

Abstract
The under-regulation of liver-specific MAT1A gene codifying for S-adenosylmethionine (SAM) synthesizing isozymes MATI/III, and the up-regulation of widely expressed MAT2A, MATII isozyme occurs in hepatocellular carcinoma (HCC). MATα1:MATα2 switch strongly contributes to the fall in SAM liver content both in rodent and human liver carcinogenesis. SAM administration to carcinogen-treated animals inhibits hepatocarcinogenesis. The opposite occurs in Mat1a-KO mice, in which chronic SAM deficiency is followed by HCC development. This review focuses upon the changes, induced by the MATα1:MATα2 switch, involved in HCC development. In association with MATα1:MATα2 switch there occurs, in HCC, global DNA hypomethylation, decline of DNA repair, genomic instability, and deregulation of different signaling pathways such as overexpression of c-MYC (avian myelocytomatosis viral oncogene homolog), increase of polyamine (PA) synthesis and RAS/ERK (Harvey murine sarcoma virus oncogene homolog/extracellular signal-regulated kinase), IKK/NF-kB (I-k kinase beta/nuclear factor kB), PI3K/AKT, and LKB1/AMPK axes. Furthermore, a decrease in MATα1 expression and SAM level induces HCC cell proliferation and survival. SAM treatment in vivo and enforced MATα1 overexpression or MATα2 inhibition, in cultured HCC cells, prevent these changes. A negative correlation of MATα1:MATα2 and MATI/III:MATII ratios with cell proliferation and genomic instability and a positive correlation with apoptosis and global DNA methylation are present in human HCC. Altogether, these data suggest that the decrease of SAM level and the deregulation of MATs are potential therapeutic targets for HCC.
Keywords: Hepatocarcinogenesis, methionine metabolism, S-adenosylmethionine (SAM), signal transduction, prognosis
Introduction
S-adenosylmethionine and folate contribute to one-carbon units’ metabolism and trafficking from the amino acids glycine, methionine, serine, and threonine of diet and endogenous compounds (choline, folate) (1). Methionine, an essential amino acid, is required for normal development and cell growth (1). Its metabolism is involved, in mammals, in three principal pathways: the methionine cycle and the transsulfuration pathway, which share the first reactions converting methionine to homocysteine (HCyst), and the polyamine (PA) biosynthesis (Figure 1). Methionine is converted to SAM by methionine adenosyltransferases (MATI/III; MATII: SAM synthetases). The liver uses daily about half of the methionine ingested to synthesize SAM using ATP. Mammalian liver cells and acinar pancreatic cells express MAT1A and MAT2A genes, which encode the MATI/III and MATII enzymes, exhibiting the two homologous catalytic subunits: α1 and α2, respectively. MAT1A is highly expressed in normal adult liver, where the α1 subunit is present both as dimer (isoenzyme MATIII) and tetramer (isoenzyme MATI) isoforms (2,3). MAT2A, exhibiting the α2 catalytic subunit, is expressed in the fetal liver and distributed ubiquitously in the adult extrahepatic human tissues, and in Ito and Kupffer liver cells, (3,4). All mammalian cells express the MAT2B gene, encoding the beta-regulatory subunit, which regulates the activity of MATII enzyme, making this molecule more susceptible to a negative feedback by SAM. The availability of the latter is essential to several biological functions, including DNA methylation, methylation of phosphatidylethanolamine, biosynthesis of phosphatidylcholine, and biosynthesis of reduced glutathione (GSH) and polyamines.
Figure 1.

Metabolic cycles involved in methionine metabolism. Substrates: Ad, adenine; Bet, betaine; Chol, choline; DMG, dimethylglycine; dSAM, decarboxylated S-adenosylmethionine; GN, glycine; GSG, reduced glutathione; HCyst, homocysteine; MTHF, 5-methyltetrahydrofolate; MeTHF, 5-methenyltetrahydrofolate; MTA, 5'-methylthioadenosine; MTR, methylthioribose; Orn, ornithine; PC, phosphatidylcholine; PE, phosphatidylethanolamine; Putr, putrescine; SAH, S-adenosylhomocysteine; SAM S-adenosylmethionine; SN, sarcosine; SPD, spermidine; SPR, spermine; THF, tetrahydrofolate. Enzymes: 1, MATI/III; 2, MATII; 3, phospholipid N-methyltransferase; 4, various phospholipases; 5, choline oxidase; 6, betaine aldehyde dehydrogenase; 7, betaine homocysteine methyltransferase; 8, glycine N-methyltransferase; 9, various methyltransferases; 10, S-adenosylhomocysteine hydroxylase; 11, methyltetrahydrofolate reductase; 12, sarcosine dehydrogenase; 13, 5,10-methenyl-tetrahydrofolate reductase; 14, cystathionine synthetase; 15, S-adenosylmethionine decarboxylase; 16, ornithine decarboxylase; 17, spermine synthetase; 18, spermidine synthetase; 19, 5-methylthioadenosine nucleosidase. The dotted arrow indicates the “salvage pathway” for methionine resynthesis.
The methionine cycle
The SAM of normal tissues is mostly used for the transmethylation of different acceptor molecules that receive methyl groups, producing SAH, which is further transformed to HCyst and adenosine (5,6). These reactions are catalyzed by specific methyltransferases, the most abundant of which, in the liver, is the glycine-N-methyltransferase (GNMT) (7). GNMT catalyzes the methylation of glycine to sarcosine, thus contributing to maintain the normal cellular pool of MeTHF that is further transformed to MTHF (Figure 1).
The intermediate product, SAH is a potent, competitive inhibitor of transmethylation reactions and, consequently, its removal is required. SAH-hydrolase (also known as adenosylhomocysteinase) hydrolyzes SAH in vivo when the products of this reaction, adenosine and HCyst, are rapidly removed. HCyst is a toxic by-product of sulfur amino acid metabolism and it is also known as an independent risk factor for cardiovascular diseases (8). MTHR (also called methionine synthetase), generates methionine by remethylating HCyst. MTHR activity in part depends on the cell growth status: it particularly increases in growing normal and cancer cells (9). In hepatocytes, methionine is also generated by the betaine/homocysteine-methyltransferase enzyme BHMT, which uses betaine (trimethylglycine) as methyl donor and MTHF, in the presence of vitamin B12 (9) (Figure 1).
HCyst can be also converted to cysteine via the transsulfuration pathway that utilizes methionine for GSH synthesis (10,11): a reaction catalyzed by cystathionine β-synthetase (CBS) produces cystathionine, which, after cleavage by γ-cystathionase, releases cysteine used for GSH synthesis. GSH protects the cells from oxidative stress by reducing ROS (10) (Figure 1). Thus, in the transsulfuration pathway CBS and γ-cystathionase enzymes catalyze the production of H2S, a molecule that by favoring the dilatation of aorta and mesenteric arteries, reduces blood pressure (11). The decrease of γ-cystathionase, is responsible for hypercystathioninemia and plasma H2S reduction, and may contribute to portal vein hypertension (12), which complicates liver cirrhosis.
SAM is the major aminopropyl group donor for the PA synthetic pathway (10). The first step of the latter produces decarboxylated SAM (dSAM), which donates the aminopropyl group to putrescine to produce spermidine (SPD) and 5‘-methylthioadenosine (MTA). SPD obtains an additional propylamino group from dSAM, forming spermine (SPM) and MTA. The latter is used to regenerate methionine by the salvation pathway, whose first step is catalyzed by 5-methylthioadenosine nucleosidase (10) (Figure 1).
PA biosynthesis is essential for the growth of normal and cancer cells. Low SAM and MTA levels characterize the development of preneoplastic and neoplastic liver. MTA accumulation, following the administration of exogenous SAM, is probably partially responsible for the inhibition of PA and DNA synthesis, the decrease of cell growth, as well as the inhibition of liver cancer promotion (13). However, several other mechanisms (14,15) may account for the inhibitory effect of SAM on the regenerative and neoplastic liver growth (see further).
Deregulation of methionine metabolism in hepatocellular carcinoma (HCC)
The downregulation of MAT1A gene characterizes alcoholic hepatitis, cirrhosis and HCC (16,17). This largely depends, at the transcriptional level, on CpG methylation of MAT1A promoter and histone H4 deacetylation, and, at a post-transcriptional level, on MAT1A mRNA interaction with AUF1 protein that enhances its decay (18-20). In contrast, MAT2A gene is upregulated in HCC due to the hypomethylation of its promoter and histone H4 acetylation, and the interaction of MAT2A mRNA with HuR protein, which increases its stability (18-20). This situation (MATα1:MATα2 switch) is responsible for the decrease in SAM/SAH ratio in cirrhosis and HCC. Various trans-activating factors such as Sp1, c-Myb (avian myeloblastosis viral oncogene homolog), nuclear factor kappa B (NF-kB), and AP-1 are involved in MAT2A transcriptional upregulation in HCC (21).
MATα2 has been found to regulate expression of BCL-2 at different levels in human colon cancer cell line RKO and in liver cancer cell line HepG2 (22). In both cell lines MATα2 activates BCL-2 gene transcription by binding to its promoter. It also directly interacts with BCL-2 protein enhancing its stability. These MATα2 effects involve the ubiquitin-conjugating enzyme 9 required for the sumoylation of MATα2 at K340, K372 and K394, necessary for MATα2 stability.
Mat1a-KO mice exhibit lower expression of the mitochondrial chaperon PHB1 (23). In HCC and CCA (cholangiocarcinoma) cell lines, PHB1 positively regulates MAT1A (24). Both MATα1 and PHB1 (prohibitin 1) form heterodimers with MAX to repress the E-box driven promoter activity (24). This results in the negative regulation of the transcription factors c-MYC, MAFG and c-MAF, and of their oncogenic activity (24).
Interestingly, miRNAs deregulation is implicated in the decrease in MAT1A expression in HCC (25). The individual knockdown of miR-664, miR-485-3p, and miR-495 provokes MAT1A expression in Hep3B and HepG2 liver tumor cells, whereas stable overexpression of miRNAs-664/485-3p/495 decreases Hep3B cell tumorigenesis in nude mice. The opposite occurs by miRNAs-664/485-3p/495 knockdown (25). These findings clearly indicate that the upregulation of these miRNAs may contribute to hepatocarcinogenesis by inhibiting MAT1A expression.
The mechanisms regulating MAT2B expression are not well known. MAT2B promoter is activated by Sp1 (26). The upregulation of two MAT2B dominant splicing variants, V1 and V2, is present in HCC. MAT2B V1 promoter expression is stimulated by TNFα (tumor necrosis factor α) and leptin, and inhibited by SAM through mechanisms involving ERK and AKT signaling (21). MATβ2 protein regulates many other proteins by physical interaction (27-29). Among these proteins, GIT1 is activated by MATβ2 (30). The latter also activates the MEK1/2/ERK1/2 signaling pathway, thus promoting liver and colon cancer cells proliferation (30).
SAM levels and SAM/SAH ratio regulate numerous important liver functions, including proliferation, regeneration, differentiation and sensitivity to liver injury. The SAM/SAH ratio controls the in vivo methylation reactions; its decrease lessens the methylation capacity (19). SAH-hydrolase deficiency is indeed responsible of a rare genetic disease characterized by SAM and methionine plasma accumulation and inhibition of transmethylation reactions and, consequently, by the reduction of SAM/SAH ratio (31).
MATα1:MATα2 switch and increase in SAM decarboxylation for PA synthesis concur to the severe SAM decrease that characterize liver injury and HCC (32).The strong involvement of MATα1:MATα2 switch and decrease in SAM content in liver carcinogenesis was confirmed by the observation that the Mat1a-KO mice, characterized by chronic SAM deficiency not compensated by Mat2a induction, undergo hepatomegaly, at 3 months of age, followed by steatosis of 25–50% of hepatocytes, at 8 months, and infiltration of mononuclear cells in periportal areas and HCC, at 18 months (33).
Mechanism of the SAM antitumor effect
SAM, a naturally occurring nontoxic and non-mutagenic compound that is produced by liver cells (34,35). Different observations show a decrease in SAM liver content during acute and chronic ethanol intoxication (36,37). Exogenous SAM load, during ethanol injury, reconstitutes the hepatocytes SAM content, and prevents fatty liver accumulation and ethanol-induced glutathione decrease (36). SAM administration to hepatocytes isolated from fatty liver of choline-deficient rats stimulates phosphatidylcholine synthesis through the transmethylation pathway, thus restoring lipoproteins secretion (38). SAM has been shown to favor the assembly of very low-density lipoproteins (39). SAM administration also counteracts the toxic effect of acetaldehyde and/or peroxides produced during ethanol intoxication, contributing to maintain a high GSH liver pool, and prevents the inhibition of (Na+,K+)ATPase activity induced by ethanol intoxication (32). Therefore, the maintenance of the SAM physiological levels may function as therapeutic tool in patients with nonalcoholic steatohepatitis and alcoholic liver cirrhosis (40).
Hepatocarcinogenesis induced by different carcinogens and experimental models, in rats fed adequate diet, is characterized by a fall in liver SAM content and SAM/SAH ratio (13,41,42), that persists in dysplastic nodules (DN) and HCC several weeks after cessation of carcinogen administration (41-44). SAM decrease and no change in SAH occur in human HCC and, at a lower extent, in the surrounding cirrhotic liver (45). The administration of exogenous SAM during carcinogen-induced rat liver carcinogenesis prevents the development of preneoplastic and neoplastic lesions (40-44). Interestingly, SAM intravenous infusion inhibits orthotropic HCC development induced by injection of the H4IIE human HCC cells in rat liver parenchyma (46). However, SAM infusion for 24 days does not affect the size of already established tumors, probably because of the prevention of SAM accumulation by the compensatory induction of hepatic GNMT (46). A SAM and MTA anti-proliferative effect has also been described for colon carcinogenesis, where both compounds reduce chronic inflammation, a main risk factor for this type of cancer (47). MAT2A upregulation occurs in human colon cancer. Its silencing in in vitro growing colon cancer cells induces apoptosis (47).
SAM treatment of rats with preneoplastic and neoplastic liver lesions induces decrease in labeling index and apoptosis of preneoplastic cells (41-44). Also, transfection of MAT1A or culture in the presence of SAM inhibits the proliferation of human HCC cell lines (48). Accordingly, HuH7 cell transfectants, stably overexpressing MAT1A, exhibit higher SAM levels and apoptosis, and lower growth rate, microvessel density, CD31 and Ki-67 staining, than control tumor cells (49).
PA synthesis
Hepatocarcinogenesis is associated with a sharp increase in ornithine decarboxylase (ODC) activity and PA synthesis (50,51). Early studies on HCC chemoprevention by SAM have shown a great decrease of PA synthesis, associated with the inhibition of ODC, in preneoplastic liver lesions developing in rats treated with exogenous SAM (50,51) (Figure 2). ODC inhibition could be attributed to the accumulation of MTA, end-product of PA synthesis that could also arise from the spontaneous splitting of SAM at physiologic temperature and pH (52). However, only moderate accumulation of MTA occurs during SAM treatment, probably because of the activation of the “salvage pathway”, which utilizes MTA for methionine synthesis (41) (Figure 1). Moreover, SAM is a stronger inhibitor of DNA synthesis and rat hepatocarcinogenesis than MTA (41).
Figure 2.

Effects of SAM treatment during hepatocarcinogenesis. SAM is involved in DNA methylation and stabilization of the DNA repair enzyme APEX1. SAM antioxidant activity reduces genomic instability. The inhibition by SAM of LKB1/AMPK axis increases cytoplasmic concentration of HuR, which stabilizes p53 and USP7 mRNAs. Through the control of the LKB1/AMPK axis, SAM impedes the production of IL6 and cytokines and the activation of iNOS and eNOS, thus limiting the oxidative damage. SAM also controls cell growth and survival by inducing PPA2 expression that phosphorylates and inactivates AKT and its targets. Moreover, PPA2 activation and DUSP1 stabilization inhibit RAS/ERK pathway. Finally, SAM affects cell cycle by inhibiting c-MYC expression and polyamine synthesis. SAM, S-adenosylmethionine. Adapted with permission from Frau et al., 2013.
SAM antioxidative action
The observation that SAM treatment of CCl4-intoxicated rats preserves a high GSH pool (53) suggests the possibility that this SAM antioxidative effect is involved in HCC chemoprevention. Indeed, the protection of DNA from oxidative damage by antioxidants was known to prevent tumor development in different tissues, including liver (54-56). An antioxidative effect, attributed to MTA (57), could be exerted by sulfoxide and sulfone derivatives of MTA oxidation by microsomal monooxygenases (58). However, SAM exerts an antitumor action independent of MTA (41). Indeed, higher SAM levels and no change in MTA content occurs in stable MAT1A transfectants of in vitro growing liver tumor HuH7 cells, which are less tumorigenic in vivo than untransfected Huh7 cells (49).
DNA and protein methylation
A further implication of a SAM chemopreventive effect is the observation that the deficit of SAM, during hepatocarcinogenesis, is associated with global DNA hypomethylation (41) and consequent genomic instability (59). The presence of AP (apurinic/apyrimidinic) sites represents the most frequent DNA lesion in cancer cells (60). SAM, but not MTA, reverses global DNA hypomethylation (41) (Figure 2). Indeed, the restraint of preneoplastic foci development in rat liver, induced by SAM, accompanies the complete recovery of DNA hypomethylation (19), and is prevented by the hypomethylating compound 5-azacytidine (61).
Alterations of MATs expression in HCC also interfere with protein methylation. In most rat tissues are present, at the C-terminal end of the protein implicated in cytoplasmic retention and nuclear localization of MATI/III, two partially overlapping areas (62). The nuclear accumulation of the active enzyme was implicated in histone H3K27 tri-methylation, an epigenetic modification associated with DNA methylation, therefore indicating the need of active MATI/III to ensure the SAM supply necessary for the methylation reactions. Interestingly, MATα2 may also interact with chromatin-related proteins, involved in histone modification, chromatin remodeling, transcription regulation, and nucleo-cytoplasmic transport to deliver SAM locally on chromatin (63,64). This requires MATβ2 (therefore MATII isozyme that contains both MATα2 and MATβ2). This mechanism may also regulate MAFK. The latter is a member of MAF oncoproteins that interacts with both MATα2 and MATβ2 (63,64). MAFK forms diverse heterodimers to bind MAF recognition elements of DNA, thus operating as a transcription activator/repressor (64). However, the oncogenic role of MAFK and its targets in HCC are not known.
SAM and signal transduction
Pioneering observations on the impact of SAM on signal transduction showed that the treatment of rats with SAM, during the development of preneoplastic liver nodules, inhibits the expression of c-myc, H-ras and K-ras and PA synthesis (43) (Figure 2). Further studies have shown that various signaling pathways are involved in SAM antitumor effect. The treatment with SAM of rats, during the development of preneoplastic foci, prevents NF-kB activation (65) and induces the overexpression of the onco-suppressor PP2A (protein phosphatase 2) gene, which dephosphorylates and inactivates AMPK, pAKT, and pERK (66,67) (Figure 2). Accordingly, rat and human HCCs with highest pAKT and pERK expression and proliferation rates exhibit low SAM content and PP2A expression (68).
SAM may also control the MAPK (V-MAF avian musculoaponeurotic fibrosarcoma oncogene family, protein K) pathway. It has been in fact observed (69) that SAM may induce a decrease in ERK1/2 activity by interfering with DUSP1, a specific ERK inhibitor (Figure 2). SAM treatment increases DUSP1 expression through multiple mechanisms, including increased transcription and stability of its mRNA and protein, and inhibition of proteasomal chymotrypsin-like and caspase-like activities (69). ERK1/2 upregulation is associated with low DUSP1 expression in fast growing DN and HCC induced in F344 rats, genetically susceptible to hepatocarcinogenesis, and human HCC with poorer prognosis (based on patient’s survival length) (70,71). This can partly depend on DUSP1 Ser296 phosphorylation by ERK1/2, followed by DUSP1 ubiquitination, by SKP2-CKS1 ubiquitin ligase, and proteasomal degradation (70,71) (Figure 3). Notably, DUSP1 mRNA and protein levels are sharply decreased in the livers of Mat1a-KO mice as well as in cultured mouse and human hepatocytes (69). SAM administration to Mat1a-KO mice induces an increase in Dusp1 mRNA and protein levels, and a decrease in Erk activity. Further, SAM prevents DUSP1 mRNA and protein fall in cultured mouse and human hepatocytes probably by inhibiting its proteasomal degradation (69).
Figure 3.
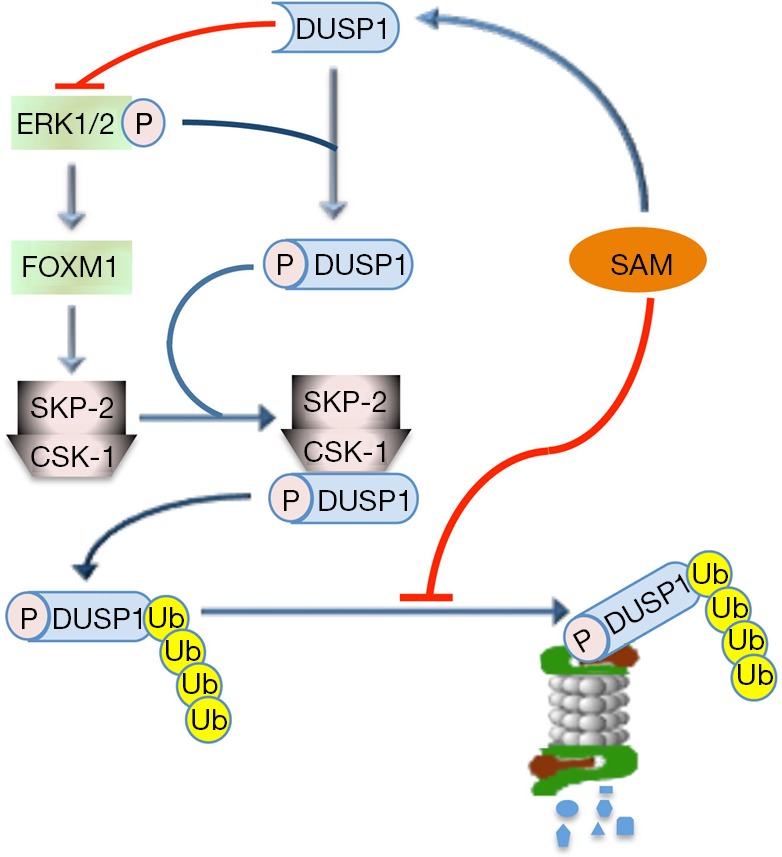
Interference of SAM with ERK1/2 inhibition by DUSP1. ERK1/2 inhibition by DUSP1 is controlled by DUSP1 phosphorylation at the Ser296 residue, followed by its ubiquitination by the SKP2–CKS1 ubiquitin ligase and proteasomal degradation. A control is also operated by FOXM1, an ERK1/2 target, that activates SKP2-CKS1. SAM enhances DUSP1 inhibitory effect by increasing DUSP1 mRNA transcription, and contributing to the increase in DUSP1 protein at post-translational levels, probably through inhibition of its proteasomal degradation. SAM, S-adenosylmethionine. Adapted with permission from Frau et al., 2013.
A suppressive effect of SAM on malignant transformation through ERK1/2 inhibition is also suggested by the finding that the TNF-α/HIF-1α (HIF-1α, hypoxia-inducible factor 1, alpha subunity) axis sustains the expression of FOXM1 (72), which mediates the ERK1/2 effects on cell cycle, cell survival, and angiogenesis (73). It was indeed found that hypoxia reduces SAM levels of HCC cells by promoting HIF-1α binding to MAT2A promoter (74).
Activation of the RAS/ERK pathway, produced by growth factors in different cell lines including HCC cells, may be limited by the arginine methylation of RAF protein by PRMT5 (protein arginine methyltransferase 5) (5). The amplitude and length of ERK activation by growth factors is increased by the expression of RAF mutants that cannot be methylated (75). However, PRMT5 activates cell cycle progression through the G1 phase and PI3K/AKT, while it suppresses JNK/c-Jun signaling in lung cancer (76). PRMT5 localization may explain these apparent discrepancies. PRMT5 and p44/MET50/WD45/WDR77 cytoplasmic localization supports prostate cancer cell growth (77). In contrast, nuclear PRMT5 localization in normal prostate epithelium, inhibits cell growth in a methyltransferase activity-independent manner (77).
SAM could also protect JAK/STAT signaling in HCV-induced liver damage. HCV protein weakens JAK-STAT signaling by the inhibition of STAT1 methylation, which favors STAT1 binding by its inhibitor PIAS1 (78). The restoration of STAT1 methylation by SAM and betaine recover IFNα antiviral effect in the cell culture (78).
The role of the DNA repair protein, APEX
This protein is involved in base excision repair and, as a redox co-activator of transcription factors, contributes to the regulation of EGR-1, p53, and AP-1 (79). The stimulation of APEX1 (apurinic endonuclease) gene transcription by ROS, contributes to the defense against genomic instability (80). The livers of 1-month old Mat1a-KO mice exhibit higher genomic instability than the livers of wild type mice, whereas Apex1 mRNA and protein levels undergo 20% and 50% decreases, respectively. Significant increase in AP sites and under-expression of the APEX1 targets Bax, Fas, and p21 accompany these changes (81). Decrease in MAT1A mRNA, associated with increase in APEX1 and c-MYC mRNAs occurs in cultured human and mouse hepatocytes, in which, however, APEX1 protein level decreases by 60% (81). SAM inhibits APEX1 transcription, but stabilizes APEX1 protein thus preventing APEX1 protein level decrease in cultured hepatocytes (81) (Figure 2). These interesting findings indicate that APEX1 stabilization by SAM contributes to SAM chemopreventive effect and may in part explain why chronic SAM deficiency predisposes to HCC.
The mechanism of APEX1 stabilization by SAM is not known. Recent reports suggest that ubiquitin-9 is involved in APEX1 protein degradation in HeLa cells (82). SAM inhibits chymotrypsin-like and caspase-like activities of 26S proteasome and causes degradation of some proteasomal subunits (83). Furthermore, SAM and MTA induce a decrease of CDC2 (cell division cycle 2) expression, which is upregulated in several cancers, resulting in reduced ubiquitin-9 phosphorylation and expression (83).
Nitric oxide (NO)
NO is the product of L-arginine conversion to L-citrulline catalyzed by the calcium-independent, inducible iNOS of hepatocytes, Kupffer and stellate cells, and cholangiocytes, and the calcium-dependent eNOS of endothelial cells (84). NO provokes DNA mutations in hepatocytes and vasodilatation, thus providing transformed cells with adequate amounts of metabolites and oxygen. During early stages of hepatocarcinogenesis, inflammatory cytokines and growth factors activate iNOS (84), thus inducing an overproduction of reactive nitrogen species that may damage DNA. iNOS inhibition by aminoguanidine causes a decrease in NF-kB and RAS/ERK expression and HCC cell growth and apoptosis (85). During hepatocarcinogenesis, AMPK activates eNOS thus causing additional NO production, which may further activate AMPK (86), and inactivates MATI/III (87) (Figure 2).
The role of the LKB1/AMPK axis in hepatocarcinogenesis is supported by the observation that the LKB1/AMPK activation is necessary for the survival of SAM-deficient cells isolated from HCC of Mat1a-KO mice (88). LKB1 may also regulate AKT-mediated cell survival independently of PI3K, AMPK, and mTORC2 (mechanistic target of rapamycin complex 2) (88). In SAM-deficient cells, such as neoplastic hepatocytes, LKB1 controls apoptosis by provoking the cytoplasmic localization of p53. The de-ubiquitinylating enzyme USP7 (HAUSP) has an important role. USP7 contributes to the stability of mouse double minute (MDM), a negative p53 regulator, impairing its ubiquitination and degradation (89). LKB1 contributes to the phosphorylation of cytosolic p53 (90). p53 hyperphosphorylation, and its cytoplasmic retention, blocks the negative regulation of p53 by MDM2. Furthermore, LKB1 induces the cytosolic translocation of HuR, an RNA-binding protein that increases the half-life of target mRNA, such as cyclin A2, and cell proliferation; SAM blocks this process (90) (Figure 2). In complex, present knowledge indicates that LKB1 controls the apoptotic response through the phosphorylation and cytoplasmic retention of p53, the regulation of the de-ubiquitination enzyme USP7, and the nucleo-cytoplasmic shuttling of HuR. Furthermore, AMPK upregulation results in activation of PFK-2, a key enzyme for glycolysis (91), which contributes to the glycolytic metabolism of cancer cells (Figure 2).
Notably, cytoplasmic localization of p53 and p-LKB1 (Ser428) has been documented in the NASH-HCC present in Mat1a-KO mice and in human HCC derived from both ASH and NASH (88). However, these results contrast with the observation of LKB1 loss in cancer cells, including HCC (89). LKB1 is considered a suppressor gene and LKB1-activated AMPK inhibits the AKT pathway by triggering the tumor suppressor complex TSC2/TSC1 (92). Furthermore, the deregulation of the AMPKa2 catalytic subunit is associated with poor HCC differentiation and patients’ prognosis (93). The inactivation of AMPK fosters hepatocarcinogenesis through the destabilization of p53 in a p53 deacetylase (SIRTUIN-1)-dependent way (93). These contradictory findings remain unexplained.
An important aspect of the SAM antitumor effect deals with its effect on the PI3K/AKT axis and the LKB1/AMPK/PFK-2 axis (Figure 2). Previous work (94) suggested that during the development of preneoplastic foci the glucose used for the synthesis of triacylglycerol and pyruvate synthesis decreases in rat liver, whereas there occurs a rise of the production of reducing equivalents and pentose phosphates that favors DNA synthesis and detoxification reactions. The reduction of DNA synthesis, in SAM-treated rats, is accompanied by the partial reversion of carbohydrate metabolism to that present in normal liver (94). The SAM effect could impact on the metabolism of neoplastic cells, characterized by active glycolysis even in aerobiosis [Warburg effect (95)]. The respiratory activity of neoplastic liver cells, comparable to that of normal cells in absence of glucose, is highly restrained after glucose addition (96-98). This mainly depends on a decreased availability of intracellular ADP, largely used for synthesis of glycolytic ATP, that limits oxygen consumption (97). Warburg hypothesized that glycolytic metabolism was somehow involved in carcinogenesis (95). In accordance with this hypothesis, the inhibition of aerobic glycolysis in neoplastic cells by 2-DG, a competitive inhibitor acting at the level of hexokinase, causes strong inhibition of protein synthesis in AH-130 rat hepatocarcinoma, characterized by high glycolytic activity, but not in normal cells in which ATP is mainly produced during mitochondrial oxygen consumption (98). These observations suggested that neoplastic cells, unlike normal ones, use glycolytic energy, in aerobiosis, for protein synthesis. In agreement with these observations, recent findings implicate glycolysis in signal transduction in cancer cells. It was demonstrated (99) that glycolysis inhibitors, including 2-DG, strongly inhibit the YAP/TAZ signaling, which is active in cells that incorporate glucose and produce lactic acid, such as mammary and liver tumors. Mechanistically it was found that PFK-1 (phosphofructokinase 1), which regulates the first step of glycolysis, binds the YAP/TAZ transcriptional cofactors TEADs and promotes their cooperation with YAP/TAZ (99). We have recently shown the implication of YAP/TAZ in the acquisition of stemness properties by HCC cells (100). Furthermore, CHIP (carboxyl terminus of Hsc70-interacting protein), a U-box E3 ligase, suppresses ovarian carcinomas progression by inhibiting aerobic glycolysis. PFK-2, was identified as a target of CHIP-mediated degradation indicating that Warburg effect is regulated by CHIP through the degradation of PFK-2 during tumor progression (101).
Oncogenes are largely involved in the glycolytic metabolism of cancer cells. MYC and AKT activate hexokinase II, MYC and HIF-1α activate glucose transport, pyruvate kinase and lactate dehydrogenase; pyruvate kinase is also activated by RAS, and AKT activates glucose transport (102-104). Moreover, HSF-1α and MYC trigger pyruvate dehydrogenase kinase that, by activating pyruvate dehydrogenase, impedes the synthesis of acetyl-CoA (102), thus contributing to maintain low the respiratory activity of cancer cells in the presence of glucose (96-98). The activation of glucose-6-phosphate dehydrogenase, by HSF-1α, provides pentose phosphates for nucleic acids synthesis (105,106). Interestingly many of these genes are upregulated in HCC and are sensitive to the SAM inhibitory effect (reviewed in 107,108).
Alterations of methionine metabolism as determinants of the prognosis of HCC in humans and rodents
The progressive development of altered hepatocytes foci (FAH), DN, and HCC occurs during human and rodents hepatocarcinogenesis (109). In the hepatocarcinogenesis induced in genetically susceptible F344 rats by diethylnitrosamine/2-acetylaminofluorene/partial hepatectomy treatments, according to the “resistant hepatocyte” protocol (109), the hepatocyte initiation is followed by the selective proliferation of initiated hepatocytes (promotion), leading to the development of numerous FAH, that in part progress to DN and HCCs. These treatments induce lower incidence of slow-proliferating DN and HCCs, in genetically resistant BN rats, than in susceptible F344 rats (110). Accordingly, the up-regulation of cell cycle, iNos/IKK/NF-kB axis, Ras/Erk signaling, and Mybl2, that characterize DN and HCC in F344 rats, is much lower or absent in BN rats (70,111,112).
Two different types of human HCC have been identified: one of which with better prognosis (based on survival length; HCCB), lower activation of cell cycle and signaling pathways and low genomic instability, whereas the second type exhibits poorer prognosis and extensive chromosomal instability (HCCP) (110-113). Interestingly, alterations of cell cycle and signaling pathways analogous to those of the HCCP are present in HCCs of the genetically susceptible F344 rats, whereas in the HCCs of the genetically resistant BN rats lower alterations similar to those of HCCB occur (110-113).
Gene expression profiles, performed by microarray analysis and confirmed by quantitative RT-PCR and immuno-precipitation analyses (68), revealed two different gene expression patterns: the first one comprised normal liver of F344 and BN rats and DN of BN rats, and the second one included the DN of F344 rats and HCC of both strains. A signature that predicted DN and HCC progression, was typified by highest expression of the onco-suppressors Csmd1, Dmbt1, Dusp1, and Gnmt, in DN, and Bhmt, Dmbt1, Dusp1, Gadd45g, Gnmt, Napsa, Pp2ca, and Ptpn13 in HCCs of resistant rats. Integrated gene expression results disclosed highest expression of proliferation-related CTGF, c-MYC, and PCNA, and lowest expression of BHMT, DMBT1, DUSP1, GADD45g, and GNMT, in more aggressive rat and human HCC. BHMT, DUSP1, and GADD45g expression were predictive of patients’ survival (68). These findings indicate the existence of an evolutionarily conserved gene expression signature that distinguishes HCC with different tendency to progress in rat and human. Interestingly, we found that some genes involved in the methionine cycle, such as BHMT and GNMT may contribute to the determination of HCC prognosis.
Recent results in our laboratory (20) showed that under-expression of the Mat1a gene, over-expression of Mat2b (MATα1:MATα2 switch), and low SAM levels, occurred in fast-growing HCC of F344 rats. This was associated with CpG hypermethylation and histone H4 deacetylation of Mat1A promoter, and CpG hypomethylation and histone H4 acetylation of Mat2A promoter. In low-growing HCC of BN rats, the MATα1:MATα2 ratio, CpG methylation, and histone H4 acetylation underwent low changes with respect to normal liver. A comparison between human HCCs with different prognosis showed higher MAT1A promoter methylation and lower MAT2A promoter methylation in HCCP than in HCCB. Furthermore, there occurred sharp increases of AUF1 protein, destabilizing MAT1A mRNA, and HuR protein, stabilizing MAT2A mRNA, and of Mat1α-AUF1 and Mat2α-HuR ribonucleoproteins complexes in F344 and human HCC, while these parameters underwent low/no increase in BN HCC. In human HCC, MAT1A:MAT2A expression and MATI/III:MATII activity ratios were correlated negatively with cell proliferation and genomic instability, and positively with apoptosis and DNA methylation. The MATI/III:MATII ratio predicted the length of patient survival. Forced MAT1A overexpression in HepG2 and HuH7 liver cancer cell lines induced rise in SAM level, decrease in cell proliferation, increase in apoptosis, under-expression of the cyclin D1, E2F1, IKK, NF-kB genes, and of the antiapoptotic BCL2 and XIAP genes, while and increase inexpression of the BAX and BAK proapoptotic genes occurred.
These results showed a post-transcriptional regulation of MAT1A and MAT2A by AUF1 and HuR in HCC. We also demonstrated that a low MATI/III:MATII ratio is a prognostic marker contributing to determine a phenotype susceptible to HCC and poor patients’ survival. Furthermore, it was shown that an interference of SAM with IKK/NF-kB signaling contributes to its anti-proliferative and pro-apoptotic effect in HCC.
Another experimental system, used to predict the molecular alterations present in HCCB and HCCP, is represented by the c-Myc and c-Myc/Tgf-α transgenic mice (113-116). Intriguingly, these mouse models repeat the main pathogenetic mechanisms of human HCC: c-Myc tumors, like human HCC, exhibit activated β-catenin and better prognosis, whereas c-Myc/Tgf-α tumors are like to HCC with shorter survival. In this experimental system, we evaluated the correlation between the genomic instability and DNA methylation, and the influence of methionine metabolism deregulation on these parameters and hepatocarcinogenesis (45). SAM/SAH ratio and liver-specific MatI/III progressively decreased in dysplastic and neoplastic lesions of liver of c-Myc transgenic mice and of human HCCB and HCCP. This was associated with a rise of global DNA hypomethylation in c-Myc mice and human liver lesions, and was positively correlated with genomic instability both in mice and humans, and inversely correlated with patients’ survival extent. No changes in MatI/III and DNA methylation were found in the lesions of c-Myc/Tgf-α mice and in a small human HCC subgroup with intermediate prognosis, in which the proliferative activity, similar to that of c-Myc HCC and HCCB, was associated with low apoptosis. c-Myc/Tgf-α HCCs and HCCP were characterized by high overexpression of genes implicated in PA synthesis, methionine salvage pathway and under-expression of the PA negative regulator OAZ1. These findings indicate that the alterations in the activity of MAT/I/III, and the extent of DNA hypomethylation and genomic instability are prognostic markers for human HCC. Nevertheless, a small human HCC subgroup, similar to c-Myc/Tgf-α tumors, develops in the absence of alterations in DNA methylation.
Above findings, taken together, indicate that changes in methionine and SAM metabolism strongly contribute to HCC pathogenesis and outcome. These alterations seem to be required for the development of the majority, although probably not all, human HCCs. Furthermore, these observations may have some importance for the prevention and therapy of preneoplastic liver lesions and the chemoprevention of liver tumors by SAM.
Conclusions
Following the pioneering observations on the interference of SAM with alcoholic hepatitis and experimental hepatocarcinogenesis (15,37,43), increasing evidence has shown that alterations of methionine cycle largely contribute to the development and progression of liver cancer. A large deal of research from different laboratories has demonstrated the prognostic role of these alterations and the chemopreventive effect of SAM. The chemoprevention of hepatocarcinogenesis by SAM is the result of numerous pleiotropic actions of the latter on signal transduction pathways. It was shown that SAM interferes at different levels with signal transduction mechanisms and is largely involved in the pathogenesis of liver preneoplastic and neoplastic lesions. Importantly, BHMT and GNMT genes, involved in the methionine cycle, are part of an evolutionarily conserved gene expression profile that distinguishes HCCs with different tendency to progress in the rat and human (68). The observation that MAT1A:MAT2A and MATI/III:MATII ratios correlate negatively, in human HCC, with cell proliferation and genomic instability, and positively with apoptosis and global DNA methylation suggests that MATs deregulation and consequent SAM decrease represent possible therapeutic targets for HCC.
Acknowledgements
None.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Cavuoto P, Fenech MF. A review of methionine dependency and the role of methionine restriction in cancer growth control and life-span extension. Cancer Treat Rev 2012;38:726-36. 10.1016/j.ctrv.2012.01.004 [DOI] [PubMed] [Google Scholar]
- 2.González B, Pajares MA, Hermoso JA, et al. The crystal structure of tetrameric methionine adenosyltransferase from rat liver reveals the methionine-binding site. J Mol Biol 2000;300:363-75. 10.1006/jmbi.2000.3858 [DOI] [PubMed] [Google Scholar]
- 3.Mato JM, Lu SC. Role of S-adenosyl-l-methionine in liver health and injury. Hepatology 2007;45:1306-12. 10.1002/hep.21650 [DOI] [PubMed] [Google Scholar]
- 4.Mato JM, Lu SC. Role of S-adenosylmethionine. In: Coates PM, Blackman MR, Levine M, et al. editors. Encyclopedia of Dietary Supplements 2nd ed. Dekker, New York, 2010:1-6. [Google Scholar]
- 5.Lu SC. Glutathione synthesis. Biochim Biophys Acta 2013;1830:3143-53. 10.1016/j.bbagen.2012.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mosharov E, Cranford MR, Banerjee R. The quantitatively important relationship between homocysteine metabolism and glutathione synthases by the transsulfuration pathway and its regulation by redox changes. Biochemistry 2000; 39:13005-11. 10.1021/bi001088w [DOI] [PubMed] [Google Scholar]
- 7.Huidobro C, Torano EG, Fernandez AF, et al. A DNA methylation signature associated with the epigenetic repression of N-glycinetransferase in human hepatocellular carcinoma. J Mol Med 2013;91:939-50. 10.1007/s00109-013-1010-8 [DOI] [PubMed] [Google Scholar]
- 8.Refsum H, Ueland PM, Nygard MD, et al. Homocysteine and cardiovascular disease. Annu Rev Med 1998;49:31-62. 10.1146/annurev.med.49.1.31 [DOI] [PubMed] [Google Scholar]
- 9.Kenyon SH, Waterfield CJ, Timbrell JA, et al. Methionine synthase activity and sulfur amino acid levels in rat liver tumour cells HTC and Phi-1. Biochem Pharmacol 2002;63:381-91. 10.1016/S0006-2952(01)00874-7 [DOI] [PubMed] [Google Scholar]
- 10.Mato JM, Martinez-Chantar ML, Lu SC. S-adenosylmethionine metabolism and liver disease. Ann Hepatol 2013;12:183-9. [PMC free article] [PubMed] [Google Scholar]
- 11.Ishii I, Akahoshi N, Yamada H, et al. Cystathionine gamma-lyase-deficient mice require dietary cysteine to protect against acute lethal myopathy and oxidative injury. J Biol Chem 2010;285:26358-68. 10.1074/jbc.M110.147439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo SB, Duan ZJ, Wang QM, et al. Endogenous carbon monoxide downregulates hepatic cystathionine-γ-lyase in rats with liver cirrhosis. Exp Ther Med 2015;10:2039-46. 10.3892/etm.2015.2823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garcea R, Pascale RM, Daino L, et al. Variations of ornithine decarbosylase activity and S-asenosyl-L-methionine and 5’-methylthioadenosine contents during the development of diethylnitrosamine-induced liver hyperplastic nodules and hepatocellular carcinoma. Carcinogenesis 1987;8:653-8. 10.1093/carcin/8.5.653 [DOI] [PubMed] [Google Scholar]
- 14.Lu SC, Mato JM. S-adenosylmethionine in cell growth, apoptosis and liver cancer. J Gastroenterol Hepatol 2008;23:S73-7. 10.1111/j.1440-1746.2007.05289.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcea R, Daino L, Pascale RM, et al. Inhibition of promotion and persistent nodule growth by S-adenosyl-L-methionine in rat liver carcinogenesis: role of remodeling and apoptosis. Cancer Res 1989;49:1850-6. [PubMed] [Google Scholar]
- 16.Cai J, Sun W, Hwang JJ, et al. Changes in S-adenosylmethionine synthetase in human liver cancer: molecular characterization and significance. Hepatology 1996;24:1090-7. 10.1002/hep.510240519 [DOI] [PubMed] [Google Scholar]
- 17.Ramani K, Lu SC. Methionine adenosyltransferases in liver health and diseases. Liver Res 2017;1:103-11. 10.1016/j.livres.2017.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vázquez-Chantada M, Fernandez D, Embade N, et al. Hur/methylated-Hur and AUF1 regulate the expression of methionine adenosyltransferase during liver proliferation, differentiation and carcinogenesis. Gastroenterology 2010;138:1943-53. 10.1053/j.gastro.2010.01.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frau M, Feo F, Pascale RM. Pleiotropic effects of methionine adenosyltransferases deregulation as determinant of liver cancer progression and prognosis. J Hepatol 2013;59:830-41. 10.1016/j.jhep.2013.04.031 [DOI] [PubMed] [Google Scholar]
- 20.Frau M, Tomasi ML, Simile MM, et al. Role of transcriptional and posttranscriptional regulation of methionine adenosyltransferases in liver cancer progression. Hepatology 2012;56:165-75. 10.1002/hep.25643 [DOI] [PubMed] [Google Scholar]
- 21.Ramani K, Mato JM, Lu SC. Role of methionine adenosyltransferase genes in hepatocarcinogenesis. Cancers 2011;3:1480-97. 10.3390/cancers3021480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tomasi ML, Ryoo M, Ramani K, et al. Methionine adenosyltransferase α2 sumoylation positively regulate Bcl-2 expression in human colon and liver cancer cells. Oncotarget 2015;6:37706-23. 10.18632/oncotarget.5342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Santamaría E, Avila MA, Latasa MU, et al. Functional proteomics of non-alcoholic steatohepatitis: mitochondrial proteins as targets of S-adenosylmethionine. Proc Nat Acad Sci USA 2003;100:3065-70. 10.1073/pnas.0536625100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fan W, Yang H, Liu T, et al. Prohibitin 1 suppresses liver cancer tumorigenesis in mice and human hepatocellular and cholangiocarcinoma cells. Hepatology 2017;65:1249-66. 10.1002/hep.28964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang H, Cho ME, Li TW, et al. MicroRNAs regulate methionine adenosyltransferase 1A expression in hepatocellular carcinoma. J Clin Invest 2013;123:285-98. 10.1172/JCI63861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.LeGros L, Halim AB, Chamberlin M, et al. Regulation of the human MAT2B gene encoding the regulatory beta subunit of methionine adenosyltransferase, MAT II. J Biol Chem 2001;276:24918-24. 10.1074/jbc.M102816200 [DOI] [PubMed] [Google Scholar]
- 27.Ramani K, Yang HP, Kuhlenkamp J, et al. Changes in methionine adenosyltransferase and S-adenosylmethionine during hepatic stellate cell activation. Hepatology 2010;51:986-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang H, Ara AI, Magilnick N, et al. Expression pattern, regulation and function of methionine adenosyltransferase 2β alternative splicing variants in hepatoma cells. Gastroenterology 2008;134:281-91. 10.1053/j.gastro.2007.10.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murray B, Antonyuk SV, Marina A, et al. Structure and function study of the complex that synthesizes S-adenosylmethionine. IUCrJ 2014;1:240. 10.1107/S2052252514012585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peng H, Li TW, Yang H, et al. Methionine adenosyltransferase 2B-GIT1 complex serves as scaffold to regulate Ras/Raf/mek1/2 activity in human liver and colon cancer cells. Am J Pathol 2015;185:1135-44. 10.1016/j.ajpath.2014.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matthews RP, Lorent K, Manoral-Mobias R, et al. TNFalfpha-dependent hepatic steatosis and liver degeneration caused by mutation of zebrafish S-adenosylhomocysteine hydrolase. Development 2009;136:865-75. 10.1242/dev.027565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pascale R, Daino L, Garcea R, et al. Inhibition by ethanol of rat liver plasma membrane (Na+,K+)ATPase: protective effect of S-adenosyl-L-methionine, L-methionine, and N-acetylcysteine. Toxicol Appl Pharmacol 1989;97:216-29. 10.1016/0041-008X(89)90327-X [DOI] [PubMed] [Google Scholar]
- 33.Lu SC, Alvarez L, Huang ZZ, et al. Methionine adenosyltransferase 1A knockout mice are predisposed to liver injury and exhibit increased expression of genes involved in proliferation. Proc Natl Acad Sci U S A 2001;98:5560-5. 10.1073/pnas.091016398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pezzoli C, Stramentinoli G, Galli-Kienle M, et al. Uptake and metabolism of S-adenosyl-L-methionine by isolated rat hepatocytes. Biochem Biophys Res Commun 1978;85:1031-8. 10.1016/0006-291X(78)90646-0 [DOI] [PubMed] [Google Scholar]
- 35.Giulidori P, Galli-Kienle M, Catto E, et al. Transmethylation, transsulfuration, and aminopropylation reactions of S-adenosyl-L-methionine in vivo. J Biol Chem 1984;259:4205-11. [PubMed] [Google Scholar]
- 36.Feo F, Pascale R, Garcea R, et al. Effect of the variations of S-adenosyl-L-methionine liver content on fat accumulation and ethanol metabolism in ethanol-intoxicated rats. Toxicol Appl Pharmacol 1986;83:331-41. 10.1016/0041-008X(86)90310-8 [DOI] [PubMed] [Google Scholar]
- 37.Pascale RM, Garcea R, Daino L, et al. The role of S-adenosyl-methionine in the regulation of glutathione pool and acetaldehyde production in acute ethanol intoxication. Res Comm Subst Abuse 1984;4:321-4. [Google Scholar]
- 38.Pascale R, Pirisi L, Daino L, et al. Role of phosphatidylethanolamine methylation in the synthesis of phosphatidylcholine by hepatocytes isolated from choline-deficient rats. FEBS Lett 1982;145:293-7. 10.1016/0014-5793(82)80186-5 [DOI] [PubMed] [Google Scholar]
- 39.Martínez-Uña M, Varela R, Mestre D, et al. S-adenosylmethionine increases circulating very-low density lipoprotein clearance in nonalcoholic fatty liver disease. J Hepatol 2015;62:673-81. 10.1016/j.jhep.2014.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anstee QM, Day CP. S-adenosylmethionine (SAMe) therapy in liver disease: a review of current evidence and clinical utility. J Hepatol 2012;57:1097-109. 10.1016/j.jhep.2012.04.041 [DOI] [PubMed] [Google Scholar]
- 41.Pascale RM, Simile MM, Satta G, et al. Comparative effects of l-methionine, S-adenosyl-l-methionine and 5’-methylthioadenosine on the growth of preneoplastic lesions and DNA methylation in rat liver during the early stages of hepatocarcinogenesis. Anticancer Res 1991;11:1617-24. [PubMed] [Google Scholar]
- 42.Simile MM, Saviozzi M, De Miglio MR, et al. Persistent chemopreventive effect of S-adenosyl-L-methionine on the development of liver putative preneoplastic lesions induced by thiobenzamide in diethylnitrosamine-initiated rats. Carcinogenesis 1996;17:1533-7. 10.1093/carcin/17.7.1533 [DOI] [PubMed] [Google Scholar]
- 43.Garcea R, Daino L, Pascale RM, et al. Protooncogene methylation and expression in regenerating liver and preneoplastic liver nodules induced in the rat by diethylnitrosamine: effect of variations of S-adenosylmethionine: S-adenosylhomocysteine ratio. Carcinogenesis 1989;10:1183-92. 10.1093/carcin/10.7.1183 [DOI] [PubMed] [Google Scholar]
- 44.Pascale RM, Simile MM, De Miglio MR, et al. Chemoprevention by S-adenosyl-L-methionine of rat liver carcinogenesis initiated by 1,2-dimethylhydrazine and promoted by orotic acid. Carcinogenesis 1995;16:427-30. 10.1093/carcin/16.2.427 [DOI] [PubMed] [Google Scholar]
- 45.Calvisi DF, Simile MM, Ladu S, et al. Altered methionine metabolism and global DNA methylation in liver cancer: relationship with genomic instability and prognosis. Int J Cancer 2007;121:2410-20. 10.1002/ijc.22940 [DOI] [PubMed] [Google Scholar]
- 46.Lu SC, Ramani K, Ou X, et al. S-adenosylmethionine in the chemoprevention and treatment of hepatocellular carcinoma in a rat model. Hepatology 2009;50:462-71. 10.1002/hep.22990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li TW, Yang H, Peng H, et al. Effects of S-adenosylmethionine and methylthioadenosine on inflammation-induced colon cancer in mice. Carcinogenesis 2012;33:427-35. 10.1093/carcin/bgr295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cai J, Mao Z, Hwang JJ, et al. Differential expression of methionine adenosyltransferase genes influences the rate of growth of human hepatocellular carcinoma cells. Cancer Res 1998;58:1444-50. [PubMed] [Google Scholar]
- 49.Li J, Ramani K, Sun Z, et al. Forced expression of methionine adenosyltransferase 1A in human hepatoma cells suppresses in vivo tumorigenicity in mice. Am J Pathol 2010;176:2456-66. 10.2353/ajpath.2010.090810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Feo F, Garcea R, Daino L, et al. Early stimulation of polyamine biosynthesis during promotion by phenobarbital of diethylnitrosamine-induced rat liver carcinogenesis. The effects of variations of the S-adenosyl-L-methionine cellular pool. Carcinogenesis 1985;6:1713-20. 10.1093/carcin/6.12.1713 [DOI] [PubMed] [Google Scholar]
- 51.Pascale RM, Simile MM, Gaspa L, et al. Alterations of ornithine decarboxylase gene during the progression of rat liver carcinogenesis. Carcinogenesis 1993;14:1077-80. 10.1093/carcin/14.5.1077 [DOI] [PubMed] [Google Scholar]
- 52.Wu SE, Huskey WP, Borchardt RT, et al. Chiral instability at sulfur of S-adenosylmethione. Biochemistry 1983;22:2828-32. 10.1021/bi00281a009 [DOI] [PubMed] [Google Scholar]
- 53.Corrales F, Giménez A, Alvarez L, et al. S-adenosylmethionine treatment prevents carbon tetrachloride-induced S-adenosylmethionine synthetase inactivation and attenuates liver injury. Hepatology 1992;16:1022-7. 10.1002/hep.1840160427 [DOI] [PubMed] [Google Scholar]
- 54.De Flora S, Izzotti A, D’Agostin F, et al. Mechanisms of N-acetylcysteine in the prevention of DNA damage and cancer, with special reference to smoking-related end-points. Carcinogenesis 2001;22:999-1013. 10.1093/carcin/22.7.999 [DOI] [PubMed] [Google Scholar]
- 55.Rafieian-Kopaie M, Nasri H. The possibility of cancer prevention or treatment with antioxidants: the ongoing cancer prevention researches. Int J Prev Med 2015;6:108. 10.4103/2008-7802.169077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shaban S, El-Husseny MW, Abushouk AI, et al. Effects of Antioxidant Supplements on the Survival and Differentiation of Stem Cells. Oxid Med Cell Longev 2017;2017:5032102. 10.1155/2017/5032102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Simile MM, Banni S, Angioni E, et al. 5-Methylthioadenosine administration prevents lipid peroxidation and fibrogenesis induced in rat liver by carbon-tetrachloride intoxication. J Hepatol 2001;34:386-94. 10.1016/S0168-8278(00)00078-7 [DOI] [PubMed] [Google Scholar]
- 58.Brunmark A, Cadenas E. Redox and addition chemistry of quinoid compounds and its biological implications. Free Radic Biol Med 1989;7:435-77. 10.1016/0891-5849(89)90126-3 [DOI] [PubMed] [Google Scholar]
- 59.Kokalj-Vokac N, Almeida A, Viegas-Pequignot E, et al. Specific induction of uncoiling and recombination by azacytidine in classical satellite-containing constitutive heterochromatin. Cytogenet Cell Genet 1993;63:11-5. 10.1159/000133492 [DOI] [PubMed] [Google Scholar]
- 60.Boiteux S, Guillet M. Abasic sites in DNA: repair and biological consequences in Saccharomyces cerevisiae. DNA Repair (Amst) 2004;3:1-12. 10.1016/j.dnarep.2003.10.002 [DOI] [PubMed] [Google Scholar]
- 61.Pascale R, Simile MM, Ruggiu ME, et al. Reversal by 5-azacytidine of the S-adenosyl-L-methionine-induced inhibition of the development of putative preneoplastic foci in rat liver carcinogenesis. Cancer Lett 1991;56:259-65. 10.1016/0304-3835(91)90011-6 [DOI] [PubMed] [Google Scholar]
- 62.Reytor E, Pérez-Miguelsanz J, Alvarez L, et al. Conformational signals in the C-terminal domain of methionine adenosyltransferase I/III determine its nucleocytoplasmic distribution. FASEB J 2009;23:3347-60. 10.1096/fj.09-130187 [DOI] [PubMed] [Google Scholar]
- 63.Katoh Y, Ikura T, Hoshikawa Y, et al. Methionine adenosyltransferase II serves as a transcriptional corepressor of Maf oncoprotein. Mol Cell 2011;41:554-66. 10.1016/j.molcel.2011.02.018 [DOI] [PubMed] [Google Scholar]
- 64.Igarashi K, Katoh Y. Metabolic aspects of epigenome: coupling of S-adenosylmethionine synthesis and gene regulation on chromatin by SAMIT module. Subcell Biochem 2013;61:105-18. 10.1007/978-94-007-4525-4_5 [DOI] [PubMed] [Google Scholar]
- 65.García-Román R, Salazar-González D, Rosas S, et al. The differential NF-kB modulation by S-adenosyl-L-methionine, acetylcysteine and quercetin on the promotion stage of chemical hepatocarcinogenesis. Free Radic Res 2008;42:331-43. 10.1080/10715760802005169 [DOI] [PubMed] [Google Scholar]
- 66.Millward TA, Zolnierowicz S, Hemmings BA. Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem Sci 1999;24:186-91. 10.1016/S0968-0004(99)01375-4 [DOI] [PubMed] [Google Scholar]
- 67.Eichhorn PJ, Creyghton MP, Bernards R. Protein phosphatase 2A regulatory subunits and cancer. Biochim Biophys Acta 2009;1795:1-15. [DOI] [PubMed] [Google Scholar]
- 68.Frau M, Simile MM, Tomasi ML, et al. An expression signature of phenotypic resistance to hepatocellular carcinoma identified by cross-species gene expression analysis. Cell Oncol (Dordr) 2012;35:163-73. 10.1007/s13402-011-0067-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tomasi ML, Ramani K, Lopitz-Otsoa F, et al. S-adenosylmethionine regulates dual-specificity mitogen-activated protein kinase phosphatase expression in mouse and human hepatocytes. Hepatology 2010;51:2152-61. 10.1002/hep.23530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Calvisi DF, Pinna F, Pellegrino R, et al. Ras-driven proliferation and apoptosis signalling during rat liver carcinogenesis is under genetic control. Int J Cancer 2008;123:2057-64. 10.1002/ijc.23720 [DOI] [PubMed] [Google Scholar]
- 71.Calvisi DF, Pinna F, Meloni F, et al. Dual specificity phosphatase 1 ubiquitination in extracellular signal-regulated kinase-mediated control of growth in human hepatocellular carcinoma. Cancer Res 2008;68:4192-200. 10.1158/0008-5472.CAN-07-6157 [DOI] [PubMed] [Google Scholar]
- 72.Calvisi DF, Pinna F, Ladu S, et al. Forkhead box M1B is a determinant of rat susceptibility to hepatocarcinogenesis and sustains ERK activity in human HCC. Gut 2009;58:679-87. 10.1136/gut.2008.152652 [DOI] [PubMed] [Google Scholar]
- 73.Xia L, Mo P, Huang W, et al. The TNF-a/ROS/HIF-1-induced upregulation of FoxMI expression promotes HCC proliferation and resistance to apoptosis. Carcinogenesis 2012;33:2250-9. 10.1093/carcin/bgs249 [DOI] [PubMed] [Google Scholar]
- 74.Liu Q, Liu L, Zhao Y, et al. Hypoxia induces genomic DNA demethylation through the activation of HIF-1a and transcriptional upregulation of MAT2A in hepatoma cells. Mol Cancer Ther 2011;10:1113-23. 10.1158/1535-7163.MCT-10-1010 [DOI] [PubMed] [Google Scholar]
- 75.Andreu-Pérez P, Esteve-Puig R, de Torre-Minguela C, et al. Protein arginine methyltransferase 5 regulates ERK1/2 signal transduction amplitude and cell fate through CRAF. Sci Signal 2011;4:ra58. 10.1126/scisignal.2001936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wei TY, Juan CC, Hisa JY, et al. Protein arginine methyltransferase 5 is a potential oncoprotein that upregulates G1 cyclins/cyclin-dependent kinases and the phosphoinositide 3-kinase/AKT signaling cascade. Cancer Sci 2012;103:1640-50. 10.1111/j.1349-7006.2012.02367.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gu Z, Li Y, Lee P, et al. Protein arginine methyltransferase 5 functions in opposite ways in the cytoplasm and nucleus of prostate cancer cells. PLoS One 2012;7:e44033. 10.1371/journal.pone.0044033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Duong FH, Christen V, Filipowicz M, et al. S-Adenosylmethionine and betaine correct hepatitis C virus induced inhibition of interferon signaling in vitro. Hepatology 2006;43:796-806. 10.1002/hep.21116 [DOI] [PubMed] [Google Scholar]
- 79.Tell G, Damante G, Caldwell D, et al. The intracellular localization of APE1/Ref-1: more than a passive phenomenon? Antioxid Redox Signal 2005;7:367-84. 10.1089/ars.2005.7.367 [DOI] [PubMed] [Google Scholar]
- 80.Izumi T, Brown DB, Naidu CV, et al. Two essential but distinct functions of the mammalian AP endonuclease. Proc Natl Acad Sci U S A 2005;102:5739-43. 10.1073/pnas.0500986102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tomasi ML, Iglesias-Ara A, Yang H, et al. S-adenosylmethionine regulates apurinic/apyrimidinic endonuclease 1 stability: implication in hepatocarcinogenesis. Gastroenterology 2009;136:1025-36. 10.1053/j.gastro.2008.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yan MD, Xu WJ, Lu LR, et al. Ubiquitin conjugating enzyme Ubc9 is involved in protein degradation of redox factor-1 (Ref-1). Sheng Wu Hua Xue Yu Sheng Wu Wu Li Xue Bao (Shanghai) 2000;32:63-8. [PubMed] [Google Scholar]
- 83.Tomasi ML, Tomasi I, Ramani K, et al. S-adenosylmethionine regulates ubiquitin-conjugating enzyme 9 protein expression and sumoylation in murine liver and human cancers. Hepatology 2012;56:982-93. 10.1002/hep.25701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pascale RM, Frau M, Feo F. Prognostic significance of iNOS in hepatocellular carcinoma. In: Bonavida B. editor. Nitric oxide (NO) and cancer. New York: Springer, 2010:309-28. [Google Scholar]
- 85.Calvisi DF, Pinna F, Ladu S, et al. Aberrant iNOS signalling is under genetic control in rodent liver cancer and potentially prognostic for human disease. Carcinogenesis 2008;29:1639-47. 10.1093/carcin/bgn155 [DOI] [PubMed] [Google Scholar]
- 86.Zhang J, Xi Z, Dong Y, et al. Identification of nitric oxide as an endogenous activator of the AMP-activated protein kinase in vascular endothelial cells. J Biol Chem 2008;283:27452-61. 10.1074/jbc.M802578200 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 87.Martínez-Chantar ML, Garcıa-Trevijano ER, Latasa MU, et al. Importance of a deficiency in S-adenosyl-L-methionine synthesis on the pathogenesis of liver injury. Am J Clin Nutr 2002;76:1177S-82S. 10.1093/ajcn/76.5.1177S [DOI] [PubMed] [Google Scholar]
- 88.Martínez-López N, Varela-Rey M, Fernández-Ramos D, et al. Activation of LKB1-Akt pathway independent of phosphoinositide 3-kinase plays a critical role in the proliferation of hepatocellular carcinoma from nonalcoholic steatohepatitis. Hepatology 2010;52:1621-31. 10.1002/hep.23860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer 2011;11:85-95. 10.1038/nrc2981 [DOI] [PubMed] [Google Scholar]
- 90.Martínez-Chantar ML, Vázquez-Chantada M, Garnacho M, et al. S-adenosylmethionine regulates cytoplasmic HuR via AMP-activated kinase. Gastroenterology 2006;131:223-32. 10.1053/j.gastro.2006.04.019 [DOI] [PubMed] [Google Scholar]
- 91.Tavana O, Gu W. Modulation of the p53/MDM2 interplay by HAUSP inhibitors. J Mol Cell Biol 2017;9:45-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hezel AF, Bardeesy N. LKB1; linking cell structure and tumor suppression. Oncogene 2008;27:6908-19. 10.1038/onc.2008.342 [DOI] [PubMed] [Google Scholar]
- 93.Lee CW, Wong LL, Tse EY, et al. AMPK promotes p53 acetylation via phosphorylation and inactivation of SIRT1 in liver cancer cells. Cancer Res 2012;72:4394-404. 10.1158/0008-5472.CAN-12-0429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gerbracht U, Eigenbrodt E, Simile MM, et al. Effect of S-adenosyl-L-methionine on the development of preneoplastic foci and the activity of some carbohydrate metabolizing enzymes in the liver, during experimental hepatocarcinogenesis. Anticancer Res 1993;13:1965-72. [PubMed] [Google Scholar]
- 95.Warburg O. On respiratory impairment in cancer cells. Science 1956;124:269-70. [PubMed] [Google Scholar]
- 96.Crabtree HG. The carbohydrate metabolism of certain pathological overgrowths. Biochem J 1928;22:1289-98. 10.1042/bj0221289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chance B, Hess B. Spectroscopic evidence of metabolic control. Science 1959;129:700-8. 10.1126/science.129.3350.700 [DOI] [PubMed] [Google Scholar]
- 98.Terranova T, Feo F, Gravela E, et al. The effect of 2-desoxyglucose on energy metabolism and protein synthesis of tumor cells and normal cells. Z Krebsforsch 1964;66:41-5. 10.1007/BF00525559 [DOI] [PubMed] [Google Scholar]
- 99.Enzo E, Santinon G, Pocaterra A, et al. Aerobic glycolysis tunes YAP/TAZ transcriptional activity. EMBO J 2015;34:1349-70. 10.15252/embj.201490379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Simile MM, Latte G, Demartis MI, et al. Post-translational deregulation of YAP1 is genetically controlled in rat liver cancer and determines the fate and stem-like behavior of the human disease. Oncotarget 2016;7:49194-216. 10.18632/oncotarget.10246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Shang Y, He J, Wang Y, et al. CHIP/Stub1 regulates the Warburg effect by promoting degradation of PKM2 in ovarian carcinoma. Oncogene 2017;36:4191-200. 10.1038/onc.2017.31 [DOI] [PubMed] [Google Scholar]
- 102.Mikawa T, LLeonart ME, Takaori-Kondo A, et al. Dysregulated glycolysis as an oncogenic event. Cell Mol Life Sci 2015;72:1881-92. 10.1007/s00018-015-1840-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen JQ, Russo J. Dysregulation of glucose transport, glycolysis, TCA cycle and glutaminolysis by oncogenes and tumor suppressors in cancer cells. Biochim Biophys Acta 2012;1826:370-84. [DOI] [PubMed] [Google Scholar]
- 104.Yu L, Chen X, Sun X, et al. The Glycolytic Switch in Tumors: How Many Players Are Involved? J Cancer 2017;8:3430-40. 10.7150/jca.21125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ripoli M, D'Aprile A, Quarato G, et al. Hepatitis C virus-linked mitochondrial dysfunction promotes hypoxia-inducible factor 1 alpha-mediated glycolytic adaptation. J Virol 2010;84:647-60. 10.1128/JVI.00769-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Barrero CA, Datta PK, Sen S, et al. HIV-1 Vpr modulates macrophage metabolic pathways: a SILAC-based quantitative analysis. PLoS One 2013;8:e68376. 10.1371/journal.pone.0068376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Luo J, Li YN, Wang F, et al. S-adenosylmethionine inhibits the growth of cancer cells by reversing the hypomethylation status of c-myc and H-ras in human gastric cancer and colon cancer. Int J Biol Sci 2010;6:784-95. 10.7150/ijbs.6.784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sadek KM, Lebda MA, Nasr NE, et al. Role of lncRNAs as prognostic markers of hepatic cancer and potential therapeutic targeting by S-adenosylmethionine via inhibiting PI3K/Akt signaling pathways. Environ Sci Pollut Res Int 2018. [Epub ahead of print]. 10.1007/s11356-018-2179-8 [DOI] [PubMed] [Google Scholar]
- 109.Solt DB, Medline A, Farber E. Rapid emergence of carcinogen-induced hyperplastic lesions in a new model for the sequential analysis of liver carcinogenesis. Am J Pathol 1977;88:595-618. [PMC free article] [PubMed] [Google Scholar]
- 110.Feo F, De Miglio MR, Simile MM, et al. Hepatocellular carcinoma as a complex polygenic disease. Interpretive analysis of recent developments on genetic predisposition. Biochim Biophys Acta 2006;1765:126-47. [DOI] [PubMed] [Google Scholar]
- 111.Pascale RM, Simile MM, Calvisi DF, et al. Role of HSP90, CDC37, and CRM1 as modulators of P16(INK4A) activity in rat liver carcinogenesis and human liver cancer. Hepatology 2005;42:1310-9. 10.1002/hep.20962 [DOI] [PubMed] [Google Scholar]
- 112.Frau M, Ladu S, Calvisi DF, et al. Mybl2 expression is under genetic control and contributes to determine a hepatocellular carcinoma susceptible phenotype. J Hepatol 2011;55:111-9. 10.1016/j.jhep.2010.10.031 [DOI] [PubMed] [Google Scholar]
- 113.Frau M, Biasi F, Feo F, et al. Prognostic markers and putative therapeutic targets for hepatocellular carcinoma. Mol Aspects Med 2010;31:179-93. 10.1016/j.mam.2010.02.007 [DOI] [PubMed] [Google Scholar]
- 114.Laurent-Puig P, Legoix P, Bluteau O, et al. Genetic alterations associated with hepatocellular carcinomas define distinct pathways of hepatocarcinogenesis. Gastroenterology 2001;120:1763-73. 10.1053/gast.2001.24798 [DOI] [PubMed] [Google Scholar]
- 115.Lee JS, Chu IS, Mikaelyan A, et al. Application of comparative functional genomics to identify best-fit mouse models to study human cancer. Nat Genet 2004;36:1306-11. 10.1038/ng1481 [DOI] [PubMed] [Google Scholar]
- 116.Murakami H, Sanderson ND, Nagy P, et al. Transgenic mouse model for synergistic effects of nuclear oncogenes and growth factors in tumorigenesis: interaction of c-myc and transforming growth factor alpha in hepatic oncogenesis. Cancer Res 1993;53:1719-23. [PubMed] [Google Scholar]