

Abstract
Liver cancer is the sixth most prevailing cancer worldwide. Hepatocellular carcinoma (HCC), the most common form of primary liver cancer, has a rather heterogeneous pathogenesis making it highly refractive to current therapeutic approaches. Hence, HCC patients have a poor and gloomy prognosis making liver cancer the second leading cause of global cancer-related deaths. On this basis, a more global mechanism, such as post-translational modifications (PTMs) of proteins, may provide a valuable therapeutic approach for HCC clinical management by simultaneously regulating multiple disrupted signaling pathways. In the last years, the ubiquitin-like molecule NEDD8 (Neural precursor cell-expressed developmentally downregulated-8) conjugation pathway, neddylation, was shown to be aberrant in HCC patients with a significant positive correlation found among global levels of neddylation and poorer prognosis. Even though the best-established role for NEDD8 is the activation of ubiquitin E3 ligase family of cullin-RING ligases, the putative role for other NEDD8 substrates has been explored in recent years leading to the identification of novel neddylation targets in HCC. Importantly, treatment with the small pharmacological inhibitor Pevonedistat (MLN4924) (Millennium Pharmaceuticals, Takeda Pharmaceutical), currently in clinical trials for the treatment of some types of leukemias and other advanced solid tumors, was shown to suppress the outgrowth of hepatoma cells and liver cancer in pre-clinical mouse models. Overall, considering that the neddylation inhibitor Pevonedistat was well-tolerated and displayed a significant antitumor effect in pre-clinical models, combinatory pharmacological treatment based on Pevonedistat are highly recommended to enter clinical trials targeting advanced HCC.
Keywords: Hepatocellular carcinoma (HCC), NEDD8, NAE1, Pevonedistat (MLN4924)
Introduction
Liver cancer is the sixth most prevailing cancer worldwide (1). Hepatocellular Carcinoma (HCC) is the most common form of primary liver cancer (70–85% of total liver cancer). Common risk factors for the development of HCC include chronic hepatitis B or C infection, metabolic syndrome, Type 2 diabetes and alcohol consumption (2). Whereas the prevalence of hepatitis C virus is expected to decline in the next decades, the proportion of HCC related to non-alcoholic steatohepatitis (NASH), a condition characterized by liver inflammation and injury as a result of the build-up of fat in the liver and closely related to metabolic syndrome and obesity, is anticipated to increase exponentially in the near future.
Nowadays, therapeutic options for HCC treatment rely mainly on the Barcelona Clinic Liver Cancer (BCLC) staging system (3) and include surgical, locoregional, and systemic approaches. Their application depends on tumor extension, cirrhotic state and co-morbidities of the patient. Surgical procedures for HCC include both resection and transplantation and are used mainly in very-early and early stage of HCC. On the other hand, locoregional treatments, used in combination with surgery or when surgery is not an option, are mainly used in intermediate-stage HCC and include ablation and different types of transcatheter arterial chemoembolization (TACE) (4,5). Finally, according to the BCLC staging and treatment strategy the unique first-line systemic chemotherapy agent currently approved for the treatment of unresectable and metastatic HCC is the multi-kinase inhibitor Sorafenib (2,6). In spite of alternative available therapeutic approaches for HCC, these patients have a poor and dismal prognosis, with a 5-year survival rate of less than 10%, making liver cancer the second leading cause of global cancer-related deaths (7). Indeed, tumor recurrence is a problem in the first 5 years after resection [almost 70% of cases (4)] and is made more probable by factors such as portal hypertension, multifocality, size, poor histological differentiation, satellites, and vascular invasion (8,9). Furthermore, advanced HCC is often refractive to Sorafenib treatment by mechanisms that still remain to be fully elucidated (10,11).
HCC is considered phenotypically and genetically a very heterogeneous cancer. Some of the most important genetic alterations in HCC are telomerase (TERT) promoter mutations being the most common mutations in HCC (30–60%) and also the most common form of TERT activation, a fundamental step in tumorigenesis (12,13). In addition, mutations of the tumor suppressor gene TP53 in HCC depend on etiology and can range from 18% to 50% (14). Other genes commonly mutated in HCC are Catenin beta-1 (CTNNB1) and Axis Inhibition Protein 1 (AXIN1) (Wnt signaling) and AT-Rich Interaction Domain 1A (ARID1A) (chromatin remodeling). Also, some HCC tumors seldom contain genomic amplifications of vascular endothelial growth factor A (VEGFA), TERT, MYC and/or MET genes (4).
In addition, HCC pathogenesis is characterized by disruption of a complex network of signaling pathways. The main signaling pathways associated with hepatic malignant transformation include p53, retinoblastoma (Rb), hypoxia inducible factor (HIF)-1α (15), c-Myc, Wnt/β-catenin (16-18), Ras/Raf/MEK/ERK (4,19), PI3K/Akt/mammalian target of rapamycin (mTOR) (19,20), nuclear factor κB (NFκB) and JAK/STAT (19) pathways. Likewise, growth and angiogenic factors such as hepatic growth factor (HGF), insulin-like growth factor (IGF), transforming growth factor (TGF)-β (21,22), platelet-derived growth factor (PDGF) and vascular endothelial growth factor (VEGF) (19) play a role in HCC pathogenesis. Furthermore, several studies have demonstrated oncogenic properties of other proteins in HCC, like for example Liver kinase B1 (LKB1) (23) and Hu Antigen R (HuR) (24-27), whose altered expression and signaling may lead to metabolic reprogramming, aberrant cell proliferation and apoptosis resistance. Although our knowledge of the major molecular pathways implicated in the pathogenesis of HCC has increased dramatically in the last years, one of the main difficulties when treating HCC is that many pathways are activated and inhibiting one generally drives compensation by other pathways. Thus, a more global mechanism, such as post-translational modifications (PTMs) of proteins that can simultaneously regulate multiple disrupted signaling pathways may provide a valuable therapeutic approach for HCC clinical management.
PTMs
The human genome is estimated to contain around 20,000 to 25,000 genes (28). However, there are an estimated 1,000,000 proteins (proteome) (29). Genomic recombination, initiation of transcription at different promoters, different transcription termination and alternative transcript splicing are a few ways of increasing protein diversity from the same set of genes (30). The remaining diversity is obtained from PTMs that occur during the “life cycle” of the protein. PTMs are considered key mechanisms regulating protein homeostasis and function in eukaryotic cells. These modifications extend the diversity of the proteome by inducing structural and functional changes in proteins through different mechanisms like covalent binding of functional groups, cleavage of regulatory subunits and degradation of other proteins. Protein PTMs influence enzymatic activities, protein turnover, subcellular localization, protein-protein interactions, DNA repair and cell division, among other processes, being essential to maintain normal cellular signaling, metabolism and function. The most common PTMs include phosphorylation, methylation, acetylation, glycosylation, ubiquitination and ubiquitin-like proteins (UBLs) mediated PTMs. Even though, UBLs are all structurally related, they can be classified in nine phylogenetically distinct classes comprising NEDD8, SUMO (small ubiquitin-like modifier), and others such as ISG15, FUB1, FAT10, Atg8, Atg12, Urm1, and Ufm1. UBLs regulate a strikingly diverse set of cellular processes, including nuclear transport, proteolysis, translation, autophagy, and antiviral pathways. Because UBLs mediated PTMs are necessary for normal physiology, alterations in their pathways have been associated with the development and progression of many diseases (31,32). In the liver, UBLs PTMs also influence almost all aspects of normal cell biology and aberrant modifications have been linked to different hepatic pathologies (33,34). For all these reasons, in the last few years many groups have been dedicated to better understanding how changes in protein homeostasis may drive pathogenesis in human diseases, including liver cancer, providing the basis for the discovery of several important therapies. The role of PTMs mediated by the UBL NEDD8 protein in the pathogenesis of HCC is the main topic of this review.
The NEDD8 conjugation pathway
The NEDD8 gene was initially identified in 1992 (35). The NEDD8 peptide shares 59% amino acid identity (36,37) and 80% homology with ubiquitin (38). Despite their similarity, both NEDD8 and ubiquitin have non-interchangeable functions in cells as a result of small differences in their structures as will be addressed later in this section. NEDD8 is conserved in most eukaryotes being highly expressed in the embryonic mouse brain and presenting a broad expression pattern in adult tissues (39) (Figure 1). Structurally, NEDD8 can be subdivided into a flexible carboxy-terminal tail (40) and a globular ubiquitin-fold domain (UFD) characterized by four β-sheets interspersed by one α-helix and two 310-helices. The tail ends with a Gly-Gly sequence that becomes covalently attached to protein targets and adopts different extended structures upon interaction with neddylation and/or deneddylation enzymes (41-43).
Figure 1.

NEDD8 (neural precursor cell-expressed developmentally down-regulated-8) is ubiquitously expressed in human tissues. Tissue distribution of NEDD8 by immunofluorescence staining in a tissue array from healthy adult humans. WAT, white adipose tissue.
The NEDD8 conjugation pathway, neddylation, is similar to that described for ubiquitination, resulting in the reversible covalent conjugation of a molecule of NEDD8 to a lysine residue of the substrate protein. The neddylation pathway is composed of a 3-step enzymatic cascade that involves the activities of E1 activating enzymes, E2 conjugation enzymes and E3-ligases (44,45). Briefly, the NEDD8 precursor form is first processed at the carboxy-terminal glycine residue (Gly)76 by specific proteases and after this is then adenylated by the E1 NEDD8-activating enzyme (NAE), formed by the heterodimer of amyloid-β precursor protein binding protein 1 (APPBP1) also called NEDD8 activating enzyme E1 subunit 1 (NAE1) and the catalytic subunit known as ubiquitin-activating enzyme 3 (UBA3). In an ordered process, binding of ATP, Mg2+ and NEDD8 occurs first at the adenylation site of UBA3 followed by adenosine monophosphate (AMP) attachment to the carboxy-terminal of NEDD8 (formation of NEDD8-adenylate) and the release of inorganic pyrophosphate. The carboxy-terminal of NEDD8 can then be attacked by the catalytic Cys of NAE which probably adopts a ‘closed’ formation as a result of a structural remodeling, while a second NEDD8 binds to the adenylation domain, and NAE re-adopts an ‘open’ conformation. The NEDD8-loaded NAE is later transferred to the E2 NEDD8-conjugating enzymes. The neddylation cascade has two E2 enzymes known as ubiquitin-conjugating enzyme E2M (UBE2M or UBC12) and ubiquitin conjugating enzyme E2F (UBE2F) (41,46-49). Ultimately, a substrate specific-E3 ligase transfers NEDD8 to a lysine residue in its target protein. With the exception of SMURF1 all identified NEDD8 E3-ligases belong to the RING family of E3s, including the cullins-associated ring-box proteins 1/2 (RBX1/2) and its cooperator DCN1 or DCUN1D1, murine double minute 2 (Mdm2), casitas β-lineage lymphoma (CBl) and the transcriptional co-activator TBF3 (50-60). Finally, neddylation is a reversible process through the action of isopeptidases, named deneddylases, such as the COP9 signalosome (CSN) and NEDD8 protease 1 (NEDP1, also known as SENP8 and DEN1), which free the substrate and NEDD8 (38) in order to reinitiate the neddylation conjugation cycle (Figure 2).
Figure 2.
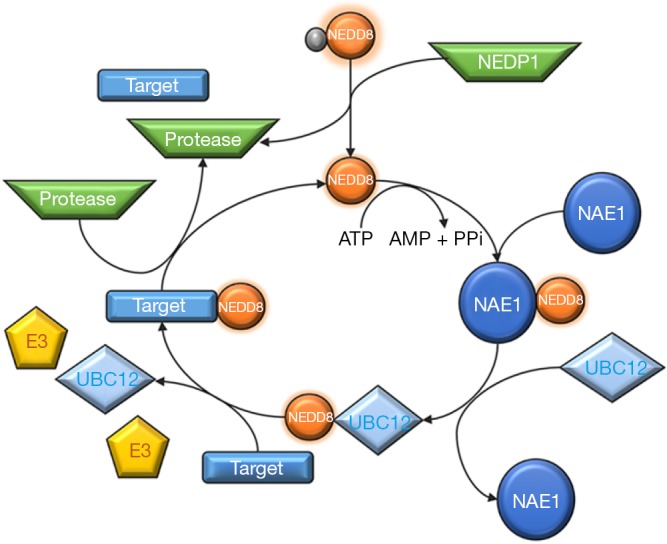
NEDD8 (neural precursor cell-expressed developmentally down-regulated-8) conjugation pathway, neddylation.
Overall, binding of NEDD8 molecules to target proteins can affect their stability, subcellular localization, conformation and function. The direct effects of NEDD8 on target proteins can be classified into three categories: those due to conformational changes, those that preclude certain interactions or those that provide a novel binding surface. Even though NEDD8 substrates are believed to be mainly mono-neddylated on a single or several conserved lysine residues, like other UBLs proteins, NEDD8 can form chains on its substrates in vitro although it is unclear whether they have a function in vivo (61). Interestingly, NEDD8 can form mixed chains with ubiquitin and can function as a chain terminator as NEDD8 is not a good ubiquitin acceptor due to the fact that Lys60 is conserved in NEDD8 but not in ubiquitin (62). Moreover, under physiological conditions, both the ubiquitination and neddylation pathway exhibit great specificity, mostly due to a single amino acid difference in the carboxy-terminal of the two UBLs, Ala72 in NEDD8 and Arg72 in ubiquitin, which are recognized by their respective E1 enzymes. On the other hand, under conditions where neddylation may be increased, as occurs in different disease stages, the ubiquitin E1 enzyme UBE1 can activate NEDD8 (36,63). In agreement, after NEDD8 overexpression, an extensive ectopic neddylation pattern dependent on UBE1 is detectable (64). Neddylation is regulated in vivo by several mechanisms, being that in humans the main regulators of neddylation are the five defective in cullin neddylation 1-like (DCNLs) proteins that bind both cullins and NEDD8 E2 enzymes (65). Their involvement in the neddylation of non-cullin proteins is not currently known and must be further investigated.
Due to the relevance of neddylation in many physiological and pathological processes found in the last years, genetically modified mice presenting disrupted neddylation were created together with the development of pharmacological inhibitors specifically targeting neddylation.
Genetically modified animal models with impaired neddylation
Germline knockout (KO) of the regulatory NAE1 enzyme in mice leads to embryonic lethality before E10.5, revealing its critical role in embryonic development. Likewise, mice with cardiomyocyte-restricted KO of NAE1 (αMHC-CreTg/+::Nae1flox/flox) exhibited myocardial hypoplasia, defective ventricular chamber maturation, heart failure, and embryonic and neonatal lethality (66), whereas Nae1CamKIIα-CreERT2 mice, in which neddylation is conditionally ablated in adult excitatory forebrain neurons, show synaptic loss, impaired neurotransmission and severe cognitive deficits (67). Alternatively, our group has recently acquired Nae1tm1b(EUCOMM)Wtsi heterozygous mice, generated at the Mouse Clinical Institute of the Institut Clinique de la Souris (ICS), CERBMgie, France. Our group has been breeding these animals in our animal facilities and hepatic NAE1, as detected by immunohistochemistry, is significantly reduced. Most probably these changes are not so significant at the mRNA level, as mice are heterozygous for NAE1, but slight changes in NAE1 mRNA hepatic levels may be associated with these dramatic effects at the protein level. Under these circumstances, the liver phenotype between NAE1 wild-type and heterozygous animals of old age (15 months old) is similar (Figure 3), with animals presenting enhanced steatosis characteristic of old animals and mild fibrosis and inflammation (68). These preliminary evidences suggest that long-term inhibition of neddylation is not associated with adverse hepatic effects, at least under ageing conditions that to our knowledge are not characterized by augmented neddylation, and that long-term therapeutic approaches targeting neddylation inhibition appear to be safe. However, we cannot exclude that mice with impaired hepatic neddylation activity may be protected against liver diseases characterized by aberrant neddylation, as we will address further ahead in this Review.
Figure 3.

Liver characterization of Nae1 (NEDD8 activating enzyme E1 subunit 1) wild-type (WT) and heterozygous mice (HE) (15 months old). Representative immunohistochemical staining and quantification for Nae1 and F4/80 inflammatory marker together with staining for lipids (Sudan red) and collagen fibers (Sirius Red) in Nae1tm1b(EUCOMM)Wtsi wt and He mice. *, P<0.05 is shown.
Pevonedistat, a neddylation pharmacological inhibitor
Pevonedistat or MLN4924 ((((1S,2S,4R)-4-{4-[(S)-2,3-Dihydro-1H-inden-1-ylamino]-7H-pyrrolo[2,3-d]pyrimidin-7-yl}-2-hydroxycyclopentyl)methyl sulfamate hydrochloride) from Takeda Oncology (previously Millennium Pharmaceuticals) was discovered as a result of iterative medicinal chemistry efforts on N6-benzyl adenosine that was originally identified as an inhibitor of NAE1 via high throughput screening. Pevonedistat is an adenosine sulfamate analogue and a first in class inhibitor of NAE1 and the NEDD8 pathway (69). Due to the fact that Pevonedistat is structurally related to AMP—a tight binding product of the NAE reaction- its action of inhibition is based on a substrate-assisted mechanism (70). The main differences between AMP and Pevonedistat are: (I) in place of the adenine base, Pevonedistat has a deazapurine base substituted with an aminoindane at N6; (II) in place of the ribose sugar, Pevonedistat has a carbocycle and the equivalent of the 29-hydroxyl group of AMP is absent; (III) in place of the phosphate, Pevonedistat has a sulfamate; and finally, (IV) in contrast to the stereochemistry of AMP, the methylene sulfamate of Pevonedistat is in a non-natural anti-relationship to the deazapurine. As previously described, NEDD8 and Mg-ATP (active magnesium adenosine triphosphate) bind to NAE where NEDD8 is adenylated before it reacts with the catalytic cysteine in UBA3 to form a thioester-linked NEDD8. A second round of NEDD8 adenylation allows the thioester linked NEDD8 to be transferred to UBC12 or UBE2F (41). During this round, Pevonedistat competes for Mg-ATP binding on NAE1 and attacks the thioester-linked NEDD8. The resultant NEDD8-Pevonedistat covalent adduct is not transferred on the E2s and subsequently blocks NEDD8 conjugation (70). Pevonedistat specificity for neddylation arises from the fact that the IC50 of this drug for NAE is in the nanomolar range compared with micromolar scale for UBE1 or other E1-activating enzymes (69).
Neddylation, a relevant PTM in the prognosis and therapy of HCC
NEDD8 plays a vital role in regulating processes such as cell growth, viability and development and hence, alterations in the neddylation conjugation pathway have been associated with a variety of human diseases, including different types of cancer, Alzheimer, and pulmonary fibrosis among others (71-73). Regarding liver disease, neddylation conjugation was shown to be aberrant both in early stages of the disease, such as during progression of the fibrosis stage of the disease (74) as well as in HCC (75,76). Furthermore, neddylation was also aberrant during intrahepatic cholangiocarcinoma progression, the second most common primary hepatic malignancy (77). Aberrant neddylation conjugation in HCC agrees with earlier evidence showing that both NEDD8 and NAE1 mRNA expression of published microarrays were augmented in a large cohort of HCC patients (78). Additionally, a significant correlation among global levels of neddylation, NAE1 protein expression and the poorest prognosis of HCC in human liver tumors has been detected (75). More importantly, treatment with Pevonedistat was shown to suppress the outgrowth of liver cancer and pre-tumoral cells in vitro (75,76) and in a xenograft ectopic mouse model of human hepatoma cells as well as in a genetically modified mouse model that spontaneously develops HCC (75,76). Importantly, we and others have observed that Pevonedistat administration in vivo was well-tolerated and displayed a significant antitumor effect (75,76). The mechanisms of action of Pevonedistat-induced tumor regression and associated protein targets in HCC have been an interest of our group and others in the last couple of years.
NEDD8 targets in HCC
Even though until recently neddylation modifications have been principally characterized in the context of its main target, the ubiquitin E3 ligase family of cullin-RING ligases (CRLs) (79,80), nowadays, it is well known that NEDD8 conjugates to a broad range of proteins besides cullins with several reports of non-cullin neddylation targets in recent years. Additional targets for neddylation have been identified and include: transcription factors and co-regulators (e.g., E2F1, NFκB, and the p53 tumor suppressor and its homologue TAp53), signaling receptors [e.g., EGFR (epidermal growth factor receptor) and TβRII (transforming growth factor β type II receptor)], components of the protein synthesis and apoptotic machineries, E-3 ligases and histones among others [see (81,82) for Review]. In recent years, the putative role for CRLs and other identified NEDD8 targets in HCC has been explored.
Cullin-RING ligases
The best-established role for NEDD8 is the activation of ubiquitin E3 ligase family of CRLs (79,80), which comprise the largest known class of ubiquitin ligases. These enzymes require NEDD8 conjugation onto the cullin subunit to be active whereas deneddylation by CSN and NEDP1 has the opposite effect on CRLs, thus inactivating their ubiquitination activity. Therefore, cullin neddylation and deneddylation are important to maintain the ubiquitination pathway and respective proteasomal degradation and cellular protein homeostasis.
Cullins are a family of hydrophobic proteins providing a scaffold for ubiquitin ligases (E3). In humans, this family is composed of seven cullins (CUL1, 2, 3, 4A, 4B, 5 and 7), whereas PARC (CUL9) and APC2 (component of the anaphase promoting complex APC) contain a cullin-homology domain (83-87). Different types of Cullins play an essential role in HCC. For example, CUL7 shows a high expression in HCC tumor tissues, especially in metastatic HCC tumor tissues, and a positive correlation was found between CUL7 and poor prognosis. Silencing of CUL7 in liver cancer cells can significantly reduce the migration, invasion, and metastatic abilities. Also, detection of epithelial-mesenchymal transition (EMT) marker expression showed that CUL7 promotes EMT of cancer cells (88). Likewise, it was reported that the CUL4A/B genes show amplification together with increased CUL4A/B expressions as detected by immunohistochemistry in human primary HCC. Statistical analysis disclosed an inverse correlation between CUL4A/B expression and tumor differentiation grade, and patient survival, but a positive correlation with hepatocyte proliferation as well as lymphatic and venous invasion. Consistently, CUL4A/B knockdown inhibited the proliferation of established HCC cells and ameliorated the motility of HCC cell lines with altered expression of EMT-associated molecules (89,90). Finally, CUL1 expression is apparently increased in HCC tissues compared with paired adjacent non-tumor tissues and more importantly CUL1 staining significantly correlates with tumor size, histology grade and tumor/node/metastasis (TNM) stage as well as with worse 5-year overall and disease-specific survival rates in HCC patients (91).
CRLs is a superfamily that controls the stability and turnover of a rapidly growing list of proteins with diverse functions including cell cycle progression [p21 p27, cyclin D/E (83,92)], DNA re-replication [Chromatin licensing and DNA replication factor 1 (CDT1) (93)], the oxidative response [nuclear factor erythroid 2-related factor 2 (NRF2) (94)] and the response to hypoxia [HIF1α (95)]. In liver cancer, RhoB, a well-known tumor suppressor, was identified as a new target for the neddylation-CRLs pathway. Specifically, CUL2–RBX1 E3 ligase, which requires NEDD8 conjugation for its activation, interacts with RhoB promoting its ubiquitination and degradation (96). By blocking cullins neddylation, the small pharmacological neddylation inhibitor, Pevonedistat, inactivates CRLs and causes the accumulation of CRLs substrates that trigger cell cycle arrest, senescence and/or apoptosis to suppress the growth of cancer cells in vitro and in vivo.
Paradoxically, Pevonedistat also triggers a pro-survival autophagy response, and abrogation of autophagy enhanced Pevonedistat-induced apoptosis in cancer cells (76,97,98). Autophagy is a highly regulated catabolic process essential for the maintenance of intracellular homeostasis. Upon autophagy activation, cytosolic components are sequestrated into double membrane organelles called autophagosomes that fuse with lysosomes for the hydrolytic degradation of their cargo. When the products of autophagy degradation (e.g., sugars, amino acids, fatty acids and nucleosides/nucleotides) are released into the cytosol, they can serve as substrates for metabolic processes (99-101). Autophagy functionality is tightly regulated by mTOR activity (102). Autophagic flux can be stimulated in response to several signals, including starvation, hypoxia or drug treatment, among others (101). Cancer cells can use autophagy to survive under adverse microenvironmental conditions, and pro-survival autophagy is normally induced during cancer treatment as a mechanism of drug resistance (103,104). Supporting this key role for autophagy in cancer cell survival after a metabolic or a therapeutic stress, there are multiple reports indicating that inhibition of autophagy, either genetically (by silencing of key autophagy-related genes, including ATG5 and ATG7) or pharmacologically (by treatment with chloroquine or hydroxychloroquine lysosomal inhibitors), can suppress cancer cell growth and impair drug resistance (103-105).
As mentioned above, mTOR is a well-known inhibitor of autophagosome formation (102), and mechanistic studies in human liver cancer cell lines have shown that the Pevonedistat-induced autophagy response was mediated by mTOR inactivation. Indeed, Pevonedistat was shown to induce inhibition of cullins activity, leading to an accumulation of the CRL/SCF E3 substrates DEPTOR (DEP domain-containing mTOR-interacting protein) and HIF-1α, two negative regulators of mTOR activity. Direct interaction of DEPTOR to mTOR and activation of the HIF-1α-REDD1 (regulated in development and DNA damage responses 1)-TSC1 (tuberous sclerosis protein 1) axis leads to the inhibition of mTOR activity and induced autophagy (76,97). Another study showed that inhibition of autophagy, using the lysosomal inhibitor chloroquine enhances the anti-tumour efficacy of Pevonedistat both in vitro and in vivo. Importantly, the authors of this study observed that the induction of apoptosis after autophagy inhibition and Pevonedistat treatment was mediated by the up-regulation of NOXA/PMAIP1 protein (phorbol-12-myristate-13-acetate-induced protein 1) and by the down-regulation of classical anti-apoptotic proteins [c-inhibitor of apoptosis (IAP)1/2, B-cell lymphoma 2 (Bcl-2), X-linked inhibitor of apoptosis protein (XIAP), and induced myeloid leukemia cell differentiation protein 1 (Mcl-1)]. They demonstrated that the induction of NOXA was mediated by oxidative stress and enhanced DNA damage (98).
Altogether, these studies suggest that a combinatory therapy of autophagy inhibitors and the neddylation inhibitor Pevonedistat could be very effective for inducing hepatoma cell death without associated drug resistance and could be effective to treat liver cancer patients.
Hu antigen R
HuR is a ubiquitously expressed protein belonging to the Elav/Hu family of RNA-binding proteins that plays major biological roles regulating gene expression by binding to and stabilizing mRNAs containing AU-rich elements, or even enhancing and repressing their translation (106,107). Despite being predominantly nuclear, HuR translocates into the cytosol where it stabilizes target mRNAs encoding important genes for cell cycle control and proliferation like cyclin A1, B1, D1 and E1, thus promoting cell growth and survival. Notably, among HuR targets there also exist proto-oncogenes like c-Myc and c-Fos. On the other hand, under situations like genotoxic stress, HuR may exert an anti-proliferative role by targeting pro-apoptotic p53 and cell cycle inhibitor p21 mRNAs, thus interfering cell growth. Regarding the role of HuR in tumorigenesis, it has been primarily associated with the main cancer traits: proliferation, angiogenesis, enhancement of cell survival, evasion of immune recognition, invasiveness and metastasis. Indeed, HuR expression is frequently increased in colon, breast, pancreatic, ovarian, prostate and many other cancers correlating with tumor malignancy (108,109).
In the liver, HuR plays a role in hepatocyte proliferation, differentiation and HCC transformation (24). Also, HuR has been found highly expressed in HCC-derived cell lines, in which it stabilizes HAUSP (Herpesvirus-associated ubiquitin-specific protease) mRNA. HAUSP is an ubiquitin specific protease that stabilizes p53 in the cytosol inducing cell cycle arrest and an apoptotic response (23). Moreover, HuR is able to regulate hepatic stellate cell activation and to raise the expression of proinflammatory and chemoattractant genes in a cholestatic liver injury model (bile duct ligation). In this way, HuR increases liver damage, oxidative stress, inflammation, macrophage infiltration and liver fibrosis development, enhancing the risk of HCC development (110). Altogether, these findings support the involvement of HuR in liver malignant transformation.
It is well known that (NFκB) can activate HuR transcription downstream of the PI3K/Akt signaling pathway and also that the ubiquitin-proteasome pathway, at the post-transcriptional level, can regulate HuR function (111). In 2012, the mechanism underlying HuR overexpression in HCC was finally unraveled by our group. Neddylation of HuR by the Mdm2 E3 ligase retains it in the nucleus and protects it from proteasomal degradation. In agreement, HuR lysine mutants (K283, 313, 326R) that are incapable of being neddylated or Mdm2 mutants, increase the cytoplasmic localization of HuR and its ubiquitination leading to proteasomal degradation (112). These data suggest that the novel Mdm2/NEDD8/HuR regulatory framework is essential for the malignant transformation of tumor cells. Finally, the neddylation inhibitor Pevonedistat was shown to exert antitumoral effects in vitro and in vivo in liver cancer, partially through HuR destabilization. Importantly, overexpression of HuR in hepatoma cells offers resistance to pharmacological neddylation inhibition while low levels of HuR sensitized cells to the treatment, suggesting that HuR levels determine the drugability of the neddylation pathway in HCC (113).
Liver kinase B1
Liver Kinase B1 [LKB1 or Serine/threonine kinase 11 (STK11)] is an ubiquitously expressed serine/threonine protein kinase originally discovered as a mutation in the familial Peutz-Jeghers Syndrome (PJS) (114), characterized by the formation of hamartomatous polyps in the gastrointestinal tract and hyperpigmented macules in the lips, oral mucosa, genitalia or palmar surfaces (115) and increased probability of developing cancers especially of the lung, ovary, breast, colon and pancreas (116). LKB1 is an upstream activator of AMP-activated protein kinase (AMPK) (117,118), a major cellular energy sensor, and the AMPK related kinases (ARKs) that are involved in a variety of processes such as cell polarity, migration and gene transcription (119,120).
Although considered an oncosuppressor in a variety of tissues, in the liver, LKB1 has been previously implicated in hepatic regeneration and HCC proliferation (23,75,121). During hepatic regeneration, HGF activates the LKB1-AMPK axis leading to the non-canonical activation of endothelial nitric oxide synthase (eNOS) and concomitant increase in the second messenger nitric oxide that facilitates HGF-induced hepatocyte proliferation (121). Furthermore, mouse hepatoma cells (SAMe-D cells) have increased phosphorylation of LKB1 as well as increased cytoplasmic p53 levels making these cells more resistant to UVC exposure-induced apoptosis. Likewise, another type of mouse hepatoma cells (OKER cells) also have increased phosphorylated LKB1 levels showing an increased RAS pathway activation due to increased methylation of RAS inhibitor genes [Suppressor Of Cytokine Signaling 1 (SOCS1) and Ras Association Domain Family Member 1 (RASSF1A)] (122). Finally, LKB1 expression is augmented in livers of HCC patients derived from alcoholic and NASH being that patients with bad prognosis have higher expression of LKB1 (75).
The mechanisms underlying LKB1 overexpression in HCC were further explored. On this basis, the levels of LKB1 and NEDD8 are positively correlated in HCC patients (75). Furthermore, when liver tumor bearing mice were treated with the neddylation inhibitor Pevonedistat the levels of LKB1 fell (75). Likewise, when pre-tumoral hepatocytes were treated either with Pevonedistat or by silencing NEDD8 using molecular approaches, LKB1 levels were also reduced (75). Liver tumor cell apoptosis induced by Pevonedistat was reduced by overexpressing LKB1 as this was able to overcome the loss of stability caused by the inhibitor. Additionally, LKB1 was detected after His-tagged NEDD8 purification, strongly suggesting that LKB1 is directly neddylated and that NEDD8 directly stabilizes LKB1 in HCC (75).
Akt
The PI3K/Akt/mTOR signaling pathway is a key regulator of cell survival and proliferation (123) and one of the main pathways implicated in liver carcinogenesis (124). It is frequently deregulated in HCC and its activation has been correlated with advanced and poor prognosis HCC (125). Akt, also known as protein kinase B (PKB), is a serine/threonine kinase and a crucial downstream effector of PI3K that regulates the function of a great variety of proteins involved in processes that include metabolism, cell growth, apoptosis and angiogenesis (126,127). Akt is probably the most frequently activated oncoprotein in human cancer (128) and is known to play a critical role in the development and progression of HCC by promoting proliferation, cell survival, tumor growth and metastasis (124). Moreover, Akt has also been described as a “Warburg kinase” because of its effect on enzymes involved in the switch to aerobic glycolysis and metabolic reprogramming, well-known hallmarks of cancer (129). On this basis, Akt has been found to promote both glycolysis (130) and oxidative phosphorylation (131). Despite having a critical role in malignant transformation and being a promising anticancer target, the mechanisms underlying Akt deregulation have not been fully elucidated.
It had been previously shown that Akt is subjected to post-translational regulatory events such as ubiquitination (132) and SUMOylation (133). In 2015, the mechanism underlying Akt overexpression in HCC was finally described by our group (75). We have demonstrated that global neddylation is highly increased in human HCC, correlating with both a poorer prognosis and Akt protein levels. As observed in human HCC and supporting the role for neddylation and Akt in liver carcinogenesis, neddylation and Akt protein levels were also found upregulated in livers and hepatocytes from HCC mouse models (134). Using different in vitro approaches, it was shown that Akt is a target of NEDD8 and that this PTM promotes Akt overexpression in the liver. Akt was detected by pull-down of histidine-NEDD8 conjugated proteins in primary hepatocytes transfected with a plasmid expressing histidine-tagged NEDD8. In addition, it was found that neddylation inhibition with the specific pharmacological inhibitor Pevonedistat or a specific siRNA against NEDD8 or the deneddylase NEDP1 led to the downregulation of Akt protein levels. Finally, a cycloheximide assay verified that neddylation stabilizes Akt since NEDD8 silencing significantly decreased its half-life. In terms of metabolism, it was observed that Akt partially mediates the neddylation inhibition-induced metabolic changes that caused ATP depletion, oxidative stress and finally apoptosis in liver cancer cells and pre-tumoral hepatocytes, supporting the major role of the kinase in liver cancer survival and progression. In this regard, Akt overexpression, which enhanced both oxidative phosphorylation and glycolysis, counteracted the metabolic switch and associated cell death induced by Pevonedistat treatment. These results were validated in vivo using two different HCC mouse models, the Prohibitin (Phb1)-deficient mouse and the HepG2 tumor xenograft, where neddylation inhibition with Pevonedistat and a NEDD8 siRNA respectively promoted cell death, tumor regression and metabolic reprogramming, as well as global neddylation and Akt downregulation (75). These findings suggest that Akt neddylation is essential for the malignant transformation of liver cancer cells and reveal that neddylation of Akt is a novel regulatory mechanism accounting for Akt stabilization.
Hypoxia inducible factor (HIF)-2α
A hypoxic microenvironment is believed to contribute to cancer progression by numerous mechanisms, including activation of angiogenesis, cell survival, motility and invasiveness of malignant cells, and increased EMT ability. In this scenario, HIF-2α, a master regulator of oxygen homeostasis, has been suggested to favour proliferation of cancer cells (135). In HCC, it has been reported that HIF-2α expression correlates with poor patient outcome (136). Interestingly, HIF-2α expression has been previously shown to be stabilized by NEDD8 conjugation in a reactive oxygen species (ROS)-dependent manner (95). In HCC, preliminary findings by Cannito et al. have shown that SerpinB3, usually induced by hypoxia, can affect the behaviour of liver cancer cells by promoting the direct and selective neddylation and stabilization of HIF-2α (137,138).
Conclusions
Herein, we have reviewed the most recent findings regarding the relevance of NEDD8 mediated PTMs in HCC pathogenesis, prognosis and reversal. In this regard, recent reports both from our group and others have clearly shown that first of all neddylation activity is augmented in HCC and second, treatment with the neddylation inhibitor Pevonedistat accounts for reduced hepatoma cell growth and tumor regression in pre-clinical mouse models. Under these circumstances, Pevonedistat can act as an anti-tumoral drug by either triggering cell cycle arrest, senescence and apoptosis to suppress the growth of cancer cells in association with a deregulation of the cell energetic metabolism. Pevonedistat has currently entered several clinical trials to treat both leukemias and advanced solid tumors and more details can be found in https://clinicaltrials.gov/ct2/results?cond=&term=MLN4924&cntry=&state=&city=&dist=. Clinical trials addressing the effects of Pevonedistat in combination with other systemic drugs, such as the multi-kinase inhibitor Sorafenib, the only systemic drug currently approved for advanced HCC therapy, or clinically-approved autophagy inhibitors such as the anti-malarial chloroquine and its derivative hydroxychloroquine, in order to overcome Pevonedistat-induced pro-survival autophagy response and associated drug-resistance to Pevonedistat, can provide a potential and novel therapeutic strategy to tackle advanced HCC. Furthermore, we provide a comprehensive revision of the main NEDD8 targets relevant for HCC, such as CRLs, HuR, LKB1, Akt and HIF-2α. Currently, there are still many open questions concerning some of these neddylation targets in HCC, namely the enzymes involved in the neddylation and deneddylation pathway of the novel above-mentioned NEDD8 substrate proteins together with the residues that are relevant for neddylation. Finally, in a near future, we believe that the application of very sensitive proteomics approaches may lead to the identification of novel neddylation targets involved in HCC pathogenesis broadening the spectrum of NEDD8 targets and increasing our current knowledge of the real importance of NEDD8 mediated modifications in HCC.
Acknowledgements
The authors would like to acknowledge Vírginia Gutiérrez-de-Juan for the immunohistochemistry and immunofluorescence micrographs showed.
Funding: This work was supported by grants from Gobierno Vasco-Departamento de Salud 2013111114 (to ML Martínez-Chantar), MINECO: SAF2017-87301-R integrado en el Plan Estatal de Investigación Cientifica y Técnica y Innovación 2017-2020 cofinanciado con Fondos FEDER (to ML Martínez-Chantar), BIOEF (Basque Foundation for Innovation and Health Research): EITB Maratoia BIO15/CA/014; Instituto de Salud Carlos III:PIE14/00031, integrado en el Plan Estatal de Investigación Cientifica y Técnica y Innovacion 2013-2016 cofinanciado con Fondos FEDER (to ML Martínez-Chantar), Asociación Española contra el Cáncer (ML Martínez-Chantar, TC Delgado), 2017 Leonardo Grant for Researchers and Cultural Creators, BBVA Foundation (M Varela-Rey). Ciberehd_ISCIII_MINECO is funded by the Instituto de Salud Carlos III. We thank MINECO for the Severo Ochoa Excellence Accreditation to CIC bioGUNE (SEV-2016-0644).
Footnotes
Conflicts of Interest: Dr. Martínez-Chantar advises for Mitotherapeutix LLC. The other authors have no conflicts of interest to declare.
References
- 1.Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet 2012;379:1245-55. 10.1016/S0140-6736(11)61347-0 [DOI] [PubMed] [Google Scholar]
- 2.Gerbes A, Zoulim F, Tilg H, et al. Gut roundtable meeting paper: selected recent advances in hepatocellular carcinoma. Gut 2018;67:380-8. 10.1136/gutjnl-2017-315068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Forner A, Gilabert M, Bruix J, et al. Treatment of intermediate-stage hepatocellular carcinoma. Nat Rev Clin Oncol 2014;11:525-35. 10.1038/nrclinonc.2014.122 [DOI] [PubMed] [Google Scholar]
- 4.Bruix J, Han KH, Gores G, et al. Liver cancer: Approaching a personalized care. J Hepatol 2015;62:S144-56. 10.1016/j.jhep.2015.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Llovet JM, Real MI, Montana X, et al. Arterial embolisation or chemoembolisation versus symptomatic treatment in patients with unresectable hepatocellular carcinoma: a randomised controlled trial. Lancet 2002;359:1734-9. 10.1016/S0140-6736(02)08649-X [DOI] [PubMed] [Google Scholar]
- 6.Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008;359:378-90. 10.1056/NEJMoa0708857 [DOI] [PubMed] [Google Scholar]
- 7.Lepage C, Bossard N, Dejardin O, et al. Trends in net survival from rectal cancer in six European Latin countries: results from the SUDCAN population-based study. Eur J Cancer Prev 2017;26 Trends in cancer net survival in six European Latin Countries: the SUDCAN study:S48-S55. [DOI] [PubMed]
- 8.Imamura H, Matsuyama Y, Tanaka E, et al. Risk factors contributing to early and late phase intrahepatic recurrence of hepatocellular carcinoma after hepatectomy. J Hepatol 2003;38:200-7. 10.1016/S0168-8278(02)00360-4 [DOI] [PubMed] [Google Scholar]
- 9.Berzigotti A, Reig M, Abraldes JG, et al. Portal hypertension and the outcome of surgery for hepatocellular carcinoma in compensated cirrhosis: a systematic review and meta-analysis. Hepatology 2015;61:526-36. 10.1002/hep.27431 [DOI] [PubMed] [Google Scholar]
- 10.van Malenstein H, Dekervel J, Verslype C, et al. Long-term exposure to sorafenib of liver cancer cells induces resistance with epithelial-to-mesenchymal transition, increased invasion and risk of rebound growth. Cancer Lett 2013;329:74-83. 10.1016/j.canlet.2012.10.021 [DOI] [PubMed] [Google Scholar]
- 11.Villanueva A, Llovet JM. Second-line therapies in hepatocellular carcinoma: emergence of resistance to sorafenib. Clin Cancer Res 2012;18:1824-6. 10.1158/1078-0432.CCR-12-0151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nault JC, Mallet M, Pilati C, et al. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat Commun 2013;4:2218. 10.1038/ncomms3218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dhanasekaran R, Bandoh S, Roberts LR. Molecular pathogenesis of hepatocellular carcinoma and impact of therapeutic advances. F1000Res 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hussain SP, Schwank J, Staib F, et al. TP53 mutations and hepatocellular carcinoma: insights into the etiology and pathogenesis of liver cancer. Oncogene 2007;26:2166-76. 10.1038/sj.onc.1210279 [DOI] [PubMed] [Google Scholar]
- 15.Luo D, Wang Z, Wu J, et al. The role of hypoxia inducible factor-1 in hepatocellular carcinoma. Biomed Res Int 2014;2014:409272. 10.1155/2014/409272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zucman-Rossi J, Benhamouche S, Godard C, et al. Differential effects of inactivated Axin1 and activated beta-catenin mutations in human hepatocellular carcinomas. Oncogene 2007;26:774-80. 10.1038/sj.onc.1209824 [DOI] [PubMed] [Google Scholar]
- 17.Guichard C, Amaddeo G, Imbeaud S, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet 2012;44:694-8. 10.1038/ng.2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lachenmayer A, Alsinet C, Savic R, et al. Wnt-pathway activation in two molecular classes of hepatocellular carcinoma and experimental modulation by sorafenib. Clin Cancer Res 2012;18:4997-5007. 10.1158/1078-0432.CCR-11-2322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Calvisi DF, Ladu S, Gorden A, et al. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology 2006;130:1117-28. 10.1053/j.gastro.2006.01.006 [DOI] [PubMed] [Google Scholar]
- 20.Villanueva A, Chiang DY, Newell P, et al. Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology 2008;135:1972-83, 83 e1-11. [DOI] [PMC free article] [PubMed]
- 21.Roberts AB, Wakefield LM. The two faces of transforming growth factor beta in carcinogenesis. Proc Natl Acad Sci U S A 2003;100:8621-3. 10.1073/pnas.1633291100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giannelli G, Bergamini C, Fransvea E, et al. Laminin-5 with transforming growth factor-beta1 induces epithelial to mesenchymal transition in hepatocellular carcinoma. Gastroenterology 2005;129:1375-83. 10.1053/j.gastro.2005.09.055 [DOI] [PubMed] [Google Scholar]
- 23.Martinez-Lopez N, Varela-Rey M, Fernandez-Ramos D, et al. Activation of LKB1-Akt pathway independent of phosphoinositide 3-kinase plays a critical role in the proliferation of hepatocellular carcinoma from nonalcoholic steatohepatitis. Hepatology 2010;52:1621-31. 10.1002/hep.23860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vazquez-Chantada M, Fernandez-Ramos D, Embade N, et al. HuR/methyl-HuR and AUF1 regulate the MAT expressed during liver proliferation, differentiation, and carcinogenesis. Gastroenterology 2010;138:1943-53. 10.1053/j.gastro.2010.01.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hung CM, Huang WC, Pan HL, et al. Hepatitis B virus X upregulates HuR protein level to stabilize HER2 expression in hepatocellular carcinoma cells. Biomed Res Int 2014;2014:827415. 10.1155/2014/827415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cao C, Sun J, Zhang D, et al. The long intergenic noncoding RNA UFC1, a target of MicroRNA 34a, interacts with the mRNA stabilizing protein HuR to increase levels of beta-catenin in HCC cells. Gastroenterology 2015;148:415-26 e18. [DOI] [PubMed]
- 27.Zhu H, Berkova Z, Mathur R, et al. HuR Suppresses Fas Expression and Correlates with Patient Outcome in Liver Cancer. Mol Cancer Res 2015;13:809-18. 10.1158/1541-7786.MCR-14-0241 [DOI] [PubMed] [Google Scholar]
- 28.International Human Genome Sequencing C. Finishing the euchromatic sequence of the human genome. Nature 2004;431:931-45. 10.1038/nature03001 [DOI] [PubMed] [Google Scholar]
- 29.Jensen ON. Modification-specific proteomics: characterization of post-translational modifications by mass spectrometry. Curr Opin Chem Biol 2004;8:33-41. 10.1016/j.cbpa.2003.12.009 [DOI] [PubMed] [Google Scholar]
- 30.Ayoubi TA, Van De Ven WJ. Regulation of gene expression by alternative promoters. FASEB J 1996;10:453-60. 10.1096/fasebj.10.4.8647344 [DOI] [PubMed] [Google Scholar]
- 31.Skaug B, Chen ZJ. SUMO, Ubiquitin, UBL Proteins: Implications For Human Diseases - Fifth International Conference. IDrugs 2010;13:224-7. [PubMed] [Google Scholar]
- 32.Hu H, Sun SC. Ubiquitin signaling in immune responses. Cell Res 2016;26:457-83. 10.1038/cr.2016.40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tomasi ML, Tomasi I, Ramani K, et al. S-adenosyl methionine regulates ubiquitin-conjugating enzyme 9 protein expression and sumoylation in murine liver and human cancers. Hepatology 2012;56:982-93. 10.1002/hep.25701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dawson SP. Hepatocellular carcinoma and the ubiquitin-proteasome system. Biochim Biophys Acta 2008;1782:775-84. 10.1016/j.bbadis.2008.08.003 [DOI] [PubMed] [Google Scholar]
- 35.Kumar S, Tomooka Y, Noda M. Identification of a set of genes with developmentally down-regulated expression in the mouse brain. Biochem Biophys Res Commun 1992;185:1155-61. 10.1016/0006-291X(92)91747-E [DOI] [PubMed] [Google Scholar]
- 36.Whitby FG, Xia G, Pickart CM, et al. Crystal structure of the human ubiquitin-like protein NEDD8 and interactions with ubiquitin pathway enzymes. J Biol Chem 1998;273:34983-91. 10.1074/jbc.273.52.34983 [DOI] [PubMed] [Google Scholar]
- 37.Rao-Naik C, delaCruz W, Laplaza JM, et al. The rub family of ubiquitin-like proteins. Crystal structure of Arabidopsis rub1 and expression of multiple rubs in Arabidopsis. J Biol Chem 1998;273:34976-82. 10.1074/jbc.273.52.34976 [DOI] [PubMed] [Google Scholar]
- 38.Kamitani T, Kito K, Nguyen HP, et al. Characterization of NEDD8, a developmentally down-regulated ubiquitin-like protein. J Biol Chem 1997;272:28557-62. 10.1074/jbc.272.45.28557 [DOI] [PubMed] [Google Scholar]
- 39.Hori T, Osaka F, Chiba T, et al. Covalent modification of all members of human cullin family proteins by NEDD8. Oncogene 1999;18:6829-34. 10.1038/sj.onc.1203093 [DOI] [PubMed] [Google Scholar]
- 40.Choi YS, Jeon YH, Ryu KS, et al. 60th residues of ubiquitin and Nedd8 are located out of E2-binding surfaces, but are important for K48 ubiquitin-linkage. FEBS Lett 2009;583:3323-8. 10.1016/j.febslet.2009.09.034 [DOI] [PubMed] [Google Scholar]
- 41.Walden H, Podgorski MS, Huang DT, et al. The structure of the APPBP1-UBA3-NEDD8-ATP complex reveals the basis for selective ubiquitin-like protein activation by an E1. Mol Cell 2003;12:1427-37. 10.1016/S1097-2765(03)00452-0 [DOI] [PubMed] [Google Scholar]
- 42.Reverter D, Wu K, Erdene TG, et al. Structure of a complex between Nedd8 and the Ulp/Senp protease family member Den1. J Mol Biol 2005;345:141-51. 10.1016/j.jmb.2004.10.022 [DOI] [PubMed] [Google Scholar]
- 43.Shen LN, Liu H, Dong C, et al. Structural basis of NEDD8 ubiquitin discrimination by the deNEDDylating enzyme NEDP1. EMBO J 2005;24:1341-51. 10.1038/sj.emboj.7600628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liakopoulos D, Doenges G, Matuschewski K, et al. A novel protein modification pathway related to the ubiquitin system. EMBO J 1998;17:2208-14. 10.1093/emboj/17.8.2208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Osaka F, Kawasaki H, Aida N, et al. A new NEDD8-ligating system for cullin-4A. Genes Dev 1998;12:2263-8. 10.1101/gad.12.15.2263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leyser HM, Lincoln CA, Timpte C, et al. Arabidopsis auxin-resistance gene AXR1 encodes a protein related to ubiquitin-activating enzyme E1. Nature 1993;364:161-4. 10.1038/364161a0 [DOI] [PubMed] [Google Scholar]
- 47.Gong L, Yeh ET. Identification of the activating and conjugating enzymes of the NEDD8 conjugation pathway. J Biol Chem 1999;274:12036-42. 10.1074/jbc.274.17.12036 [DOI] [PubMed] [Google Scholar]
- 48.Walden H, Podgorski MS, Schulman BA. Insights into the ubiquitin transfer cascade from the structure of the activating enzyme for NEDD8. Nature 2003;422:330-4. 10.1038/nature01456 [DOI] [PubMed] [Google Scholar]
- 49.Huang DT, Ayrault O, Hunt HW, et al. E2-RING expansion of the NEDD8 cascade confers specificity to cullin modification. Mol Cell 2009;33:483-95. 10.1016/j.molcel.2009.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kamura T, Conrad MN, Yan Q, et al. The Rbx1 subunit of SCF and VHL E3 ubiquitin ligase activates Rub1 modification of cullins Cdc53 and Cul2. Genes Dev 1999;13:2928-33. 10.1101/gad.13.22.2928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kamura T, Koepp DM, Conrad MN, et al. Rbx1, a component of the VHL tumor suppressor complex and SCF ubiquitin ligase. Science 1999;284:657-61. 10.1126/science.284.5414.657 [DOI] [PubMed] [Google Scholar]
- 52.Skowyra D, Koepp DM, Kamura T, et al. Reconstitution of G1 cyclin ubiquitination with complexes containing SCFGrr1 and Rbx1. Science 1999;284:662-5. 10.1126/science.284.5414.662 [DOI] [PubMed] [Google Scholar]
- 53.Xirodimas DP, Saville MK, Bourdon JC, et al. Mdm2-mediated NEDD8 conjugation of p53 inhibits its transcriptional activity. Cell 2004;118:83-97. 10.1016/j.cell.2004.06.016 [DOI] [PubMed] [Google Scholar]
- 54.Oved S, Mosesson Y, Zwang Y, et al. Conjugation to Nedd8 instigates ubiquitylation and down-regulation of activated receptor tyrosine kinases. J Biol Chem 2006;281:21640-51. 10.1074/jbc.M513034200 [DOI] [PubMed] [Google Scholar]
- 55.Yang Y, Kitagaki J, Dai RM, et al. Inhibitors of ubiquitin-activating enzyme (E1), a new class of potential cancer therapeutics. Cancer Res 2007;67:9472-81. 10.1158/0008-5472.CAN-07-0568 [DOI] [PubMed] [Google Scholar]
- 56.Yang X, Zhou J, Sun L, et al. Structural basis for the function of DCN-1 in protein Neddylation. J Biol Chem 2007;282:24490-4. 10.1074/jbc.C700038200 [DOI] [PubMed] [Google Scholar]
- 57.Kurz T, Chou YC, Willems AR, et al. Dcn1 functions as a scaffold-type E3 ligase for cullin neddylation. Mol Cell 2008;29:23-35. 10.1016/j.molcel.2007.12.012 [DOI] [PubMed] [Google Scholar]
- 58.Rabut G, Le Dez G, Verma R, et al. The TFIIH subunit Tfb3 regulates cullin neddylation. Mol Cell 2011;43:488-95. 10.1016/j.molcel.2011.05.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scott DC, Monda JK, Bennett EJ, et al. N-terminal acetylation acts as an avidity enhancer within an interconnected multiprotein complex. Science 2011;334:674-8. 10.1126/science.1209307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xie P, Zhang M, He S, et al. The covalent modifier Nedd8 is critical for the activation of Smurf1 ubiquitin ligase in tumorigenesis. Nat Commun 2014;5:3733. 10.1038/ncomms4733 [DOI] [PubMed] [Google Scholar]
- 61.Ohki Y, Funatsu N, Konishi N, et al. The mechanism of poly-NEDD8 chain formation in vitro. Biochem Biophys Res Commun 2009;381:443-7. 10.1016/j.bbrc.2009.02.090 [DOI] [PubMed] [Google Scholar]
- 62.Singh RK, Zerath S, Kleifeld O, et al. Recognition and cleavage of related to ubiquitin 1 (Rub1) and Rub1-ubiquitin chains by components of the ubiquitin-proteasome system. Mol Cell Proteomics 2012;11:1595-611. 10.1074/mcp.M112.022467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hjerpe R, Thomas Y, Chen J, et al. Changes in the ratio of free NEDD8 to ubiquitin triggers NEDDylation by ubiquitin enzymes. Biochem J 2012;441:927-36. 10.1042/BJ20111671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hjerpe R, Thomas Y, Kurz T. NEDD8 overexpression results in neddylation of ubiquitin substrates by the ubiquitin pathway. J Mol Biol 2012;421:27-9. 10.1016/j.jmb.2012.05.013 [DOI] [PubMed] [Google Scholar]
- 65.Kurz T, Ozlu N, Rudolf F, et al. The conserved protein DCN-1/Dcn1p is required for cullin neddylation in C. elegans and S. cerevisiae. Nature 2005;435:1257-61. 10.1038/nature03662 [DOI] [PubMed] [Google Scholar]
- 66.Zou J, Li J, Ma W, et al. Abstract 14914: Inactivation of Neddylation Activating Enzyme 1 (NAE1) in Mice and Rats Leads to Cardiac Developmental and Functional Defects. Circulation 2016;134:A14914. [Google Scholar]
- 67.Vogl AM, Brockmann MM, Giusti SA, et al. Neddylation inhibition impairs spine development, destabilizes synapses and deteriorates cognition. Nat Neurosci 2015;18:239-51. 10.1038/nn.3912 [DOI] [PubMed] [Google Scholar]
- 68.Ogrodnik M, Miwa S, Tchkonia T, et al. Cellular senescence drives age-dependent hepatic steatosis. Nat Commun 2017;8:15691. 10.1038/ncomms15691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Soucy TA, Smith PG, Milhollen MA, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009;458:732-6. 10.1038/nature07884 [DOI] [PubMed] [Google Scholar]
- 70.Brownell JE, Sintchak MD, Gavin JM, et al. Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: the NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic in situ. Mol Cell 2010;37:102-11. 10.1016/j.molcel.2009.12.024 [DOI] [PubMed] [Google Scholar]
- 71.Deng Q, Zhang J, Gao Y, et al. MLN4924 protects against bleomycin-induced pulmonary fibrosis by inhibiting the early inflammatory process. Am J Transl Res 2017;9:1810-21. [PMC free article] [PubMed] [Google Scholar]
- 72.Li L, Wang M, Yu G, et al. Overactivated neddylation pathway as a therapeutic target in lung cancer. J Natl Cancer Inst 2014;106:dju083. 10.1093/jnci/dju083 [DOI] [PubMed] [Google Scholar]
- 73.Chen Y, Neve RL, Liu H. Neddylation dysfunction in Alzheimer's disease. J Cell Mol Med 2012;16:2583-91. 10.1111/j.1582-4934.2012.01604.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zubiete-Franco I, Fernandez-Tussy P, Barbier-Torres L, et al. Deregulated neddylation in liver fibrosis. Hepatology 2017;65:694-709. 10.1002/hep.28933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Barbier-Torres L, Delgado TC, Garcia-Rodriguez JL, et al. Stabilization of LKB1 and Akt by neddylation regulates energy metabolism in liver cancer. Oncotarget 2015;6:2509-23. 10.18632/oncotarget.3191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Luo Z, Yu G, Lee HW, et al. The Nedd8-activating enzyme inhibitor MLN4924 induces autophagy and apoptosis to suppress liver cancer cell growth. Cancer Res 2012;72:3360-71. 10.1158/0008-5472.CAN-12-0388 [DOI] [PubMed] [Google Scholar]
- 77.Gao Q, Yu GY, Shi JY, et al. Neddylation pathway is up-regulated in human intrahepatic cholangiocarcinoma and serves as a potential therapeutic target. Oncotarget 2014;5:7820-32. 10.18632/oncotarget.2309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Roessler S, Jia HL, Budhu A, et al. A unique metastasis gene signature enables prediction of tumor relapse in early-stage hepatocellular carcinoma patients. Cancer Res 2010;70:10202-12. 10.1158/0008-5472.CAN-10-2607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wu JT, Lin HC, Hu YC, et al. Neddylation and deneddylation regulate Cul1 and Cul3 protein accumulation. Nat Cell Biol 2005;7:1014-20. 10.1038/ncb1301 [DOI] [PubMed] [Google Scholar]
- 80.Sakata E, Yamaguchi Y, Miyauchi Y, et al. Direct interactions between NEDD8 and ubiquitin E2 conjugating enzymes upregulate cullin-based E3 ligase activity. Nat Struct Mol Biol 2007;14:167-8. 10.1038/nsmb1191 [DOI] [PubMed] [Google Scholar]
- 81.Abidi N, Xirodimas DP. Regulation of cancer-related pathways by protein NEDDylation and strategies for the use of NEDD8 inhibitors in the clinic. Endocr Relat Cancer 2015;22:T55-70. 10.1530/ERC-14-0315 [DOI] [PubMed] [Google Scholar]
- 82.Enchev RI, Schulman BA, Peter M. Protein neddylation: beyond cullin-RING ligases. Nat Rev Mol Cell Biol 2015;16:30-44. 10.1038/nrm3919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hochstrasser M. There's the rub: a novel ubiquitin-like modification linked to cell cycle regulation. Genes Dev 1998;12:901-7. 10.1101/gad.12.7.901 [DOI] [PubMed] [Google Scholar]
- 84.Pan ZQ, Kentsis A, Dias DC, et al. Nedd8 on cullin: building an expressway to protein destruction. Oncogene 2004;23:1985-97. 10.1038/sj.onc.1207414 [DOI] [PubMed] [Google Scholar]
- 85.Skaar JR, Pagano M. Control of cell growth by the SCF and APC/C ubiquitin ligases. Curr Opin Cell Biol 2009;21:816-24. 10.1016/j.ceb.2009.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Schreiber A, Stengel F, Zhang Z, et al. Structural basis for the subunit assembly of the anaphase-promoting complex. Nature 2011;470:227-32. 10.1038/nature09756 [DOI] [PubMed] [Google Scholar]
- 87.Chang LF, Zhang Z, Yang J, et al. Molecular architecture and mechanism of the anaphase-promoting complex. Nature 2014;513:388-93. 10.1038/nature13543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang D, Yang G, Li X, et al. Inhibition of Liver Carcinoma Cell Invasion and Metastasis by Knockdown of Cullin7 In Vitro and In Vivo. Oncol Res 2016;23:171-81. 10.3727/096504016X14519995067562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pan Y, Wang B, Yang X, et al. CUL4A facilitates hepatocarcinogenesis by promoting cell cycle progression and epithelial-mesenchymal transition. Sci Rep 2015;5:17006. 10.1038/srep17006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yuan J, Jiang B, Zhang A, et al. Accelerated hepatocellular carcinoma development in CUL4B transgenic mice. Oncotarget 2015;6:15209-21. 10.18632/oncotarget.3829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu W, Wang Y, Zhang C, et al. Cullin1 is up-regulated and associated with poor patients' survival in hepatocellular carcinoma. Int J Clin Exp Pathol 2015;8:4001-7. [PMC free article] [PubMed] [Google Scholar]
- 92.Bloom J, Amador V, Bartolini F, et al. Proteasome-mediated degradation of p21 via N-terminal ubiquitinylation. Cell 2003;115:71-82. 10.1016/S0092-8674(03)00755-4 [DOI] [PubMed] [Google Scholar]
- 93.Lin JJ, Milhollen MA, Smith PG, et al. NEDD8-targeting drug MLN4924 elicits DNA rereplication by stabilizing Cdt1 in S phase, triggering checkpoint activation, apoptosis, and senescence in cancer cells. Cancer Res 2010;70:10310-20. 10.1158/0008-5472.CAN-10-2062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li W, Kong AN. Molecular mechanisms of Nrf2-mediated antioxidant response. Mol Carcinog 2009;48:91-104. 10.1002/mc.20465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ryu JH, Li SH, Park HS, et al. Hypoxia-inducible factor alpha subunit stabilization by NEDD8 conjugation is reactive oxygen species-dependent. J Biol Chem 2011;286:6963-70. 10.1074/jbc.M110.188706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Xu J, Li L, Yu G, et al. The neddylation-cullin 2-RBX1 E3 ligase axis targets tumor suppressor RhoB for degradation in liver cancer. Mol Cell Proteomics 2015;14:499-509. 10.1074/mcp.M114.045211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhao Y, Xiong X, Jia L, et al. Targeting Cullin-RING ligases by MLN4924 induces autophagy via modulating the HIF1-REDD1-TSC1-mTORC1-DEPTOR axis. Cell Death Dis 2012;3:e386. 10.1038/cddis.2012.125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chen P, Hu T, Liang Y, et al. Synergistic inhibition of autophagy and neddylation pathways as a novel therapeutic approach for targeting liver cancer. Oncotarget 2015;6:9002-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science 2000;290:1717-21. 10.1126/science.290.5497.1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal 2014;20:460-73. 10.1089/ars.2013.5371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yin Z, Pascual C, Klionsky DJ. Autophagy: machinery and regulation. Microb Cell 2016;3:588-96. 10.15698/mic2016.12.546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jung CH, Ro SH, Cao J, et al. mTOR regulation of autophagy. FEBS Lett 2010;584:1287-95. 10.1016/j.febslet.2010.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rybstein MD, Bravo-San Pedro JM, Kroemer G, et al. The autophagic network and cancer. Nat Cell Biol 2018;20:243-51. 10.1038/s41556-018-0042-2 [DOI] [PubMed] [Google Scholar]
- 104.Galluzzi L, Bravo-San Pedro JM, Demaria S, et al. Activating autophagy to potentiate immunogenic chemotherapy and radiation therapy. Nat Rev Clin Oncol 2017;14:247-58. 10.1038/nrclinonc.2016.183 [DOI] [PubMed] [Google Scholar]
- 105.Bingel C, Koeneke E, Ridinger J, et al. Three-dimensional tumor cell growth stimulates autophagic flux and recapitulates chemotherapy resistance. Cell Death Dis 2017;8:e3013. 10.1038/cddis.2017.398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Brennan CM, Steitz JA. HuR and mRNA stability. Cell Mol Life Sci 2001;58:266-77. 10.1007/PL00000854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hinman MN, Lou H. Diverse molecular functions of Hu proteins. Cell Mol Life Sci 2008;65:3168-81. 10.1007/s00018-008-8252-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Abdelmohsen K, Gorospe M. Posttranscriptional regulation of cancer traits by HuR. Wiley Interdiscip Rev RNA 2010;1:214-29. 10.1002/wrna.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Srikantan S, Gorospe M. HuR function in disease. Front Biosci (Landmark Ed) 2012;17:189-205. 10.2741/3921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Woodhoo A, Iruarrizaga-Lejarreta M, Beraza N, et al. Human antigen R contributes to hepatic stellate cell activation and liver fibrosis. Hepatology 2012;56:1870-82. 10.1002/hep.25828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kang MJ, Ryu BK, Lee MG, et al. NF-kappaB activates transcription of the RNA-binding factor HuR, via PI3K-AKT signaling, to promote gastric tumorigenesis. Gastroenterology 2008;135:2030-42, 2042.e1-3. [DOI] [PubMed]
- 112.Embade N, Fernandez-Ramos D, Varela-Rey M, et al. Murine double minute 2 regulates Hu antigen R stability in human liver and colon cancer through NEDDylation. Hepatology 2012;55:1237-48. 10.1002/hep.24795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Barbier-Torres L, Fernandez-Ramos D, Martinez-Chantar ML. The levels of the RNA binding protein Hu antigen R determine the druggability of the neddylation pathway in liver cancer. RNA & DISEASE 2016;3:e1123. [Google Scholar]
- 114.Hemminki A, Markie D, Tomlinson I, et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 1998;391:184-7. 10.1038/34432 [DOI] [PubMed] [Google Scholar]
- 115.McGarrity TJ, Amos C. Peutz-Jeghers syndrome: clinicopathology and molecular alterations. Cell Mol Life Sci 2006;63:2135-44. 10.1007/s00018-006-6080-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Giardiello FM, Brensinger JD, Tersmette AC, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology 2000;119:1447-53. 10.1053/gast.2000.20228 [DOI] [PubMed] [Google Scholar]
- 117.Hawley SA, Boudeau J, Reid JL, et al. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol 2003;2:28. 10.1186/1475-4924-2-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Woods A, Johnstone SR, Dickerson K, et al. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol 2003;13:2004-8. 10.1016/j.cub.2003.10.031 [DOI] [PubMed] [Google Scholar]
- 119.Alessi DR, Sakamoto K, Bayascas JR. LKB1-dependent signaling pathways. Annu Rev Biochem 2006;75:137-63. 10.1146/annurev.biochem.75.103004.142702 [DOI] [PubMed] [Google Scholar]
- 120.Monteverde T, Muthalagu N, Port J, et al. Evidence of cancer-promoting roles for AMPK and related kinases. FEBS J 2015;282:4658-71. 10.1111/febs.13534 [DOI] [PubMed] [Google Scholar]
- 121.Varela-Rey M, Fernandez-Ramos D, Martinez-Lopez N, et al. Impaired liver regeneration in mice lacking glycine N-methyltransferase. Hepatology 2009;50:443-52. 10.1002/hep.23033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Martinez-Lopez N, Garcia-Rodriguez JL, Varela-Rey M, et al. Hepatoma cells from mice deficient in glycine N-methyltransferase have increased RAS signaling and activation of liver kinase B1. Gastroenterology 2012;143:787-98 e1-13. [DOI] [PMC free article] [PubMed]
- 123.Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front Oncol 2014;4:64. 10.3389/fonc.2014.00064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhou Q, Lui VW, Yeo W. Targeting the PI3K/Akt/mTOR pathway in hepatocellular carcinoma. Future Oncol 2011;7:1149-67. 10.2217/fon.11.95 [DOI] [PubMed] [Google Scholar]
- 125.Zhou L, Huang Y, Li J, et al. The mTOR pathway is associated with the poor prognosis of human hepatocellular carcinoma. Med Oncol 2010;27:255-61. 10.1007/s12032-009-9201-4 [DOI] [PubMed] [Google Scholar]
- 126.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell 2007;129:1261-74. 10.1016/j.cell.2007.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Testa JR, Tsichlis PN. AKT signaling in normal and malignant cells. Oncogene 2005;24:7391-3. 10.1038/sj.onc.1209100 [DOI] [PubMed] [Google Scholar]
- 128.Altomare DA, Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene 2005;24:7455-64. 10.1038/sj.onc.1209085 [DOI] [PubMed] [Google Scholar]
- 129.Robey RB, Hay N. Is Akt the "Warburg kinase"?-Akt-energy metabolism interactions and oncogenesis. Semin Cancer Biol 2009;19:25-31. 10.1016/j.semcancer.2008.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Elstrom RL, Bauer DE, Buzzai M, et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res 2004;64:3892-9. 10.1158/0008-5472.CAN-03-2904 [DOI] [PubMed] [Google Scholar]
- 131.Gottlob K, Majewski N, Kennedy S, et al. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev 2001;15:1406-18. 10.1101/gad.889901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yang WL, Wang J, Chan CH, et al. The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science 2009;325:1134-8. 10.1126/science.1175065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Li R, Wei J, Jiang C, et al. Akt SUMOylation regulates cell proliferation and tumorigenesis. Cancer Res 2013;73:5742-53. 10.1158/0008-5472.CAN-13-0538 [DOI] [PubMed] [Google Scholar]
- 134.Ko KS, Tomasi ML, Iglesias-Ara A, et al. Liver-specific deletion of prohibitin 1 results in spontaneous liver injury, fibrosis, and hepatocellular carcinoma in mice. Hepatology 2010;52:2096-108. 10.1002/hep.23919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gordan JD, Bertout JA, Hu CJ, et al. HIF-2alpha promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell 2007;11:335-47. 10.1016/j.ccr.2007.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bangoura G, Liu ZS, Qian Q, et al. Prognostic significance of HIF-2alpha/EPAS1 expression in hepatocellular carcinoma. World J Gastroenterol 2007;13:3176-82. 10.3748/wjg.v13.i23.3176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Cannito S, Turato C, Paternostro C, et al. Hypoxia up-regulates SERPINB3 through HIF-2alpha in human liver cancer cells. Oncotarget 2015;6:2206-21. 10.18632/oncotarget.2943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Cannito S, Villano G, Turato C, et al. editors. SerpinB3 up-regulates hypoxia inducible factors-1α and -2α in liver cancer cells through different mechanisms. Barcelona: The International Liver Congress, 2016. [Google Scholar]