored at 25°C using an Oxygraph Clark‐type oxygen electrode (Hansatech Instruments) as previously described (Wang et al, 2011). Reactions were initiated by adding Ero1α proteins to a final concentration of 1 or 2 μM into buffer B (100 mM Tris‐HAc, pH 8.0, 50 mM NaCl, 2 mM EDTA) containing 1 mM dithiothreitol (DTT) or 20 μM PDI proteins and 10 mM glutathione (GSH), respectively. For phosphorylated Ero1α assay, reactions were initiated by adding 1 mM DTT or 20 mM GSH into buffer B with an in vitro kinase assay mixture containing 0.5 μM Ero1α or 1 μM Ero1α and 20 μM PDI, respectively. Reaction rates were calculated by measuring the slope of the linear phase of the oxygen consumption curve.
Cell culture and transfection
HeLa cells were cultured in Dulbecco's modified Eagle's medium (Gibco), and HepG2 cells were cultured in RPMI 1640 medium (Gibco) supplemented with 10% fetal bovine serum (Gibco), 100 units/ml penicillin, and 100 μg/ml streptomycin (Invitrogen) at 37°C with 5% CO2. Plasmid transfection was accomplished by using Lipofectamine 2000 (Invitrogen) or ViaFect (Promega) according to the manufacturer's instructions. After 24–48 h, the transfected cells and culture medium were harvested for further analysis. For hypoxia treatment, cells were placed in a hypoxic chamber (BioSpherix) with 0.1% oxygen concentration for different period of time.
RNA interference
The pSUPER.retro.puro vector (Oligoengine) expressing the short‐hairpin RNA (shRNA) targeting FAM20C sequence 5′‐GAGCTGTACTCCAGACACA‐3′ was constructed according to the manufacturer's instructions. siERP44 with sequence 5′‐UUAAUUGCCGAGCUACUUCAUUCUG‐3′ was obtained from GENEWIZ. HeLa cells were transfected with pSUPER plasmid by Lipofectamine 2000 or siRNA by RNAi‐MAX (Invitrogen) on day 1, and co‐transfected with the other plasmid on day 3. Cells and culture medium were harvested on day 4 for further analysis.
Mass spectrometry (MS)
Sample preparation
To identify the Fam20C interactome, HeLa cells were transfected with pcDNA4.0 or pcDNA4.0‐Fam20C‐Flag for 48 h, then harvested, and lysed. The cell extracts were incubated with anti‐Flag M2 affinity gel (Sigma‐Aldrich) overnight at 4°C with occasional vortexing. The immunoprecipitates were washed five times with ice‐cold PBS. The proteins were then separated by SDS–PAGE and stained with Coomassie Brilliant Blue for MS analysis. For phosphorylation site identification, HeLa cells were transfected with pcDNA3.1‐Ero1α‐HA for 24 h. The cell extracts were incubated with anti‐HA (HA‐7; Sigma‐Aldrich) overnight, followed by addition of Agarose A+G (Beyotime). The immunoprecipitates were washed for five times, and the proteins were separated by SDS–PAGE gels, stained by Coomassie Brilliant Blue, and immunoblotted with anti‐HA in parallel.
In‐gel digestion of proteins
All gel bands excluding Fam20C and immunoglobulin chains were excised and cut into small plugs. After destaining, the gel plugs were reduced (10 mM DTT in 25 mM NH4HCO3 for 45 min at 56°C) and further alkylated (40 mM iodoacetamide in 25 mM NH4HCO3 for 45 min at room temperature in the dark). The plugs were washed twice with 50% acetonitrile in 25 mM NH4HCO3, dried using a SpeedVac, and digested with trypsin in 25 mM NH4HCO3 overnight at 37°C, followed by addition of formic acid to a 1% final concentration to terminate the reaction.
LC‐MS/MS analysis
The digestion peptides were separated on a 75 μm ID × 20 cm C18 column packed with reversed‐phase silica (Reprosil‐Pur C18‐AQ, 3 μm; Dr. Maisch GmbH). A linear acetonitrile gradient was used to elute the bounded peptides, which were directly electrosprayed at a 2.0 kV voltage directly into a Q Exactive mass spectrometer (Thermo Fisher Scientific) equipped with a nano‐liquid chromatography electrospray ionization source. In the data‐dependent acquisition mode, the MS data were acquired at a high resolution of 70,000 (m/z 200) across a mass range of 300–1,600 m/z, followed by 20 tandem mass events on the top 20 precursor ions, which were isolated and fragmented in the HCD collision cell. Subsequently, MS/MS spectra were acquired at a resolution of 17,500 at m/z 200. The dynamic exclusion time was 40 s.
Protein identification
Database searches were performed on Proteome Discovery software, version 1.4 using SEQUEST HT search engine for protein identification and Percolator for false discovery rate (FDR, < 1%) analysis against a UniProt human protein database. The search parameters were set as follows: 10 ppm mass tolerance for precursor ions; 0.02 Da mass tolerance for product ions; two missed cleavage sites allowed for trypsin digestion. For Fam20C interactome identification, proteins (unique peptides ≥ 2) were selected and protein interaction was defined as significant if the MS area ratio of Fam20C overexpressed group (B) to control group (A) is ≥ 3. A total of 1,876 proteins were identified, of which 349 are localized in the ER and Golgi based on DAVID GO term analysis. For phosphorylation site identification, we selected phosphorylation for serine, threonine or tyrosine and methionine oxidation as variable modifications, and the cysteine carbamidomethylation as a fixed modification. The peptides confidence was set as high for peptides filter. The tandem mass spectra of the matched phosphorylated peptides were manually checked for their validity.
Subcellular fractionation
Subcellular fractionation was performed as described (Wu et al, 2018). Briefly, the harvested HeLa cells were suspended in 20 mM tricine (pH 7.8) and 250 mM sucrose containing protease inhibitor cocktail and phosphatase inhibitor cocktail and kept on ice for 20 min. The cells were homogenized before centrifugation at 3,000 × g for 10 min to remove the nucleus and residual cells. The postnuclear supernatant was loaded on 30% Percoll and centrifuged at 144,200 × g for 30 min in a Beckman SW41 Ti rotor. Fractionated samples were collected from top to bottom gently and subjected to SDS–PAGE and immunoblotting.
Immunoblotting
Harvested cells were lysed in radio immunoprecipitation assay (RIPA) lysis buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 0.25% deoxycholic acid, 1% NP‐40, 1 mM EDTA, Millipore) containing 1 mM phenylmethanesulfonyl fluoride (PMSF), phosphatase inhibitor cocktail (Roche), and protease inhibitor cocktail (Roche) for 30 min. The homogenate was centrifuged at 13,000 × g for 15 min to remove cell debris. Protein quantification was performed using a BCA Kit (Beyotime). Culture medium was enriched by Concanavalin A (ConA, Sigma‐Aldrich) at 4°C overnight, and the precipitates were washed three times before adding the loading buffer. Protein samples were analyzed by SDS–PAGE and then transferred to polyvinylidene difluoride (PVDF) membranes (Millipore) using a semi‐dry transfer apparatus. The membranes were blocked in 5% milk (5% BSA for p‐Ero1α immunoblotting), incubated with antibodies and visualized by using a Chemi‐Scope Mini imaging system (Clinx Science) with enhanced chemiluminescence (Thermo Fisher Scientific).
The following antibodies were used for immunoblotting: mouse monoclonal anti‐Ero1α (2G4, 1:1,000; Millipore), anti‐PDI (RL90, 1:2,000; Abcam), anti‐calnexin (610523, 1:500; BD Transduction), anti‐GM130 (610822, 1:500; BD Transduction), anti‐HIF1α (NB100‐105, 1:500; Novusbio), anti‐Flag (M2, 1:2,000; Sigma‐Aldrich), anti‐myc (9E10, 1:5,000; Sigma‐Aldrich), anti‐HA (HA‐7, 1:5,000; Sigma‐Aldrich), anti‐GAPDH (GAPDH‐71.1, 1:50,000; Sigma‐Aldrich); rabbit polyclonal anti‐ERp44 (D17A6, 1:2,000; Cell Signaling Technology), anti‐IRE1α (14C10, 1:1,000; Cell Signaling Technology), anti‐p‐IRE1α (EPR5253, 1:1,000; Abcam), anti‐PERK (C33E10, 1:1,000; Cell Signaling Technology), anti‐eIF2α (D7D3, 1:1,000; Cell Signaling Technology), anti‐p‐eIF2α (D7D3, 1:1,000; Cell Signaling Technology), goat anti‐rabbit IgG (1:10,000; Sigma‐Aldrich), and goat anti‐mouse IgG (1:10,000, Sigma‐Aldrich). Anti‐p‐Ero1α recognizing pSer145 was purified from rabbit serum on immunization with a synthesized keyhole limpet hemocyanin (KLH)‐conjugated peptide DESL(pS)EETQKAVLQC, and anti‐Fam20C was purified from rabbit serum on immunization with a synthesized KLH‐conjugated peptide CHSVVDDDLDTEHRAASAR.
Immunofluorescence
HepG2 cells were fixed with 4% formaldehyde for 15 min, permeabilized with 0.1% Triton X‐100 for 5 min and blocked with 1% BSA for 30 min at room temperature. The cells were incubated with primary antibody and secondary antibody, each for 1 h. The antibodies used for immunofluorescence were as follows: anti‐p‐Ero1α (1:50), anti‐HA (1:500), anti‐Flag (1:1,000), anti‐PDI (1:500), anti‐GM130 (1:250), goat anti‐rabbit Alexa Fluor 488 (1:1,000; Invitrogen), goat anti‐mouse Alexa Fluor 568 (1:1,000; Invitrogen). Hoechst 33258 (1:10,000; Sigma‐Aldrich) was used to stain the nuclear DNA.
J‐chain folding assay
J‐chain folding assay was conducted as described (Wang et al, 2014). Myc‐tagged human immunoglobulin J‐chain (JcM) was co‐expressed with Ero1α‐HA or Fam20C‐Flag in HeLa cells for 24 h. Harvested cells were incubated with 10 mM DTT in DMEM at 37°C for 5 min to fully reduce JcM protein, instantly washed twice with ice‐cold PBS to remove residual DTT, and re‐suspended in DMEM at 20°C. Aliquots were taken and immediately blocked with 20 mM N‐ethylmaleimide (NEM) at different time points. Cells were lysed in RIPA buffer containing 1 mM PMSF, phosphatase and protease inhibitor cocktails and 40 mM NEM. To observe the redox state of J‐chain, the supernatants were resolved by non‐reducing SDS–PAGE and visualized by immunoblotting with anti‐myc.
Determination of ER redox with superfolded‐roGFP‐iEER
HeLa cells were transfected with superfolded‐roGFP‐iEER or co‐transfected with Ero1α‐myc or Fam20C‐Flag for 48 h. Harvested cells were washed twice in Hanks’ balanced salt solution (HBSS; Gibco) and seeded onto a flat‐bottom 96‐well plate. The fluorescence intensities at 525 nm were measured with excitation at 390 and 465 nm, and the fluorescence ratio from excitation at 390 versus 465 nm was calculated.
λ‐phosphatase assay
ConA precipitates from conditioned medium of HeLa cells transiently expressing Ero1α were treated with 100 or 200 units λ‐phosphatase (λ‐PP; New England Biolabs) in 50 μl buffer (50 mM HEPES, pH 7.5, 100 mM NaCl, 2 mM DTT, and 0.01% Brij 35) with 10 mM MnCl2 at 30°C for 1 h, followed by immunoblotting.
Mouse mammary gland protein and RNA isolation
Mammary glands were taken from virgin or lactating C57BL/6 female mice at 14–17 weeks and homogenized in RIPA buffer containing 1 mM PMSF, phosphatase, and protease inhibitors cocktails with gentle stir in a microhomogenizer (Bio‐gen Pro200). The homogenate was centrifuged at 13,000 × g for 15 min, and the supernatant was mixed with a reducing loading buffer and analyzed by SDS–PAGE and immunoblotting. Total RNA was isolated from lactating or non‐lactating mammary glands using TRIzol reagent (Invitrogen).
Quantitative real‐time polymerase chain reaction (RT–qPCR)
RNA products were reverse‐transcribed to cDNA using GoScript Reverse Transcription System (Promega). RT–qPCR was performed using SYBR Select Master Mix (Applied Biosystems) and QuantStudio 7 Flex machine (Applied Biosystems) according to the manufacturer's instructions. The expression level of the target genes was calculated as 2−ΔΔCT and normalized to that of GAPDH. All primers used are listed in Table EV1.
Generation of FAM20C knockout and ERO1A knockout HeLa cell lines by CRISPR/Cas9 genome editing
The 20‐nt guide sequences targeting human FAM20C and ERO1A were designed using the clustered regularly interspaced short palindromic repeats (CRISPR) design tool at http://www.genome-engineering.org/crispr and cloned into the expression vector pSpCas9(BB)‐2A‐GFP (PX458) containing human codon optimized Cas9, the RNA components, and GFP (a gift from Feng Zhang, Addgene plasmid # 48138).
The guide sequences targeting exon 1 of human FAM20C and human ERO1A are shown below.
FAM20C
5′‐GGGCTGCGCGCACGAACAGC‐3′ (clone 3 and 5)
ERO1A
5′‐CAAACAAGAATCCCCAGCCG‐3′ (clone 10 and 12)
HeLa cells were transfected with pSpCas9(BB)‐2A‐GFP vector containing the single‐guide RNAs (sgRNAs) by Viafect for 48 h, and GFP‐positive cells were single‐cell‐sorted by fluorescence‐activated cell sorting (BD Influx) into a 96‐well plate format into DMEM supplemented with 10% FBS. Expanded single clones were screened for Fam20C and Ero1α by protein immunoblotting. Genomic DNA (gDNA) was purified from clones using the TransDirect Animal Tissue PCR Kit (TransGen), and the region surrounding the protospacer adjacent motif (PAM) was amplified using the following primers.
FAM20C.
Forward: 5′‐CGCGCGGCCATGAAGATGAT‐3′
Reverse: 5′‐TTCTCCAGCGAGTGGGACGAGAGGT‐3′
ERO1A.
Forward: 5′‐GATCGCTGAGAGGCAGGA‐3′
Reverse: 5′‐AGAAGCACCTCTGTGCCG‐3′
PCR products were purified using a gel extraction kit (Omega) and cloned into pEASY‐T5 Zero cloning vector (TransGen). To determine the indels of individual alleles, eight to 16 bacterial colonies were expanded and the plasmid DNA was purified and sequenced (Invitrogen).
Circular dichroism spectroscopy
Circular dichroism spectrum of 0.2 mg/ml recombinant Ero1α WT or Ero1α S145E in 50 mM sodium phosphate buffer (pH 7.0) was measured from 190 to 260 nm at 25°C by Chirascan Plus (Applied Photophysics). Scanning speed was 2.5 s/dot.
Intrinsic fluorescence assay
Fluorescence spectrum of 2 μM recombinant Ero1α WT or Ero1α S145E in protein store buffer (50 mM Tris–HCl, pH 7.6, 150 mM NaCl, 2 mM EDTA) from 305 to 405 nm was measured with excitation at 295 nm at 25°C using Hitachi F7000. Scanning speed was 60 nm/min.
Ero1α activation assay
Recombinant Ero1α C99A/C104A/C166A or Ero1α C99A/C104A/C166A/S145E was incubated with 50 mM potassium ferricyanide for 1 h at 25°C to prepare the oxidized Ero1α protein. After chromatograph, 1 μM monomeric oxidized Ero1α was mixed with 10 μM reduced PDI, and aliquots were taken at indicated times and immediately quenched with 20 mM NEM.
Ethics
Animal experiments were conducted with the approval of the Institutional Biomedical Research Ethics Committee of the Institute of Biophysics, Chinese Academy of Science.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Vizcaino et al, 2016) partner repository with the dataset identifier PXD009333.
Author contributions
LW conceived the project. JZ, JX, CCW, and LW designed research; JZ, QZ, XEW, JY, XC, JW, and XW performed research; JZ, JX, and LW analyzed data; JZ, CCW, and LW wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Dataset EV1
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Acknowledgements
We acknowledge Junjie Hu and Yanfang Wu for assisting in subcellular fractionation, Likun Wang for providing a hypoxic chamber, Yuhao Gao, Junjie Hou, Jingqi Fang, Yunpeng Zhai, Mengmeng Zhang, and Yan Teng for technical assistance. This work was supported by National Key R&D Program of China (2016YFA0500200, 2017YFA0504000), National Natural Science Foundation of China (31571163, 31771261, 31370758), and Youth Innovation Promotion Association CAS to LW and XW.
The EMBO Journal (2018) 37: e98699
References
- Anelli T, Alessio M, Bachi A, Bergamelli L, Bertoli G, Camerini S, Mezghrani A, Ruffato E, Simmen T, Sitia R (2003) Thiol‐mediated protein retention in the endoplasmic reticulum: the role of ERp44. EMBO J 22: 5015–5022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anelli T, Bergamelli L, Margittai E, Rimessi A, Fagioli C, Malgaroli A, Pinton P, Ripamonti M, Rizzuto R, Sitia R (2012) Ero1 alpha regulates Ca2+ fluxes at the endoplasmic reticulum‐mitochondria interface (MAM). Antioxid Redox Signal 16: 1077–1087 [DOI] [PubMed] [Google Scholar]
- Appenzeller‐Herzog C, Riemer J, Christensen B, Sorensen ES, Ellgaard L (2008) A novel disulphide switch mechanism in Ero1 alpha balances ER oxidation in human cells. EMBO J 27: 2977–2987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker KM, Chakravarthi S, Langton KP, Sheppard AM, Lu H, Bulleid NJ (2008) Low reduction potential of Ero1 alpha regulatory disulphides ensures tight control of substrate oxidation. EMBO J 27: 2988–2997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battle DM, Gunasekara SD, Watson GR, Ahmed EM, Saysell CG, Altaf N, Sanusi AL, Munipalle PC, Scoones D, Walker J, Viswanath Y, Benham AM (2013) Expression of the endoplasmic reticulum oxidoreductase Ero1 alpha in gastro‐intestinal cancer reveals a link between homocysteine and oxidative protein folding. Antioxid Redox Signal 19: 24–35 [DOI] [PubMed] [Google Scholar]
- Birk J, Meyer M, Aller I, Hansen HG, Odermatt A, Dick TP, Meyer AJ, Appenzeller‐Herzog C (2013) Endoplasmic reticulum: reduced and oxidized glutathione revisited. J Cell Sci 126: 1604–1617 [DOI] [PubMed] [Google Scholar]
- Cabibbo A, Pagani M, Fabbri M, Rocchi M, Farmery MR, Bulleid NJ, Sitia R (2000) ERO1‐L, a human protein that favors disulfide bond formation in the endoplasmic reticulum. J Biol Chem 275: 4827–4833 [DOI] [PubMed] [Google Scholar]
- Chin K, Kang G, Qu J, Gardner LB, Coetzee WA, Zito E, Fishman GI, Ron D (2011) The sarcoplasmic reticulum luminal thiol oxidase ERO1 regulates cardiomyocyte excitation‐coupled calcium release and response to hemodynamic load. FASEB J 25: 2583–2591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P (2002) The origins of protein phosphorylation. Nat Cell Biol 4: E127–E130 [DOI] [PubMed] [Google Scholar]
- Cui J, Xiao J, Tagliabracci VS, Wen J, Rahdar M, Dixon JE (2015) A secretory kinase complex regulates extracellular protein phosphorylation. eLife 4: e06120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaunay‐Moisan A, Ponsero A, Toledano MB (2017) Reexamining the function of glutathione in oxidative protein folding and secretion. Antioxid Redox Signal 27: 1178–1199 [DOI] [PubMed] [Google Scholar]
- Fass D, Thorpe C (2018) Chemistry and enzymology of disulfide cross‐linking in proteins. Chem Rev 118: 1169–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilady SY, Bui M, Lynes EM, Benson MD, Watts R, Vance JE, Simmen T (2010) Ero1 alpha requires oxidizing and normoxic conditions to localize to the mitochondria‐associated membrane (MAM). Cell Stress Chaperones 15: 619–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross E, Kastner DB, Kaiser CA, Fass D (2004) Structure of Ero1p, source of disulfide bonds for oxidative protein folding in the cell. Cell 117: 601–610 [DOI] [PubMed] [Google Scholar]
- Gross E, Sevier CS, Heldman N, Vitu E, Bentzur M, Kaiser CA, Thorpe C, Fass D (2006) Generating disulfides enzymatically: reaction products and electron acceptors of the endoplasmic reticulum thiol oxidase Ero1p. Proc Natl Acad Sci USA 103: 299–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarsten O (1883) Zur frage ob caseín ein einheitlicher stoff sei. Hoppe Seylers Z Physiol Chem 7: 227–273 [Google Scholar]
- Hogg PJ (2003) Disulfide bonds as switches for protein function. Trends Biochem Sci 28: 210–214 [DOI] [PubMed] [Google Scholar]
- Hoseki J, Oishi A, Fujimura T, Sakai Y (2016) Development of a stable ERroGFP variant suitable for monitoring redox dynamics in the ER. Biosci Rep 36: e00316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba K, Masui S, Iida H, Vavassori S, Sitia R, Suzuki M (2010) Crystal structures of human Ero1 alpha reveal the mechanisms of regulated and targeted oxidation of PDI. EMBO J 29: 3330–3343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa HO, Xu A, Ogura E, Manning G, Irvine KD (2012) The Raine syndrome protein Fam20C is a Golgi kinase that phosphorylates bio‐mineralization proteins. PLoS ONE 7: e42988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Sideris DP, Sevier CS, Kaiser CA (2012) Balanced Ero1 activation and inactivation establishes ER redox homeostasis. J Cell Biol 196: 713–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukita K, Tamura Y, Tanaka T, Kajiwara T, Kutomi G, Saito K, Okuya K, Takaya A, Kanaseki T, Tsukahara T, Hirohashi Y, Torigoe T, Furuhata T, Hirata K, Sato N (2015) Cancer‐associated oxidase ERO1‐alpha regulates the expression of MHC class I molecule via oxidative folding. J Immunol 194: 4988–4996 [DOI] [PubMed] [Google Scholar]
- Kutomi G, Tamura Y, Tanaka T, Kajiwara T, Kukita K, Ohmura T, Shima H, Takamaru T, Satomi F, Suzuki Y, Torigoe T, Sato N, Hirata K (2013) Human endoplasmic reticulum oxidoreductin 1‐alpha is a novel predictor for poor prognosis of breast cancer. Cancer Sci 104: 1091–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Mongillo M, Chin KT, Harding H, Ron D, Marks AR, Tabas I (2009) Role of ERO1‐alpha‐mediated stimulation of inositol 1,4,5‐triphosphate receptor activity in endoplasmic reticulum stress‐induced apoptosis. J Cell Biol 186: 783–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino M, Stoilova T, Giorgi C, Bachi A, Cattaneo A, Auricchio A, Pinton P, Zito E (2015) SEPN1, an endoplasmic reticulum‐localized selenoprotein linked to skeletal muscle pathology, counteracts hyperoxidation by means of redox‐regulating SERCA2 pump activity. Hum Mol Genet 24: 1843–1855 [DOI] [PubMed] [Google Scholar]
- May D, Itin A, Gal O, Kalinski H, Feinstein E, Keshet E (2005) Ero1‐L alpha plays a key role in a HIF‐1‐mediated pathway to improve disulfide bond formation and VEGF secretion under hypoxia: implication for cancer. Oncogene 24: 1011–1020 [DOI] [PubMed] [Google Scholar]
- Mezghrani A, Fassio A, Benham A, Simmen T, Braakman I, Sitia R (2001) Manipulation of oxidative protein folding and PDI redox state in mammalian cells. EMBO J 20: 6288–6296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu Y, Zhang L, Yu J, Wang CC, Wang L (2016) Novel roles of the non‐catalytic elements of yeast protein‐disulfide isomerase in its interplay with endoplasmic reticulum oxidoreductin 1. J Biol Chem 291: 8283–8294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollak AJ, Haghighi K, Kunduri S, Arvanitis DA, Bidwell PA, Liu G, Singh VP, Gonzalez DJ, Sanoudou D, Wiley SE, Dixon JE, Kranias EG (2017) Phosphorylation of serine96 of histidine‐rich calcium‐ binding protein by the Fam20C kinase functions to prevent cardiac arrhythmia. Proc Natl Acad Sci USA 114: 9098–9103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raine J, Winter RM, Davey A, Tucker SM (1989) Unknown syndrome: microcephaly, hypoplastic nose, exophthalmos, gum hyperplasia, cleft‐palate, low set ears, and osteosclerosis. J Med Genet 26: 786–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramming T, Appenzeller‐Herzog C (2012) The physiological functions of mammalian endoplasmic oxidoreductin 1: on disulfides and more. Antioxid Redox Signal 16: 1109–1118 [DOI] [PubMed] [Google Scholar]
- Sevier CS, Qu H, Heldman N, Gross E, Fass D, Kaiser CA (2007) Modulation of cellular disulfide‐bond formation and the ER redox environment by feedback‐regulation of Ero1. Cell 129: 333–344 [DOI] [PubMed] [Google Scholar]
- Sevier CS, Kaiser CA (2008) Ero1 and redox homeostasis in the endoplasmic reticulum. Biochim Biophys Acta‐Mol Cell Res 1783: 549–556 [DOI] [PubMed] [Google Scholar]
- Tagliabracci VS, Engel JL, Wen J, Wiley SE, Worby CA, Kinch LN, Xiao J, Grishin NV, Dixon JE (2012) Secreted kinase phosphorylates extracellular proteins that regulate biomineralization. Science 336: 1150–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliabracci VS, Pinna LA, Dixon JE (2013) Secreted protein kinases. Trends Biochem Sci 38: 121–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliabracci VS, Wiley SE, Guo X, Kinch LN, Durrant E, Wen J, Xiao J, Cui J, Nguyen KB, Engel JL, Coon JJ, Grishin N, Pinna LA, Pagliarini DJ, Dixon JE (2015) A single kinase generates the majority of the secreted phosphoproteome. Cell 161: 1619–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Kutomi G, Kajiwara T, Kukita K, Kochin V, Kanaseki T, Tsukahara T, Hirohashi Y, Torigoe T, Okamoto Y, Hirata K, Sato N, Tamura Y (2016) Cancer‐associated oxidoreductase ERO1‐alpha drives the production of VEGF via oxidative protein folding and regulating the mRNA level. Br J Cancer 114: 1227–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Kutomi G, Kajiwara T, Kukita K, Kochin V, Kanaseki T, Tsukahara T, Hirohashi Y, Torigoe T, Okamoto Y, Hirata K, Sato N, Tamura Y (2017) Cancer‐associated oxidoreductase ERO1‐alpha promotes immune escape through up‐regulation of PD‐L1 in human breast cancer. Oncotarget 8: 24706–24718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu BP, Weissman JS (2002) The FAD‐ and O(2)‐dependent reaction cycle of Ero1‐mediated oxidative protein folding in the endoplasmic reticulum. Mol Cell 10: 983–994 [DOI] [PubMed] [Google Scholar]
- Tu BP, Weissman JS (2004) Oxidative protein folding in eukaryotes: mechanisms and consequences. J Cell Biol 164: 341–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vavassori S, Cortini M, Masui S, Sannino S, Anelli T, Caserta IR, Fagioli C, Mossuto MF, Fornili A, van Anken E, Degano M, Inaba K, Sitia R (2013) A pH‐regulated quality control cycle for surveillance of secretory protein assembly. Mol Cell 50: 783–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitu E, Kim S, Sevier CS, Lutzky O, Heldman N, Bentzur M, Unger T, Yona M, Kaiser CA, Fass D (2010) Oxidative activity of yeast Ero1p on protein disulfide isomerase and related oxidoreductases of the endoplasmic reticulum. J Biol Chem 285: 18155–18165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaino JA, Csordas A, Del‐Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez‐Riverol Y, Reisinger F, Ternent T, Xu QW, Wang R, Hermjakob H (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res 44: 11033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wang L, Vavassori S, Li S, Ke H, Anelli T, Degano M, Ronzoni R, Sitia R, Sun F, Wang CC (2008) Crystal structure of human ERp44 shows a dynamic functional modulation by its carboxy‐terminal tail. EMBO Rep 9: 642–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Li S, Sidhu A, Zhu L, Liang Y, Freedman RB, Wang CC (2009) Reconstitution of human Ero1‐L alpha/protein‐disulfide isomerase oxidative folding pathway in vitro. Position‐dependent differences in role between the a and a′ domains of protein‐disulfide isomerase. J Biol Chem 284: 199–206 [DOI] [PubMed] [Google Scholar]
- Wang L, Zhu L, Wang CC (2011) The endoplasmic reticulum sulfhydryl oxidase Ero1 beta drives efficient oxidative protein folding with loose regulation. Biochem J 434: 113–121 [DOI] [PubMed] [Google Scholar]
- Wang L, Zhang L, Niu Y, Sitia R, Wang CC (2014) Glutathione peroxidase 7 utilizes hydrogen peroxide generated by Ero1 alpha to promote oxidative protein folding. Antioxid Redox Signal 20: 545–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wang X, Wang CC (2015) Protein disulfide‐isomerase, a folding catalyst and a redox‐regulated chaperone. Free Radic Biol Med 83: 305–313 [DOI] [PubMed] [Google Scholar]
- Wu Y, Li X, Jia J, Zhang Y, Li J, Zhu Z, Wang H, Tang J, Hu J (2018) Transmembrane E3 ligase RNF183 mediates ER stress‐induced apoptosis by degrading Bcl‐xL. Proc Natl Acad Sci USA 115: E2762–E2771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao J, Tagliabracci VS, Wen J, Kim SA, Dixon JE (2013) Crystal structure of the Golgi casein kinase. Proc Natl Acad Sci USA 110: 10574–10579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Li DF, Wang X, Liang J, Sitia R, Wang CC, Wang X (2016) Crystal structure of the ERp44‐peroxiredoxin 4 complex reveals the molecular mechanisms of thiol‐mediated protein retention. Structure 24: 1755–1765 [DOI] [PubMed] [Google Scholar]
- Zhang L, Niu Y, Zhu L, Fang J, Wang X, Wang L, Wang CC (2014) Different interaction modes for protein‐disulfide isomerase (PDI) as an efficient regulator and a specific substrate of endoplasmic reticulum oxidoreductin‐1alpha (Ero1alpha). J Biol Chem 289: 31188–31199 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Dataset EV1
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Vizcaino et al, 2016) partner repository with the dataset identifier PXD009333.