

Abstract
Oligonucleotide therapies are currently experiencing a resurgence driven by advances in backbone chemistry and discoveries of novel therapeutic pathways that can be uniquely and efficiently modulated by the oligonucleotide drugs. A quarter of a century has passed since oligonucleotides were first applied in living mammalian brain to modulate gene expression. Despite challenges in delivery to the brain, multiple oligonucleotide-based compounds are now being developed for treatment of human brain disorders by direct delivery inside the blood brain barrier (BBB). Notably, the first new central nervous system (CNS)-targeted oligonucleotide-based drug (nusinersen/Spinraza) was approved by US Food and Drug Administration (FDA) in late 2016 and several other compounds are in advanced clinical trials. Human testing of brain-targeted oligonucleotides has highlighted unusual pharmacokinetic and pharmacodynamic properties of these compounds, including complex active uptake mechanisms, low systemic exposure, extremely long half-lives, accumulation and gradual release from subcellular depots. Further work on oligonucleotide uptake, development of formulations for delivery across the BBB and relevant disease biology studies are required for further optimization of the oligonucleotide drug development process for brain applications.
Nucleic acid-based therapies, seemingly deprioritized in the early 2010s when major pharmaceutical companies, including Roche (Basel, Switzerland), Pfizer (New York), Abbott (now AbbVie; Deerfield, IL, USA) and Merck (Kenilworth, NJ, USA) shuttered their oligonucleotide programs, are once again becoming more mainstream in drug development. Oligonucleotide-based drugs nusinersen (Spinraza), mipomersen (Kynamro) and eteplirsen (Exondys 51) have been recently approved by the FDA for the treatment of spinal muscular atrophy, homozygous familial hypercholesterolemia and Duchenne muscular dystrophy, respectively (http://www.accessdata.fda.gov/scripts/cder/daf/index.cfm). With a growing cadre of oligonucleotide-focused companies (Fig. 1), hundreds of clinical trials of diverse oligonucleotide types are being developed to target disease. This resurgence is mostly due to discoveries of novel oligonucleotide chemistries, delivery agents and greater understanding of mechanisms of action of oligonucleotides, as well as improved knowledge of disease mechanisms, all of which has opened opportunities to address novel therapeutic targets previously deemed undruggable.
Figure 1.
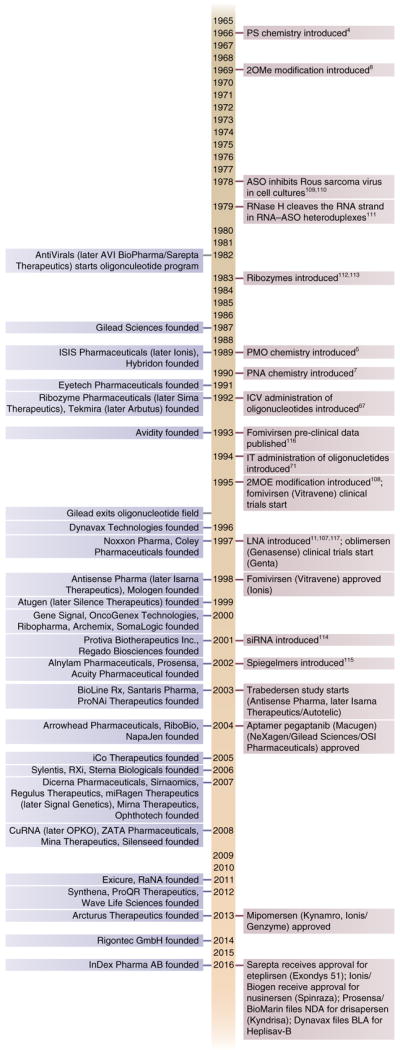
Selected milestones from the history of ODN drug development. The list shows on the left selected companies mentioned in the text, having compounds in clinical trials or employing novel mechanisms of action of ODNs; on the right are shown key milestones in the development of ODN therapies.
Notably, despite difficulties in delivering oligonucleotides to the CNS, interest is growing in developing these treatments for disorders of the nervous system. Possible reasons for this interest include high target specificity of oligonucleotides, an ability to address previously inaccessible drug targets, limited systemic exposure and toxicity, as well as extended half-life permitting infrequent dosing. In addition to downregulation of disease-relevant genes, oligonucleotides offer multiple approaches for precise gene upregulation and splice editing, goals that are not easily achieved by traditional small-molecule therapies or monoclonal antibodies. At the same time, oligonucleotide effects are reversible; thus, oligonucleotides do not have many of the problems associated with the more permanent alterations used in gene therapy, another possible approach to modulating gene activity or correcting mutations. An additional advantage of oligonucleotide drugs, which makes them particularly suited to the treatment of orphan diseases, is their simplified development cycle, due to uniform class chemistry and a more straightforward candidate selection process.
There are, however, substantial obstacles for the application of oligonucleotides in CNS disorders. First and foremost, oligonucleotides do not readily cross the BBB. Multiple studies have shown that fewer than 1% of systemically delivered oligonucleotides reach the brain1,2. The current solution to this problem is intracerebroventricular (ICV) or intrathecal (IT) delivery. As these methods are invasive and require office visits burdensome for patients, a large amount of work, now largely in the investigational stage, is also being carried out to develop chemical modifications and/or vehicles that will facilitate BBB crossing with systemic, intranasal or oral administration (see below and ref. 3).
Another difficulty encountered in the application of oligonucleotides to CNS indications is their unconventional pharmacokinetic (PK) and pharmacodynamic (PD) properties, which remain poorly studied but have far-reaching implications for dosing regimens and efficacy in the clinic. Furthermore, as a consequence of their engagement of novel disease targets, the mechanisms of oligonucleotide action are not yet fully understood. To illustrate the complexity of the molecular processes that could be modulated by oligonucleotides, we review examples of CNS disorders that can be addressed by oligonucleotide drugs.
Chemically, oligonucleotide drugs fall into two categories: double- stranded RNA (dsRNA)-based drugs (e.g., short interfering RNA: siRNA) and single-stranded DNA (ssDNA)-based drugs (e.g., oligodeoxyribonucleotides: ODNs). Initially, these two groups were associated with the two mechanisms of oligonucleotide action known at the time, RNA interference (RNAi) and RNase-H-mediated degradation. The corresponding terms ‘RNAi therapy’ and ‘antisense therapy’ were perceived as describing drugs employing these two mechanisms. However, in recent years, this association has been gradually fading with the discovery of additional novel biological mechanisms that can be addressed by oligonucleotides (e.g., ODNs that bind receptors or work through steric hindrance mechanisms to affect splicing or inhibit microRNA (miRNA)). As the usage of these terms is becoming more inclusive, their meaning has essentially merged and expanded to denote all oligonucleotide-based compounds. In the following review, we use ‘oligonucleotide-based compounds/therapies’ as they do not imply any particular mechanism. At the same time, there is a point in discussing oligonucleotide drugs as a group because, even if they might not share the mechanisms of action, they do share many manufacturing, quality control, cell uptake, PK, PD and toxicological aspects.
The therapeutic oligonucleotide field is now becoming quite large, with over 200 clinical trials registered at ClinicalTrials.gov and over 10,000 patients treated with oligonucleotide drugs by the end of 2016. In the following review, we place greatest emphasis on late-stage aspects of ssDNA ODN-based drug development for disorders of the CNS.
Advances in ODN chemistry
A major breakthrough in the development of oligonucleotide drugs came in 1966 (Fig. 1) with the introduction of the phosphorothioate backbone modification4, which replaces one of the oxygens in the phosphate residue with sulfur (Fig. 2). Phosphorothioate modification increases nuclease resistance and facilitates association with carrier proteins in the blood, leading to slower excretion through kidneys, longer half-life and increased absorption3. Other backbone modifications, including phosphorodiamidate morpholino (PMO)5, tetramethyl phosphoryl guanidine6 and peptide nucleic acids (PNAs)7, were proposed, alone or in combination with the sugar modifications (Fig. 2). These modifications decrease the charge on the oligonucleotide backbone, which may facilitate their uptake into cells.
Figure 2.
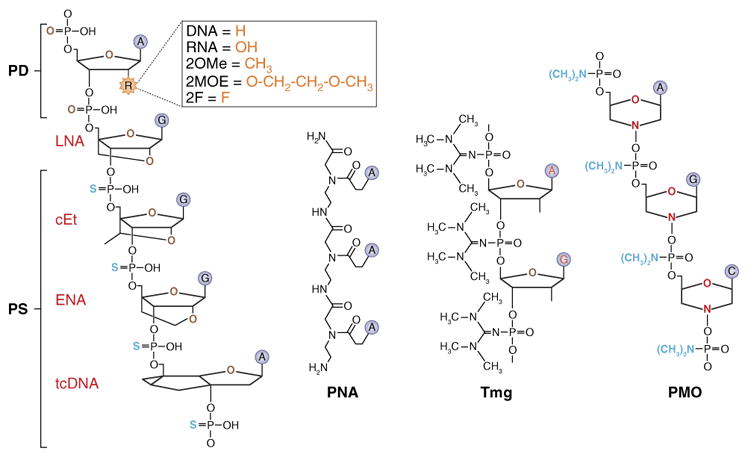
Oligonucleotide modifications. Backbone modifications include the following: PD, phosphodiester bond; PS, phosphorothioate (one of the oxygens in the phosphate moiety is replaced with sulfur)4; PNA, peptide nucleic acid (entire sugar phosphate backbone is replaced with polyamide linkage)7; Tmg, internucleotide phosphate modified with a tetramethyl phosphoryl guanidine group6; PMO, phosphorodiamidate morpholino oligonucleotide, or morpholino (backbone composed of methylenemorpholine rings and phosphorodiamidate linkages)5. Sugar modifications include the following: DNA, hydrogen at the 2′ position of the sugar moiety; RNA, OH group at the 2′ position of the sugar moiety; 2OMe, 2′-O-methyl (a methyl substitution in the 2′ position of the sugar moiety)8; 2MOE, 2′-O-methoxyethyl (a methoxyethyl substitution in the 2′ position of the sugar moiety)107; 2F, 2′-fluoro substitution in the 2′ position of the sugar moiety; LNA, locked nucleic acid (an extra bridge between 2′ oxygen and 4′ carbon of the sugar moiety)10,11; cEt, constrained ethyl (an ethyl bridge between 2′ oxygen and 4′ carbon of the sugar moiety); ENA, 2′-O,4′-C-ethylene-bridged nucleic acid (an ethylene bridged at the furanose sugar ring at 2′-O and 4′-C ends); tcDNA, tricyclo-DNA (3 additional C-atoms between C(5′) and C(3′) of the sugar moiety)108; ZNAs, zip nucleic acids, oligonucleotides conjugated with oligospermine chains; XNAs, polymers formed from building blocks not found in nature that mimic many of the properties of RNA and DNA.
Modifications of the backbone, although highly useful in improving in vivo characteristics of the oligonucleotide drugs, introduce multiple chiral centers into oligonucleotide molecules. As a consequence, the standard synthesis methods produce mixtures of hundreds of thousands of stereoisomers, only some of which are capable of efficient interaction with DNA or RNA. Wave Life Sciences (Cambridge, MA, USA) has developed a method allowing the synthesis of stereopure oligonucleotides, which could increase the potency of the oligonucleotide drugs.
Modifications of the sugar moiety have further improved RNA/DNA binding affinity, and bioavailability, and decreased immunostimulatory effects and toxicity of oligonucleotide drugs. Several oligonucleotides with these chemistries are now in clinical trials for CNS disorders (see below and Table 1). 2′-modified phosphorothioate oligonucleotides (Fig. 2) may be particularly adaptable for CNS disorders, given their long half-life, with effects in the brain lasting up to 6 months following a single injection3,8–10. In another type of sugar moiety modification, locked nucleic acids (LNAs), a bridge is introduced that connects the 2′ oxygen and 4′ carbon. This modification substantially elevates the melting temperature of the LNA–DNA and LNA–RNA hybrids, thus allowing the creation of shorter ODN-based compounds with increased bioavailability and reduced manufacturing costs11. A recently proposed tricyclo-DNA, a conformationally constrained oligonucleotide analog, has three additional C-atoms between C(5′) and C(3′) of the sugar (Fig. 2). This modification increases stability, hydrophobicity and RNA affinity, and improves tissue uptake and BBB permeability12.
Table 1.
Selected therapeutic oligonucleotide modalities in development for CNS indications
| Drug | Company/partner | Indication | Current phase | Modality | Target | Route of administration | Orphan |
|---|---|---|---|---|---|---|---|
| IONIS-SOD1Rx | Biogen/Ionis Pharmaceuticals | Amyotrophic lateral sclerosis | Phase 1/2 | ODN, knockdown | SOD1 | Intrathecal | Yes |
| ISIS-HTTRx | Roche/Ionis Pharmaceuticals | Huntington’s disease | Phase 2/3 | ODN, knockdown | Huntingtin (HTT) | Intrathecal | Yes |
| ATL1102 (ISIS 107248) | Antisense Therapeutics (Melbourne, Australia)/Ionis Pharmaceuticals | Multiple sclerosis | Phase 2 | ODN, knockdown | Integrin alpha-4 beta-1/VLA-4 | Inhaled, subcutaneous | No |
| Patisiran | Alnylam Pharmaceuticals | Familial amyloid neuropathy | Phase 3 | siRNA, knockdown | TTR | IV | Yes |
| Nusinersen | Biogen/Ionis Pharmaceuticals | Spinal muscular atrophy | Approved | ODN, exon skipping | Survival of motor neuron (SMN) | Intrathecal | Yes |
Currently, multiple ODN types are being developed for the same disease, with different modifications frequently combined in one molecule. For example, ODNs inducing (survival of motor neuron 2) SMN2 exon 7 inclusion, currently proposed for spinal muscular atrophy (SMA), include phosphorothioate 2MOE13,14, phosphorothioate 2OMe15 and PMO chemistries16,17. Because of the complex sequence-structure-activity relationships of the oligonucleotide drugs, it is not known whether any of these chemistries are preferable for CNS applications.
Mechanisms of action
Current oligonucleotide-based drugs employ multiple mechanisms of action that are frequently not mutually exclusive. These compounds also have the advantage of being able to address biological processes less amenable to interventions by small molecules. We briefly describe these mechanisms below, then give detailed disease examples of specific oligonucleotide therapies in the “ODNs in CNS diseases” section.
RNA knockdown
Knockdown of mRNA via oligonucleotide molecules historically was one of the first mechanisms engaged by oligonucleotide drugs. Although small-molecule inhibitors of multiple protein functions were known, oligonucleotides provided higher target specificity and a possibility to downregulate the expression of ‘non-druggable’ proteins or proteins with unknown function. These aspects fueled the interest in oligonucleotides as therapeutic agents, despite their large molecular size and inherent delivery problems.
In the case of dsRNA oligonucleotides, mRNA knockdown is mediated by RISC (RNA-induced silencing complex) complex. The first siRNAs were easily degraded by nucleases and poorly absorbed by the cells, which necessitated the use of liposome carriers. Since that time, less toxic and more bioavailable carriers, such as biodegradable nanopolymers, and sophisticated modifications of the siRNA molecule itself, including phosphorothioate, 2OMe, LNA, as well as conjugation with carrier molecules (e.g., polyethylene glycol (PEG), peptides, lipids and glycosides), have been developed18. Furthermore, multiple approaches to introduce siRNA in vivo, including plasmid and viral expression of short hairpin RNAs in target cells, have been proposed, although the synthetic molecules represent the shorter route to the clinic. Although these efforts resulted in multiple dsRNA drugs entering clinical trials in recent years19, we do not address the achievements of the dsRNA field here owing to space constraints and instead concentrate on the more clinically advanced ssDNA–ODN drugs. The therapeutic siRNA field has recently been reviewed19–21.
ODNs are widely used to achieve highly specific gene knockdown, most often through interaction with the RNase H mechanism. RNase H recognizes an RNA–DNA heteroduplex and cleaves its RNA strand, releasing an intact ODN. The mRNA cleavage products downstream of the initial cleavage site contain intact polyA tails and are bound by the polyA-binding protein. They are further degraded by cytoplasmic 5′ to 3′ exoRNase XRN1 (ref. 22). The mRNA cleavage products upstream from the initial cleavage site are further degraded by the exosome complex containing Dis3 or cytoplasmic Dis3L1 nucleases. In the case of nuclear long-noncoding RNAs (lncRNAs) and pre-mRNAs, the downstream cleavage products are degraded by nuclear exoRNase XRN2 (ref. 22). The upstream cleavage products of the lncRNAs are degraded in the 3′ to 5′ direction by the exosome complex containing the nuclear exoRNase Dis3 (ref. 22).
As RNase H is not efficient on nucleotides with most types of 2′ sugar modifications, for knockdown applications these modifications are usually arranged in ‘gapmer’ configuration (with a central ‘gap’ of 8–10 unmodified nucleotides and 3- to 5-nucleotide-long 2′-modified ‘wings’)3.
Both RISC and RNase H mechanisms can be employed for allele-specific knockdown when they are targeted to sequences that differentiate the alleles. For example, Miller et al.23 used single-nucleotide polymorphisms (SNPs) known to segregate with the mutant Machado–Joseph disease (also known as spinocerebellar ataxia type 3) allele, which contains CAG repeat expansion, to design an siRNA. SNP sites were used because of the difficulty designing the oligonucleotides to the repeat sequence itself. The siRNA treatment reduced the expression of the mutant, but not wild-type (WT) allele of ATXN3 (ref. 23).
Another mechanism that can be used for RNA knockdown is the induction of aberrant exon skipping, which leads to RNA degradation through nonsense-mediated decay (NMD). For example, Ward et al.24 have shown that transfection of ODNs that do not support RNase H degradation and are complementary to constitutive exons in Stat3 and Sod1 transcripts induces exon skipping and generation of premature termination codon (PTC)-containing mRNAs, which are then degraded by the NMD pathway. PTC-containing mRNAs were recognized by the ATP-dependent RNA helicase Upf1 (up frameshift 1), cleaved by the Smg6 nonsense-mediated mRNA decay factor and finally degraded by the cytoplasmic exonuclease Xrn1. Transfection with siRNA against the nuclear 5′-to-3′ exonuclease Xrn2 did not attenuate the ODN-induced Stat3 mRNA degradation. In addition to exon skipping, ODN treatment caused intron retention and reduction of chromatin-associated Stat3 mRNA24.
Exon skipping via modulation of pre-mRNA splicing
Although small-molecule drugs inducing point mutation read-through are known—for example, ataluren (also known as PTC124) is currently in clinical trials for Duchenne muscular dystrophy25 and cystic fibrosis26—they have low target specificity and may thus be associated with an elevated risk for side effects. Oligonucleotides, on the other hand, can be designed to specifically modulate splicing of a single protein, reducing the risk of off-target toxicity. In this mechanism, oligonucleotides are designed to bind in the area of the mutated exons, causing their exclusion from the final spliced mRNA version. The exact mechanisms of the oligonucleotide-induced exon skipping are not completely understood, but probably include interfering with the function of splicing machinery or splicing repressors (reviewed in ref. 27). Frequently, the resulting truncated proteins retain some or most of the functions of the original protein and thus lead to a positive therapeutic effect.
Several clinical trials have been initiated for exon-skipping ODNs in recent years. In 2016, the clinical data for nusinersen (http://bit.ly/2iWikcg) and eteplirsen (http://bit.ly/2iXl3ob) resulted in approvals for SMA and DMD, respectively. An NDA application for drisapersen in DMD was rejected by the FDA (https://www.drugs.com/history/kyndrisa.html) (see “ODNs in CNS diseases” below, reviewed in ref. 28).
Transcriptional regulation by blocking miRNA
In recent years, miRNAs have emerged as important regulators of gene expression and attractive therapeutic targets (reviewed in ref. 29). These miR-NAs are small non-coding regulatory RNAs, initially transcribed as long primary miRNA transcripts (pri-miRNA) containing a double- stranded hairpin structure. The pri-miRNAs are processed into 70-nt stem-loop structures known as precursor miRNAs (pre-miR-NAs) by a complex consisting of an RNase Drosha and a dsRNA binding protein DGCR8 (Di George syndrome critical region 8). Pre-miRNAs are then further processed by an RNase Dicer, to generate RNA duplexes with 2-base-pair overhangs (ds-miRNAs). Ds-miRNAs are loaded onto RNA-induced silencing complex (RISC) where the strand with the less stable 5′ end associates with AGO. AGO unwinds the duplex, removes the ‘passenger’ strand and retains the guide strand. The ‘seed’ region of the guide strand (positions 2 to 8 at the 5′ end) pairs to its responsive elements in mRNAs. Multiple proteins can associate with the miRNA–Ago–mRNA complex, determining the type of regulation to be carried out by the miRNA, with mRNA degradation being one of the possibilities (reviewed in refs. 29,30). Synthetic oligonucleotides imitating ds-miRNAs can enter the miRNA processing pathway as miRNA mimics and guide the regulation of their target mRNA. Alternatively, synthetic oligonucleotides, termed Antagomirs (or Antimirs), can be designed to block a disease-relevant endogenous miRNA31. For example, miRagen Therapeutics (now Signal Genetics; Boulder, CO, USA) is targeting miR-155, an miRNA increased in the spinal cord of patients with amyotrophic lateral sclerosis (ALS), with an ODN (MRG-107), as a novel therapy for the disease. Development is now in the Investigational New Drug (IND)-enabling phase (http://miragentherapeutics.com).
Transcriptional upregulation by modulation of epigenetic factors
lncRNAs, notably from the natural antisense transcript (NAT) class, can function as fine modulators of ongoing transcription for a single gene or a small subset of related genes32. NAT-mediated regulation is present in many gene loci and is potentially applicable to multiple genetic disorders. Many NATs act as gene-specific repressors, likely through tethering and/or scaffolding of general-purpose epigenetic complexes to a specific gene locus. The tethering can occur by the nascent lncRNA at the time of transcription33 or independently of transcription, by pairing of NATs with DNA or mRNA sequences34.
As demonstrated in our studies (by C.W. et al.), depleting NAT molecules, or blocking their interaction with epigenetic proteins using ODNs (termed AntagoNATs) leads to upregulation of the associated protein-coding genes34–38. AntagoNATs shown to upregulate SCN1A expression improve disease phenotype in a mouse model of Dravet syndrome and represent a disease-modifying treatment for this condition38. The AntagoNAT technology, developed at the Scripps Research Institute and its spin-out CuRNA (Jupiter, FL, USA), is currently being advanced by the Miami-based company OPKO Health (http://www.opko.com/therapeutics/opko-curna/).
A different but likely related mechanism for mediating transcriptional regulation is via polycomb-associated long non-coding RNAs. This approach, pioneered by Jeannie Lee and RaNA Therapeutics (Cambridge, MA, USA), is capable of selectively upregulating individual genes by sterically blocking interaction of PRC2 with regulatory lncRNA39. A novel lncRNA specific to the SMN2 locus was found to recruit PRC2 complex to SMN2 gene leading to its repression. ODNs capable of blocking this recruitment are now being developed at RaNA. This technology is also being used for other CNS- disease-associated genes (http://ranarx.com/).
Transcriptional upregulation by interactions at promoter regions
Small-activating RNAs (saRNA) are double-stranded synthetic RNAs complementary to promoter regions. Treatment with saRNA leads to upregulation of target gene expression40,41. Although the exact mechanism of gene upregulation by saRNA is unknown, it could be related to the function of RNA polymerase II (RNAP II). It has been proposed that saRNAs may be involved in the regulation of the transcription elongation step through their interaction with Ago2 (ref. 42). Ago2, mostly known for its role in the RISC complex, can serve as an RNA-programmable homology search engine. It is proposed that saRNAs program Ago2 to find and associate with target gene promoters. The saRNA–Ago2 complex then interacts with other proteins to form RNA-induced transcriptional activation complex (RITA), which interacts with RNAP II and stimulates both the initiation and elongation steps of transcription43. Besides saRNA and Ago2, RITA includes RHA and CTR9 proteins. Formation of RITA is accompanied by monoubiquitination of histone 2B and increased methylation of histone 3 at lysine 4 at the saRNA-targeted promoters. Furthermore, higher accumulation of RNAP II phosphorylated at Ser2 (elongating form) and lower accumulation of RNAP II phosphorylated at Ser5 (pausing form) is observed throughout the transcribed region of the target gene after saRNA treatment43. saRNA-induced gene upregulation is observed after a 24- to 48-h delay and persists for extended periods of time.
It has also been proposed that targeting promoter regions using exogenous saRNAs can induce the formation of nucleosome-depleted regions with exposed regulatory sites including TATA box, CpG islands, proximal enhancers and proximal promoters leading to increased initiation of transcription44. Additionally, saRNA may induce transcriptional activation through interaction with heterogeneous nuclear ribonucleoprotein A2/B1 (ref. 44).
The saRNA technology is currently being developed by a London-based company MiNA Therapeutics. MiNA has created a bioinformatics platform to design saRNA for targeted gene activation (http://minatx.com/5/29/16). A first-in-human clinical study of MTL-CEBPA in patients with advanced hepatocellular carcinoma was initiated in 2016 (ClinicalTrials.gov ID# NCT02716012). MTL-CEBPA is a dsRNA, designed to activate the CEBPA gene, that is delivered in amphoteric iminolipid nanoparticles.
Nucleic acid aptamers
Oligonucleotide aptamers are compounds selected to bind to a specific target molecule, frequently a protein, through mechanisms other than base-pairing. An example of a successful aptamer is pegaptanib (Macugen), developed by NeXagen (Boulder, CO, USA) and later Gilead Sciences (Foster City, CA, USA) and OSI Pharmaceuticals (New York), which was approved in 2004 for the treatment of age-related macular degeneration (http://www.accessdata.fda.gov/drugsatfda_docs/nda/2004/21-756_Macugen.cfm).
A novel aptamer chemistry, L-ribonucleic acid aptamers (Spiegelmers) is being developed by Noxxon Pharma (Jena, Germany). Spiegelmers are highly resistant to nuclease digestion because they use L-ribose, rather than the D-ribose present in natural nucleic acids. Several Spiegelmers are now in early clinical trials for cancer and anemia of chronic disease (http://www.noxxon.com/).
Oligonucleotides can simultaneously engage several mechanisms to achieve a given outcome. For example, both RNase-H-mediated mRNA destruction and blocking of mRNA access to the ribosome will lead to reduced levels of the target protein. In an interesting combination of mechanisms, the effect of bevasiranib, a compound designed to engage the RISC-mediated RNA degradation, was also mediated through its interaction with toll-like receptor 3 (TLR3)45.
When seeking to intervene in disease with an oligonucleotide therapy, knowledge of the underlying biology and peculiarities of the affected genes plays into the choice of mechanism of action. For example, exon-skipping ODN treatment for SMA became possible due to the presence of a partially damaged duplicate of the mutated SMN1 gene that can be reactivated to assume the SMN1 function (see below).
Oligonucleotide uptake mechanisms
Oligonucleotide uptake pathways are currently poorly understood. Most of the data on the uptake mechanisms has been generated in cultured cells, and only limited information about in vivo neuronal uptake is available. However, insights gained from other cell types may provide guidance in understanding neuronal and glial uptake mechanisms. Although some amount of naked ODNs can pass through the BBB unassisted, little is known about the mechanisms of this phenomenon as well.
The time frame for the ODN uptake into cells was estimated by Koller et al.46. When mouse hepatocellular carcinoma (MHT) cells were incubated with 6 nM to 20 μM of anti-SR-B1 ODN for short periods of time (15 min to 2 h), then washed to remove the free ODN and incubated for an additional 24 h, the ODN potency was decreased compared to a continuous 24 h exposure, indicating that the rate limiting step of the uptake likely occurs at the initial entry through the cell membrane. The maximal effect of 1 μM ODN on SR-B1 expression in MHT cells occurred at ~30 h after adding the ODN to the media. Maximal effect of lower concentrations of ODN was delayed to 48–72 h46.
It has been proposed that ODNs enter cells by at least two distinct uptake pathways: productive and non-productive47 (Fig. 3). The non-productive pathway acts as a saturable sink that competes with the productive pathway for oligonucleotides. It is likely that many different non-productive sinks exist, with candidates including lysosomes, cell-surface proteins, or specific cell types. The nature of the productive pathway is also currently uncertain. Productive uptake accounted for <20% of the total ODN delivered to liver tissue46.
Figure 3.

Proposed oligonucleotide uptake mechanisms. It is proposed that ODNs enter cells by two competing pathways: productive pathway (gray outlines), which gives them access to their targets, and non-productive pathways (black outlines), which sequesters them in saturable sinks. Both productive and non-productive pathways start with ODNs being taken up by clathrin-coated endocytic vesicles. The endocytic vesicles bud from the plasma membrane, and GTPase dynamin catalyzes membrane scission. Freed cargo-containing vesicles are transported along actin- or tubulin-based cytoskeletal structures to fuse with early endosomes (EE). The fusion vesicles then undergo maturation. Internal pH of endosomes (E) drops to ~5.5 to accommodate the acid hydrolases involved in the degradation of the contents at the lysosome stage. Mature late endosomes (LE) eventually fuse with enzyme-containing transport vesicles, budded from the trans-Golgi network, to form lysosomes. This endosomal/lysosomal compartment likely corresponds to cytoplasmic phosphorothioate (PS)-bodies, large sharply defined structures intensely staining for ODNs. Productive and non-productive uptake pathways diverge during or after lysosome formation with the majority of oligonucleotides remaining in the non-productive pathway, possibly in endosomes/lysosomes, and a small amount escaping into the productive pathway, which likely involves ODN entry into the nucleus through the RAN-mediated pathway. Transfected ODNs may associate with paraspeckle proteins in the nucleus and form morphologically normal and apparently functional paraspeckle-like structures containing no NEAT1 RNA. Additionally, ODN can be sequestered into a non-productive pathway by binding to membrane-associated glycoproteins.
It is possible that both productive and non-productive pathways start with ODNs being taken up by clathrin-coated endocytic vesicles, together with other endocytosed material. However, other endocytic pathways may be also involved47. The endocytic vesicles bud from the plasma membrane, and GTPase dynamin catalyzes membrane scission. Freed cargo-containing vesicles are transported along actin- or tubulin-based cytoskeletal structures to fuse with early endosomes. The resulting fusion vesicles then undergo maturation. One of the important steps in the endosome maturation process is lowering of the internal pH to ~5.5 to accommodate the acid hydrolases involved in the degradation of the lysosomal contents. Mature late endosomes eventually fuse with enzyme-containing transport vesicles, budded from the trans-Golgi network, to form lysosomes48. This endosomal/lysosomal compartment likely corresponds to cytoplasmic phosphorothioate bodies, large, sharply defined structures, intensely staining for ODNs, observed in cells after ODN administration in vitro and in vivo49 (see ref. 34 for confocal video).
Productive and non-productive uptake pathways may diverge during or after lysosome formation with the majority of oligonucleotide remaining in the non-productive pathway, possibly in endosomes/lysosomes, and a small amount escaping into the productive pathway, which likely involves ODN entry into the nucleus. This model is supported by the fact that the share of productive uptake is not affected by knockdown of clathrin, caveolin or dynamin, and thus is likely independent from the initial stages of lysosome formation46. Nuclear import of phosphorothioate ODNs occurs at least partially through the RAN-mediated pathway50,51. Obviously, rerouting higher proportions of ODNs to the productive pathway is central to enhancing drug bioavailability and reducing off-target effects; thus, extensive effort currently aims to understand the mechanisms of both uptake processes, including identification of ODN-binding proteins capable of enhancing the functional activity of the ODNs.
Non-productive pathway proteins
Recently, an unbiased short-hairpin-RNA screen has revealed that knockdown of TSG101 and VPS28, components of ESCRT-I (endosomal sorting complexes required for transport) complex increases anti-proliferative effects of anti-miR-21, a phosphorothioate ODN. ESCRT-I complex enables membrane budding associated with the formation of intraluminal vesicles (ILVs) during endosome maturation. This process helps concentrate selected proteins and lipids in the late endosome lumen facilitating their lysosomal degradation48. Depletion of ESCRT-I in several cancer cell lines with inherently poor ODN uptake resulted in improved activity of anti-miR-21. Finally, knockdown of TSG101 in vivo increased uptake of anti-miR-21 following systemic delivery, indicating that late endosomes/lysosomes may represent one of the non-productive ODN sinks48.
Notably, when transfected, synthetic 20-mer phosphorothioate ODNs have been shown to associate with paraspeckle proteins, including P54nrb, PSF, PSPC1 and hnRNPK and form morphologically normal and apparently functional paraspeckle-like structures containing no NEAT1 RNA49. NEAT1 is an lncRNA, which normally seeds co-transcriptional paraspeckle formation. Paraspeckles may serve to concentrate and/or sequester proteins in the nucleus50. Moreover, paraspeckle proteins including P54nrb, PSPC1 and PSF have been shown to negatively affect the antisense activities of phosphorothioate- ODNs, indicating that nuclear localization alone is not sufficient for ODN participation in the productive uptake pathway49.
Ku70 and Ku80 proteins, which are involved in DNA double-strand break repair and V(D)J recombination, can inhibit ODN activity, possibly by competing with RNase H for binding to the ASO–RNA duplex51. Binding to alpha 2 macroglobulin (A2M) could mediate delivery of ODNs to non-productive compartments. Shemesh et al.52 have shown that activity of a phosphorothioate MOE ODN targeting SRB1 (scavenger receptor class B type I) was substantially improved in murinoglobulin double-knockout mice compared to WT mice. The overall plasma and tissue exposure kinetics were not different between the two mouse strains, indicating increased distribution of ODNs to productive uptake pathway52.
Productive pathway candidates
It is possible that the productive pathway requires a lysosome acidification step because it is negatively affected by chloroquine, an inhibitor of lysosome acidification as well as of autophagy. Interestingly, although chloroquine blocked the antisense effects of an ODN, it increased the total ODN amount accumulated in cells46. Additionally, experiments with bio- responsive nanovehicles show that nuclear drug uptake efficiency strongly depended on the lysosomal acidity53.
A screen for ODN-interacting proteins has identified six out of eight subunits of the TCP1 complex54. TCP1 is an ~1-mDa chaperone complex involved in folding of ~10% of cellular proteins, including actin, tubulin, cyclin E1 and histone deacetylases. A beta subunit of the TCP1 complex bound ODNs and co-localized with them in nuclear and cytoplasmic phosphorothioate-bodies. Interaction of phosphorothioate-ASOs with TCP1 proteins enhanced the knockdown activity of ASOs, indicating that TCP1 is involved in the productive uptake pathway54.
Reduction of Hsp90 protein levels decreased activity of some ODN chemistries51. Additionally, La (also known as SSB) and NPM1 proteins may increase ODN retention in the nucleus, thus facilitating ODN activity51. La is an RNA-binding chaperone protein involved in tRNA transcription and processing. NPM1 is a nucleic-acid binding protein associated with nucleolar ribonucleoprotein structures. AP2M1, ANXA2 and PC4 proteins have also been shown to enhance ODN activity51. AP2M1 is involved in endocytosis and autophagy. ANXA2 is a calcium-dependent phospholipid-binding protein involved in exocytosis of intracellular proteins to the extracellular domain, among other functions. PC4 protein is involved in transcription, chromatin organization and DNA repair.
ODNs have also been shown to bind and be taken up by scavenger receptors. Miller et al.55 have demonstrated that members of the stabilin family of scavenger receptors, including stabilin-1 (STAB1) and stabilin-2 (STAB2) were responsible for bulk clathrin-mediated endocytosis of phosphorothioate ODNs. These receptors likely belong to the productive uptake pathway as increased internalization rates in stabilin- overexpressing cell lines enhanced ODN potency for knockdown of the lncRNA Malat-1. Class A scavenger receptors have also been implicated in ODN uptake56,57.
As a consideration for the interpretation of data from the ODN-binding protein screens, 2′-modifications may affect ODN protein binding profiles. ODNs containing more hydrophobic 2′-modifications exhibit higher affinity for proteins in general. In particular, Hsp90 protein binds LNA or constrained ethyl (cEt) ODNs more efficiently than 2MOE ODNs. Furthermore, reduction of Hsp90 protein levels decreased activity of LNA or cEt ODN, but not 2MOE ODNs51. Differential ability of ODNs of different chemistry to self-assemble into nanoparticles may affect their uptake by class A scavenger receptor subtypes (SCARAs). Peptide-conjugated PMO (PPMO) and tricyclo-DNA have high propensity to form nanoparticles and are predominantly transported by SCARAs, including SCARA1, SCARA3 and SCARA5, as shown by competitive inhibition, co-localization and RNAi silencing experiments in vitro and in vivo, in SCARA1 knockout mice compared to wild-type animals. 2OMe ODNs were also transported by SCARA1, but to a lesser extent than PPMO and tcDNA56,57.
Furthermore, the oligonucleotide uptake pathways may be different in cases of free uptake (in the absence of delivery agent) and liposome-mediated delivery (transfection). After free uptake, ODNs predominately localize in the cytoplasmic phosphorothioate bodies. After transfection, ODNs localize primarily in the nucleus for extended periods of time46, accumulating in the nuclear phosphorothioate bodies58. Phosphorothioate bodies appear within minutes after the start of oligonucleotide treatment and remain stable for extended periods of time. However, during mitosis phosphorothioate bodies are reorganized and then reassembled from diffuse ODN pools in the daughter nuclei58.
Transfected ODNs are quickly released from their cationic vesicle carriers. Using transfection complexes with fluorescently labeled lipids and oligonucleotides targeted to protein kinase C-alpha, Marcusson et al.59 demonstrated that the lipids initially co-localized with the oligonucleotides on the cell surface and in punctate structures in the cytoplasm. At later times the oligonucleotides, but not lipids, began to appear in the nucleus. Knockdown of protein kinase C-alpha mRNA did not begin until the oligonucleotides entered the nucleus59.
ODN uptake mechanisms can also differ depending on the cell types involved. For example, in vivo kidney proximal tubule cells and liver Kupffer cells exhibit a strikingly higher productive and non-productive oligonucleotide uptake capacity than other cell types. Positional signaling and cell surface proteins may be involved in the regulation of the uptake process. For example, in primary hepatocytes productive uptake is substantially diminished in the first 24–36h after introduction into culture46. However, information on the uptake mechanisms in different CNS cell types is currently scarce.
Moreover, ODN uptake mechanisms can undergo disease-associated changes. It has been shown that in mdx mice (a DMD model) skeletal muscles show 4–8 times higher ODN levels (up to 10 μg/g) than in WT mice at 48 h after single intravenous (IV) injection of 50 mg/kg of 2OMe-phosphorothioate ODN. Disease-dependent differences in uptake were not observed in kidney and liver, which showed high (up to100 μg/g) and equal levels of ODN in both mouse strains60.
Assisted delivery and CNS targeting of ODNs
Although ODNs are readily taken up by brain tissues and have extended tissue half-life, development of specialized carriers or further chemical modification of ODNs can be beneficial in terms of enhancing passage through the BBB, improving distribution to a productive uptake pathway and targeting ODNs to specific cell types or diseased cells. At the current state of knowledge, this additional chemistry is likely to elevate toxicity, complicate drug approval process and increase cost. However, if optimized, it could be highly beneficial for disease treatment, thus, extensive research is being carried out in this field3.
Among modifications aimed at improving ODN delivery are cationic lipids and polymers that can destabilize the endosome membrane. Buffering peptides or polymers have been used to change intra- endosomal pH to affect endosome stability and trafficking. Small molecules that increase endocytosis of oligonucleotides or selectively permeabilize endosomal compartments, thus leading to oligonucleotide release to the cytosol, have been proposed. Different types of nanocarriers have been used to both facilitate the transport through the cell membrane and target ODNs to particular cell types3.
Multiple cell-penetrating peptides and conjugation with receptor ligands are being tested for targeted delivery of ODNs, and all of the above methods are being used to increase BBB permeability3. For example, an advanced peptide–oligonucleotide, Pip6a-PMO designed to modulate exon 7 splicing of SMN2, demonstrated potent efficacy in the CNS after systemic administration at doses an order of magnitude lower than those of naked ODNs. Animal survival was extended from 12 to 456 days, with improvement in neuromuscular junction morphology and other disease manifestations61.
Some chemical modifications, such as tricyclo-DNA, have been shown to improve trans-BBB availability of ODNs12. Purified exosomes loaded with ODN cargo are capable of crossing BBB after IV injection (reviewed in ref. 9). It is important to note, however, that most, if not all of these approaches are likely to reduce ODN specificity and increase treatment-associated adverse events by simultaneously redistributing other non-related molecules into new subcellular compartments and tissues.
Another approach to optimizing delivery of ODNs through the BBB is intranasal administration. Intranasal delivery has been previously used for delivery of small molecules with poor BBB permeability, peptides or proteins. For example, in an Alzheimer’s disease clinical trial, insulin delivered intranasally successfully reached the brain, without adversely affecting blood insulin or glucose levels. Treatment-related adverse events were minor, such as mild rhinitis62. Furthermore, an FITC (fluorescein isothiocyanate)-labeled ODN GRN163 administered to rats intranasally every 2 min as 6 μ1 drops into alternating sides of the nasal cavity over 22 min was observed in brain tumor cells at all time points studied, with a peak at 4 h after delivery. When delivered intranasally daily for 12 days, GRN163 significantly prolonged the median survival of rats from 35 days in the control group to 75.5 days in the GRN163-treated group63. Moreover, an anti-miR-210 LNA ODN delivered ICV or intranasally in rats produced similar decreases in hypoxia-induced neonatal brain injury and improvements of neurological function later in life64. Finally, intranasal administration of a sertraline-conjugated siRNA inhibited serotonin transporter (SERT) expression and function and produced antidepressant-like effects in mice65.
An alternative method of enhancing intranasal delivery has been proposed by Bleier et al.66. It involves introducing a semipermeable window in the BBB by creating an opening using endoscopic surgery, then engrafting autologous nasal mucosa. This procedure is currently used in the clinic to remove brain tumors endoscopically, accessing the skull base through the nose. After surgery, the opening in the skull base is sealed with mucosal grafts harvested from the patient’s nasal septum. The engrafted nasal mucosa, which lacks restrictive BBB qualities, may be subsequently dosed with ODNs, peptides, proteins or other large molecules. This procedure can be further enhanced by carriers that improve residence time and permeation of the mucosa.
PK and PD characteristics
The complex multi-stage and multi-compartment uptake and clearance mechanisms of the ODN drugs lead to their very unusual PK and PD properties, which bear little resemblance to those of small molecules. Consequently, ODNs cannot fully benefit from the existing pre-clinical development tools that are largely optimized for small compounds. At the same time, the information on PK of ODN drugs, which may lead to the development of tools fundamental for determining dose and regimen for chronic administration, is scarce. In this section, we review the available data, mostly centered on 2MOE ODNs because of their advanced position in clinical development. However, the distribution and metabolism of other oligonucleotide chemistries may be somewhat different, as seen from the diverging side-effect profiles of 2OMe and PMO ODNs reported in their respective clinical trials for Duchenne muscular dystrophy (http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/PeripheralandCentralNervousSystemDrugsAdvisoryCommittee/UCM473737.pdf accessed 5/29/16; http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/PeripheralandCentralNervousSystemDrugsAdvisoryCommittee/UCM473737.pdf accessed 5/29/16).
Following subcutaneous (SC), IV, ICV or IT administration, ODNs are rapidly absorbed from the injection site into the circulation or into cerebrospinal fluid (CSF). Below, we consider absorption and distribution profiles following direct CNS administration of ODNs.
Application of ODNs to the brain
In the early 1990s, one of us (C.W. et al.)67–71 was involved in the first successful attempts to apply ODNs directly into CSF (ICV or IT) to target specific mRNAs in the brain of living rodents. At that time, we suggested that the CNS could be viewed as a protected environment because of the demonstration that CSF contains negligible nuclease activity to degrade ODNs and, moreover, that it is an immune- privileged zone; oligonucleotides are therefore highly stable in this body fluid69,70. Over the following quarter century, many academic and industry-based investigators have applied a variety of different types of oligonucleotides, injected ICV, IT or into specific brain sites of experimental animals. However, it is only in the past few years that this approach has matured to the point where clinical trials for human brain disorders are being pursued in a concerted manner (see below).
After IT administration, ODNs are distributed from CSF within minutes to hours, mainly through uptake into CNS tissues and, to a lesser degree, through transfer to systemic circulation72. Similar to plasma, elimination of ODNs from CSF follows a two-phase course. Seven days after a single IT bolus injection of either 1, 3 or 7 mg of a phosphorothioate 2MOE ODN in adult cynomolgus monkeys, the greatest ODN accumulation was observed in large and small cell bodies of the gray matter, consistent with neuronal and glial cell uptake73. Interestingly, when the same 2MOE ODN was administered by IP bolus injection, the half maximum effective concentration (EC50) value in the liver was more than tenfold higher than in the brain, possibly reflecting a lower ratio of productive to non- productive uptake in the liver73.
The existence of productive and non-productive pathways substantially complicates the PD characteristics of ODNs. Counterintuitively, ICV bolus injection of a phosphorothioate MOE ODN resulted in a 4–5 times lower EC50 than a one-week osmotic pump infusion73. Furthermore, although infusion of ODN resulted in a larger amount taken into the liver cells, little increase in ODN activity was associated with this higher concentration, suggesting that the saturation point of the non-productive pathway was not being reached and only small amounts of ODN were available for the productive pathway. Addition of a nonsense ODN resulted in lower overall amount of active ODN in liver but paradoxically greater target RNA silencing73.
However, a different situation was observed after systemic administration in dystrophin-deficient mdx mice treated with different dosing regimens of exon-skipping phosphorothioate 2OMe ODN over a period of 8 weeks74. In this study, higher levels of ODN, more pronounced exon skipping and increased protein were observed in muscle after low daily doses compared with large weekly doses. After receiving a high loading dose (1,250 mg/kg) in the first week, mice treated with maintenance injections twice weekly for 8 weeks showed higher preservation of therapeutic effects than mice receiving fewer or no maintenance injections74. These differences could be due to impaired barrier function and lower reliance on active uptake mechanisms in mdx mice.
Another unusual aspect of ODN pharmacology is the delayed protein effect. For example, huntingtin (Htt) protein reduction was observed 2 weeks later than downregulation of Htt mRNA in Huntington’s disease (HD) mouse model75. At the same time, protein expression did not return to vehicle-treated levels until 4 months after the end of treatment. Motor function improved after a two-month lag in 6-month-old mice. Improvement in motor performance persisted over 6 months after the end of treatment and 2 months past the return of the protein expression to saline levels75.
Furthermore, SMN2 splicing correction in mouse spinal cord and brain after a single ICV bolus injection of 100 μg of a 2MOE ODN also resulted in prolonged pharmacological activity, which was maintained for at least 36 weeks after dosing and past complete ODN elimination73. Notably, duration of action of the ODN in the liver after IP bolus injection was much shorter, with minimal SMN2 splicing correction observed 8 weeks after dosing73. SMN2 splicing correction in the spinal cord and brain 71 days after the end of the 30-μg/day ICV infusion was comparable to the level observed 2 days after the end of the infusion73.
Such persistent effects of the ODNs could be partially explained by their long half-life in the peripheral tissues and extremely long half-life in the CNS (due to its low nuclease levels and immune- privileged status). For example, nusinersen, a 2MOE ODN, was detectable in motor neurons 1 year after a 7-day ICV infusion73. Tissue half-life of ICV-infused 2MOE ODNs were 71–206 days in the brain and 145–191 days in the spinal cord73. Meanwhile, half-life of 2OMe phosphorothioate ODNs in rodent peripheral tissues was around 10–65 days after IV injection60,74,75. Slower rates of degradation and clearance from the CNS, together with the fact that the CNS represents a relatively small contained volume, allow using decreased doses of ODNs for IT administration, thus reducing the possibility of adverse events.
Importantly, an antidote (a fully complementary oligonucleotide) can be designed to counteract the effects of an ODN drug. Such antidote reversed SMN2 exon 7 inclusion in SMA mice initially treated with splice-modulating 2MOE phosphorothioate ODN73.
More than 85% of phosphorothioate ODNs escaping into systemic circulation are bound to plasma proteins, mainly albumin, in all species studied3,72. This may result in different relative potency of ODNs observed in vitro and in vivo. For example, a PMO ODN, which exhibited the same potency as a 2MOE phosphorothioate ODN in vitro in SMA patient fibroblasts, was three- to fivefold less potent for SMN2 exon 7 inclusion in vivo. This could be due to a 4–8 times lower accumulation of the PMO ODN in CNS tissues compared with that of 2MOE phosphorothioate ODN73.
In vivo, minimal nuclease cleavage of ODNs is observed with the main degradation product being 3′ n–1 oligonucleotides. Because of the decreased binding to plasma and tissue proteins, the shorter endonucleolytic products are rapidly eliminated in urine72. Kidney is the primary route of excretion of phosphorothioate ODNs, although this is a relatively slow process and mainly involves nuclease degradation products3.
Differences in PK and toxicology among species
The limited clinical trial experience with direct ODN administration to the CNS shows that although the PK of 2MOE ODNs is generally similar for different sequences and species76, there could be some species- and ODN-sequence-specific differences in PK and toxicologic profiles. Insights gained from the studies using systemic administration of ODNs indicate that animal species used in toxicology studies are possibly more sensitive to ODN treatment77–81.
The unusual PK and PD properties of the ODNs could be partly responsible for the difficulties surrounding two exon-skipping ODNs being developed for the treatment of DMD: drisapersen, which was rejected by the FDA, and eteplirsen, which ultimately received accelerated approval in 2016. For both these drugs, the time frames required to observe statistically significant accumulation of their target protein (months) and to demonstrate slowing of disease progression (years) are strikingly different from those commonly used in the trials for small-molecule compounds. At the same time, the clinical trials for ODNs cannot be simply extended to the required duration without modifying the current trial requirements and rules optimized for small molecules82. As study duration is extended, placebo controls, and especially sham controls common in cases of IT injections, become less feasible because of difficulties with maintaining blinding of the trial. Furthermore, subject recruitment can be negatively influenced by the prospect of receiving placebo treatment for extended periods. Possible solutions may involve multi-stage approval and more extensive use of surrogate markers.
In addition to the recent approval of nusinersen, multiple other ODN compounds are currently in different stages of clinical development (Table 1). In the following sections, we briefly review the current status of these studies below, starting with the more advanced clinical programs.
Exon-skipping ODNs
Multiple CNS-targeted ODN compounds are currently in different stages of clinical development (Table 1). We briefly review the current status of these studies below, starting with the more advanced clinical programs. Exon-skipping ODNs have been applied to treat a variety of splicing disorders. These include spinal muscular atrophy (SMA), spinocerebellar ataxia type 3 (SCA3) and ataxia telangiectasia (AT).
SMA
SMA is a severe genetic disorder characterized by degeneration of spinal motor neurons, progressive muscle weakness beginning in infancy, and early death. The disease is caused by reduced amounts of SMN1 protein. A known modifier of SMA severity is the copy number of the SMN2 gene. SMN2 sequence is highly similar to SMN1 but has a mutation that leads to exon 7 omission and production of a truncated protein, which is normally rapidly degraded.
Nusinersen, a phosphorothioate 2MOE ODN (also known as Spinraza, ISIS 396443, ISIS-SMNRx and ASO–10-27) is designed to target a site in intron 7, called ISS-N1. ISS-N1 normally recruits splicing repressors heterogeneous nuclear ribonuclear proteins (hnRNP) A1 and A2, which inhibit exon 7 inclusion. Hybridization of nusinersen to ISS-N1 blocks hnRNP recruitment, which leads to exon 7 inclusion and increased production of the full-length SMN2 protein83.
Nusinersen has shown good safety and tolerability profiles in open-label phase 2 clinical trials after IT delivery in infants and children with SMA. Immunogenic response to nusinersen was not observed over 9–14 months after treatment84. Nusinersen study represents the first experience of IT ODN delivery in children 2–14 years of age. The ODN was administered IT as a 5-mL bolus over 1 to 3 min at the L3–L4 disc space; 5 to 6 mL of cerebrospinal fluid was collected before drug injection. Additional lumbar punctures were conducted for follow-up collection of CSF for safety and PK analyses85. Most adverse events observed in nusinersen studies were attributed to lumbar puncture, including headache, back pain and post-lumbar puncture syndrome. Adverse events were more frequent in older children, children with type 3 SMA and with the use of 21- or 22-gauge needle (compared with a 24-gauge or smaller). Lumbar puncture was complicated by scoliosis, common in SMA patients85.
The dose range selected for the phase 2 study was expected to produce the tissue concentration of ODN of 1–10 μg/g in spinal cord tissues, shown to elicit a 50–90% SMN2 exon 7 inclusion in animal models. Preliminary efficacy data showed increases in motor function scores at the highest dose used (9 mg/kg) in treated patients compared with values from natural history studies. As no dose-limiting toxicity was observed, Chiriboga et al.84 proposed to use higher doses in the future studies.
PK data from the clinical trial were consistent with results from animal studies and other ODN clinical trials81. Peak plasma levels were observed after a few hours followed by a slow decline lasting for ~20 h and an extended final elimination period of 7 days. Nusinersen was detectable in plasma for over 24 h after a dose, and at 7 days after a dose in the CSF. Furthermore, in the CSF of the 9-mg group, detectable levels of nusinersen were observed 29 days after dose (2.33 ± 0.928 ng/ml). At the 9- to 14-month evaluation, CSF nusinersen concentrations were still above the limit of quantification (1 ng/ml). Apparent terminal half-life in CSF was estimated to be 132–166 days (4.5–6 months). For comparison, nusinersen half-life in CNS tissues in mice was estimated at 145–191 days and in monkeys at 139 ± 54 days. This extremely long CNS half-life may justify infrequent administration of nusinersen (e.g., every 4–6 months)84. No correlation was observed between age or total body weight of patients and CSF concentrations of nusinersen, consistent with literature reports indicating that CSF volume is similar in children older than 2 years. Thus, no dose scaling was proposed for this pediatric population84. Consistent with delayed effects observed for many ODNs, doubling of SMN2 protein levels in the CSF was observed at 9–14 months after a dose, compared to baseline, in the 6- and 9-mg groups. The 9-mg group demonstrated improved functional assay scores compared to baseline at 3 months after a dose, and further improvement at 9–14 months during the extension study84.
A phase 3 clinical trial sponsored by Ionis Pharmaceuticals (formerly ISIS Pharmaceuticals; Carlsbad, CA, USA) and Biogen (Cambridge, MA, USA) was initiated in 2014. The randomized, double-blind, sham-procedure-controlled, 13-month study evaluated the efficacy and safety of a 12-mg dose of nusinersen in 110 infants with SMA with a primary endpoint of survival. The preliminary results announced in 2016 demonstrated that children receiving nusinersen experienced a highly statistically significant improvement in motor function compared to those who did not receive treatment. Additionally, the phase 2 open-label study of nusinersen in infants with SMA showed continued increases in mean event-free survival and muscle function scores, as well as achievement of motor milestones normally not seen in infants with type 1 SMA, such as sitting, standing and walking. In December 2016, FDA approved nusinersen.
Interestingly, although SMA is considered a motor neuron disease, considerable amelioration of disease symptoms is observed after systemic administration of splice switching 2MOE ODNs. Systemic treatment completely eliminated necrosis in mice with mild SMA and resulted in a 21-fold increase in life span and improved motor neuron counts in mice with severe SMA. Meanwhile, increasing SMN levels specifically in motor neurons ameliorated the phenotype of SMA mice only partially, including a limited increase in survival86. Peripheral rescue of SMA phenotype could be explained by peripheral production of trophic factors supporting survival of central motor neurons. After systemic ODN treatment in neonatal mice, all gastrointestinal abnormalities observed in the mouse model of SMA, including reduction of vascular density, overabundance of enteric neurons and increased macrophage infiltration, were restored to near-normal levels87. Additionally, IP injection of 80 μg/g of 2MOE ODN twice at the age of onset in a mouse model of the intermediate forms of SMA extended survival and rescued the delay in neuromuscular junction maturation88.
Besides exon 7 retention, a new therapeutic target for SMA, an intronic repressor Element1 (E1), located upstream of SMN2 exon 7, has been described. A single ICV injection of E1-blocking MAO ODN resulted in an increase of SMN protein and extension of life span from an average of 13 days in the control to 54 days in ODN-treated SMNΔ7 mice. These changes were accompanied by large weight gains and correction of the neuronal pathology. In an intermediate SMA mouse model, ODN treatment extended life span by ~700%89.
SCA3
This disorder, also known as Machado–Joseph disease (MJD), is caused by polyglutamine expansion in the ATXN3, resulting in production of a toxic mutant protein. As ATXN3 is involved in deubiquitination and proteasomal degradation of proteins, its long-term silencing might not be desirable. Evers et al.90 described targeted removal of the polyglutamine repeat using 2OMe ODN-mediated exon skipping. Because omitting the CAG-repeat-containing exon 10 in the ATXN3 gene disrupts the reading frame, both exons 9 and 10 had to be skipped simultaneously. Single ICV co-injection of 20 μg per ODN resulted in significant exon skipping in mice after 7 days. The generated novel truncated version of ATXN3 retained its normal ubiquitin binding function and did not show any toxicity in vivo90.
AT
This progressive autosomal recessive neurodegenerative disorder, also referred to as Louis–Bar syndrome, results from mutations in the ATM (ataxia telangiectasia mutated) gene. Sequencing of a patient’s DNA has identified a novel mutation which created a new donor (5′) splice site 405 bp upstream of the exon 12 acceptor (3′) splice site. The mutation results in the inclusion of a 212-bp non-coding pseudoexon with a premature stop codon. Treatment with 1–2 μM of a PMO ODN, designed to mask the mutant donor splice site, corrects ATM splicing and restores ATM kinase activity to 50% of the WT levels in patient lymphoblasts. AT patients with 5–20% of functional ATM have a mild phenotype, and ATM heterozygotes do not show any sign of disease91.
mRNA-knockdown ODNs
ODNs that work through mRNA knockdown have a longer track record in the clinic than those that intervene in splicing. Thus far, they have been applied to treat a variety of CNS disorders, including amyotrophic lateral sclerosis (ALS), Huntington’s disease (HD), focal ischemia, familial amyloid polyneuropathy (FAP), leptomeningeal amyloidosis and MECP2 duplication syndrome. We discuss work with these ODNs below.
ALS
This rapidly progressive disorder, also called Lou Gehrig’s disease, attacks neurons controlling voluntary muscles. The exact causes of ALS are not known, but several genes causing ALS in subsets of patients have been identified.
Mutations in SOD1 account for ~2% of all ALS cases and are characterized by high penetrance and dominant inheritance. SOD1 mutations generate toxic protein, therefore reduction in SOD1 levels is considered a desirable therapeutic target. Ionis and Biogen are currently evaluating IONIS-SOD1Rx, a 2MOE ODN, in a phase 1/2a clinical study in patients with SOD1-ALS started in 2016 (ClinicalTrials.gov ID# NCT02623699). In the phase 1 randomized, placebo-controlled trial of IONIS-SOD1Rx (ClinicalTrials.gov ID# NCT01041222), started in 2010, 21 participants received IT infusions of ODN using an external pump over 11.5 h at increasing doses (0.15 mg, 0.50 mg, 1.50 mg, 3.00 mg). Subjects were allowed to re-enroll in subsequent cohorts. Safety and tolerability assessments were made during the infusion and periodically over the following 28 days. No dose-limiting toxicities were identified. The most common adverse events were related to post-lumbar puncture syndrome76. There were two adverse events potentially related to the study drug: a mild paresthesia in the 1-mg group and a report of palpitations in the 3-mg group, both of which resolved spontaneously. There were no clinically significant changes in CSF clinical chemistry or CSF cytokines76.
Another frequent cause of ALS is GGGGCC repeat expansions in C9ORF72 (chromosome 9 open reading frame 72). The mechanism of the C9ORF72-related disease likely involves the toxic gain-of-function effects of the GGGGCC expansion-containing RNA (GGGGCCexp RNA), including its increased expression and accumulation in the intranuclear foci, together with hnRNPA1 and Pur-α proteins. Such accumulation may alter RNA metabolism, dysregulate expression of genes involved in membrane excitability, including DPP6, induce sequestration of GGGGCCexp-RNA-binding protein ADARB2, diminish neuronal capacity to fire continuous spikes upon depolarization and increase susceptibility to excitotoxicity92,93. ODNs that degrade only the repeat-containing RNAs but preserve the levels of healthy C9ORF72 RNAs would represent a desirable option, because a chronic 50% reduction of C9ORF72 in mice did not result in overt symptoms, while its complete inactivation produced splenomegaly, enlarged lymph nodes, and mild social interaction deficits without motor dysfunction92. Donnelly et al.92 have developed such ODNs, targeting the intronic region downstream of the repeat or exon 2. These ODNs effectively reduce C9ORF72 RNA levels in C9ORF72 patient fibroblasts and iPSCNs (induced pluripotent stem cell motor neurons)94. The ODN treatment results in a sustained decrease in RNA foci and dipeptide-repeat proteins, and ameliorates behavioral deficits95.
Apart from SOD1 and hexanucleotide repeat expansions of C9ORF72, ODNs have been designed against another two targets in ALS pathogenesis: acetylcholinesterase (AChE) and miR-155. AChE, an enzyme involved in cholinergic synapses in the CNS and peripheral nervous system, has been linked with motor unit denervation and neurotoxicity in ALS. ALS (G93A-SOD1) mice treated with AChE-downregulating ODN mEN101 at the presymptomatic, but not at the symptomatic, stage, show prolonged survival and attenuated motor-neuron loss96. The microRNA miR-155 has been found to be significantly upregulated in the CSF of ALS patients. When G93A-SOD1 ALS mice were treated for two weeks by ICV infusion of a phosphorothioate 2MOE/2F ODN against miR-155, survival was extended by ten days compared with controls97.
Huntington’s
HD is caused by expansion of the CAG trinucleotide sequence in the HTT gene. The expanded mRNA variants produce a toxic protein that progressively destroys neurons in the brain. IONIS-HTTRx is a 2MOE ODN developed by Ionis and Roche (Basel, Switzerland) for the treatment of HD. A phase 1/2a clinical study in patients with HD was initiated in 2015. It is a randomized, placebo-controlled, dose escalation study to evaluate the safety, tolerability, PK and PD of multiple ascending doses of IONIS-HTTRx, administered IT.
In preclinical studies, continuous ICV infusion of IONIS-HTTRx (10, 25 or 50 μg/day) for two weeks into the right lateral ventricle of the BACHD mouse model of HD mediated a sustained reversal of disease phenotype that persisted longer than the HTT knockdown at 16 weeks after the end of the infusion. Suppression of mutant HTT for 8 weeks was required before reversal in phenotype was apparent75.
Another approach to HD treatment is allele-specific HTT knockdown, which is potentially safer than the reduction of total HTT levels. ODNs targeted to HD-associated SNPs selectively silenced the mutant HTT mRNA throughout the CNS for 16 weeks or more after a single ICV injection of a 300-μg dose in an acute humanized mouse model of HD, Hu97/18 (ref. 98). The cEt ODNs employed showed greater potency than 2MOE ODNs. Mutant HTT reduction of 25–35% was still observed at 36 weeks after injection98.
Focal ischemia
Inhibition of expression of N-methyl-D-aspartate receptor 1 (NMDA-R1) can prevent the neurotoxicity elicited by NMDA during ischemic conditions and minimize brain damage by reducing the volume of the focal ischemic infarctions. Treatment with LNA ODNs to NMDA-R1 protected cortical neurons from excitotoxicity, reduced focal ischemic infarctions after occlusion of the middle cerebral artery in rats and prevented NMDA neurotoxicity in vitro68.
FAP
In patients with FAP and other transthyretin (TTR) amyloidosis diseases, both the mutant and WT TTR build up as fibrils in CNS, peripheral nerves, heart, gastrointestinal system, eyes, kidneys, thyroid and bone marrow. Thus, lowering the expression levels of both mutant and WT alleles could slow the disease progression. IONIS-TTRRx is a 2MOE ODN designed to reduce the production of TTR for the treatment of all forms of TTR amyloidosis. IONIS-TTRRx is currently in a phase 3 study, NEURO-TTR, initiated in 2013. It is a randomized, double-blind, 15-month study of 300 mg/week of IONIS-TTRRx or placebo. The study will evaluate the effects of IONIS-TTRRx on neurological dysfunction and quality of life. The results are expected in 2017. Ionis and GlaxoSmithKline are preparing an new drug application (NDA) filing (http://ir.ionispharma.com/phoenix.zhtml?c=222170&p=irol-newsArticle&ID=2172683, accessed 5/28/16). After 3 months of IONIS-TTRRx treatment in the open-label extension study, initiated for FAP patients who have completed the phase 3 study, an average reduction in TTR protein of 76% compared to patients’ baseline TTR levels was observed.
Preliminary data from an ongoing phase 2 study in patients with TTR amyloid cardiomyopathy showed evidence of cardiac disease stabilization after a 12-month treatment with IONIS-TTRRx. However, a planned phase 3 outcome study, CARDIO-TTR, of IONIS-TTRRx in patients with amyloid cardiomyopathy was placed on clinical hold by the FDA as a result of safety findings in the ongoing NEURO-TTR study (http://ir.ionispharma.com/phoenix.zhtml?c=222170&p=irol-newsArticle&ID=2172683, accessed 5/28/16).
Another approach for the treatment of FAP is taken by Alnylam (Cambridge, MA, USA), which is developing patisiran, an siRNA compound. Preliminary results from its ongoing phase 2 open-label extension studies with patisiran were announced in July 2016. Data from the patisiran study showed improvement or no change in the mean neuropathy impairment score (mNIS+7) after 24 months of dosing in patients with FAP. There was a positive correlation between the degree of serum TTR knockdown and changes in mNIS+7. A total of 225 patients with FAP were enrolled in the phase 3 study of patisiran between December 2013 and January 2016 (http://www.alnylam.com/product-pipeline/hereditary-attr-amyloidosis-with-polyneuropathy/).
Another dsRNA compound from Alnylam, revusiran, which was in clinical trials for hereditary ATTR amyloidosis with cardiomyopathy (hATTR-CM) was discontinued in October 2016. Although initial 12-month data from the revusiran study showed knockdown of serum TTR and stable 6-min walk distance (6-MWD) results in hATTR-CM patients, there were reports of new-onset or worsening peripheral neuropathy in the trial. Additionally, there was an imbalance of mortality in the revusiran arm compared with the placebo. The Data Monitoring Committee did not find conclusive evidence of these effects being drug-related, but concluded that the benefit–risk profile for revusiran no longer supports continued dosing (http://www.alnylam.com/product-pipeline/hereditary-attr-amyloidosis-with-polyneuropathy/).
Leptomeningeal amyloidosis
Leptomeningeal amyloidosis associated with mutations in TTR is a rare fatal form of amyloidosis characterized by dementia and intracerebral hemorrhage. Mice transgenic for a human amyloidosis-associated mutation Ile84Ser were treated with an ICV infusion of a TTR-specific ODN (50 μg or 75 μg per day) for 4 weeks. This treatment significantly reduced human TTR expression in choroid plexus, which is believed to be the source of the amyloid protein in leptomeningeal amyloidosis99.
MECP2 duplication syndrome
This disease is characterized by autism, intellectual disability, motor dysfunction, anxiety, epilepsy, recurrent respiratory tract infections and early death. In a conditional Mecp2-overexpressing mouse model, reduction of Mecp2 levels reversed the behavioral, molecular and electrophysiological deficits associated with Mecp2 overexpression. ODN treatment also induced phenotypic improvement in adult symptomatic transgenic Mecp2 duplication mice (MECP2-TG). Furthermore, ODN treatment of lymphoblastoid cells from MECP2 duplication patients reduced MECP2 levels in a dose-dependent manner100.
mRNA-upregulating ODNs
ODNs can also increase transcript production by a variety of mechanisms. We discuss two CNS conditions below where this mechanism has been applied.
Dravet syndrome (DS)
DS is caused by heterozygous loss-of- function mutations in the SCN1A gene, which encodes the pore-forming alpha subunit of the voltage-gated sodium channel Nav1.1. Clinically, DS is characterized by seizure onset in the first year of life, febrile seizures resistant to anticonvulsants, progressive psychomotor retardation and high incidence of sudden unexpected death. Upregulation of the remaining healthy SCN1A allele represents a desirable therapeutic target in DS. Such upregulation has been achieved by blocking the transcriptional inhibitory activity of a regulatory lncRNA from the SCN1A locus (SCN1ANAT) using 2OMe ODNs termed AntagoNATs. In non-human primates and a knock-in mouse model of Dravet, IT injection of AntagoNATs induced upregulation of Scn1a. A once-weekly injection of 20 μg of AntagoNAT for 4 weeks led to significant improvements in seizure frequency and duration, as well as normalization of excitability of hippocampal interneurons in Dravet mice38.
Angelman syndrome
Angelman syndrome is characterized by intellectual disability, developmental delay, seizures and ataxia. It is caused by maternal deficiency of the imprinted gene UBE3A (ubiquitin protein ligase E3A). The paternal copy of UBE3A is usually intact, but silenced by a nucleus-localized lncRNA, UBE3A-ATS. Ube3a-ATS reduction in mice has been shown to increase paternal UBE3A expression. ICV treatment with anti-Ube3a-ATS ODN led to reduction in Ube3a-ATS expression and upregulation of Ube3a in mice. No significant changes in body weight, microglial activation marker (AIF1) or astrocyte marker (GFAP) expression were observed one month after single ICV injection. Notably, a blocking ODN did not affect paternal UBE3A expression, indicating that RNase H degradation of the UBE3A-ATS transcript is required for paternal UBE3A derepression101.
Notably, knockdown of Ube3a-ATS is complicated by the fact that Snrpn, Snord116 and Snord115 small nucleolar RNAs are processed from the same precursor transcript. Mutations affecting these small nucleolar RNAs are known to cause Prader–Willi syndrome. Interestingly, expression of these RNAs was not affected by the ODN treatment, possibly due to a very fast rate of Snord splicing101.
Combination ODN approaches
In a final category, ODNs have been combined to address disease targets by different mechanisms. An example of such therapeutic application is Alzheimer’s disease (AD).
AD is a progressive brain disorder that severely affects learning and memory. Accumulation of toxic aggregates of Abeta peptide is considered a hallmark of AD. Although the exact genetic causes of AD remain unclear, multiple proteins have been proposed as potential therapeutic targets for the treatment of this disorder.
In one of the early ODN applications, an ODN against Abeta reversed learning and memory deficits of aged SAMP8 mice, a model of AD that overproduces Abeta1. In another attempt to use ODNs to decrease Abeta accumulation, a phosphorothioate ODN corresponding to the 17–30 region of Abeta (OL-1) was administered ICV three times at two-week intervals to SAMP8 mice. OL-1 did not change mRNA levels, suggesting that its major effect is blocking at the level of translation. Treatment with OL-1 reversed learning and memory deficits in T-maze foot-shock-avoidance tests and lowered oxidative stress in SAMP8 mice. In a second study, OL-1 was injected systemically three times at two-week intervals in the tail vein of SAMP8 mice. OL-1 was shown to cross the BBB, perhaps partially due to impairment of the BBB observed in AD models. OL-1 administered IV also improved learning and memory in both T-maze foot-shock-avoidance and novel object-place recognition tests2.
Other therapeutic targets for AD are now being investigated. It has been shown that the balance of ApoER2 exon 19 splicing is deregulated in postmortem brain tissue from AD patients and in a transgenic mouse model of AD. Blocking putative binding sites of SRSF1, a regulator of exon 19 splicing, with a 2MOE ODN results in increased exon 19 inclusion. A single ICV dose of the ODN administered 1 or 2 days after birth by ICV injection in AD mice corrected ApoER2 splicing starting already at 1 week after injection. Splicing correction lasted up to 6 months and was accompanied by improvements in synaptic function, learning and memory102.
MicroRNA-33 is proposed as another therapeutic target for the treatment of AD. Inhibition of microRNA-33 increased lipidation of brain ApoE and reduced Abeta levels by inducing ABCA1. A four-week ICV infusion of 30 μg/d of anti-miR-33, a 2MOE ODN from Regulus Therapeutics (San Diego, CA, USA), resulted in the brain-specific inhibition of microRNA-33 and markedly decreased Abeta levels in the cortex of APP/PS1 mice103,104.
Glycogen synthase kinase 3 beta (GSK-3β) represents yet another AD target. To suppress the level of GSK-3β, SAMP8 mice were treated with an anti-GSK-3β ODN (GAO). The treatment decreased GSK-3β levels in the cortex and improved performance in aversive T-maze and object recognition tests105.
Low expression of another target, brain-derived neurotrophic factor (BDNF), has been associated with AD and other disorders of the nervous system. Our work (C.W. and colleagues)36 has shown that interfering with function of a BDNF NAT, a regulatory lncRNA in the BDNF locus, using ICV administration of LNA ODNs (AntagoNATs) results in increased expression of biologically active BDNF protein in mouse brain and proliferation and differentiation of neurons in mouse neurosphere cultures36.
A final drug target in AD is pathological accumulation of tau protein. This could be potentially reversible via ODNs that knockdown tau expression. Indeed, such ODNs (2MOE PS gapmers) have recently been shown to selectively decrease human tau mRNA and protein in mice expressing human tau with P301S mutation, which led to dissolution of the pre-existing phosphorylated tau and thioflavin S pathology. Furthermore, hippocampal volume loss, neuronal death, nesting deficits and mouse survival were normalized after ODN treatment. In non-human primates, tau ODN-induced reduction of tau mRNA and protein in both the CNS tissue and CSF persisted for at least 6 weeks after lumbar IT injection. These results support the feasibility of a tau-lowering therapy in AD, even after pathological tau deposition has begun106. Another possible drug target in AD is the pathological accumulation of tau protein, potentially reversible by ODNs that knockdown tau expression. Indeed, such ODNs (2MOE PS gapmers) were shown to selectively decrease human tau mRNA and protein in mice expressing human tau with P301S mutation, which led to dissolution of the pre-existing phosphorylated tau and Thioflavin S pathology. Furthermore, hippocampal volume loss, neuronal death, nesting deficits and mouse survival were normalized after ODN treatment. In nonhuman primates, tau ODN-induced reduction of tau mRNA and protein in the CNS tissue and CSF persisted for at least 6 weeks after lumbar IT injection. These results support the feasibility of a tau-lowering therapy in AD even after pathological tau deposition has begun118.
Conclusions
Overall, the therapeutic oligonucleotide field has made great progress in recent years, including substantial achievements in the CNS area. As initially suggested, already a quarter of a century ago, because of their unique properties, ODNs represent viable drug candidates for the therapy of CNS disorders. In fact, CNS can be viewed as a protected environment for exogenously applied oligonucleotides, and show striking stability in CSF. High target specificity and a relatively simple design/selection process, as well as adaptability to diverse therapeutic targets, ensure continued interest in developing oligonucleotide drugs, especially for rare genetic disorders and other diseases with known genetic causes. Discovery of regulatory long-noncoding RNAs has created an opportunity to selectively upregulate the expression of most genes. In addition, exon skipping methods allow targeted mutation correction. As shown above, ODNs with diverse mechanisms of action are already being developed to address the underlying genetic defects in several CNS diseases. The advantages of the ODN approach over gene therapy in genetic diseases include reversibility of the treatment and lack of need for the viral vectors with their inherent liabilities.
Although long half-lives and gradual release from tissue depots ensure the possibility of infrequent dosing, direct CNS administration using IT or ICV routes could be a challenge for chronic treatment due to the possibility of infection and tissue damage during the injection procedure (Table 2). Medium-term (months to years) exposure to IT-administered ODNs did not reveal any serious toxicity; however, the possibility of complications after long-term IT or ICV use cannot be excluded. Oral or inhalation-ready formulation would greatly reduce these risks. Although development of such formulations is underway, substantial further efforts are required to bring alternative administration routes and/or carriers for efficient trans-BBB uptake of oligonucleotides to the clinic, to overcome the potential adverse effects of direct CNS delivery.
Table 2.
Comparison of different oligonucleotide delivery routes
| Route | Delivery site | Convenience to patients | Probability of adverse events | Applicability to CNS delivery of naked ODNs |
|---|---|---|---|---|
| Oral (PO) | By mouth | ++++++ | + | − |
| Intranasal | Inhalation or application of gel/liquid formulation inside the nose | ++++++ | + | ++ |
| Subcutaneous injection (SC) | Intra-skin injection | ++++ | ++ | − |
| Intraperitoneal injection (IP) | Injection into body cavity | ++ | +++ | − |
| Intramuscular injection (IM) | Injection deep into the muscle | +++ | +++ | N.A. |
| Intravenous injection (IV) | Injection directly into vein | +++ | ++++ | − |
| Intrathecal injection (IT) | Injection through brain/spinal cord sheath, usually between lumbar vertebrae | ++ | +++++ | ++++++ |
| Intracerebroventricular injection (ICV) | Injection deep inside the brain, into a ventricle, requires brain surgery to install a catheter | + | ++++++++ | ++++++ |
+, high; N.A., not applicable; −, low
Furthermore, extensive CNS-focused studies of ODN PK and PD characteristics are urgently needed. Given class similarities of different ODNs, PK and PD data from IND filings, if made available by the FDA, would facilitate development of PK models that in turn will improve dosing regimen and clinical study designs for future ODN therapies.
Acknowledgments
RNA-related work in C.W.’s laboratory is currently funded by NIH grants DA035592, NS071674 and AA023781.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare competing financial interests: details are available in the online version of the paper.
References
- 1.Banks WA, et al. Delivery across the blood-brain barrier of antisense directed against amyloid beta: reversal of learning and memory deficits in mice overexpressing amyloid precursor protein. J Pharmacol Exp Ther. 2001;297:1113–1121. [PubMed] [Google Scholar]
- 2.Farr SA, Erickson MA, Niehoff ML, Banks WA, Morley JE. Central and peripheral administration of antisense oligonucleotide targeting amyloid-β protein precursor improves learning and memory and reduces neuroinflammatory cytokines in Tg2576 (AβPPswe) mice. J Alzheimer’s Dis. 2014;40:1005–1016. doi: 10.3233/JAD-131883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Juliano RL. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016;44:6518–6548. doi: 10.1093/nar/gkw236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eckstein F. Nucleoside phosphorothioates. J Am Chem Soc. 1970;92:4718–4723. doi: 10.1021/ja00718a039. [DOI] [PubMed] [Google Scholar]
- 5.Stirchak EP, Summerton JE, Weller DD. Uncharged stereoregular nucleic acid analogs: 2. Morpholino nucleoside oligomers with carbamate internucleoside linkages. Nucleic Acids Res. 1989;17:6129–6141. doi: 10.1093/nar/17.15.6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kupryushkin MS, Pyshnyi DV, Stetsenko DA. Phosphoryl guanidines: a new type of nucleic acid analogues. Acta Naturae. 2014;6:116–118. [PMC free article] [PubMed] [Google Scholar]
- 7.Nielsen PE, Egholm M, Berg RH, Buchardt O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science. 1991;254:1497–1500. doi: 10.1126/science.1962210. [DOI] [PubMed] [Google Scholar]
- 8.Bobst AM, Rottman F, Cerutti PA. Effect of the methylation of the 2′-hydroxyl groups in polyadenylic acid on its structure in weakly acidic and neutral solutions and on its capability to form ordered complexes with polyuridylic acid. J Mol Biol. 1969;46:221–234. doi: 10.1016/0022-2836(69)90418-5. [DOI] [PubMed] [Google Scholar]
- 9.Evers MM, Toonen LJ, van Roon-Mom WM. Antisense oligonucleotides in therapy for neurodegenerative disorders. Adv Drug Deliv Rev. 2015;87:90–103. doi: 10.1016/j.addr.2015.03.008. [DOI] [PubMed] [Google Scholar]
- 10.Cook PD, Guinosso CJ. 2′-O-modified nucleosides and phosphoramidites. 5,914,396. US patent no. 1995
- 11.Wahlestedt C, et al. Potent and nontoxic antisense oligonucleotides containing locked nucleic acids. Proc Natl Acad Sci USA. 2000;97:5633–5638. doi: 10.1073/pnas.97.10.5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goyenvalle A, et al. Functional correction in mouse models of muscular dystrophy using exon-skipping tricyclo-DNA oligomers. Nat Med. 2015;21:270–275. doi: 10.1038/nm.3765. [DOI] [PubMed] [Google Scholar]
- 13.Hua Y, et al. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature. 2011;478:123–126. doi: 10.1038/nature10485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Passini MA, et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci Transl Med. 2011;3:72ra18. doi: 10.1126/scitranslmed.3001777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Williams JH, et al. Oligonucleotide-mediated survival of motor neuron protein expression in CNS improves phenotype in a mouse model of spinal muscular atrophy. J Neurosci. 2009;29:7633–7638. doi: 10.1523/JNEUROSCI.0950-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Porensky PN, et al. A single administration of morpholino antisense oligomer rescues spinal muscular atrophy in mouse. Hum Mol Genet. 2012;21:1625–1638. doi: 10.1093/hmg/ddr600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou H, et al. A novel morpholino oligomer targeting ISS-N1 improves rescue of severe spinal muscular atrophy transgenic mice. Hum Gene Ther. 2013;24:331–342. doi: 10.1089/hum.2012.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ho W, Zhang XQ, Xu X. Biomaterials in siRNA Delivery: A Comprehensive Review. Adv Healthc Mater. 2016;5:2715–2731. doi: 10.1002/adhm.201600418. [DOI] [PubMed] [Google Scholar]
- 19.Ozcan G, Ozpolat B, Coleman RL, Sood AK, Lopez-Berestein G. Preclinical and clinical development of siRNA-based therapeutics. Adv Drug Deliv Rev. 2015;87:108–119. doi: 10.1016/j.addr.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qiu Y, Lam JK, Leung SW, Liang W. Delivery of RNAi therapeutics to the airways-from bench to bedside. Molecules. 2016;21:E1249. doi: 10.3390/molecules21091249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim HJ, Kim A, Miyata K, Kataoka K. Recent progress in development of siRNA delivery vehicles for cancer therapy. Adv Drug Deliv Rev. 2016;104:61–77. doi: 10.1016/j.addr.2016.06.011. [DOI] [PubMed] [Google Scholar]
- 22.Lima WF, De Hoyos CL, Liang XH, Crooke ST. RNA cleavage products generated by antisense oligonucleotides and siRNAs are processed by the RNA surveillance machinery. Nucleic Acids Res. 2016;44:3351–3363. doi: 10.1093/nar/gkw065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller VM, et al. Allele-specific silencing of dominant disease genes. Proc Natl Acad Sci USA. 2003;100:7195–7200. doi: 10.1073/pnas.1231012100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ward AJ, Norrbom M, Chun S, Bennett CF, Rigo F. Nonsense-mediated decay as a terminating mechanism for antisense oligonucleotides. Nucleic Acids Res. 2014;42:5871–5879. doi: 10.1093/nar/gku184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haas M, et al. European Medicines Agency review of ataluren for the treatment of ambulant patients aged 5 years and older with Duchenne muscular dystrophy resulting from a nonsense mutation in the dystrophin gene. Neuromuscul Disord. 2015;25:5–13. doi: 10.1016/j.nmd.2014.11.011. [DOI] [PubMed] [Google Scholar]
- 26.Kerem E, et al. Ataluren for the treatment of nonsense-mutation cystic fibrosis: a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir Med. 2014;2:539–547. doi: 10.1016/S2213-2600(14)70100-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shimizu-Motohashi Y, Miyatake S, Komaki H, Takeda S, Aoki Y. Recent advances in innovative therapeutic approaches for Duchenne muscular dystrophy: from discovery to clinical trials. Am J Transl Res. 2016;8:2471–2489. [PMC free article] [PubMed] [Google Scholar]
- 28.Arechavala-Gomeza V, Khoo B, Aartsma-Rus A. Splicing modulation therapy in the treatment of genetic diseases. Appl Clin Genet. 2014;7:245–252. doi: 10.2147/TACG.S71506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pereira P, Queiroz JA, Figueiras A, Sousa F. Current progress on microRNAs-based therapeutics in neurodegenerative diseases. Wiley Interdiscip Rev RNA. 2016 doi: 10.1002/wrna.1409. [DOI] [PubMed]
- 30.Catalanotto C, Cogoni C, Zardo G. MicroRNA in control of gene expression: an overview of nuclear functions. Int J Mol Sci. 2016;17:E1712. doi: 10.3390/ijms17101712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng CJ, et al. MicroRNA silencing for cancer therapy targeted to the tumour microenvironment. Nature. 2015;518:107–110. doi: 10.1038/nature13905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wahlestedt C. Targeting long non-coding RNA to therapeutically upregulate gene expression. Nat Rev Drug Discov. 2013;12:433–446. doi: 10.1038/nrd4018. [DOI] [PubMed] [Google Scholar]
- 33.Nakagawa S, Kageyama Y. Nuclear lncRNAs as epigenetic regulators-beyond skepticism. Biochim Biophys Acta. 2014;1839:215–222. doi: 10.1016/j.bbagrm.2013.10.009. [DOI] [PubMed] [Google Scholar]
- 34.Magistri M, Faghihi MA, St Laurent G, III, Wahlestedt C. Regulation of chromatin structure by long noncoding RNAs: focus on natural antisense transcripts. Trends Genet. 2012;28:389–396. doi: 10.1016/j.tig.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Katayama S, et al. Antisense transcription in the mammalian transcriptome. Science. 2005;309:1564–1566. doi: 10.1126/science.1112009. [DOI] [PubMed] [Google Scholar]
- 36.Modarresi F, et al. Inhibition of natural antisense transcripts in vivo results in gene-specific transcriptional upregulation. Nat Biotechnol. 2012;30:453–459. doi: 10.1038/nbt.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Halley P, et al. Regulation of the apolipoprotein gene cluster by a long noncoding RNA. Cell Rep. 2014;6:222–230. doi: 10.1016/j.celrep.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hsiao J, et al. Upregulation of haploinsufficient gene expression in the brain by targeting a long non-coding RNA improves seizure phenotype in a model of Dravet syndrome. EBioMedicine. 2016;9:257–277. doi: 10.1016/j.ebiom.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao J, et al. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol Cell. 2010;40:939–953. doi: 10.1016/j.molcel.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Janowski BA, et al. Activating gene expression in mammalian cells with promoter-targeted duplex RNAs. Nat Chem Biol. 2007;3:166–173. doi: 10.1038/nchembio860. [DOI] [PubMed] [Google Scholar]
- 41.Chu Y, Yue X, Younger ST, Janowski BA, Corey DR. Involvement of argonaute proteins in gene silencing and activation by RNAs complementary to a non-coding transcript at the progesterone receptor promoter. Nucleic Acids Res. 2010;38:7736–7748. doi: 10.1093/nar/gkq648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Portnoy V, et al. saRNA-guided Ago2 targets the RITA complex to promoters to stimulate transcription. Cell Res. 2016;26:320–335. doi: 10.1038/cr.2016.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang B, et al. Small-activating RNA can change nucleosome positioning in human fibroblasts. J Biomol Screen. 2016;21:634–642. doi: 10.1177/1087057116637562. [DOI] [PubMed] [Google Scholar]
- 44.Hu J, et al. Promoter-associated small double-stranded RNA interacts with heterogeneous nuclear ribonucleoprotein A2/B1 to induce transcriptional activation. Biochem J. 2012;447:407–416. doi: 10.1042/BJ20120256. [DOI] [PubMed] [Google Scholar]
- 45.Kleinman ME, et al. Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature. 2008;452:591–597. doi: 10.1038/nature06765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koller E, et al. Mechanisms of single-stranded phosphorothioate modified antisense oligonucleotide accumulation in hepatocytes. Nucleic Acids Res. 2011;39:4795–4807. doi: 10.1093/nar/gkr089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Juliano RL, Ming X, Nakagawa O. Cellular uptake and intracellular trafficking of antisense and siRNA oligonucleotides. Bioconjug Chem. 2012;23:147–157. doi: 10.1021/bc200377d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wagenaar TR, et al. Identification of the endosomal sorting complex required for transport-I (ESCRT-I) as an important modulator of anti-miR uptake by cancer cells. Nucleic Acids Res. 2015;43:1204–1215. doi: 10.1093/nar/gku1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shen W, Liang XH, Crooke ST. Phosphorothioate oligonucleotides can displace NEAT1 RNA and form nuclear paraspeckle-like structures. Nucleic Acids Res. 2014;42:8648–8662. doi: 10.1093/nar/gku579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Naganuma T, Hirose T. Paraspeckle formation during the biogenesis of long non-coding RNAs. RNA Biol. 2013;10:456–461. doi: 10.4161/rna.23547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liang XH, et al. Hsp90 protein interacts with phosphorothioate oligonucleotides containing hydrophobic 2′-modifications and enhances antisense activity. Nucleic Acids Res. 2016;44:3892–3907. doi: 10.1093/nar/gkw144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shemesh CS, et al. Pharmacokinetic and pharmacodynamic investigations of ION-353382, a model antisense oligonucleotide: using alpha-2-macroglobulin and murinoglobulin double-knockout mice. Nucleic Acid Ther. 2016;26:223–235. doi: 10.1089/nat.2016.0607. [DOI] [PubMed] [Google Scholar]
- 53.Zhu JY, et al. Efficient nuclear drug translocation and improved drug efficacy mediated by acidity-responsive boronate-linked dextran/cholesterol nanoassembly. Biomaterials. 2015;52:281–290. doi: 10.1016/j.biomaterials.2015.02.048. [DOI] [PubMed] [Google Scholar]
- 54.Liang XH, Shen W, Sun H, Prakash TP, Crooke ST. TCP1 complex proteins interact with phosphorothioate oligonucleotides and can co-localize in oligonucleotide-induced nuclear bodies in mammalian cells. Nucleic Acids Res. 2014;42:7819–7832. doi: 10.1093/nar/gku484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miller CM, et al. Stabilin-1 and Stabilin-2 are specific receptors for the cellular internalization of phosphorothioate-modified antisense oligonucleotides (ASOs) in the liver. Nucleic Acids Res. 2016;44:2782–2794. doi: 10.1093/nar/gkw112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ezzat K, et al. Self-assembly into nanoparticles is essential for receptor mediated uptake of therapeutic antisense oligonucleotides. Nano Lett. 2015;15:4364–4373. doi: 10.1021/acs.nanolett.5b00490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ezzat K, et al. Scavenger receptor-mediated uptake of cell-penetrating peptide nanocomplexes with oligonucleotides. FASEB J. 2012;26:1172–1180. doi: 10.1096/fj.11-191536. [DOI] [PubMed] [Google Scholar]
- 58.Lorenz P, Baker BF, Bennett CF, Spector DL. Phosphorothioate antisense oligonucleotides induce the formation of nuclear bodies. Mol Biol Cell. 1998;9:1007–1023. doi: 10.1091/mbc.9.5.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marcusson EG, Bhat B, Manoharan M, Bennett CF, Dean NM. Phosphorothioate oligodeoxyribonucleotides dissociate from cationic lipids before entering the nucleus. Nucleic Acids Res. 1998;26:2016–2023. doi: 10.1093/nar/26.8.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heemskerk H, et al. Preclinical PK and PD studies on 2′-O-methyl-phosphorothioate RNA antisense oligonucleotides in the mdx mouse model. Mol Ther. 2010;18:1210–1217. doi: 10.1038/mt.2010.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hammond SM, et al. Systemic peptide-mediated oligonucleotide therapy improves long-term survival in spinal muscular atrophy. Proc Natl Acad Sci USA. 2016;113:10962–10967. doi: 10.1073/pnas.1605731113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Craft S, et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch Neurol. 2012;69:29–38. doi: 10.1001/archneurol.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hashizume R, et al. New therapeutic approach for brain tumors: Intranasal delivery of telomerase inhibitor GRN163. Neuro-oncol. 2008;10:112–120. doi: 10.1215/15228517-2007-052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ma Q, et al. Inhibition of microRNA-210 provides neuroprotection in hypoxic-ischemic brain injury in neonatal rats. Neurobiol Dis. 2016;89:202–212. doi: 10.1016/j.nbd.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ferrés-Coy A, et al. Therapeutic antidepressant potential of a conjugated siRNA silencing the serotonin transporter after intranasal administration. Mol Psychiatry. 2016;21:328–338. doi: 10.1038/mp.2015.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bleier BS, Kohman RE, Feldman RE, Ramanlal S, Han X. Permeabilization of the blood-brain barrier via mucosal engrafting: implications for drug delivery to the brain. PLoS One. 2013;8:e61694. doi: 10.1371/journal.pone.0061694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wahlestedt C, Yee F, Yoo H, Koob GF, Heilig M. In: Zalcman S, Scheller R, Tsien R, editors. Molecular Neurobiology; Proceedings of the second NIMH Conference; 1992. [Google Scholar]
- 68.Wahlestedt C, et al. Antisense oligodeoxynucleotides to NMDA-R1 receptor channel protect cortical neurons from excitotoxicity and reduce focal ischaemic infarctions. Nature. 1993;363:260–263. doi: 10.1038/363260a0. [DOI] [PubMed] [Google Scholar]
- 69.Wahlestedt C, Pich EM, Koob GF, Yee F, Heilig M. Modulation of anxiety and neuropeptide Y-Y1 receptors by antisense oligodeoxynucleotides. Science. 1993;259:528–531. doi: 10.1126/science.8380941. [DOI] [PubMed] [Google Scholar]
- 70.Wahlestedt C. Antisense oligonucleotide strategies in neuropharmacology. Trends Pharmacol Sci. 1994;15:42–46. doi: 10.1016/0165-6147(94)90107-4. [DOI] [PubMed] [Google Scholar]
- 71.Standifer KM, Chien CC, Wahlestedt C, Brown GP, Pasternak GW. Selective loss of delta opioid analgesia and binding by antisense oligodeoxynucleotides to a delta opioid receptor. Neuron. 1994;12:805–810. doi: 10.1016/0896-6273(94)90333-6. [DOI] [PubMed] [Google Scholar]
- 72.Geary RS, Norris D, Yu R, Bennett CF. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv Drug Deliv Rev. 2015;87:46–51. doi: 10.1016/j.addr.2015.01.008. [DOI] [PubMed] [Google Scholar]
- 73.Rigo F, et al. Pharmacology of a central nervous system delivered 2′-O-methoxyethyl-modified survival of motor neuron splicing oligonucleotide in mice and nonhuman primates. J Pharmacol Exp Ther. 2014;350:46–55. doi: 10.1124/jpet.113.212407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Verhaart IE, et al. Dose-dependent pharmacokinetic profiles of 2′-O-methyl phosphorothioate antisense oligonucleotidesin mdx mice. Nucleic Acid Ther. 2013;23:228–237. doi: 10.1089/nat.2012.0398. [DOI] [PubMed] [Google Scholar]
- 75.Kordasiewicz HB, et al. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron. 2012;74:1031–1044. doi: 10.1016/j.neuron.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Miller TM, et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol. 2013;12:435–442. doi: 10.1016/S1474-4422(13)70061-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shen L, et al. Effects of repeated complement activation associated with chronic treatment of cynomolgus monkeys with 2′-o-methoxyethyl modified antisense oligonucleotide. Nucleic Acid Ther. 2016;26:236–249. doi: 10.1089/nat.2015.0584. [DOI] [PubMed] [Google Scholar]
- 78.Shen L, et al. Mechanistic understanding for the greater sensitivity of monkeys to antisense oligonucleotide-mediated complement activation compared with humans. J Pharmacol Exp Ther. 2014;351:709–717. doi: 10.1124/jpet.114.219378. [DOI] [PubMed] [Google Scholar]
- 79.Henry SP, et al. Considerations for the characterization and interpretation of results related to alternative complement activation in monkeys associated with oligonucleotide-based therapeutics. Nucleic Acid Ther. 2016;26:210–215. doi: 10.1089/nat.2015.0593. [DOI] [PubMed] [Google Scholar]
- 80.Voit T, et al. Safety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): an exploratory, randomised, placebo-controlled phase 2 study. Lancet Neurol. 2014;13:987–996. doi: 10.1016/S1474-4422(14)70195-4. [DOI] [PubMed] [Google Scholar]
- 81.Engelhardt JA. Comparative renal toxicopathology of antisense oligonucleotides. Nucleic Acid Ther. 2016;26:199–209. doi: 10.1089/nat.2015.0598. [DOI] [PubMed] [Google Scholar]
- 82.Merlini L, Sabatelli P. Improving clinical trial design for Duchenne muscular dystrophy. BMC Neurol. 2015;15:153. doi: 10.1186/s12883-015-0408-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rigo F, Hua Y, Krainer AR, Bennett CF. Antisense-based therapy for the treatment of spinal muscular atrophy. J Cell Biol. 2012;199:21–25. doi: 10.1083/jcb.201207087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chiriboga CA, et al. Results from a phase 1 study of nusinersen (ISIS-SMN(Rx)) in children with spinal muscular atrophy. Neurology. 2016;86:890–897. doi: 10.1212/WNL.0000000000002445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Haché M, et al. Intrathecal injections in children with spinal muscular atrophy: nusinersen clinical trial experience. J Child Neurol. 2016;31:899–906. doi: 10.1177/0883073815627882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hua Y, et al. Motor neuron cell-nonautonomous rescue of spinal muscular atrophy phenotypes in mild and severe transgenic mouse models. Genes Dev. 2015;29:288–297. doi: 10.1101/gad.256644.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sintusek P, et al. Histopathological defects in intestine in severe spinal muscular atrophy mice are improved by systemic antisense oligonucleotide treatment. PLoS One. 2016;11:e0155032. doi: 10.1371/journal.pone.0155032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bogdanik LP, et al. Systemic, postsymptomatic antisense oligonucleotide rescues motor unit maturation delay in a new mouse model for type II/III spinal muscular atrophy. Proc Natl Acad Sci USA. 2015;112:E5863–E5872. doi: 10.1073/pnas.1509758112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Osman EY, et al. Morpholino antisense oligonucleotides targeting intronic repressor Element1 improve phenotype in SMA mouse models. Hum Mol Genet. 2014;23:4832–4845. doi: 10.1093/hmg/ddu198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Evers MM, et al. Ataxin-3 protein modification as a treatment strategy for spinocerebellar ataxia type 3: removal of the CAG containing exon. Neurobiol Dis. 2013;58:49–56. doi: 10.1016/j.nbd.2013.04.019. [DOI] [PubMed] [Google Scholar]
- 91.Cavalieri S, Pozzi E, Gatti RA, Brusco A. Deepintronic ATM mutation detected by genomic resequencing and corrected in vitro by antisense morpholino oligonucleotide (AMO) Eur J Hum Genet. 2013;21:774–778. doi: 10.1038/ejhg.2012.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Donnelly CJ, et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 2013;80:415–428. doi: 10.1016/j.neuron.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lagier-Tourenne C, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci USA. 2013;110:E4530–E4539. doi: 10.1073/pnas.1318835110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sareen D, et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med. 2013;5:208ra149. doi: 10.1126/scitranslmed.3007529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jiang J, et al. Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron. 2016;90:535–550. doi: 10.1016/j.neuron.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gotkine M, et al. Presymptomatic treatment with acetylcholinesterase antisense oligonucleotides prolongs survival in ALS (G93A-SOD1) mice. BioMed Res Int. 2013;2013:845345. doi: 10.1155/2013/845345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Koval ED, et al. Method for widespread microRNA-155 inhibition prolongs survival in ALS-model mice. Hum Mol Genet. 2013;22:4127–4135. doi: 10.1093/hmg/ddt261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Southwell AL, et al. In vivo evaluation of candidate allele-specific mutant huntingtin gene silencing antisense oligonucleotides. Mol Ther. 2014;22:2093–2106. doi: 10.1038/mt.2014.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Benson MD, et al. Suppression of choroid plexus transthyretin levels by antisense oligonucleotide treatment. Amyloid. 2010;17:43–49. doi: 10.3109/13506129.2010.483121. [DOI] [PubMed] [Google Scholar]
- 100.Sztainberg Y, et al. Reversal of phenotypes in MECP2 duplication mice using genetic rescue or antisense oligonucleotides. Nature. 2015;528:123–126. doi: 10.1038/nature16159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Meng L, et al. Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature. 2015;518:409–412. doi: 10.1038/nature13975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hinrich AJ, et al. Therapeutic correction of ApoER2 splicing in Alzheimer’s disease mice using antisense oligonucleotides. EMBO Mol Med. 2016;8:328–345. doi: 10.15252/emmm.201505846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kim J, et al. microRNA-33 regulates ApoE lipidation and amyloid-β metabolism in the brain. J Neurosci. 2015;35:14717–14726. doi: 10.1523/JNEUROSCI.2053-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jan A, et al. Direct intracerebral delivery of a miR-33 antisense oligonucleotide into mouse brain increases brain ABCA1 expression. [Corrected] Neurosci Lett. 2015;598:66–72. doi: 10.1016/j.neulet.2015.05.007. [DOI] [PubMed] [Google Scholar]
- 105.Farr SA, et al. Antisense oligonucleotide against GSK-3β in brain of SAMP8 mice improves learning and memory and decreases oxidative stress: Involvement of transcription factor Nrf2 and implications for Alzheimer disease. Free Radic Biol Med. 2014;67:387–395. doi: 10.1016/j.freeradbiomed.2013.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.DeVos SL, et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci Transl Med. 2017;9:eaag0481. doi: 10.1126/scitranslmed.aag0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Obika S, et al. Synthesis of 2′-O,4′-C-methyleneuridine and -cytidine. Novel bicyclic nucleosides having a fixed C3′-endo sugar puckering. Tetrahedr Lett. 1997;38:8735–8738. [Google Scholar]
- 108.Steffens R, Leumann CJ. Synthesis and thermodynamic and biophysical properties of tricyclo-DNA. J Am Chem Soc. 1999;121:3249–3255. [Google Scholar]
- 109.Zamecnik PC, Stephenson ML. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc Natl Acad Sci USA. 1978;75:280–284. doi: 10.1073/pnas.75.1.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Stephenson ML, Zamecnik PC. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc Natl Acad Sci USA. 1978;75:285–288. doi: 10.1073/pnas.75.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Donis-Keller H. Site specific enzymatic cleavage of RNA. Nucleic Acids Res. 1979;7:179–192. doi: 10.1093/nar/7.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Guerrier-Takada C, Gardiner K, Marsh T, Pace N, Altman S. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell. 1983;35:849–857. doi: 10.1016/0092-8674(83)90117-4. [DOI] [PubMed] [Google Scholar]
- 113.Bass BL, Cech TR. Specific interaction between the self-splicing RNA of Tetrahymena and its guanosine substrate: implications for biological catalysis by RNA. Nature. 1984;308:820–826. doi: 10.1038/308820a0. [DOI] [PubMed] [Google Scholar]
- 114.Elbashir SM, et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 115.Wlotzka B, et al. In vivo properties of an anti-GnRH Spiegelmer: an example of an oligonucleotide-based therapeutic substance class. Proc Natl Acad Sci USA. 2002;99:8898–8902. doi: 10.1073/pnas.132067399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Azad RF, Driver VB, Tanaka K, Crooke RM, Anderson KP. Antiviral activity of a phosphorothioate oligonucleotide complementary to RNA of the human cytomegalovirus major immediate-early region. Antimicrob Agents Chemother. 1993;37:1945–1954. doi: 10.1128/aac.37.9.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kumar R, et al. The first analogues of LNA (locked nucleic acids): phosphorothioate-LNA and 2′-thio-LNA. Bioorg Med Chem Lett. 1998;8:2219–2222. doi: 10.1016/s0960-894x(98)00366-7. [DOI] [PubMed] [Google Scholar]
- 118.DeVos SL, et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci Transl Med. 2017;9:374. doi: 10.1126/scitranslmed.aag0481. [DOI] [PMC free article] [PubMed] [Google Scholar]