

Abstract
The impact of LMO2 expression on cell lineage decisions during T‐cell leukemogenesis remains largely elusive. Using genetic lineage tracing, we have explored the potential of LMO2 in dictating a T‐cell malignant phenotype. We first initiated LMO2 expression in hematopoietic stem/progenitor cells and maintained its expression in all hematopoietic cells. These mice develop exclusively aggressive human‐like T‐ALL. In order to uncover a potential exclusive reprogramming effect of LMO2 in murine hematopoietic stem/progenitor cells, we next showed that transient LMO2 expression is sufficient for oncogenic function and induction of T‐ALL. The resulting T‐ALLs lacked LMO2 and its target‐gene expression, and histologically, transcriptionally, and genetically similar to human LMO2‐driven T‐ALL. We next found that during T‐ALL development, secondary genomic alterations take place within the thymus. However, the permissiveness for development of T‐ALL seems to be associated with wider windows of differentiation than previously appreciated. Restricted Cre‐mediated activation of Lmo2 at different stages of B‐cell development induces systematically and unexpectedly T‐ALL that closely resembled those of their natural counterparts. Together, these results provide a novel paradigm for the generation of tumor T cells through reprogramming in vivo and could be relevant to improve the response of T‐ALL to current therapies.
Keywords: cancer initiation, epigenetic priming, mouse models, oncogenes, stem cells
Subject Categories: Cancer, Development & Differentiation, Immunology
Introduction
The identification of the cell‐of‐origin from which acute lymphoblastic leukemia (ALL) initially arises is of great importance, both for our understanding of the basic biology of tumors and for the translation of this knowledge to the prevention, treatment, and precise prognosis of ALL (Visvader, 2011). Traditionally, the identity of the cell‐of‐origin was extrapolated from the immunophenotypic characterization of a leukemic cell. However, several transcriptome studies have shown that the molecular characteristics of leukemic cells do not correspond, in many cases, to what they seem to be according to their immunophenotype (Lim et al, 2009; Gilbertson, 2011). For this reason, extrapolating the identity of the cancer cell‐of‐origin from the ALL phenotype, without appropriate functional lineage tracing, can lead to the wrong conclusions (Molyneux et al, 2010).
Lmo2 is one of the most frequent drivers of childhood T‐ALL (Van Vlierberghe et al, 2006; Liu et al, 2017). LMO2 serves as a T‐cell oncogene, recurrently translocated in T‐ALL, and is implicated in leukemogenesis among X‐linked severe combined immunodeficiency (SCID) patients, who received retroviral IL2Rγc gene therapy (Hacein‐Bey‐Abina et al, 2003, 2008; Pike‐Overzet et al, 2007; Howe et al, 2008). Aberrant expression of LMO2 in hematopoietic stem/progenitor cells (HSC/PC) or in immature T cells (present in the thymus) leads to thymocyte self‐renewal, early lymphoid precursor's accumulation, and transformation to T‐ALL (McCormack et al, 2010; Treanor et al, 2011; Cleveland et al, 2013; Chambers & Rabbitts, 2015). Moreover, LMO2 was recently identified as one of the six transcription factors required for reprogramming committed murine blood cells into induced hematopoietic stem cells (Riddell et al, 2014). Notably, in addition to T‐ALL, LMO2 is expressed in hematologic cancer of the B‐cell lineage including DLBCL (Natkunam et al, 2007; Cubedo et al, 2012) and BCP‐ALL (de Boer et al, 2011; Malumbres et al, 2011; Deucher et al, 2015). Induction of pluripotency in blood cells and LMO2 expression in B‐cell malignancies suggest that LMO2 might exert leukemogenic potential in specific hematopoietic cell lineages other than the T‐cell lineage. Besides that, a significant proportion of human T‐ALL displays rearrangements of immunoglobulin heavy‐chain genes, which additionally supports this hypothesis (Mizutani et al, 1986; Szczepanski et al, 1999; Meleshko et al, 2005). However, despite frequent alterations of Lmo2 in hematologic tumors, its impact on lineage organization during leukemogenesis and the importance of the cell‐of‐origin for heterogeneity and aggressiveness of Lmo2‐driven tumors have remained unclear. By using in vivo genetic lineage tracing, we show that Lmo2 expression in HSC/PC as well as a precursor and mature B cells causes reprogramming and induction of T‐ALL. Thereby the differentiation state of the tumor cell‐of‐origin influences the frequency and latency of T‐ALL. These findings unveil a novel role of Lmo2 expression and demonstrate that Lmo2 promotes tumorigenesis in a manner contrasting that of other traditional oncogenes, which are persistently active in fully evolved tumor cells (Weinstein, 2002).
Results
Generation of a targeted mouse line conditionally expressing Lmo2 in HSCs
Cell type‐specific conditional activation of Lmo2 is a powerful tool for investigating the cell‐of‐origin of T‐ALL. To achieve this aim, the Lmo2 cDNA was targeted to the ubiquitously expressed Rosa26 locus (Mao et al, 1999) where the green fluorescent protein (eGFP) was linked to the mouse Lmo2 cDNA via an internal ribosomal entry site (IRES). In the absence of Cre, neither Lmo2 nor eGFP is expressed (Appendix Fig S1A and B).
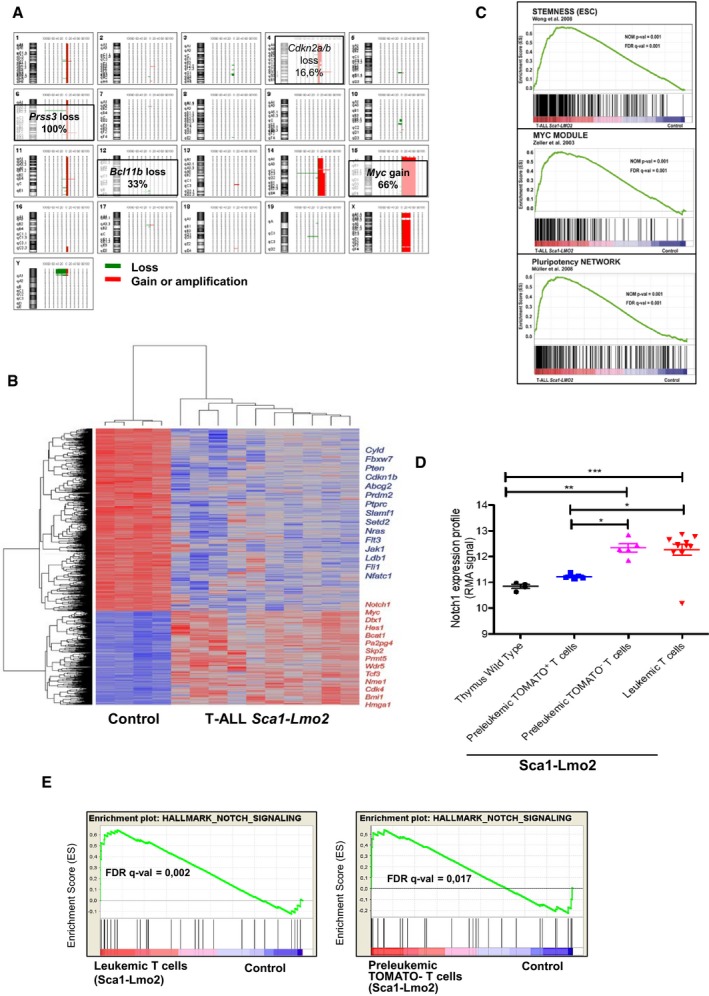
Two sets of observations suggest a reprogramming effect of non‐T‐cell lineage cells by LMO2. First, LMO2 expression due to retroviral insertion and transactivation in CD34+ HSCs of X‐SCID patients caused T‐ALL but no other hematopoietic tumors (Hacein‐Bey‐Abina et al, 2008; Howe et al, 2008). And second, Lmo2 expression in murine blood cells negatively regulated erythroid differentiation (Visvader, 2011) and gives rise to induced pluripotent stem (iPS) cells (Batta et al, 2014; Riddell et al, 2014). We thus aimed to model the capability of Lmo2 to reprogram HSCs. Therefore, we initially crossed the Rosa26‐Lmo2 mice with a Sca1‐Cre mouse strain (Mainardi et al, 2014), in order to initiate Lmo2 expression in HSCs and maintain its expression in all hematopoietic cells (Appendix Fig S1C). Young Rosa26‐Lmo2 + Sca1‐Cre mice showed regular hematopoietic cell differentiation in the bone marrow, peripheral blood, spleen, and thymus (Appendix Figs S1C–E and S2A–D). Rosa26‐Lmo2 + Sca1‐Cre mice had a shorter lifespan than their wild‐type (WT) littermates [Fig 1A; P < 0.0001; log‐rank (Mantel–Cox) test] due to the development of T‐ALL (96.7%; 30/31) that manifested as thymoma, splenomegaly, and disrupted thymic, liver, and splenic architectures (Fig 1B; Appendix Fig S3A and B). Fluorescent activating cell sorting (FACS) analysis of leukemic cells revealed an immature CD8+CD4+/− cell surface phenotype (Fig 1C; Appendix Fig S3C) with Lmo2 expression in the tumor T cells (Fig 1D) and clonal immature T‐cell receptor (TCR) rearrangement (Fig 1E). We also performed whole‐exome sequencing (WES) of these Lmo2+ T‐ALLs (n = 9; Table 1), which were derived from thymuses of diseased Rosa26‐Lmo2 + Sca1‐Cre mice. We detected 23 somatic mutations, including six mutations in genes recorded in the cancer gene list (Table 1; Table EV1). Briefly, we identified recurrent Notch1 single‐nucleotide variations (SNVs; 3/9) and Notch1 indels (4/9), Kras SNVs (3/9), and Nras SNVs (1/9; Table 1). This model corroborated previous findings, especially the observation from the SCID‐X1 gene therapy trial, where integration of γC vector occurred close or in the LMO2 locus and Lmo2 expression was maintained throughout the progeny of the targeted cell (Hacein‐Bey‐Abina et al, 2003, 2008; Pike‐Overzet et al, 2007; Howe et al, 2008). However, in our model Lmo2 expression was maintained constitutively, not only in HSC/PC but also in precursor and mature T cells (McCormack et al, 2010). Thus, a definite conclusion about an exclusive reprogramming effect of Lmo2 in murine HSC/PC in contrast to its expression in T‐cell precursors and mature T cells was limited.
Figure 1. T‐ALL development in Rosa26‐Lmo2 + Sca1‐Cre mice.
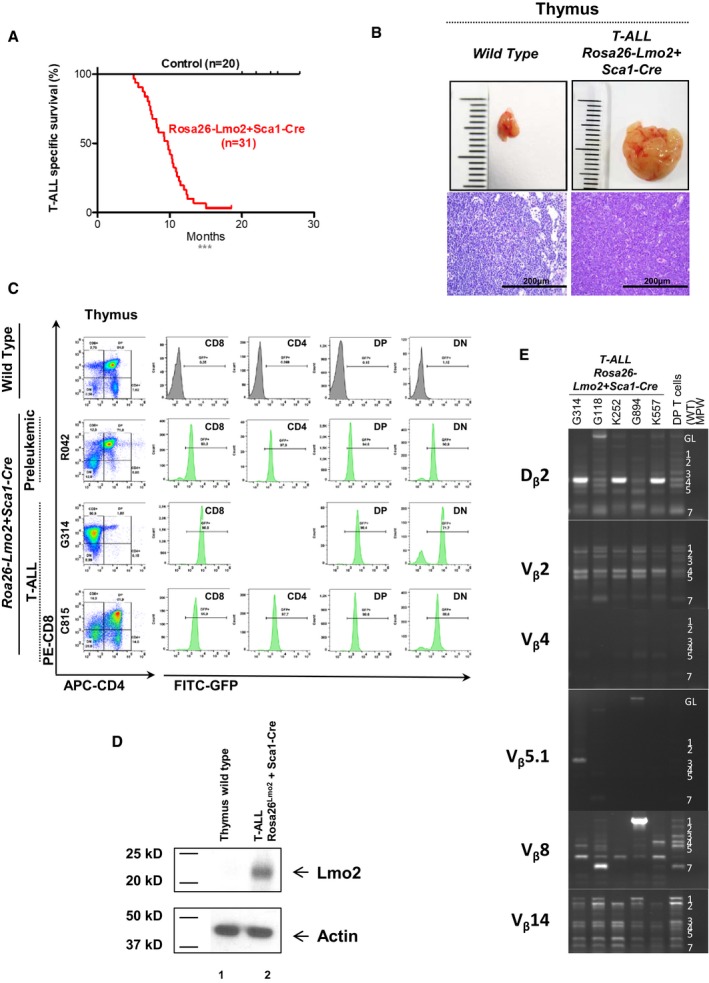
- Leukemia‐specific survival of Rosa26‐Lmo2 + Sca1‐Cre mice (red line, n = 31), showing a significantly (log‐rank ***P < 0.0001) shortened lifespan compared to control littermate WT mice (black line, n = 20) as a result of T‐ALL development.
- An example of thymomas observed in the Rosa26‐Lmo2 + Sca1‐Cre mice studied. A thymus from a control littermate WT mouse is shown for reference. Hematoxylin and eosin staining showing infiltration of the thymus in Rosa26‐Lmo2 + Sca1‐Cre leukemic mice. Images are photographed at 400× magnification (scale bars: 200 μm).
- GFP expression in the pre‐leukemic and leukemic cells from Rosa26‐Lmo2 + Sca1‐Cre mice, respectively. A control littermate WT mouse is shown for reference.
- Western blot analysis for Lmo2 and actin in T cells from the thymus of a wild‐type mouse (1) and from the thymus of a Rosa26‐Lmo2 + Sca1‐Cre leukemic mouse (2). Tumoral cells of Rosa26‐Lmo2 + Sca1‐Cre T‐ALL showed expression of the Lmo2 protein.
- TCR clonality in Rosa26‐Lmo2 + Sca1‐Cre mice. PCR analysis of TCR gene rearrangements in infiltrated thymuses of diseased Rosa26‐Lmo2 + Sca1‐Cre leukemic mice. Sorted DP T cells from the thymus of healthy mice served as a control for polyclonal TCR rearrangements. Leukemic thymus shows an increased clonality within their TCR repertoire (indicated by the code number of each Rosa26‐Lmo2 + Sca1‐Cre mouse analyzed).
Table 1.
Recurrent mutations in mouse models and human Lmo2+ T‐ALL
Lmo2 functions as a “hit‐and‐run” oncogene in T‐ALL development
We next addressed these limitations and modeled the scenario of HSC/PC restricted Lmo2 expression in vivo in a mouse strain where Lmo2 expression was initiated and maintained only in HSC/PC by placing Lmo2–TdTomato cDNA (Shaner et al, 2004) under the control of the stem‐cell‐specific Sca1 promoter (Sca1‐Lmo2; Appendix Fig S4A). All T‐cell subsets in the thymus contained a mosaic of Lmo2 expression (Appendix Fig S4B). Sca1‐Lmo2 mice showed the regular distribution of hematopoietic populations in early post‐gestational development, with TdTomato expression in all hematopoietic cell lineages (Appendix Fig S4C–G). By 3 months, a decrease in the double‐positive (DP) T‐cell population was accompanied by an increase in pre‐leukemic double‐negative (DN) T cells and CD8 T cells (Appendix Fig S4H). Lmo2 expression was detected by quantitative polymerase chain reaction (qPCR); enhanced expression of Cdkn2a was observed in the thymus in transgenic mice, consistent with the induction of Lmo2‐dependent oncogenic stress (Appendix Fig S4I). The Sca1 promoter is active in subsets of T‐cell precursors, and thus, both Lmo2‐expressing and Lmo2‐non‐expressing precursor T cells coexisted in the thymus (Appendix Fig S4J). Studying whether the T‐ALL cases are Lmo2‐Tomato‐positive or Lmo2‐Tomato‐negative has allowed identifying whether the Lmo2 expression is needed for the survival of T‐ALL cells (Tomato+) or it serves as an earlier reprogramming event in leukemogenesis (Tomato−).
Sca1‐Lmo2 mice had a shorter lifespan than their wild‐type (WT) littermates due to a highly disseminated form of T‐ALL, consisting of a clonally immature CD8 or CD4 single‐positive/DP‐like population (Fig 2A–C; Appendix Fig S5A–E), as reported for human T‐ALL (Van Vlierberghe et al, 2006) and Rosa26‐Lmo2 + Sca1‐Cre mice (Fig 1). Histological thymus sections were characterized by infiltrates of highly proliferative tumors and CD3 and TdT positivity (Fig 2B). Surprisingly, all Sca1‐Lmo2 T‐ALL cases studied (18 out of 21) were TdTomato −. Because there is evidence to suggest that the immunogenicity and cytotoxicity of the fluorescent marker potentially may confound the interpretation of in vivo experimental data (Ansari et al, 2016), we next formally excluded the possibility that the cells that were originally marked with the fluorescent marker cannot be accurately traced over time by showing that tumors had lost their Lmo2 expression by three different complementary approaches: immunohistochemistry (Fig 2C) and both real‐time PCR and Western blot in sorted‐purified leukemic Sca1‐Lmo2 cells (Fig 2D and E). This observation indicates that an early expression of the Lmo2 oncogene in HSC/PC has the potential to induce aggressive T‐ALL without any need for its perpetual expression to develop T‐ALL.
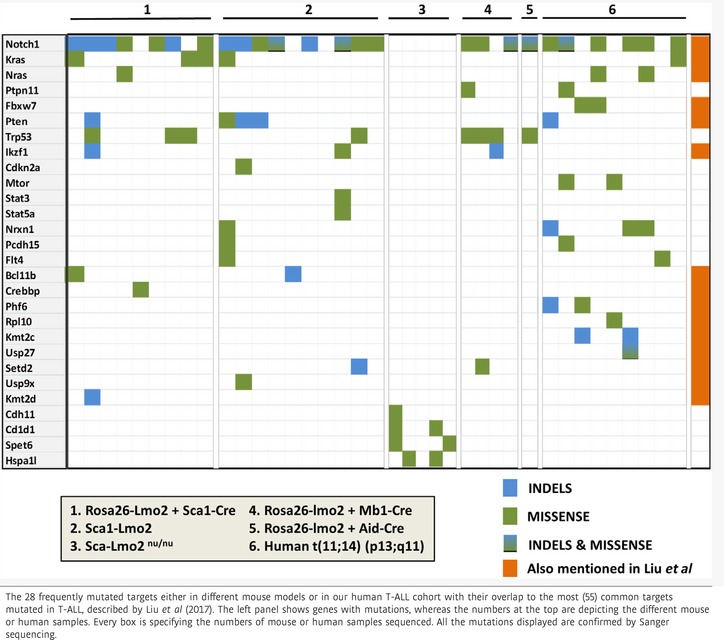
Figure 2. Reprogramming of HS/PCs cells to aggressive malignant mature T cells.
- T‐ALL‐specific survival of Sca1‐Lmo2 mice (red line, n = 21), showing a significantly (log‐rank ***P < 0.0001) shortened lifespan compared to control littermate WT mice (blue line, n = 20) as a result of mature T‐cell malignancies.
- An example of thymomas observed in 100% (21/21) of the Sca1‐Lmo2 mice studied. A thymus from a control littermate WT mouse is shown for reference. Hematoxylin and eosin staining of tumor‐bearing thymuses from Sca1‐Lmo2 mice shows infiltrate of medium‐sized, relatively uniform lymphoid cells that have a high nuclear/cytoplasmic ratio and immature chromatin with a starry‐sky appearance. Immunohistochemistry shows that tumor T cells from Sca1‐Lmo2 thymomas are defined by the presence of TdT (a marker of T‐cell identity) and CD3 (a marker of immature lymphoid cells), and the absence of Pax5 and Lmo2. Images are representative of ≥ 3 replicates. Images are photographed at 300× magnifications (scale bars: 100 μm).
- Flow cytometric analysis of T‐cell subsets in the thymuses of diseased Sca1‐Lmo2 mice. Representative plots of cell subsets from the thymuses are shown. These exhibited the accumulation of DP, CD8, or CD4 single‐positive tumoral T cells. Thymuses from a control littermate WT mouse and a pre‐leukemic Sca1‐Lmo2 mouse are shown for reference. Flow cytometric images are representative of 17 mice analyzed. Tracking of the TdTomato marker for Lmo2 transgene expression in the thymomas of Sca1‐Lmo2 mice shows that tumor T cells are TdTomato‐negative in 100% (17/17) of the Sca1‐Lmo2 mice studied. However, a mosaic of Lmo2 expression remains present within non‐tumoral T‐cell populations (not denoted as tumoral populations). Three plots of cell subsets from the thymuses of diseased Sca1‐Lmo2 mice are shown and are representative of the analysis of 17 diseased Sca1‐Lmo2 mice. TdTomato expression in the thymuses of a control littermate WT mouse and a pre‐leukemic Sca1‐Lmo2 mouse is shown for reference.
- Relative expression of Lmo2 in sorted‐purified leukemic Sca1‐Lmo2 cells compared to control thymus wild‐type cells. Leukemic cells of both Rosa26‐Lmo2 + Mb1‐Cre and Rosa26‐Lmo2 + Sca1‐Cre mice were used as a positive control. The fold change in each group, calculated as 2 sample, was compared. Bars represent the mean and the standard deviation of three replicates.
- Western blot analysis for Lmo2 and actin in T cells from a wild‐type thymus (1), in sorted‐purified leukemic Sca1‐Lmo2 cells (2, 3) and from the thymus of a Rosa26‐Lmo2 + Mb1‐Cre leukemic mouse (4).
Tumor T cells in Sca1‐Lmo2 mice display genetic signatures analogous to human malignant T cells
In human and mouse T‐ALL, genomic gains and losses reflect genomic instability (Maser et al, 2007; Hacein‐Bey‐Abina et al, 2008; Howe et al, 2008; De Keersmaecker et al, 2010). We analyzed DNA from leukemic cells using array‐comparative genomic hybridization (aCGH). Twelve Sca1‐Lmo2 leukemias were analyzed and revealed copy number loss of Cdkn2a/b (2/12) and Bcl11b (4/12), similar to human T‐ALL (Diccianni et al, 1997; Gutierrez et al, 2011), as well as c‐Myc amplification (8/12; Fig 3A). Hence, T‐cell progenitors lacking in Lmo2 expression were genomically unstable and were clonally selected; moreover, they have acquired additional aberrations. To explore the relevance of our findings for human T‐ALL, we analyzed molecular expression signatures in thymic Lmo2‐negative T‐ALL cells (Sca1‐Lmo2; Fig 3B). We observed upregulation of the Notch1 pathway (Weng et al, 2004) and c‐Myc transcriptional targets (Weng et al, 2006), as well as downregulation of Fbxw7, Pten (O'Neil et al, 2007; Palomero et al, 2007; Thompson et al, 2007; Van Vlierberghe & Ferrando, 2012), Cyld, and Cdkn1b (Komuro et al, 1999; Dohda et al, 2007; Espinosa et al, 2010; D'Altri et al, 2011; Fig 3B and C). Hence, similar oncogenic pathways were deregulated in murine T‐ALL arising from Lmo2‐negative T cells, which is consistent with human T‐ALL but not with B‐ALL. Thus, we can exclude that the correlation between murine and human T‐ALL reflects a transformed state.
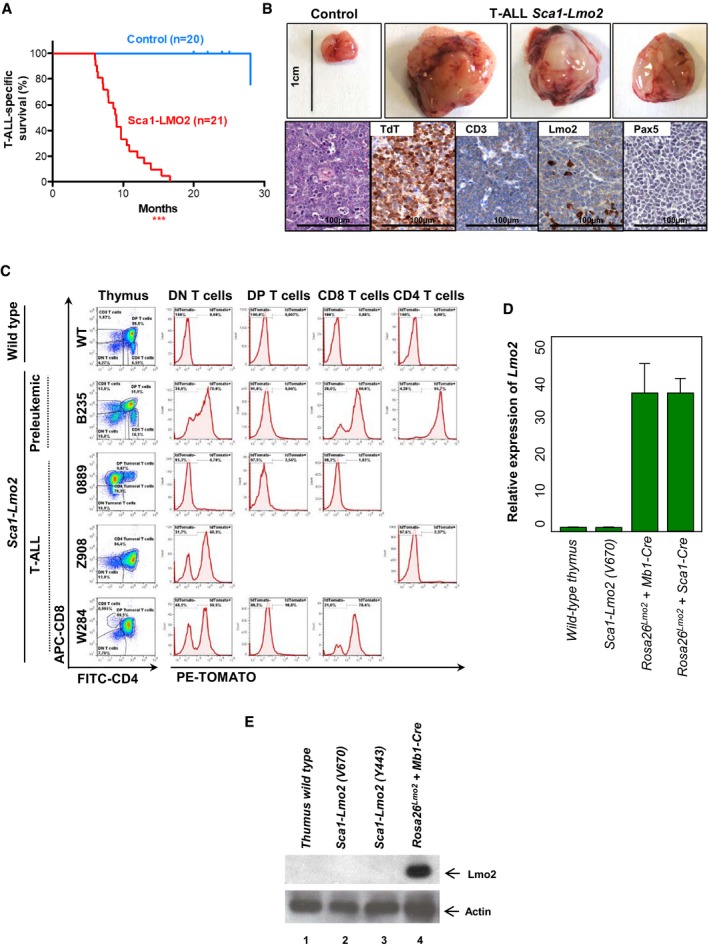
Figure 3. Molecular identity of tumor cells in Sca1‐Lmo2 T‐ALL .
- Overview of chromosomal imbalances mapped by 4x180k oligonucleotide aCGH in 12 T‐ALL cases in Sca1‐Lmo2 mice. The 20 chromosome ideograms of T‐ALL Sca1‐Lmo2 mice are shown with DNA deletions drawn as green lines and amplifications or gains as red lines. Selected chromosomal alterations are highlighted.
- Genes significantly induced or repressed within tumor T cells of Sca1‐Lmo2 mice in comparison with WT littermates, as determined by significance analysis of microarrays using FDR 1%. Each row represents a separate gene, and each column denotes a separate mRNA sample. The level of expression of each gene in each sample is represented using a red–blue color scale (upregulated genes are displayed in red and downregulated genes in blue). Selected genes are highlighted.
- GSEA of the transcriptional signatures within tumor T cells compared with control WT littermates. Gene expression data from Sca1‐Lmo2 tumor T cells showed significant enrichment in embryonic stem cell genes (Wong et al, 2008) (GSEA FDR q‐value = 0.001), Myc target genes (Zeller et al, 2003) (GSEA FDR q‐value = 0.001), and pluripotency genes (Muller et al, 2008) (GSEA FDR q‐value=0.001).
- Notch1 expression profile in control WT T cells, pre‐leukemic tomato+ T cells, pre‐leukemic tomato− T cells, and leukemic T cells. The statistical test used was Mann–Whitney U‐test: wild‐type thymus versus leukemic T cells (***P < 0.001), wild‐type thymus versus pre‐leukemic tomato− T cells (**P = 0.0031), pre‐leukemic tomato+ T cells versus leukemic T cells (*P = 0.0127), and pre‐leukemic tomato+ T cells versus pre‐leukemic tomato− T cells (*P = 0.0159). Error bars represent the mean ± SEM.
- GSEA of the Notch signaling in leukemic and tomato− cells (GSEA FDR q‐value = 0.002, 0.017, respectively).
Gene sets pertaining to stem cell identity were highly enriched in Lmo2‐negative T‐ALL (Fig 3C), suggesting that the stem‐cell‐specific transcriptional program remains activated in the absence of Lmo2 expression. However, gene expression analysis identified upregulation of the Notch1 pathway in thymic pre‐leukemic tomato− versus tomato+ cells, comparable to the leukemic T cells (Fig 3D and E; Appendix Fig S6A–E; Table EV2). Thus, these data suggest that Lmo2 initiates a reprogramming‐like mechanism in HSC/PC, while the T‐ALL is maintained independently of Lmo2 expression.
To further explore the relevance of our findings to human T‐ALL, we performed WES of 10 Lmo2‐negative tumors from the thymuses of diseased Sca1‐Lmo2 mice (Table 1). We detected 40 somatic mutations, including 10 mutations in genes recorded in the cancer gene list (Table EV1); primarily, we observed recurrent Notch1 (SNVs 5/10, indels 5/10) and KRas (SNVs 1/10) mutations (Table 1), consistent with Rosa26‐Lmo2 + Sca1‐Cre and human LMO2 + T‐ALL pathogenesis (Table 1). However, we did not identify Lmo2 target genes and/or pathways that could replace Lmo2 function in Lmo2‐negative T‐ALL. WES of a corresponding human LMO2+ T‐ALL cohort (n = 9) in which translocation t(11;14)(p13;q11) was confirmed by fluorescent in situ hybridization (FISH) analysis (Table EV3; Appendix Fig S7) corroborated the relevance of these mutations. Briefly, we found 34 somatic alterations, including eight genes recorded in the cancer gene list, and confirmed recurrent NOTCH1 (SNVs 6/9, indels 1/9), KRAS (SNVs 1/9), NRAS (SNVs 2/9), FBXW7 (SNVs 2/9), and MTOR (SNVs 2/9) mutations (Table 1; Table EV1). Human LMO2+ and Sca1‐Lmo2 T‐ALL showed highly recurrent SNVs and indels in NOTCH1 (p.L1585P) and KRAS (p.G12D/V; Table 1), targeting the same amino acid. Thus, our data suggest that transient Lmo2 expression in murine HSC/PCs is sufficient for induction of human‐like T‐ALL without the need for sustained Lmo2 expression in the T‐ALL bulk.
Secondary genomic alterations take place within the thymus during T‐ALL development
In an attempt to exclude the impact of Lmo2 expression in thymic precursor cells and to analyze specifically the reprogramming potential of Lmo2 expression in HSC/PC, we crossed Sca1‐Lmo2 mice with thymus‐deficient nu/nu mice. Sca1‐Lmo2 + nu/nu mice had a similar lifespan compared to Sca1‐Lmo2 (Fig 4A). Leukemic Sca1‐Lmo2 + nu/nu mice (n = 8/10) had enlarged spleens and succumbed to a highly disseminated form of leukemia that infiltrated both hematopoietic and non‐hematopoietic tissues (Fig 4B and C; Appendix Fig S8A). In contrast to Sca1‐Lmo2 T‐ALL, Lmo2 was expressed in Sca1‐Lmo2 + nu/nu leukemia (Fig 4C) and expression array data showed enrichment in human early T‐cell precursor (ETP) ALL genes (Fig 4D), in agreement with human ETP ALL cases that commonly showed LMO2/LYL1 deregulation (Liu et al, 2017). In addition, these leukemias showed enrichment in pluripotency, stemness (Fig 4D; Appendix Fig S8B and C), underscoring a reprogramming effect of Lmo2 in HSC/PC. These results suggest that Lmo2 is able to reprogram HSC/PC before entering the thymus. Next, we performed WES of five Sca1‐Lmo2 + nu/nu mice with leukemia and observed 14 somatic alterations (SNVs) in Cdh11 (1/5), Cd1d1 (2/5), Sept6 (2/5), and Hspa1l (1/5; Table 1; Table EV1). We did not observe Notch1 or Ras indels/SNVs, in contrast to T‐ALL from Rosa26‐Lmo2 + Sca1‐Cre and Sca1‐tomato‐IRES‐Lmo2 mice and human LMO2+ T‐ALL (Table 1). Hence, Lmo2 is able to reprogram the cellular identity of HSC/PC into a tumorigenic one, but the thymus is indispensable to retain the T‐ALL phenotype.
Figure 4. Leukemia development in Sca1‐Lmo2 + nu/nu mice.
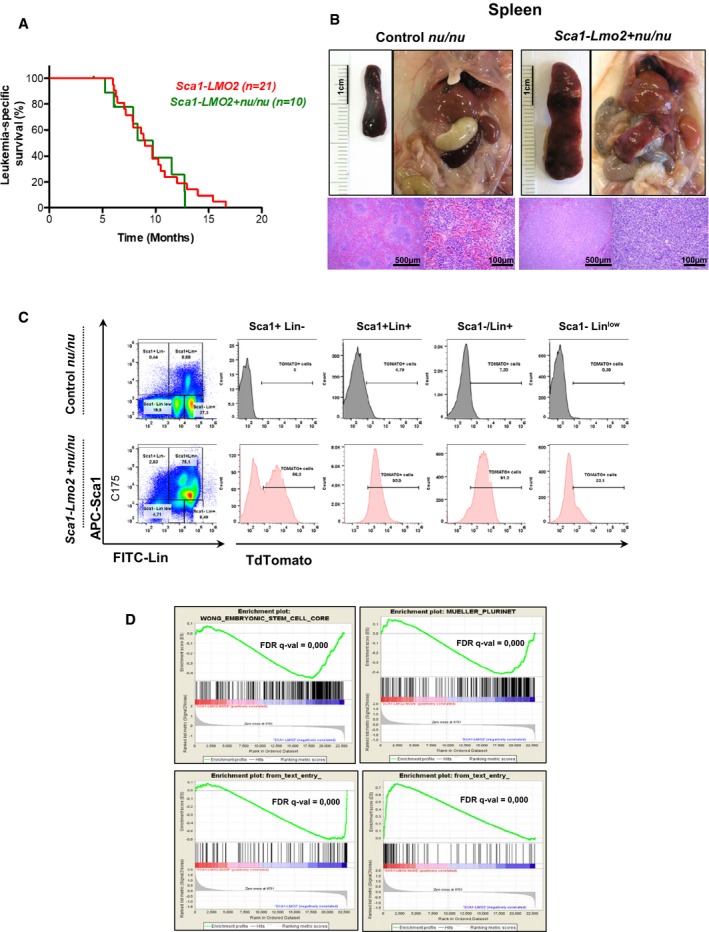
- Leukemia‐specific survival of Sca1‐Lmo2 + nu/nu mice (green line, n = 10), showing a similar shortened lifespan compared to Sca1‐Lmo2 mice (red line, n = 21) as a result of leukemia development.
- An example of splenomegaly observed in Sca1‐Lmo2 + nu/nu mice studied pointing out by an arrowhead. Hematoxylin and eosin staining showing infiltration of spleen from Sca1‐Lmo2 + nu/nu leukemic mice. A spleen from a control littermate nu/nu mouse is shown for reference.
- TdTomato expression in the leukemic cells from Sca1‐Lmo2 + nu/nu mouse. A control littermate nu/nu mouse is shown for reference.
- GSEA of the transcriptional signatures within tumor cells of Sca1‐Lmo2 + nu/nu mice compared to tumor T cells of Sca1‐Lmo2 mice. Gene expression data from Sca1‐Lmo2 + nu/nu tumor cells showed significant enrichment of embryonic stem cell genes (GSEA FDR q‐value = 0.000), pluripotency genes (GSEA FDR q‐value = 0.000), genes upregulated in human ETP T‐ALL (GSEA FDR q‐value = 0.000), and genes downregulated in human ETP T‐ALL (GSEA FDR q‐value = 0.000).
B‐cell‐restricted Lmo2 expression reprograms B cells into T‐ALL
We next asked whether the reprogramming potential of LMO2 is restricted to the HSC/PC compartment or this ability applies to precursor and mature non‐T‐cell lineage cells. Lmo2 is expressed in other types of hematologic cancer including diffuse large B‐cell lymphoma (DLBCL; Natkunam et al, 2007; Cubedo et al, 2012) and B‐cell precursor acute lymphoblastic leukemia (BCP‐ALL; de Boer et al, 2011; Malumbres et al, 2011; Deucher et al, 2015), and a significant proportion of human T‐ALL exhibits rearrangement of immunoglobulin heavy‐chain genes (Mizutani et al, 1986; Szczepanski et al, 1999; Meleshko et al, 2005). Thus, we next address the effects of Lmo2 expression in B cells. We initially used pro‐B cells as targets for reprogramming because they carry genomic rearrangements of genes encoding VDJ regions of immunoglobulin heavy‐chain locus that serve as natural genetic barcodes and they have weak barriers for reprogramming (Riddell et al, 2014). To this aim, we crossed the Rosa26‐Lmo2 mice with an Mb1‐Cre mouse strain (Hobeika et al, 2006). The resulting strain deletes the stop cassette upon B‐lineage commitment at the pro‐B‐cell level via the Cre recombinase, driven by the promoter from Mb1 locus encoding the immunoglobulin‐associated alpha chain Cd79a. FACS analysis confirmed uniform and efficient GFP expression at the pro‐B stage, and therefore all subsequent stages of B‐cell differentiation (Appendix Fig S9A). B cells from Rosa26‐Lmo2 + Mb1‐Cre mice showed a developmental pattern comparable to that of B cells from their control littermates (Appendix Fig S9B), which indicated that induction of Lmo2 at the pro‐B‐cell stage has a minimal effect on B‐cell development. GFP expression was not detected outside the B‐cell lineage in Rosa26‐Lmo2 + Mb1‐Cre mice as the frequency of GFP+ cells within both the BM myeloid progenitors and thymus T cells was undetectable (Appendix Fig S9A). These results also indicated that forced expression of Lmo2 was not able to reprogram committed progenitors of B cells into normal T lymphocytes. Importantly, Rosa26‐Lmo2 + Mb1‐Cre mice do not develop B‐cell malignancies. However, Rosa26‐Lmo2 + Mb1‐Cre mice showed a shorter lifespan than their wild‐type (WT) littermates (Fig 5A) due to the development of aggressive T‐cell malignancies (5/27; 18.5%). Malignant T cells were primarily either double‐positive for CD4/CD8 or single‐positive for CD8 or single‐positive for CD4 (Fig 5B), with Lmo2 expression in the tumor T cells (Fig 2D and E). These mice also showed infiltration of malignant cells into the spleen, liver, and thymus, resulting in disruption of normal architecture (Appendix Fig S9C). The latency of these Rosa26‐Lmo2 + Mb1‐Cre T‐ALL was higher than the latency of Rosa26‐Lmo2 + Sca1‐Cre T‐ALLs (Fig 5A), suggesting that the cell‐of‐origin impacts the disease malignancy.
Figure 5. T‐ALL development through Lmo2 expression in B cells.
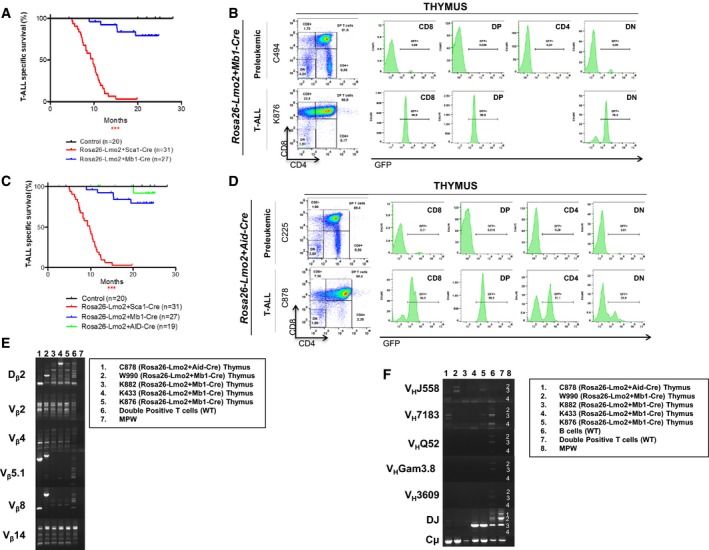
- Leukemia‐specific survival of Rosa26‐Lmo2 + Mb1‐Cre mice (blue line, n = 27), showing a significantly (log‐rank ***P < 0.0328) shortened lifespan compared to control littermate WT mice (black line, n = 20) as a result of T‐ALL development. The latency of Rosa26‐Lmo2 + Mb1‐Cre T‐ALL is higher than that of Rosa26‐Lmo2 + Sca1‐Cre T‐ALL.
- GFP expression in the pre‐leukemic and leukemic cells from Rosa26‐Lmo2 + Mb1‐Cre mice, respectively.
- Leukemia‐specific survival of Rosa26‐Lmo2 + Aid‐Cre mice (green line, n = 19), not showing a significantly (log‐rank ***P < 0.3173) shortened lifespan compared to control littermate WT mice (black line, n = 20). The latency of Rosa26‐Lmo2 + Aid‐Cre T‐ALL is higher than that of Rosa26‐Lmo2 + Sca1‐Cre and Rosa26‐Lmo2 + Mb1‐Cre T‐ALLs.
- GFP expression in the pre‐leukemic and leukemic cells from Rosa26‐Lmo2 + Aid‐Cre mice, respectively.
- TCR clonality in Rosa26‐Lmo2 + Aid‐Cre and Rosa26‐Lmo2 + Mb1‐Cre mice. PCR analysis of TCR gene rearrangements in infiltrated thymuses of diseased Rosa26‐Lmo2 + Aid‐Cre and Rosa26‐Lmo2 + Mb1‐Cre leukemic mice. Sorted DP T cells from the thymus of healthy mice served as a control for polyclonal TCR rearrangements. Leukemic thymus shows an increased clonality within their TCR repertoire (indicated by the code number of each mouse analyzed).
- BCR clonality in Rosa26‐Lmo2 + Aid‐Cre and Rosa26‐Lmo2 + Mb1‐Cre mice. PCR analysis of BCR gene rearrangements in infiltrated thymuses of diseased Rosa26‐Lmo2 + Aid‐Cre and Rosa26‐Lmo2 + Mb1‐Cre leukemic mice. Sorted CD19+ B cells (B cells) from spleens of healthy mice serve as a control for polyclonal BCR rearrangements. DP T cells from the thymus of healthy mice served as a negative control. Leukemic thymus shows an increased clonality within their BCR repertoire (indicated by the code number of each mouse analyzed).
Due to the increased expression of Lmo2 in DLBCL (Natkunam et al, 2007; Cubedo et al, 2012), we therefore next crossed conditional Rosa26‐Lmo2 mice with the Aid‐Cre strain, which expresses Cre recombinase in germinal center (GC) B cells (Crouch et al, 2007). Upon reaching immunological maturity, Rosa26‐Lmo2 + Aid‐Cre mice were injected with T‐cell‐dependent antigen sheep red blood cells (SRBC) to induce Lmo2 expression in GC cells. FACS analysis confirmed uniform and efficient GFP expression at GC stage (Appendix Fig S9D) and therefore within the subsequent stages of B‐cell differentiation (Appendix Fig S9E). GFP expression was not detected in bone marrow progenitor B cells, bone marrow myeloid cells, and thymus T cells from pre‐leukemic Rosa26‐Lmo2 + Aid‐Cre mice (Appendix Fig S9E). These results also indicated that forced expression of Lmo2 within GC B cells was not able to contribute to normal T‐cell development. However, Lmo2 expression in GC cells did not result in DLBCL or other types of B‐cell malignancies. However, 5% (1/19) of Rosa26‐Lmo2 + Aid‐Cre mice developed aggressive T‐cell malignancy (Fig 5C). Malignant T cells were primarily CD8+CD4+/− (Fig 5D). These mice showed infiltration of malignant cells into the spleen, liver, kidney, and lung, resulting in disruption of normal architecture (Appendix Fig S9F). Similarly, the latency of the Rosa26‐Lmo2 + Aid‐Cre T‐ALL was even higher than the latency of Rosa26‐Lmo2 + Sca1‐Cre and Rosa26‐Lmo2 + Mb1‐Cre T‐ALLs (Fig 5C), reinforcing the evidence that the cell‐of‐origin influences the disease malignancy.
T‐ALL leukemia, which originated from either pro‐B or GC cells, showed clonal TCR rearrangements (Fig 5E) and a significant similarity to Rosa26‐Lmo2 + Sca1‐Cre tumors at the genomic level due to the presence of recurrent Notch1 (SNVs (3/4), indels (1/4) in Rosa26‐Lmo2 + Mb1‐Cre; SNVs (1/1), indels (1/1) in Rosa‐Lmo2 + Aid‐Cre) mutations (Table 1; Table EV1). In line with a B‐cell origin, Rosa26‐Lmo2 + Mb1‐Cre and Rosa26‐Lmo2 + Aid‐Cre T‐ALL also showed clonal genomic rearrangements of genes encoding VDJ regions of immunoglobulin heavy‐chain locus (Fig 5F). These results show that B‐cell‐restricted Lmo2 expression can induce T‐ALL in mice, a disease that never appears in control WT littermates. Furthermore, we show that the differentiation state of the cell‐of‐origin influences the frequency of T‐ALL. Together, these data support a novel function of Lmo2 in mice, where the cell‐of‐origin differentiation state does not dictate the Lmo2 tumor cell identity. Lmo2 expression in non‐T‐cell lineage cells including HSC/PC and B cells causes reprogramming with a common final path to T‐ALL development.
Transcriptomics landscape of Lmo2‐driven T‐ALL
To elucidate the differential transcriptomics landscape among different mouse models employed in this study, we next performed paired‐end RNA‐seq on Rosa26‐Lmo2 + Sca1‐Cre (n = 3), Sca1‐Lmo2 (n = 4), Rosa26‐Lmo2 + Mb1‐Cre (n = 2), Rosa26‐Lmo2 + Aid‐Cre (n = 1), and WT‐thymus (n = 4) mice. The 500 genes with the highest variance among the difference murine models were depicted (Fig 6A) with their corresponding FPKM values (Table EV5). Next, gene set enrichment analysis (GSEA) of Rosa26‐Lmo2 + Sca1‐Cre and Sca1‐Lmo2 mouse‐based gene signatures, against a human T‐ALL childhood expression set with healthy controls (Mootha et al, 2003; Subramanian et al, 2005), was performed (Fig 6B). The upregulated Rosa26‐Lmo2 + Sca1‐Cre signature shows a significant enrichment in the human T‐ALL group which is in accordance with the human T‐ALL situation wherein the expression of LMO2 is present throughout in tumor cells.
Figure 6. Comparison of RNA‐seq data from depicted mouse models compared to a human cohort of T‐ALL .
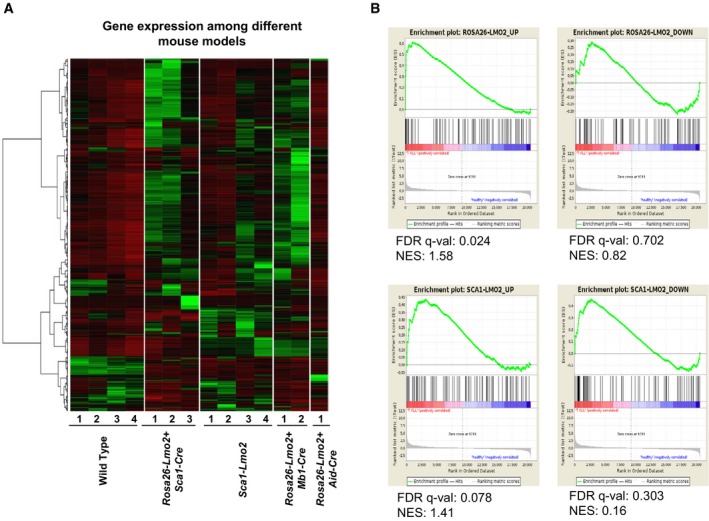
- Gene expression of Rosa26‐Lmo2 + Sca1‐Cre, Sca1‐Lmo2, Rosa26‐Lmo2 + Mb1‐Cre, and Rosa26‐Lmo2 + Aid‐Cre with WT thymus as comparison. The 500 genes with the highest variance among the murine groups were chosen, and their corresponding FPKM values transformed to standard scores for visualization. [Row clustering was conducted with the ward.D method.]
- Gene set enrichment analysis (GSEA) of Rosa26‐Lmo2 + Sca1‐Cre and Sca1‐Lmo2 mouse‐based gene signatures, against a human T‐ALL childhood expression set with healthy controls. Mouse‐based signatures consist of the 100 most up‐ and downregulated human homologue genes, as identified in a differential gene expression analysis between Rosa26‐Lmo2 + Sca1‐Cre versus WT and Sca1‐Lmo2 versus WT, respectively.
Discussion
Understanding the stepwise events taking place during tumor cell evolution is difficult, because of many genetic alterations that become clonally selected by the time of clinically manifested T‐ALL (Nowell, 1976). In principle, leukemogenesis is a process whereby a normal cell acquires novel but aberrant (malignant) identity in order to propagate a clonal population. This is only possible if the oncogenic event(s) have an inherent reprogramming capacity and the leukemia‐initiating cell has the necessary plasticity (Sanchez‐Garcia, 2015). Several prior studies have been involved in studying aberrations in HSC/PC as an important driver for myeloid and B‐cell hematopoietic neoplasms through a reprogramming mechanism (Perez‐Caro et al, 2009; Vicente‐Duenas et al, 2012a,c; Green et al, 2014; Rodriguez‐Hernandez et al, 2017), but to our knowledge, there is no evidence that a similar mechanism may be relevant for T‐ALL. Two alternative explanations can be contemplated to interpret the close association existing between the LMO2 oncogene and human T‐ALL development: on the one side, the classical interpretation that considers that the role of LMO2 as the T‐ALL‐initiating genetic alteration takes place in a committed/differentiated target T cell. Under this hypothesis, LMO2 is required for the immortalization of this committed target T cell that will later suffer additional genetic alterations with time which will further deregulate its behavior, leading to the characteristic clinical features of full‐blown T‐ALL. Therefore, this traditional model considers that the phenotype of the tumor cells mirrors that of the normal T cell that originally gave rise to the tumor. However, there is another possible way of interpreting the specific relationship between LMO2 and T‐ALL, and it is to consider that the LMO2 oncogene is directly capable of imposing the phenotypic characteristics of the tumor in a non‐T target cell. In fact, Rag‐1 expression has been detected in early progenitors in both mice and humans (Boiers et al, 2013, 2018), therefore providing a mechanistic possibility for translocations to happen at very early hematopoietic developmental stages. If this second option is true, and LMO2 can indeed impose a T‐cell program in a non‐T target cell, it would be difficult, however, to prove this fact in human tumors, since the deconvolution of the sequential events in the evolution of the leukemia becomes almost impossible due to the clonal and sub‐clonal accumulation of genetic alterations by the time of the clinical presentation of the full‐blown T‐ALL. This clonality implies that in human T‐ALL, in spite of its cellular heterogeneity, all leukemic cells carry the same LMO2 initiating oncogenic genetic lesions, and this would seem to suggest a homogenous mode of action for LMO2 within all cancer cells. However, there are other findings strongly pointing toward a reprogramming effect of non‐T‐cell lineage cells by LMO2. First, LMO2 ectopic activation caused by retroviral insertion in the CD34+ HSCs of X‐SCID patients specifically triggered T‐ALL development, but no other hematopoietic tumors (Hacein‐Bey‐Abina et al, 2008; Howe et al, 2008), although it is considered that LMO2 expression in BM progenitors is not relevant per se (Ruggero et al, 2016). And second, Lmo2 expression in murine blood cells cooperates in the generation of iPS cells (Batta et al, 2014; Riddell et al, 2014). However, in order to prove that, for T‐ALL development, LMO2 expression does not need to be maintained beyond the initial step of reprogramming, one would require an experimental system capable of limiting the expression of LMO2 to the target cell‐of‐origin compartment, since otherwise it would be impossible to discard a function for LMO2 in posterior tumor development, as exemplified by the Rosa26‐Lmo2 + Sca1‐Cre model. Such a “cell‐of‐origin‐restricted” system would therefore allow us to prove, if these was indeed the case, that the oncogenes, like LMO2, capable of initiating T‐ALL formation, might however be dispensable for posterior tumor progression and/or maintenance. In this study, we provide experimental evidence illustrating that HSC/PC and B cells are uniquely sensitive to transformation by Lmo2 oncogene. However, within the hematopoietic system, not all cells are equally permissive to transformation. Restricted Cre‐mediated activation of Lmo2 in different stages of B‐cell development induced T‐ALL. However, the differentiation state of the B cell‐of‐origin influences the latency, but it provides thoroughly and unexpectedly a T‐cell phenotype. This is a novel phenomenon in contrast to the previous assumptions; i.e., the phenotype of the leukemia cells is identical to the cell‐of‐origin (Vicente‐Duenas et al, 2013). These results indicate that the T‐ALL cell‐of‐origin must possess sufficient plasticity to allow the tumoral reprogramming to take place or, at least, to be initiated. Thus, Lmo2 has the power and capacity to switch from a B‐cell fate to a T‐cell neoplasia, although Lmo2 does not seem to contribute to the generation of normal T lymphocytes. This finding contrast with the role play by Pax5, whose deletion of this master regulator of the B‐cell lineage reprograms B cells into functional T lymphocytes which only occasionally gives rise to T‐cell malignancies (Rolink et al, 1999; Cobaleda et al, 2007). This mechanism of Lmo2‐dependent reprogramming has been reported in other contexts outside of malignancy, like Lmo2 in iHSCs (Riddell et al, 2014). Thus, the data presented here suggest a more general role for LMO2 to shape the epigenome or to be involved in chromatin remodeling early on in T‐ALL disease and it would not be surprising that other important drivers for human T‐ALL, like SCL, LMO1, or HOX11/TLX1, contribute to the neoplasm through a similar reprogramming mechanism.
The multiple genetic hits required for T‐ALL development can be related to the fact that the changes, which are necessary for reprogramming mature cells to pluripotent phenotype, are inherently disfavored developmentally. In this case, biological barriers try to prevent cells from changing their identity in order to avoid the risk of malignant transformation. Evidence to support the inherent resistance of cells to reprogramming by an oncogene to a tumor phenotype comes from recent studies of stem‐cell‐based animal models of human cancer. For instance, in a stem‐cell‐based transgenic model of multiple myeloma, the loss of p53 accelerated the appearance of disease by allowing the MafB oncogene to drive a much more efficient malignant reprogramming (Vicente‐Duenas et al, 2012b,c). Something similar happens in the case of mucosa‐associated lymphoid tissue (MALT) lymphoma that is driven by the MALT1 oncogene (Vicente‐Duenas et al, 2012a). In a stem‐cell‐based model of CML, restoration of p53 activity slowed the progression of the disease and extended the survival of leukemic animals by inducing the apoptotic death of primitive leukemic cells (Velasco‐Hernandez et al, 2013). Similarly, a significant proportion of T‐ALL in all our murine models carried p53 loss‐of‐function mutations (Table EV4) facilitating pathological reprogramming to a malignant T‐cell phenotype.
We propose that in human T‐ALL, genetic alterations of LMO2 may act in a hit‐and‐run fashion in early precursors, while evolved tumor cells are reliant on alternative oncogenic mechanisms. The presence of Lmo2 is necessary for the early stages of transformation, but the final tumor phenotype is determined by the niche. In the present results, the final phenotype of the T‐ALL is defined by the thymus environment (Fig 7). This may provide an explanation for the failure of some modern targeted therapies to clear tumor stem cells, despite being effective agents against evolved tumor cells. As a consequence, treatment strategies targeting oncogenic pathways that are active in both the early and late stages of tumor development may be needed to eradicate completely T‐ALL. These findings on the mechanisms of cellular commitment to a tumoral fate by LMO2, therefore, have important implications for understanding and therapeutically targeting T‐ALL tumor cells and to regenerative medicine, since it will be essential to have full control over the potential malignancy of reprogrammed cells.
Figure 7. A model by which ectopic expression of Lmo2 reprograms HS/PCs and B cells into tumor T cells.
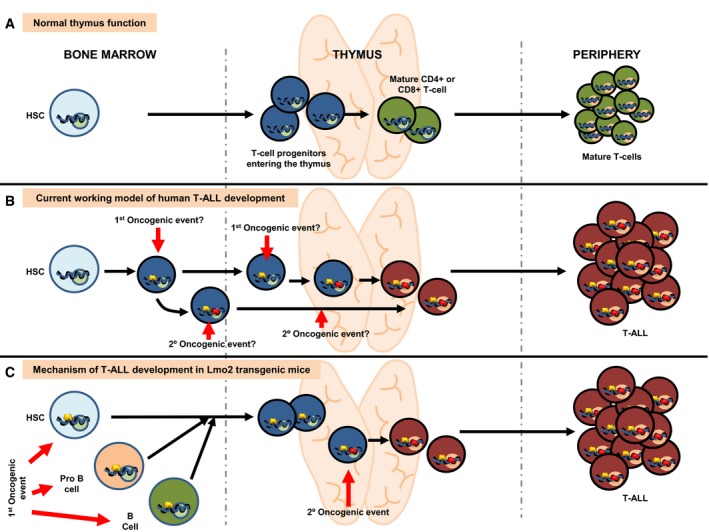
- Normal lymphoid development in human and mice. Blue circles represent normal gene regulatory events (activating or repressing) happening during T lymphocyte development. Green circles represent normal gene regulation events happening during terminal differentiation.
- Current working model for the development of T‐ALL in humans. The existence of dormant alterations previous to the terminal differentiation is unknown. The nature of both the cancer cell‐of‐origin and the cellular place where the second hit is taking place is therefore unknown.
- Mechanism of T‐ALL development in Sca1‐Lmo2 transgenic mice. Open yellow circles represent latent epigenetic regulatory events caused by Sca1‐driven expression of Lmo2. These epigenetic marks do not interfere with normal T‐cell development but become active (either activating or repressing) in the process of terminal differentiation when the second hit appears within the thymus, thus leading to the appearance of tumor T cells. According to this model, tumor T cell is the result of a cell reprogramming process that can be initiated even in committed B cells (see text for details).
Materials and Methods
Detailed methods can be found in the Appendix Supplementary Methods available online.
Generation of mouse strains
All animal work was conducted in accordance with national and international guidelines on animal care and was approved by the Bioethics Committee of the University of Salamanca and the Bioethics Subcommittee of Consejo Superior de Investigaciones Cientificas (CSIC). The Rosa26‐Lmo2 mice were bred to Sca1‐Cre (Mainardi et al, 2014), Mb1‐Cre (Hobeika et al, 2006), or Aid‐Cre mice (Crouch et al, 2007) to generate Rosa26‐Lmo2 + Sca1‐Cre, Rosa26‐Lmo2 + Mb1‐Cre, and Rosa26‐Lmo2 + Aid‐Cre mice, respectively. The Sca1‐Lmo2 vector was generated by inserting the TdTomato–IRES‐mouse Lmo2 cassette into the ClaI site of the pLy6E vector (Miles et al, 1997), and the transgene (2 ng/μl) was injected into CBAxC57BL/6J fertilized eggs.
Upon clinical manifestations of disease, mice were sacrificed and subjected to standard necropsy procedures. All major organs were examined under the dissecting microscope. Tissue samples were taken from homogenous portions of the resected organ and fixed immediately after excision. Differences in survival of transgenic and control WT mice were analyzed using the log‐rank (Mantel–Cox) test.
Array‐comparative genomic hybridization (aCGH)
Whole‐genome analysis was conducted using Mus musculus whole‐genome 4x180k oligonucleotide aCGH (AMADID 27411; Agilent Technologies) following the standard protocol. Copy number‐altered regions were detected using ADM‐2 (set as 6) statistics with a minimum number of five consecutive probes.
Gene expression microarray analysis of murine tumors
Tumoral and normal thymuses were harvested from 10 Sca1‐Lmo2 mice and 4 control littermate WT mice, respectively. Samples were analyzed using Affymetrix Mouse Gene 1.0 ST arrays. A cutoff of FDR < 0.05 was used for the differential expression calculations. All analyses were performed using R and Bioconductor software.
Mouse and human exome library preparation and next‐generation sequencing
Mouse exome library preparation was performed using the Agilent SureSelectXT Mouse All Exon kit with modifications. Furthermore, 2 × 100 bp sequencing with a 6‐bp index read was performed using the TruSeq SBS Kit v3 on the HiSeq 2500 (Illumina). Fastq files were generated using BcltoFastq 1.8.4 (Illumina). BWA version 0.7.4 was used to align sequence data to the mouse reference genome (GRCm38.71). Human translocation t(11;14)(q13;q11) bone marrow samples were obtained after informed consent within the AIEOP‐ BFM‐ ALL study and processed via the Macrogen Europe platform, Heidelberg.
RNA sequencing
RNA sequencing libraries were generated from 500 ng of total RNA using the TruSeq RNA sample prep kit (Illumina) from the blast cells obtained different mouse models employed in the study, including cells from healthy thymus as a control (wt). Later, the libraries were subjected to 2 × 100 bp paired‐end sequencing using HiSeq 2000 instrument (Illumina). The RNA‐seq data were aligned against the mouse reference genome (mm10/GRCm38.83) with TopHat 2.1.0 (Kim et al, 2013), and FPKM values per gene were calculated with the R/Bioconductor package bamsignals (Mammana & Helmuth, 2016). Genes with FPKM values > 1 in less than three samples were excluded from the analysis. We selected the 500 genes with the highest variance over all samples, transformed the corresponding FPKM values into standard scores (Table EV5), and visualized the results with the R/Bioconductor package gplots’ heatmap.2 function (row dendrogram, clustering method “ward.D”; Warnes et al, 2016).
Differential analysis between the mouse groups Rosa26‐Lmo2 + Sca1‐Cre and Sca1‐Lmo2 against the wild‐type group was conducted with DESeq2 (Love et al, 2014), with a minimum adjusted P‐value of 0.05. Signatures of the 100 most significantly up‐ and downregulated human homologue genes per differential analysis were then tested for enrichment with the Broad Institute's GSEA tool (Table EV6; Mootha et al, 2003; Subramanian et al, 2005), against a human childhood T‐ALL set with corresponding wild‐type thymus control samples (Ng et al, 2014).
Statistical analysis
Differences between the transgenic and control littermate wild‐type mice in the percentage and an absolute number of thymocytes were analyzed by analysis of variance (ANOVA) followed by the Kruskal–Wallis and Dunn's multiple comparison tests. Differences in survival of transgenic and control WT mice were analyzed using the log‐rank (Mantel–Cox) test. P‐values < 0.05 were considered statistically significant. Statistical analysis and data representation were performed using the GraphPad Prism 5.00 software (San Diego, California, USA).
Accession numbers
The mouse RNA‐seq and gene expression microarray data have been deposited in NCBI's Gene Expression Omnibus (Edgar et al, 2002) and are accessible through GEO Series accession number GSE83572.
Author contributions
Conception and design of the project: IG‐R, CV‐D, AB, IS‐G, and JH; development of methodology: IG‐R, SB, SGT‐D, AM‐L, GR‐H, IG‐H, SP, YN, AO, VD, BP, OB, DA‐L, JDLR, RJ, MBGC, FJGC, and CV‐D; data acquisition: IG‐R, SB, CW, AM‐L, GR‐H, IG‐H, SP, YN, AO, VD, BP, OB, DA‐L, JDLR, RJ, MBGC, FJGC, CV‐D, ISL, and IS‐G; management of patient samples: SB, MD, MSt, MSc, WW, OH, AB, and JH; analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): IG‐R, SB, AM‐L, GR‐H, IG‐H, CW, SP, YN, AO, VD, BP, OB, DA‐L, JDLR, RJ, MBGC, FJGC, CV‐D, ISL, IS‐G, AB, and JH; writing, review, and/or revision of the manuscript: IG‐R, SGT‐D, AM‐L, GR‐H, IG‐H, CW, SP, YN, AO, VD, BP, OB, DA‐L, JDLR, RJ, MBGC, FJGC, CV‐D, ISL, IS‐G, AB, and JH; administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): IGS, IG‐H, YN, DA‐L, IS‐G, SB, JH, and AB; and study supervision IG‐R, CV‐D, IS‐G, JH, and AB.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Table EV6
Review Process File
Acknowledgements
We are indebted to all members of our groups for useful discussions and for their critical reading of the manuscript. J.H. has been supported by the German Cancer Aid (Project 110997 and Translational Oncology Program 70112951), the German Jose Carreras Leukemia Foundation (DJCLS 02R/2016), Deutsches Konsortium für Translationale Krebsforschung (DKTK), Joint funding (Targeting MYC L*10), the Kinderkrebsstiftung (2016/17), and the “Elterninitiative Kinderkrebsklinik e.V. Düsseldorf”. SG has been supported by a scholarship of the Hochschule Bonn‐Rhein‐Sieg. AB has been supported by the German Children's Cancer Foundation and the Federal Ministry of Education and Research, Bonn, Germany. Research in ISG group is partially supported by FEDER and by MINECO (SAF2012‐32810, SAF2015‐64420‐R, and Red de Excelencia Consolider OncoBIO SAF2014‐57791‐REDC), Instituto de Salud Carlos III (PIE14/00066), ISCIII‐ Plan de Ayudas IBSAL 2015 Proyectos Integrados (IBY15/00003), by Junta de Castilla y León (BIO/SA51/15, CSI001U14, UIC‐017, and CSI001U16), Fundacion Inocente Inocente, and by the ARIMMORA project (European Union's Seventh Framework Programme (FP7/2007‐2013) under grant agreement no. 282891). ISG Lab is a member of the EuroSyStem and the DECIDE Network funded by the European Union under the FP7 program. AB and ISG have been supported by the German Carreras Foundation (DJCLS R13/26). IGR was supported by BES‐Ministerio de Economía y Competitividad (BES‐2013‐063789). AML and GRH were supported by FSE‐Conserjería de Educación de la Junta de Castilla y León (CSI001‐13, CSI001‐15). Research in CVD group is partially supported by FEDER, “Miguel Servet” Grant (CP14/00082—AES 2013‐2016) from the Instituto de Salud Carlos III (Ministerio de Economía y Competitividad), “Fondo de Investigaciones Sanitarias/Instituto de Salud Carlos III” (PI17/00167), and by the Lady Tata International Award for Research in Leukaemia 2016–2017.
The EMBO Journal (2018) 37: e98783
Contributor Information
Carolina Vicente‐Dueñas, Email: cvd@usal.es.
Arndt Borkhardt, Email: Arndt.Borkhardt@med.uni-duesseldorf.de.
Julia Hauer, Email: Julia.Hauer@med.uni-duesseldorf.de.
Isidro Sánchez‐García, Email: isg@usal.es.
References
- Ansari AM, Ahmed AK, Matsangos AE, Lay F, Born LJ, Marti G, Harmon JW, Sun Z (2016) Cellular GFP toxicity and immunogenicity: potential confounders in in vivo cell tracking experiments. Stem Cell Rev 12: 553–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batta K, Florkowska M, Kouskoff V, Lacaud G (2014) Direct reprogramming of murine fibroblasts to hematopoietic progenitor cells. Cell Rep 9: 1871–1884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer J, Yeung J, Ellu J, Ramanujachar R, Bornhauser B, Solarska O, Hubank M, Williams O, Brady HJ (2011) The E2A‐HLF oncogenic fusion protein acts through Lmo2 and Bcl‐2 to immortalize hematopoietic progenitors. Leukemia 25: 321–330 [DOI] [PubMed] [Google Scholar]
- Boiers C, Carrelha J, Lutteropp M, Luc S, Green JC, Azzoni E, Woll PS, Mead AJ, Hultquist A, Swiers G, Perdiguero EG, Macaulay IC, Melchiori L, Luis TC, Kharazi S, Bouriez‐Jones T, Deng Q, Ponten A, Atkinson D, Jensen CT et al (2013) Lymphomyeloid contribution of an immune‐restricted progenitor emerging prior to definitive hematopoietic stem cells. Cell Stem Cell 13: 535–548 [DOI] [PubMed] [Google Scholar]
- Boiers C, Richardson SE, Laycock E, Zriwil A, Turati VA, Brown J, Wray JP, Wang D, James C, Herrero J, Sitnicka E, Karlsson S, Smith AJH, Jacobsen SEW, Enver T (2018) A human IPS model implicates embryonic B‐myeloid fate restriction as developmental susceptibility to B Acute lymphoblastic leukemia‐associated ETV6‐RUNX1. Dev Cell 44: 362–377 e367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers J, Rabbitts TH (2015) LMO2 at 25 years: a paradigm of chromosomal translocation proteins. Open Biol 5: 150062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleveland SM, Smith S, Tripathi R, Mathias EM, Goodings C, Elliott N, Peng D, El‐Rifai W, Yi D, Chen X, Li L, Mullighan C, Downing JR, Love P, Dave UP (2013) Lmo2 induces hematopoietic stem cell‐like features in T‐cell progenitor cells prior to leukemia. Stem Cells 31: 882–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobaleda C, Jochum W, Busslinger M (2007) Conversion of mature B cells into T cells by dedifferentiation to uncommitted progenitors. Nature 449: 473–477 [DOI] [PubMed] [Google Scholar]
- Crouch EE, Li Z, Takizawa M, Fichtner‐Feigl S, Gourzi P, Montano C, Feigenbaum L, Wilson P, Janz S, Papavasiliou FN, Casellas R (2007) Regulation of AID expression in the immune response. J Exp Med 204: 1145–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubedo E, Gentles AJ, Huang C, Natkunam Y, Bhatt S, Lu X, Jiang X, Romero‐Camarero I, Freud A, Zhao S, Bacchi CE, Martinez‐Climent JA, Sanchez‐Garcia I, Melnick A, Lossos IS (2012) Identification of LMO2 transcriptome and interactome in diffuse large B‐cell lymphoma. Blood 119: 5478–5491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Altri T, Gonzalez J, Aifantis I, Espinosa L, Bigas A (2011) Hes1 expression and CYLD repression are essential events downstream of Notch1 in T‐cell leukemia. Cell Cycle 10: 1031–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Keersmaecker K, Real PJ, Gatta GD, Palomero T, Sulis ML, Tosello V, Van Vlierberghe P, Barnes K, Castillo M, Sole X, Hadler M, Lenz J, Aplan PD, Kelliher M, Kee BL, Pandolfi PP, Kappes D, Gounari F, Petrie H, Van der Meulen J et al (2010) The TLX1 oncogene drives aneuploidy in T cell transformation. Nat Med 16: 1321–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deucher AM, Qi Z, Yu J, George TI, Etzell JE (2015) BCL6 expression correlates with the t(1;19) translocation in B‐lymphoblastic leukemia. Am J Clin Pathol 143: 547–557 [DOI] [PubMed] [Google Scholar]
- Diccianni MB, Batova A, Yu J, Vu T, Pullen J, Amylon M, Pollock BH, Yu AL (1997) Shortened survival after relapse in T‐cell acute lymphoblastic leukemia patients with p16/p15 deletions. Leuk Res 21: 549–558 [DOI] [PubMed] [Google Scholar]
- Dohda T, Maljukova A, Liu L, Heyman M, Grander D, Brodin D, Sangfelt O, Lendahl U (2007) Notch signaling induces SKP2 expression and promotes reduction of p27Kip1 in T‐cell acute lymphoblastic leukemia cell lines. Exp Cell Res 313: 3141–3152 [DOI] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE (2002) Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30: 207–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa L, Cathelin S, D'Altri T, Trimarchi T, Statnikov A, Guiu J, Rodilla V, Ingles‐Esteve J, Nomdedeu J, Bellosillo B, Besses C, Abdel‐Wahab O, Kucine N, Sun SC, Song G, Mullighan CC, Levine RL, Rajewsky K, Aifantis I, Bigas A (2010) The Notch/Hes1 pathway sustains NF‐kappaB activation through CYLD repression in T cell leukemia. Cancer Cell 18: 268–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbertson RJ (2011) Mapping cancer origins. Cell 145: 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green MR, Vicente‐Duenas C, Romero‐Camarero I, Long Liu C, Dai B, Gonzalez‐Herrero I, Garcia‐Ramirez I, Alonso‐Escudero E, Iqbal J, Chan WC, Campos‐Sanchez E, Orfao A, Pintado B, Flores T, Blanco O, Jimenez R, Martinez‐Climent JA, Criado FJ, Cenador MB, Zhao S et al (2014) Transient expression of Bcl6 is sufficient for oncogenic function and induction of mature B‐cell lymphoma. Nat Commun 5: 3904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez A, Kentsis A, Sanda T, Holmfeldt L, Chen SC, Zhang J, Protopopov A, Chin L, Dahlberg SE, Neuberg DS, Silverman LB, Winter SS, Hunger SP, Sallan SE, Zha S, Alt FW, Downing JR, Mullighan CG, Look AT (2011) The BCL11B tumor suppressor is mutated across the major molecular subtypes of T‐cell acute lymphoblastic leukemia. Blood 118: 4169–4173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacein‐Bey‐Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, Lim A, Osborne CS, Pawliuk R, Morillon E, Sorensen R, Forster A, Fraser P, Cohen JI, de Saint Basile G, Alexander I, Wintergerst U, Frebourg T, Aurias A, Stoppa‐Lyonnet D et al (2003) LMO2‐associated clonal T cell proliferation in two patients after gene therapy for SCID‐X1. Science 302: 415–419 [DOI] [PubMed] [Google Scholar]
- Hacein‐Bey‐Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E, Clappier E, Caccavelli L, Delabesse E, Beldjord K, Asnafi V, MacIntyre E, Dal Cortivo L, Radford I, Brousse N, Sigaux F, Moshous D, Hauer J, Borkhardt A, Belohradsky BH et al (2008) Insertional oncogenesis in 4 patients after retrovirus‐mediated gene therapy of SCID‐X1. J Clin Invest 118: 3132–3142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobeika E, Thiemann S, Storch B, Jumaa H, Nielsen PJ, Pelanda R, Reth M (2006) Testing gene function early in the B cell lineage in mb1‐cre mice. Proc Natl Acad Sci USA 103: 13789–13794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe SJ, Mansour MR, Schwarzwaelder K, Bartholomae C, Hubank M, Kempski H, Brugman MH, Pike‐Overzet K, Chatters SJ, de Ridder D, Gilmour KC, Adams S, Thornhill SI, Parsley KL, Staal FJ, Gale RE, Linch DC, Bayford J, Brown L, Quaye M et al (2008) Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID‐X1 patients. J Clin Invest 118: 3143–3150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komuro H, Valentine MB, Rubnitz JE, Saito M, Raimondi SC, Carroll AJ, Yi T, Sherr CJ, Look AT (1999) p27KIP1 deletions in childhood acute lymphoblastic leukemia. Neoplasia 1: 253–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH, Asselin‐Labat ML, Gyorki DE, Ward T, Partanen A, Feleppa F, Huschtscha LI, Thorne HJ, kConFab , Fox SB, Yan M, French JD, Brown MA, Smyth GK, Visvader JE et al (2009) Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med 15: 907–913 [DOI] [PubMed] [Google Scholar]
- Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, McCastlain K, Edmonson M, Pounds SB, Shi L, Zhou X, Ma X, Sioson E, Li Y, Rusch M, Gupta P, Pei D, Cheng C, Smith MA, Auvil JG et al (2017) The genomic landscape of pediatric and young adult T‐lineage acute lymphoblastic leukemia. Nat Genet 49: 1211–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainardi S, Mijimolle N, Francoz S, Vicente‐Duenas C, Sanchez‐Garcia I, Barbacid M (2014) Identification of cancer initiating cells in K‐Ras driven lung adenocarcinoma. Proc Natl Acad Sci USA 111: 255–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres R, Fresquet V, Roman‐Gomez J, Bobadilla M, Robles EF, Altobelli GG, Calasanz MJ, Smeland EB, Aznar MA, Agirre X, Martin‐Palanco V, Prosper F, Lossos IS, Martinez‐Climent JA (2011) LMO2 expression reflects the different stages of blast maturation and genetic features in B‐cell acute lymphoblastic leukemia and predicts clinical outcome. Haematologica 96: 980–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammana A, Helmuth J (2016) bamsignals: Extract read count signals from bam files. R package version 1.8.0.
- Mao X, Fujiwara Y, Orkin SH (1999) Improved reporter strain for monitoring Cre recombinase‐mediated DNA excisions in mice. Proc Natl Acad Sci USA 96: 5037–5042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maser RS, Choudhury B, Campbell PJ, Feng B, Wong KK, Protopopov A, O'Neil J, Gutierrez A, Ivanova E, Perna I, Lin E, Mani V, Jiang S, McNamara K, Zaghlul S, Edkins S, Stevens C, Brennan C, Martin ES, Wiedemeyer R et al (2007) Chromosomally unstable mouse tumours have genomic alterations similar to diverse human cancers. Nature 447: 966–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack MP, Young LF, Vasudevan S, de Graaf CA, Codrington R, Rabbitts TH, Jane SM, Curtis DJ (2010) The Lmo2 oncogene initiates leukemia in mice by inducing thymocyte self‐renewal. Science 327: 879–883 [DOI] [PubMed] [Google Scholar]
- Meleshko AN, Lipay NV, Stasevich IV, Potapnev MP (2005) Rearrangements of IgH, TCRD and TCRG genes as clonality marker of childhood acute lymphoblastic leukemia. Exp Oncol 27: 319–324 [PubMed] [Google Scholar]
- Miles C, Sanchez MJ, Sinclair A, Dzierzak E (1997) Expression of the Ly‐6E.1 (Sca‐1) transgene in adult hematopoietic stem cells and the developing mouse embryo. Development 124: 537–547 [DOI] [PubMed] [Google Scholar]
- Mizutani S, Ford AM, Wiedemann LM, Chan LC, Furley AJ, Greaves MF, Molgaard HV (1986) Rearrangement of immunoglobulin heavy chain genes in human T leukaemic cells shows preferential utilization of the D segment (DQ52) nearest to the J region. EMBO J 5: 3467–3473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molyneux G, Geyer FC, Magnay FA, McCarthy A, Kendrick H, Natrajan R, Mackay A, Grigoriadis A, Tutt A, Ashworth A, Reis‐Filho JS, Smalley MJ (2010) BRCA1 basal‐like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell 7: 403–417 [DOI] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D et al (2003) PGC‐1alpha‐responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34: 267–273 [DOI] [PubMed] [Google Scholar]
- Muller FJ, Laurent LC, Kostka D, Ulitsky I, Williams R, Lu C, Park IH, Rao MS, Shamir R, Schwartz PH, Schmidt NO, Loring JF (2008) Regulatory networks define phenotypic classes of human stem cell lines. Nature 455: 401–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natkunam Y, Zhao S, Mason DY, Chen J, Taidi B, Jones M, Hammer AS, Hamilton Dutoit S, Lossos IS, Levy R (2007) The oncoprotein LMO2 is expressed in normal germinal‐center B cells and in human B‐cell lymphomas. Blood 109: 1636–1642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng OH, Erbilgin Y, Firtina S, Celkan T, Karakas Z, Aydogan G, Turkkan E, Yildirmak Y, Timur C, Zengin E, van Dongen JJ, Staal FJ, Ozbek U, Sayitoglu M (2014) Deregulated WNT signaling in childhood T‐cell acute lymphoblastic leukemia. Blood Cancer J 4: e192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowell PC (1976) The clonal evolution of tumor cell populations. Science 194: 23–28 [DOI] [PubMed] [Google Scholar]
- O'Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, Hardwick J, Welcker M, Meijerink JP, Pieters R, Draetta G, Sears R, Clurman BE, Look AT (2007) FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma‐secretase inhibitors. J Exp Med 204: 1813–1824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomero T, Sulis ML, Cortina M, Real PJ, Barnes K, Ciofani M, Caparros E, Buteau J, Brown K, Perkins SL, Bhagat G, Agarwal AM, Basso G, Castillo M, Nagase S, Cordon‐Cardo C, Parsons R, Zuniga‐Pflucker JC, Dominguez M, Ferrando AA (2007) Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T‐cell leukemia. Nat Med 13: 1203–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Caro M, Cobaleda C, Gonzalez‐Herrero I, Vicente‐Duenas C, Bermejo‐Rodriguez C, Sanchez‐Beato M, Orfao A, Pintado B, Flores T, Sanchez‐Martin M, Jimenez R, Piris MA, Sanchez‐Garcia I (2009) Cancer induction by restriction of oncogene expression to the stem cell compartment. EMBO J 28: 8–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike‐Overzet K, van der Burg M, Wagemaker G, van Dongen JJ, Staal FJ (2007) New insights and unresolved issues regarding insertional mutagenesis in X‐linked SCID gene therapy. Mol Ther 15: 1910–1916 [DOI] [PubMed] [Google Scholar]
- Riddell J, Gazit R, Garrison BS, Guo G, Saadatpour A, Mandal PK, Ebina W, Volchkov P, Yuan GC, Orkin SH, Rossi DJ (2014) Reprogramming committed murine blood cells to induced hematopoietic stem cells with defined factors. Cell 157: 549–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez‐Hernandez G, Hauer J, Martin‐Lorenzo A, Schafer D, Bartenhagen C, Garcia‐Ramirez I, Auer F, Gonzalez‐Herrero I, Ruiz‐Roca L, Gombert M, Okpanyi V, Fischer U, Chen C, Dugas M, Bhatia S, Linka RM, Garcia‐Suquia M, Rascon‐Trincado MV, Garcia‐Sanchez A, Blanco O et al (2017) Infection exposure promotes ETV6‐RUNX1 precursor B‐cell leukemia via impaired H3K4 demethylases. Cancer Res 77: 4365–4377 [DOI] [PubMed] [Google Scholar]
- Rolink AG, Nutt SL, Melchers F, Busslinger M (1999) Long‐term in vivo reconstitution of T‐cell development by Pax5‐deficient B‐cell progenitors. Nature 401: 603–606 [DOI] [PubMed] [Google Scholar]
- Ruggero K, Al‐Assar O, Chambers JS, Codrington R, Brend T, Rabbitts TH (2016) LMO2 and IL2RG synergize in thymocytes to mimic the evolution of SCID‐X1 gene therapy‐associated T‐cell leukaemia. Leukemia 30: 1959–1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez‐Garcia I (2015) How tumour cell identity is established? Semin Cancer Biol 32: 1–2 [DOI] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY (2004) Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol 22: 1567–1572 [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP (2005) Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczepanski T, Pongers‐Willemse MJ, Langerak AW, Harts WA, Wijkhuijs AJ, van Wering ER, van Dongen JJ (1999) Ig heavy chain gene rearrangements in T‐cell acute lymphoblastic leukemia exhibit predominant DH6‐19 and DH7‐27 gene usage, can result in complete V‐D‐J rearrangements, and are rare in T‐cell receptor alpha beta lineage. Blood 93: 4079–4085 [PubMed] [Google Scholar]
- Thompson BJ, Buonamici S, Sulis ML, Palomero T, Vilimas T, Basso G, Ferrando A, Aifantis I (2007) The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. J Exp Med 204: 1825–1835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treanor LM, Volanakis EJ, Zhou S, Lu T, Sherr CJ, Sorrentino BP (2011) Functional interactions between Lmo2, the Arf tumor suppressor, and Notch1 in murine T‐cell malignancies. Blood 117: 5453–5462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Vlierberghe P, van Grotel M, Beverloo HB, Lee C, Helgason T, Buijs‐Gladdines J, Passier M, van Wering ER, Veerman AJ, Kamps WA, Meijerink JP, Pieters R (2006) The cryptic chromosomal deletion del(11)(p12p13) as a new activation mechanism of LMO2 in pediatric T‐cell acute lymphoblastic leukemia. Blood 108: 3520–3529 [DOI] [PubMed] [Google Scholar]
- Van Vlierberghe P, Ferrando A (2012) The molecular basis of T cell acute lymphoblastic leukemia. J Clin Invest 122: 3398–3406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco‐Hernandez T, Vicente‐Duenas C, Sanchez‐Garcia I, Martin‐Zanca D (2013) p53 restoration kills primitive leukemia cells in vivo and increases survival of leukemic mice. Cell Cycle 12: 122–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente‐Duenas C, Fontan L, Gonzalez‐Herrero I, Romero‐Camarero I, Segura V, Aznar MA, Alonso‐Escudero E, Campos‐Sanchez E, Ruiz‐Roca L, Barajas‐Diego M, Sagardoy A, Martinez‐Ferrandis JI, Abollo‐Jimenez F, Bertolo C, Penuelas I, Garcia‐Criado FJ, Garcia‐Cenador MB, Tousseyn T, Agirre X, Prosper F et al (2012a) Expression of MALT1 oncogene in hematopoietic stem/progenitor cells recapitulates the pathogenesis of human lymphoma in mice. Proc Natl Acad Sci USA 109: 10534–10539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente‐Duenas C, Gonzalez‐Herrero I, Garcia Cenador MB, Garcia Criado FJ, Sanchez‐Garcia I (2012b) Loss of p53 exacerbates multiple myeloma phenotype by facilitating the reprogramming of hematopoietic stem/progenitor cells to malignant plasma cells by MafB. Cell Cycle 11: 3896–3900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente‐Duenas C, Romero‐Camarero I, Gonzalez‐Herrero I, Alonso‐Escudero E, Abollo‐Jimenez F, Jiang X, Gutierrez NC, Orfao A, Marin N, Villar LM, Criado MC, Pintado B, Flores T, Alonso‐Lopez D, De Las Rivas J, Jimenez R, Criado FJ, Cenador MB, Lossos IS, Cobaleda C et al (2012c) A novel molecular mechanism involved in multiple myeloma development revealed by targeting MafB to haematopoietic progenitors. EMBO J 31: 3704–3717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente‐Duenas C, Romero‐Camarero I, Cobaleda C, Sanchez‐Garcia I (2013) Function of oncogenes in cancer development: a changing paradigm. EMBO J 32: 1502–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visvader JE (2011) Cells of origin in cancer. Nature 469: 314–322 [DOI] [PubMed] [Google Scholar]
- Warnes GR, Bolker B, Bonebakker L, Gentleman R, Liaw WHA, Lumley T, Maechler M, Magnusson A, Moeller S, Schwartz M, Venables B (2016) gplots: Various R programming tools for plotting data. R package version 3.0.1
- Weinstein IB (2002) Cancer. Addiction to oncogenes–the Achilles heal of cancer. Science 297: 63–64 [DOI] [PubMed] [Google Scholar]
- Weng AP, Ferrando AA, Lee W, Morris JP IV, Silverman LB, Sanchez‐Irizarry C, Blacklow SC, Look AT, Aster JC (2004) Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306: 269–271 [DOI] [PubMed] [Google Scholar]
- Weng AP, Millholland JM, Yashiro‐Ohtani Y, Arcangeli ML, Lau A, Wai C, Del Bianco C, Rodriguez CG, Sai H, Tobias J, Li Y, Wolfe MS, Shachaf C, Felsher D, Blacklow SC, Pear WS, Aster JC (2006) c‐Myc is an important direct target of Notch1 in T‐cell acute lymphoblastic leukemia/lymphoma. Genes Dev 20: 2096–2109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong DJ, Liu H, Ridky TW, Cassarino D, Segal E, Chang HY (2008) Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell 2: 333–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeller KI, Jegga AG, Aronow BJ, O'Donnell KA, Dang CV (2003) An integrated database of genes responsive to the Myc oncogenic transcription factor: identification of direct genomic targets. Genome Biol 4: R69 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Table EV6
Review Process File