

Abstract
Supranormal contractile properties are frequently associated with cardiac diseases. Anesthetic agents, including propofol, can depress myocardial contraction. We tested the hypothesis that fropofol, a propofol derivative, reduces force development in cardiac muscles via inhibition of cross-bridge cycling and may therefore have therapeutic potential. Force and intracellular Ca2+ concentration ([Ca2+]i) transients of rat trabecular muscles were determined. Myofilament ATPase, actin-activated myosin ATPase, and velocity of actin filaments propelled by myosin were also measured. Fropofol dose dependently decreased force without altering [Ca2+]i in normal and pressure-induced hypertrophied-hypercontractile muscles. Similarly, fropofol depressed maximum Ca2+-activated force (Fmax) and increased the [Ca2+]i required for 50% of Fmax (Ca50) at steady state without affecting the Hill coefficient in both intact and skinned cardiac fibers. The drug also depressed cardiac myofibrillar and actin-activated myosin ATPase activity. In vitro actin sliding velocity was significantly reduced when fropofol was introduced during rigor binding of cross-bridges. The data suggest that the depressing effects of fropofol on cardiac contractility are likely to be related to direct targeting of actomyosin interactions. From a clinical standpoint, these findings are particularly significant, given that fropofol is a nonanesthetic small molecule that decreases myocardial contractility specifically and thus may be useful in the treatment of hypercontractile cardiac disorders.—Ren, X., Schmidt, W., Huang, Y., Lu, H., Liu, W., Bu, W., Eckenhoff, R., Cammarato, A., Gao, W. D. Fropofol decreases force development in cardiac muscle.
Keywords: excitation contraction coupling, myofilament protein, intracellular calcium, fropofol, in vitro motility
Hypercontractility of the heart can lead to significant clinical morbidity and, in severe cases, sudden cardiac death. For example, hypertrophic cardiomyopathy (HCM) is a well-recognized disease characterized by significant thickening and stiffening of the ventricular walls. The enlarged septum can cause left ventricular outflow tract obstruction, whereas concentric hypertrophy and altered compliance can lead to heart failure (1, 2). Recent studies have indicated that, in some cases of HCM, pathologic increases in contractile parameters [i.e., Ca2+ sensitivity, maximum Ca2+-activated force (Fmax), and actomyosin ATPase activity] may be the driving force for impaired energy metabolism and altered Ca2+ handling, which promote secondary effects, such as myocardial hypertrophy, myofilament disarray, interstitial fibrosis, and arrhythmias (3–5). These secondary effects often perpetuate and exacerbate cardiac functional decline. Common pathologic triggers, such as hypertension, increase cardiac afterload and stimulate an increased, or hypercontractile response, accompanied by compensatory hypertrophic growth (6), to maintain cardiac output (7). The hypertrophied heart usually presents with significant cardiac diastolic dysfunction, as a result of a persistent hypercontractile state, and impaired relaxation (8, 9). In this case, combinatorial treatment of the underlying hypertension and subsequent hypercontractility may prove advantageous for improving patient outcomes.
Successful treatment strategies for these heart diseases have met considerable challenges. For example, pharmacologic interventions to lessen contractility and relieve symptoms in patients with HCM have focused on decreasing intracellular Ca2+ availability through competitive β-adrenergic receptor inhibition or Ca2+ channel blockade. However, these treatments are not well supported by clinical evidence and often are not very effective (10). Therapies for patients with hypertrophy-associated diastolic dysfunction have also been largely unsuccessful. Therapeutics that reduce hypertension and interstitial fibrosis, and improve filling or maintain fluid balance have not shown mortality benefits (11). The limited success of these treatments underscores the notion that increased contractility in hypertrophied myocardium may best be attenuated via direct modulation of the myofilament proteins. For example, in some HCMs, enhanced force/power generation is directly attributable to the increase in force generated by a single myosin cross-bridge (12). Furthermore, phosphorylation of myosin binding protein C and regulatory myosin light chain has been shown to increase force by altering actomyosin binding kinetics (13). These recent findings have stimulated interest in using alternative, novel agents, especially small molecules that directly target myofilament proteins, to dampen contraction in hopes of circumventing disease progression.
General anesthetics, both inhalational and intravenous, are small molecules and known myocardial depressants (14). We have recently shown that both isoflurane and propofol depress myocardial contraction in isolated cardiac trabeculae (15, 16). Moreover, we have demonstrated that these agents act directly on myofilament proteins, specifically myosin heavy chain, myosin regulatory light chain, and actin, to decrease Fmax and increase intracellular Ca2+ concentration ([Ca2+]i) required for 50% of Fmax (Ca50) (16). We found that isoflurane and propofol dock in cavities close to the ATP binding pocket in the myosin head and subdomain 3 of actin. These regions are crucial for proper actin–myosin cross-bridge formation and cycling, and therefore, any changes near or within them may significantly affect force generation. Recently, we have found that fropofol, a small molecule propofol derivative, which does not demonstrate anesthetic efficacy in vivo, auspiciously depressed twitch force development in cardiac muscle (17). Therefore, in this study we extended these initial findings and further characterized the mechanical and biochemical effects of fropofol in cardiac muscle. Its depressing effect on in vivo muscle contraction was quantified, and we determined that the drug likely targets actin–myosin interactions, notably during active force development. Our findings warrant testing of fropofol in disease models (HCM) for which increased contractility is a root cause.
MATERIALS AND METHODS
Animals care and studies
LBN/F1 rats (250–300 g; Harlan Laboratories, Indianapolis, IN, USA) were used in these experiments. Animal care and experimental protocols were approved by the Animal Care and Use Committee of The Johns Hopkins University School of Medicine.
To model a hypercontractile myocardial phenotype, rats were injected 1 time with monocrotaline (MCT; 60 mg/kg, i.p.) to induce pulmonary hypertension and thus enhancing contractility in the right ventricle (RV) (18). One week after the injection, rats were placed in a Plexiglas box and anesthetized with isoflurane 1–3%, weighed, and placed on a heating pad to maintain body temperature at ∼37°C. Temperature was monitored with a rectal thermometer. The rats were spontaneously breathing, and inhalational anesthesia was provided via a nose cone with O2 as the carrier (2.0 L/min). Heart rate was monitored with echocardiographic recording. Echocardiography was performed and right hearts were imaged. Echocardiographic images were obtained with a Vevo 2100 ultrasound machine (VisualSonics, Toronto, ON, Canada) and a 30 MHz transducer, which was mounted on the Vevo Imaging Station (VisualSonics) for improved micromanipulation. A right parasternal long-axis view was used to visualize the RV, and RV function was determined. RV free-wall thickness and RV internal diameter were measured at the midlevel parasternal long-axis view and at the left ventricular midpapillary muscle level in the short-axis view, respectively. Serial echocardiography was repeated every half week. The animals were euthanized at ∼2.5 wk after injection.
Determination of force and [Ca2+]i in intact trabeculae preparations
Each rat was anesthetized via injection with pentobarbital (100 mg/kg, i.p.). The heart was exposed by midsternotomy, rapidly excised, and perfused in retrograde fashion with Krebs-Henseleit (K-H) solution equilibrated with a 95% O2/5% CO2 gas mixture in a dissection dish. The dissecting K-H solution was composed of (mM) NaCl 120, NaHCO3 20, KCl 5, MgCl2 1.2, glucose 10, and CaCl2 0.5 (pH 7.35–7.45 at room temperature, 21–22°C) and contained 20 mM 2,3-butanedione monoxime. Trabecular muscles from the RV were dissected and mounted between a force transducer and a motor arm, superfused with K-H solution at a rate of ∼10 ml/min, and stimulated at 0.5 Hz. The perfusion K-H solution did not contain 2,3-butanedione monoxime and was gassed with 95% O2/5% CO2 to maintain pH 7.35–7.4. Force was measured by a force transducer system (KG7; Scientific Instruments, Heidelberg, Germany) and expressed in millinewtons per square millimeter of cross-sectional area. The cross-sectional area of the muscle was calculated as width × thickness and corrected by multiplying 0.785 because of an ellipse-shaped cross-section of the muscle (i.e., π × width/2 × thickness/2). The muscles underwent isometric contractions with the resting muscle length set at which maximum twitch force developed: the corresponding resting force was usually ∼15% of the maximum force development at this length, which corresponded to a resting sarcomere length of 2.20–2.30 μm, as determined by laser diffraction (19) and was maintained throughout the experiments.
[Ca2+]i was measured by using the free-acid form of Fura-2 (20). Fura-2 potassium salt was microinjected iontophoretically into 1 cell and allowed to diffuse throughout the whole muscle (via gap junctions) without affecting force development. The tip of the electrode (∼0.2 μm in diameter) was filled with Fura-2 salt (1 mM), and the remainder of the electrode was filled with 150 mM KCl. After a successful impalement of the electrode into a superficial cell in nonstimulated muscle, a hyperpolarizing current of 5–10 nA was passed continuously for ∼15 min. Sometimes, multiple injection sites were needed, as the duration of each injection was limited to <10 min to achieve the desired Fura-2 loading. Fura-2 epifluorescence was measured by excitation at 380 and 340 nm. Fluorescent light was collected at 510 nm by a photomultiplier tube (R1527; Hamamatsu Photonics, Hamamatsu, Japan). Output from the photomultiplier was collected and digitized. [Ca2+]i was calculated by the following equation (after subtraction of the autofluorescence) (Eq. 1):
where R is the observed ratio of fluorescence (340:380 nm), K′d is the apparent dissociation constant, Rmax is the ratio of 340:380 nm at saturating [Ca2+], and Rmin is the ratio of 340:380 nm at 0 [Ca2+]. The values of K′d, Rmax, and Rmin were determined by in vivo calibrations, as described in Gao et al. (21).
Steady-state activation of cardiac muscles
Ryanodine (1.0 μM) was used to enable steady-state activation. After the muscles were exposed to ryanodine for 15 min, different levels of tetanization were induced briefly (∼4–8 s) by stimulating the muscles at 10 Hz in various extracellular Ca2+ concentrations ([Ca2+]o; 0.5–20 mM). The steady-state force–[Ca2+]i relations were fit with a function of the Hill equation (Eq. 2):
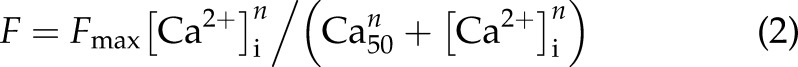 |
where F is the steady-state force at various [Ca2+]i, and n is the Hill coefficient.
After the steady-state experiments, the same trabeculae were immediately skinned by 15–20 min of exposure to Triton X-100 (1%) in relaxing solution containing (mM) KCl 100, HEPES 25, K2EGTA 10, creatine phosphate sodium salt (Na2CrP) 15, Na2ATP 5, MgCl2 5.15, and leupeptin 0.5 (pH 7.2 with KOH). The activating solution contained (mM) Ca2+-EGTA 10, KCl 100, HEPES 25, Na2CrP 15, Na2ATP 5, MgCl2 4.75, and leupeptin 0.5 (pH 7.2). Different Ca2+ concentrations were achieved by mixing the activating and relaxing solutions in different ratios. The readiness of the skinned preparation was confirmed by the loss of pink color, clear visibility of the sarcomeres, and instantaneous force development upon exposure to activating solutions. Diastolic sarcomere length was determined by direct visualization under ×100 magnification and was set at ∼2.2 μm. Resting force was usually 10–15% of maximum activated force at this sarcomere length. Force–Ca2+ relations were obtained by exposing the skinned muscles to activating solutions with various [Ca2+]o, and were fit with the Hill equation (Eq. 3):
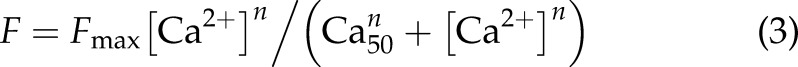 |
where F is the steady-state force at various [Ca2+], and n is the Hill coefficient.
We also determined the steady-state force–Ca2+ relationship in intact muscles and then again after skinning in the presence of fropofol (100 μM). Purified fropofol (MW 180.1314) was dissolved in K-H solution. The experiments were performed at room temperature (20–22°C).
Force measurements skeletal muscle preparations
To investigate the possibility that the effects of fropofol are cardiac muscle specific, we conducted parallel experiments in skeletal muscle, also a striated muscle, using plantar interossei muscles, each of which are located between the metatarsals and arise from a single metatarsal. Rats were anesthetized via injection with pentobarbital (100 mg/kg, i.p.), and either the right or left rear paw was excised and fixed rapidly in a dissection dish with the plantar side up. The paw was immersed and dissected in K-H solution with 2,3-butanedione monoxime at room temperature (21–22°C). After dissection, the skeletal muscle was mounted between a force transducer and a motor arm, superfused with K-H solution at a rate of 10 ml/min, and stimulated at 0.5 Hz with 0.5 mM [Ca2+]o. Force was measured by a force transducer system (KG7; Scientific Instruments) and expressed in millinewtons per square millimeter of cross-sectional area. Throughout the experiments, resting muscle was maintained at a length that enabled maximum stimulated force to be achieved, which translated to a resting force of ∼15% of total force development.
Determination of myofibrillar ATPase
Myofibrils were prepared from cardiac ventricles as described in Dai et al. (22), with careful use of protease inhibitors. Assays were performed under standard incubation conditions with varying Ca2+; ionic strength was maintained by using stability constants compiled by Fabiato (23). Assays were performed at pH 7.0 with (mM) imidazole 50, KCl 50, and MgATP 2. Inorganic phosphate liberation was measured by using a microtiter plate version of the standard assay as described by Rarick et al. (24). Protein concentration was determined by a variation of the Lowry method (Bio-Rad, Hercules, CA, USA). In the final assay conditions, myofibrillar protein concentration was diluted in buffer to a concentration of 0.2 mg/ml, and protein concentration was determined again for final calculations. Mg-ATPase activity was calculated in nanomoles of inorganic phosphate liberated per milligram of myofibrillar protein per minute.
Rat cardiac myosin purification
Rat cardiac myosin purification was performed as published (25, 26) with slight modifications. Approximately 8–12 rat hearts were obtained as previously described. All solutions were kept on ice, and all steps were performed in a cold room. Hearts were minced with a small coffee grinder (3–4 pulses of 10–15 s each) prerinsed with a minimal amount of extraction buffer [KCl 0.3, K2HPO4 0.15, and Na4PO7 0.01 (M); MgCl2 1, ATP 1, DTT 2, and PMSF 0.23 (pH 6.8) (mM)]. The tissue was weighed and transferred to a glass beaker containing 10 ml/g extraction buffer. After homogenization with a glass rod, myosin was extracted for 90 min with stirring. The homogenate was spun at 12,000 g for 10 min at 4°C to remove large particulate residue and then centrifuged for 4 h at 140,000 g. The supernatant was collected and diluted with 20 volumes of ice cold water (2 mM DTT); precipitated myosin was left overnight to settle.
The supernatant was siphoned off and the sediment (myosin) centrifuged at 12,000 g for 10 min. Half of the precipitated myosin was resuspended in ∼2.5 ml/g 1× high-salt buffer (0.5 M KCl, 0.01 M imidazole, and 1 mM DTT (pH 6.8)], and the other half was redissolved in 2× high-salt buffer. Both solutions were then centrifuged at 100,000 g for 45 min to remove aggregated proteins. Myosin in 1× high-salt buffer was collected and immediately stored on ice at 4°C. Myosin in 2× high-salt buffer was diluted 1:1 in glycerol and placed at −20°C for long-term storage. Protein concentration was measured via a Bradford-based colorimetric assay (CB-X Protein Assay; G-Biosciences, St. Louis, MO, USA).
Actin purification and labeling
Unlabeled filamentous actin (F-actin) was employed to remove enzymatically inactive myosin (“dead heads”) from solution and to prevent interaction with fluorescently labeled F-actin during in vitro motility experiments. Monomeric actin (G-actin) was purified from rabbit skeletal acetone powder (Pel-Freez Biologicals, Rogers, AR, USA), per established methods (27). The concentration of G-actin was determined via absorbance at 290 nm, and 2 mg of sucrose was added per milligram of actin. Approximately 150 µg was aliquoted, lyophilized, and stored at −80°C. To polymerize G- to F-actin, aliquots were resuspended in low-salt buffer [KCl 25, imidazole 25, MOPS 25, MgCl2 2, and DTT 10 (pH 7.4) (mM)] and left to polymerize at room temperature for ∼1 h.
Cardiac myosin-based motility of rabbit skeletal F-actin, was visualized via conjugation of F-actin to Alexa-568 phalloidin. First, lyophilized rabbit skeletal G-actin was solubilized in double distilled H2O to 10 mg/ml (∼240 µM) and then diluted in low-salt buffer to 6.6 µM and mixed with equal volume of 6.6 µM Alexa-568 phalloidin (Thermo Fisher Scientific, Waltham, MA, USA). Alexa-568 phalloidin-labeled F-actin was left to equilibrate at room temperature for ∼1 h and diluted a second time to 1 µM and stored at 4°C.
In vitro motility
Movement of Alexa-568 phalloidin-labeled F-actin was assessed via a standard in vitro motility assay (28). In brief, cardiac myosin was introduced into a flow cell and allowed 2 min to bind to a nitrocellulose-coated coverslip. The surface was blocked with 2 mg/ml bovine serum albumin, myosin dead heads were nonreversibly bound to unlabeled F-actin, and enzymatically active myosin was bound to ∼10 nM Alexa568-phalloidin–labeled rabbit skeletal F-actin in the presence or absence of fropofol. Motility of labeled F-actin at 30°C was initiated via introduction of a motility buffer containing (mM) KCl 25, MgCl2 4, EGTA 1, imidazole 25 (pH 7.2), DTT 10, ATP 1, dextrose 2, with 17 U/ml glucose oxidase, 125 U/ml catalase, and 0.5% methyl cellulose. Imaging was performed on an IX73 microscope (Olympus America, Hauppauge, NY, USA) and Alexa568-phalloidin was excited by using an X-CITE 120 LED lamp and 531/40 nm filter. Emitted light was captured at 593/40 nm and detected on a Hamamatsu Flash 4LT EMCCD camera at 0.5–2 frames per second. Videos were recorded with HCI imaging software, converted to multipage TIFFs, and imported into ImageJ (National Institutes of Health, Bethesda, MD, USA). Average filament velocities were determined via manual tracking (29) of ∼20–25 individual filaments. Of note, flow cells were partitioned into 2 separate chambers so that motility with and without fropofol could be assessed in parallel.
Statistical analysis
Paired Student’s t tests, Student’s t tests, and ANOVAs (1- and 2-way) were used for statistical analysis of the data. A value of P < 0.05 was considered to indicate significant differences between groups. Unless otherwise indicated, pooled data are expressed as means ± sem.
RESULTS
Effect of fropofol on force development and [Ca2+]i in normal cardiac muscle
We first investigated the effect of fropofol on force development and [Ca2+]i in trabeculae from the right hearts of normal rats (Fig. 1). Fropofol significantly decreased twitch force development in a dose-dependent manner between 20 and 200 µM with a ∼28% maximum reduction relative to baseline (Fig. 1A, B) at room temperature (22°C). The reduction in force was not caused by decreased [Ca2+]i, as amplitudes of [Ca2+]i transient were not affected at any dose tested. Resting force, resting [Ca2+]i, and twitch and [Ca2+]i transient dynamics were also unaffected by fropofol (Fig. 1C–E). We also found that fropofol depressed force development in trabecular muscles at body temperature in a dose-dependent manner (37°C; results not shown). However, because of the rapid metabolism of Fura-2 at this temperature, measurement of [Ca2+]i became unreliable, especially for steady-state activations. Therefore, we performed the bulk of the studies at 22°C.
Figure 1.

Fropofol decreases twitch force development in normal cardiac muscle. A) Raw recordings of force development (left) and corresponding intracellular Ca2+ transients (right) from a normal trabecular muscle in the presence of 0, 50, and 200 µM fropofol. Fropofol dose dependently reduced force development, but intracellular Ca2+ remained unchanged. B) Pooled data of trabecular force development and intracellular Ca2+ transient amplitudes in the presence of different doses of fropofol. Force decreased in a dose-dependent manner as concentrations of fropofol increased. Force became significantly less than that at baseline at doses >20 μM. *P < 0.01, vs. baseline, by paired Student’s t test. Maximal depression of force (∼28%) was achieved at 200 µM. Intracellular Ca2+ transient amplitudes remained unchanged at all doses of fropofol. C) Effect of fropofol on diastolic force and intracellular Ca2+ levels. Fropofol had no effect on diastolic force and intracellular Ca2+. D) Effect of fropofol on time to peak force and time from peak to half relaxation. Fropofol did not affect force dynamics. E) Effect of fropofol on the time course of intracellular Ca2+ transients. Both the time to peak and the time to half relaxation were not affected. Temperature, 22°C; external Ca2+, 1.0 mM; stimulation rate, 0.5 Hz (n = 8–11).
To uncover the potential for fropofol to specifically affect cardiac muscle contraction, we investigated whether the drug elicited a similar response in skeletal muscle. Unlike in cardiac preparations, fropofol (0–200 µM) affected neither stimulated nor resting forces in rat plantar interossei muscle. Therefore, we focused on examining fropofol’s effects on cardiac tissue and, specifically, how it influenced myocardium characterized by supranormal contractile properties.
Effect of fropofol on force development and [Ca2+]i in hypercontractile cardiac muscle
We examined the potential for fropofol to decrease force production in trabecular muscles from rats with cardiac hypercontractility. Muscles were dissected from the RVs of MCT-injected rats. One dose of injected MCT (60 mg/kg, i.p.) increased RV size and wall thickness (RV hypertrophy) and increased contractility after 2.5 to 3 wk (Fig. 2 and Table 1). RV wall thickness increased significantly at 2.5 wk and continued to trend upward until 4 wk after MCT injection. RV diameter also increased significantly at 3 wk postinjection and remained dilated thereafter; at 4 wk, RV ejection fraction and fractional shortening were significantly depressed (results not shown). RV trabeculae from injected rats had 2–3 times higher twitch and resting forces than normal muscles at the same external Ca2+ concentration (Fig. 3). The increased force was most likely caused by changes in myofilament properties, given that [Ca2+]i transient amplitudes were only slightly elevated (∼20% vs. normal muscles). Fropofol decreased force development in these muscles in a dose-dependent manner, with significant depression occurring at 20 µM and maximum depression (35% decrease) at 200 μM. Amplitudes of [Ca2+]i transient were not affected at any dose tested (Fig. 3A, B). Unlike what was found with control RV trabeculae (Fig. 1C), fropofol significantly reduced resting force with no change in resting [Ca2+]i (Fig. 3C) and significantly accelerated relaxation of force (Fig. 3D). Fropofol did not, however, affect the time to peak, or the time from peak to half amplitude, for twitch force and [Ca2+]i transients (Fig. 3D, E).
Figure 2.

MCT injection increases cardiac muscle contractility. Changes in RV wall fractional shortening after intra-abdominal MCT injection. RV free-wall fractional shortening were significantly elevated at 2.5 wk after MCT injection (n = 4–7 animals examined by transthoracic echocardiography at each time point). *P < 0.05, increase vs. baseline.
TABLE 1.
Changes in RV wall thickness and chamber diameter after intraperitoneal MCT injection
| Parameter | Week |
||||
|---|---|---|---|---|---|
| 0 | 2.5 | 3 | 3.5 | 4 | |
| Heart rate (beats/min) | 367 ± 7 | 359 ± 12 | 345 ± 10 | 375 ± 15 | 255 ± 9 |
| Diastolic wall thickness (mm) | 0.49 ± 0.01 | 0.53 ± 0.02* | 0.59 ± 0.02* | 0.61 ± 0.02* | 0.59 ± 0.04* |
| Systolic wall thickness (mm) | 0.92 ± 0.03 | 0.99 ± 0.03* | 1.06 ± 0.05* | 1.07 ± 0.04* | 1.02 ± 0.05 |
| RVID (mm) | 2.46 ± 0.08 | 2.60 ± 0.06 | 2.82 ± 0.19* | 3.05 ± 0.25* | 3.03 ± 0.18* |
n = 3–10 in each group. RVID, right ventricular internal diameter. *P < 0.05, paired Student’s t test vs. baseline (wk 0).
Figure 3.

Fropofol decreases force development in hypercontractile muscles. A) Raw recordings of force development (left) and corresponding intracellular Ca2+ transients (right) of a trabecular muscle from the RV of MCT-injected rats in the presence of 0, 50, and 200 µM fropofol. Fropofol decreased force in a dose-dependent manner, but intracellular Ca2+ remained unchanged. B) Pooled data of force development and amplitudes of intracellular Ca2+ transient in the presence of different doses of fropofol. Force became significantly lower than that at baseline at doses >20 μM. *P < 0.01 vs. baseline, paired Student’s t test. Maximum depression of force (∼35%) was achieved at 200 µM. Intracellular Ca2+ transient amplitudes remained unchanged at all doses of fropofol. C) Effect of fropofol on diastolic force and intracellular Ca2+ levels. Significant decreases in diastolic force were seen at fropofol doses higher than 20 µM. *P < 0.05 vs. baseline, paired Student’s t test. There were no changes in diastolic Ca2+ levels. D) Effect of fropofol on time to peak force and time from peak to half relaxation. Fropofol accelerated relaxation of force. E) Effect of fropofol on time course of intracellular Ca2+ transients. Both the time to peak and the time to half relaxation were not affected. Temperature, 22°C; external Ca2+, 1.0 mM; stimulation rate, 0.5 Hz (n = 9–11).
Effect of fropofol on steady-state force–[Ca2+]i relationships in normal and hypercontractile muscles
The data described above suggest that fropofol decreases myofilament responsiveness to Ca2+. To directly determine potential changes in myofilament Ca2+-responsiveness caused by fropofol, we determined the steady-state force–[Ca2+]i relationship in the presence and absence of fropofol in both control and hypercontractile muscles. In normal trabeculae, Fmax was 84.5 ± 2.5 mN/mm2, and Ca50 was 0.62 ± 0.11 μM (Fig. 4A). Fmax decreased in muscles treated with fropofol (100 µM) to 49.6 ± 4.4 mN/mm2 (P < 0.01 vs. baseline) and Ca50 increased to 0.95 ± 0.17 μM (P < 0.05). The Hill coefficient was unchanged (4.24 ± 0.811 vs. 3.01 ± 0.45; P > 0.05). Next, we chemically skinned the muscles to eliminate potential confounding cytosolic factors and found that fropofol significantly decreased Fmax from 81.5 ± 5.2 to 44.8 ± 3.8 mN/mm2 (P < 0.01) and increased Ca50 from 1.63 ± 0.2 to 2.31 ± 0.41 µM (P < 0.05; Fig. 4B). The Hill coefficient was not affected (2.16 ± 0.14 vs. 1.13 ± 0.07). Thus, the depressing effect of fropofol on the steady-state force–[Ca2+]i relationship persisted in skinned muscles and suggests direct action on the myofilaments.
Figure 4.

Relationship between steady-state force and intracellular Ca2+ in the presence and absence of fropofol (100 µM) in intact and skinned cardiac trabecular muscles. The steady-state forces were plotted against corresponding intracellular Ca2+ transients. A) In normal intact trabeculae, fropofol significantly reduced Fmax and increased Ca50 (n = 10). B) The same muscles in which steady-state relations were first obtained were chemically skinned and activated with various Ca2+ concentrations in the absence and presence of fropofol. Note that the effect of fropofol on Fmax and Ca50 persisted in these skinned muscles (n = 9). C) In intact trabeculae from MCT-injected rats, fropofol significantly reduced Fmax and increased Ca50 (n = 10). D) The same muscles in which steady-state relations were first obtained were chemically skinned and activated with various Ca2+ concentrations in the absence and presence of fropofol. The effect of fropofol on Fmax and Ca50 persisted in these skinned muscles (n = 9). Temperature, 22°C.
We next examined whether fropofol exerted a similar myofilament desensitizing effect on trabecular muscles from hypercontractile hearts. As with twitch force, these muscles generated higher Fmax during steady-state activations (Fig. 4C, D). In the intact muscles, fropofol (100 µM) significantly decreased Fmax (from 115.5 ± 4.2 to 70 ± 4.2 mN/mm2; P < 0.01) and increased Ca50 (from 0.52 ± 0.08 to 0.71 ± 0.16 µM; P < 0.05) without affecting the Hill coefficient (4.52 ± 0.76 vs. 2.84 ± 0.46; P > 0.1) (Fig. 4C). Fropofol’s effect persisted after skinning: Fmax was significantly depressed (from 132.8 ± 8.3 to 76.7 ± 5.46 mN/mm2, P < 0.01), and Ca50 was significantly increased (from 1.49 ± 0.2 to 2.12 ± 0.27; P < 0.05, Fig. 4D). The Hill coefficient was not affected (2.00 ± 0.14 vs. 1.73 ± 0.27). These data strongly support the notion that the negative impact of fropofol on myocardial contraction largely stems from a direct effect on the myofilaments.
Effect of fropofol on myofibrillar ATPase activity and the biochemical and mechanical properties of cardiac myosin and actin
To assess whether the depressing effect of fropofol on cardiac force development is the result of inhibited cross-bridge cycling, we determined myofibrillar Mg2+-ATPase activity–Ca2+ relationships in isolated working myofibrils from rat hearts. In the presence of fropofol, maximum myofibrillar Mg2+-ATPase activity was reduced from 124.86 ± 8.08 to 101.46 ± 6.93 pM Pi/min per milligram of protein (P < 0.05), and the Ca2+ required for half-maximum ATPase activity increased from 1.32 ± 0.17 to 1.83 ± 0.17 µM (Fig. 5A).
Figure 5.

Effect of fropofol on myofibrillar Mg2+-ATPase activity, actin sliding velocity, and myosin ATPase activity of rat myocardium. A) Fropofol significantly depressed the myofibrillar Mg2+-ATPase activity–Ca2+ relationship, as evidenced by a decrease in maximum Ca2+-activated ATPase activity and a rightward shift of the curve (n = 5). Temperature, 22°C. P < 0.05, multivariate ANOVA. B) Fropofol slowed actin sliding speed by 9 and 12% at 100 and 300 µM, respectively, with significance reached at 300 µM. Inset: decrease in absolute velocity of actin sliding over myosin at 300 µM fropofol (n = 24 filaments). **P < 0.01 vs. 0 fropofol, unpaired Student’s t test. C) Myosin ATPase activity was depressed at increasing doses of fropofol. At doses above 100 µM, fropofol significantly depressed actin-activated myosin ATPase activity (actin, 0.4 mg/ml), but had no effect on myosin ATPase activity in the absence of actin (n = 5). *P < 0.03, multivariate ANOVA. D) The actin-myosin ATPase relationship was also depressed by 200 µM fropofol (n = 3–5). *P < 0.05, multivariate ANOVA.
To gather additional evidence that decreased myofilament ATPase activity is caused by altered behavior of the cross-bridges themselves, and not a function of other regulatory proteins, we measured myosin ATPase activity and performed quantitative in vitro motility assays with purified rat cardiac myosin and rabbit skeletal actin in the presence of fropofol. Consistent with myofibril measurements, fropofol inhibited actin-activated myosin ATPase activity (Fig. 5C, D), but had no effect on basal myosin ATPase. Furthermore, in vitro motility of actin filaments sliding over a bed of myosin revealed that fropofol, at concentrations above 100 µM, significantly decreased average actin sliding velocity when the drug was added during rigor binding (Fig. 5B). The observation that fropofol did not affect basal myosin ATPase, but significantly decreased in vitro motility of actin filaments, suggests that its potential binding sites are more accessible during cross-bridge formation.
DISCUSSION
In this study, we investigated the mechanism by which the small molecule fropofol, a derivative of propofol, depresses force in both normal and hypercontractile myocardium. Fropofol also promotes relaxation and decreases resting force in hypercontractile muscles. The inhibitory effect on force production appears to be caused at least in part by inhibition of myosin cross-bridges. Given that current treatments with Ca2+ channel and β-adrenergic receptor blockers are suboptimal for heart diseases that involve hypercontractile states, such as HCM and hypertensive cardiomyopathy with diastolic dysfunction, fropofol may represent an effective alternative, as it appears to act directly on the myofilaments, reducing the risk for various unwanted, off-target effects.
Increased cardiac contractility (i.e., hypercontractility) was observed in both the RV and the trabecular muscles from rats after ∼2 wk of pulmonary hypertension induced by MCT injection. We found that the hypercontraction elicited during the early phase is associated with mildly increased intracellular Ca2+ and markedly increased myofilament Ca2+ sensitivity and maximum Ca2+-activated force production (Fig. 4). Potential mechanisms for this augmented myofilament Ca2+ responsiveness include changes in cross-bridge kinetics, phosphorylation of contractile proteins, and other posttranslational modifications of the myofilament proteins. Nonetheless, pressure-induced hypercontractile cardiac muscle from MCT-injected rats serves as an efficient system to demonstrate fropofol’s ability to inhibit force development in myocardium with enhanced contractile properties.
Particular anesthetics, including isoflurane and propofol, have been shown to depress myocardial contraction in isolated cardiac trabeculae (15, 16). These small molecules directly target cardiac myofilament proteins, especially myosin and actin, to exert a myofilament desensitizing and depressing effect (16), and could be ideal to inhibit myocardial contraction in hypercontractile states. Unfortunately, it is impractical to use these agents to treat cardiac hypercontractility because of their anesthesia effect. Fropofol (2-fluoro-1,3-diisopropylbenzene) is chemically identical to propofol (2,6-diisopropylphenol), except that the hydroxyl group is substituted with fluorine (17). Although fropofol has similar physicochemical properties as propofol and binds similar protein targets, it does not possess anesthetic efficacy. Therefore, fropofol would be an attractive candidate for treatment of cardiac disease.
Fropofol and propofol appeared to compete for the same binding site in the model protein, horse spleen apoferrin (17). Therefore, the 2 molecules may also share similar docking sites on myofilament proteins. We have recently identified 4 binding sites of propofol, and potentially of fropofol, located in the myosin heavy chain head domain and 1 in subdomain 3 of actin (16). Therefore, a significant impact on the actomyosin ATPase cycle, related to binding of the small molecules, is not unexpected. For example, 2 proposed myosin binding sites lie near hypervariable loop 1 and switch-2 domains (16). These myosin elements have been shown to affect ATP-binding affinity, hydrolysis, and nucleotide release rates, and when mutated, have induced significant defects in model systems (30–32). The observation that fropofol only reduces ATPase rate and sliding velocity when actin is present may be a result of increased binding of the drug during rigor, when the ATP-binding pocket is open and more fully exposed (33). Decreased ATPase activity would directly reduce the number of actively cycling, force producing cross-bridges and diminish overall force (Figs. 1, 4, and 5). However, ensemble force is dependent on 3 primary factors: 1) the number of cross-bridges formed, 2) the rate of cross-bridge cycling, and 3) the unitary force of a single, strongly bound myosin cross-bridge (12). We therefore cannot rule out the possibility that the unitary force may also be impaired.
We have shown that fropofol dose dependently depresses twitch force development in cardiac muscle (17). The current study not only corroborates our previous observations, but also offers potential mechanism(s) for depressing contraction. Results show that fropofol directly interacts with the myofilaments to inhibit myosin cross-bridge activity as evidenced by decreases in both actin sliding velocity in the in vitro motility assay and actin-activated myosin ATPase activity (Fig. 5). Therefore, fropofol’s interaction with myosin (16) likely directly contributes to the overall reduction in force. Fropofol (300 µM) was required to significantly decrease actin sliding velocity, compared to 20 µM to depress force in intact muscle, suggesting that this interaction is subjected to additional inhibitory processes that occur in vivo. For example, the possibility cannot be ruled out that fropofol may change the level(s) of myofilament protein phosphorylation, perhaps myosin binding protein C and myosin regulatory light chain, both of which are critical modulators of contraction. To potentially unveil the root causes of this discrepancy, current studies are focused on quantitating the extent of myofilament protein phosphorylation and expanding in vitro motility experiments to determine if the effect of fropofol is exacerbated when myosin contracts against a load (34) or interacts with isolated native thin filaments.
Fropofol decreased Ca2+-sensitivity in intact and skinned cardiac muscles. It also reduced myofibrillar and actomyosin ATPase rates. While the kinetics of myosin strong binding have been shown to influence Ca2+-sensitivity of thin filaments, a change in ATPase rate alone is not expected to influence thin filament Ca2+-sensitivity (35–37). Therefore, the drug may elicit effects via binding to actin subdomain 3, which contains numerous residues that favorably interact with tropomyosin. These contact sites help establish tropomyosin’s inhibitory position along thin filaments and are critical in maintaining proper Ca2+ activation (38–42). In fact, mutations in this region have recently been shown to induce significant effects on muscle function and, specifically, on thin filament Ca2+ sensitivity (42). Therefore, fropofol binding may induce a structural change in this region of actin that acts to desensitize the thin filament to Ca2+, which can further reduce myosin binding and overall force production. Future in vitro motility experiments performed under loaded conditions (34) and with reconstituted thin filaments at varying levels of Ca2+ will directly determine the plausibility of these potential effects.
Furthermore, the mild-to-moderate decrease in force, even at very high fropofol doses, suggests a limit to its inhibitory effect (i.e., a ceiling effect). Such an upper limit would prove advantageous to 1) help mitigate detrimental losses in force production and 2) increase the safety margin for drug toxicity and overdose. We have also observed that contractile inhibition was completely reversed after washout (results not shown), a characteristic that would allow successful management and treatment of potential overdose. All of these features establish fropofol as an attractive small molecule inhibitor of force generation.
Our study has highlighted fropofol’s potential as a novel candidate for treatment of cardiac pathologies typified by hypercontractility or elevated power output. The increased tension generated in muscles that contain HCM mutations (expressed as increased tension integral), caused by enhanced thin filament Ca2+ sensitivity, has recently been shown to be the primary cause of development of the disease (43). Thus, diminishing hypercontractility, especially via use of small molecule inhibitors of actomyosin cross-bridge cycling (12), may attenuate hypertrophy and other disease processes. This idea has been validated in HCM models by using MYK-461 (44). However, fropofol has several unique features. First, it does not alter intracellular Ca2+ cycling and thus, does not disturb Ca2+-dependent signaling pathways in the cell. Second, the ceiling effect of contractile inhibition ensures a less negative inotropic effect, which would make the drug safer and more tolerable for maintaining adequate cardiac output—a feature that Ca2+ desensitizers lack. Fropofol was also shown to be safe in animals at a dose of 100 mg/kg. This dose, which is 5-fold higher than the therapeutic level of propofol, did not produce any appreciable neurologic effects in the animals studied (17), however, the overall cardiovascular effects remain to be investigated. Thus, the paucity of available Ca2+ desensitizers to treat hypercontractile states in cardiomyopathies (45), especially those with diastolic dysfunction, warrants further testing of fropofol in models of sarcomeric cardiomyopathies.
In summary, fropofol depresses force development in both normal and hypercontractile cardiac muscles by decreasing myofilament Ca2+ responsiveness. The small molecule exerts its effect by direct interaction with myofilament proteins, thus potentially making it an ideal therapeutic candidate in treatment of sarcomeric cardiomyopathy characterized by hypercontractility and diastolic dysfunction.
ACKNOWLEDGMENTS
This research was supported, in part, by a Stimulating and Advancing Anesthesiology and Critical Care Medicine (ACCM) Research (StARR) Award from the Department of Anesthesiology and Critical Care Medicine (Johns Hopkins University School of Medicine; to W.D.G.); American Heart Association–Global Innovation Award (AHA-GIA) (17GRNT33670387; to W.D.G.); U.S. National Institutes of Health (NIH) National Institute of General Medical Sciences Grants GM055867 and GM008076, and NIH National Institute of Neurological Disease and Stroke Grant NS080519 (to R.E.); NIH National Heart, Lung, and Blood Institute Grant T32HL007227-38 (to W.S.) and R01HL124091 (to A.C.); and AHA Grant 17POST33630159 (to W.S.). The authors declare no conflicts of interest.
Glossary
- [Ca2+]i
intracellular Ca2+ concentration
- [Ca2+]o
extracellular Ca2+ concentration
- Ca50
[Ca2+]i required for 50% of Fmax
- Fmax
maximum Ca2+-activated force
- HCM
hypertrophic cardiomyopathy
- K-H
Krebs-Henseleit
- MCT
monocrotaline
- RV
right ventricle
AUTHOR CONTRIBUTIONS
X. Ren, W. Schmidt, and W. D. Gao designed the research; X. Ren, W. Schmidt, Y. Huang, H. Lu, W. Liu, and W. D. Gao performed the research; W. Bu, R. Eckenhoff contributed new reagents; X. Ren, W. Schmidt, Y. Huang, H. Lu, W. Liu, A. Cammarato, and W. D. Gao analyzed the data; and X. Ren, W. Schmidt, A. Cammarato, and W. D. Gao wrote the paper.
REFERENCES
- 1.Maron B. J., Bonow R. O., Cannon R. O., III, Leon M. B., Epstein S. E. (1987) Hypertrophic cardiomyopathy. Interrelations of clinical manifestations, pathophysiology, and therapy (2). N. Engl. J. Med. 316, 844–852 [DOI] [PubMed] [Google Scholar]
- 2.Maron B. J., Bonow R. O., Cannon R. O., III, Leon M. B., Epstein S. E. (1987) Hypertrophic cardiomyopathy: interrelations of clinical manifestations, pathophysiology, and therapy (1). N. Engl. J. Med. 316, 780–789 [DOI] [PubMed] [Google Scholar]
- 3.Spindler M., Saupe K. W., Christe M. E., Sweeney H. L., Seidman C. E., Seidman J. G., Ingwall J. S. (1998) Diastolic dysfunction and altered energetics in the alphaMHC403/+ mouse model of familial hypertrophic cardiomyopathy. J. Clin. Invest. 101, 1775–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guinto P. J., Haim T. E., Dowell-Martino C. C., Sibinga N., Tardiff J. C. (2009) Temporal and mutation-specific alterations in Ca2+ homeostasis differentially determine the progression of cTnT-related cardiomyopathies in murine models. Am. J. Physiol. Heart Circ. Physiol. 297, H614–H626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schober T., Huke S., Venkataraman R., Gryshchenko O., Kryshtal D., Hwang H. S., Baudenbacher F. J., Knollmann B. C. (2012) Myofilament Ca sensitization increases cytosolic Ca binding affinity, alters intracellular Ca homeostasis, and causes pause-dependent Ca-triggered arrhythmia. Circ. Res. 111, 170–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schiattarella G. G., Hill J. A. (2015) Inhibition of hypertrophy is a good therapeutic strategy in ventricular pressure overload. Circulation 131, 1435–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grossman W., Jones D., McLaurin L. P. (1975) Wall stress and patterns of hypertrophy in the human left ventricle. J. Clin. Invest. 56, 56–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zile M. R., Baicu C. F., Gaasch W. H. (2004) Diastolic heart failure: abnormalities in active relaxation and passive stiffness of the left ventricle. N. Engl. J. Med. 350, 1953–1959 [DOI] [PubMed] [Google Scholar]
- 9.Yip G. W., Fung J. W., Tan Y. T., Sanderson J. E. (2009) Hypertension and heart failure: a dysfunction of systole, diastole or both? J. Hum. Hypertens. 23, 295–306 [DOI] [PubMed] [Google Scholar]
- 10.Spoladore R., Maron M. S., D’Amato R., Camici P. G., Olivotto I. (2012) Pharmacological treatment options for hypertrophic cardiomyopathy: high time for evidence. Eur. Heart J. 33, 1724–1733 [DOI] [PubMed] [Google Scholar]
- 11.Sharma K., Kass D. A. (2014) Heart failure with preserved ejection fraction: mechanisms, clinical features, and therapies. Circ. Res. 115, 79–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spudich J. A. (2014) Hypertrophic and dilated cardiomyopathy: four decades of basic research on muscle lead to potential therapeutic approaches to these devastating genetic diseases. Biophys. J. 106, 1236–1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Colson B. A., Locher M. R., Bekyarova T., Patel J. R., Fitzsimons D. P., Irving T. C., Moss R. L. (2010) Differential roles of regulatory light chain and myosin binding protein-C phosphorylations in the modulation of cardiac force development. J. Physiol. 588, 981–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hanley P. J., ter Keurs H. E., Cannell M. B. (2004) Excitation-contraction coupling in the heart and the negative inotropic action of volatile anesthetics. Anesthesiology 101, 999–1014 [DOI] [PubMed] [Google Scholar]
- 15.Ding W., Li Z., Shen X., Martin J., King S. B., Sivakumaran V., Paolocci N., Gao W. D. (2011) Reversal of isoflurane-induced depression of myocardial contraction by nitroxyl via myofilament sensitization to Ca2+. J. Pharmacol. Exp. Ther. 339, 825–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meng T., Bu W., Ren X., Chen X., Yu J., Eckenhoff R. G., Gao W. D. (2016) Molecular mechanism of anesthetic-induced depression of myocardial contraction. FASEB J. 30, 2915–2925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Woll K. A., Weiser B. P., Liang Q., Meng T., McKinstry-Wu A., Pinch B., Dailey W. P., Gao W. D., Covarrubias M., Eckenhoff R. G. (2015) Role for the propofol hydroxyl in anesthetic protein target molecular recognition. ACS Chem. Neurosci. 6, 927–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ryan J. J., Archer S. L. (2014) The right ventricle in pulmonary arterial hypertension: disorders of metabolism, angiogenesis and adrenergic signaling in right ventricular failure. Circ. Res. 115, 176–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao W. D., Atar D., Backx P. H., Marban E. (1995) Relationship between intracellular calcium and contractile force in stunned myocardium. Direct evidence for decreased myofilament Ca2+ responsiveness and altered diastolic function in intact ventricular muscle. Circ. Res. 76, 1036–1048 [DOI] [PubMed] [Google Scholar]
- 20.Gao W. D., Murray C. I., Tian Y., Zhong X., DuMond J. F., Shen X., Stanley B. A., Foster D. B., Wink D. A., King S. B., Van Eyk J. E., Paolocci N. (2012) Nitroxyl-mediated disulfide bond formation between cardiac myofilament cysteines enhances contractile function. Circ. Res. 111, 1002–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao W. D., Backx P. H., Azan-Backx M., Marban E. (1994) Myofilament Ca2+ sensitivity in intact versus skinned rat ventricular muscle. Circ. Res. 74, 408–415 [DOI] [PubMed] [Google Scholar]
- 22.Dai T., Tian Y., Tocchetti C. G., Katori T., Murphy A. M., Kass D. A., Paolocci N., Gao W. D. (2007) Nitroxyl increases force development in rat cardiac muscle. J. Physiol. 580, 951–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fabiato A. (1981) Myoplasmic free calcium concentration reached during the twitch of an intact isolated cardiac cell and during calcium-induced release of calcium from the sarcoplasmic reticulum of a skinned cardiac cell from the adult rat or rabbit ventricle. J. Gen. Physiol. 78, 457–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rarick H. M., Tu X. H., Solaro R. J., Martin A. F. (1997) The C terminus of cardiac troponin I is essential for full inhibitory activity and Ca2+ sensitivity of rat myofibrils. J. Biol. Chem. 272, 26887–26892 [DOI] [PubMed] [Google Scholar]
- 25.Shiverick K. T., Thomas L. L., Alpert N. R. (1975) Purification of cardiac myosin: application to hypertrophied myocardium. Biochim. Biophys. Acta 393, 124–133 [DOI] [PubMed] [Google Scholar]
- 26.Uchida K., Murakami U., Hiratsuka T. (1977) Purification of cardiac myosin from rat heart proteolytic cleavage and its inhibition. J. Biochem. 82, 469–476 [PubMed] [Google Scholar]
- 27.Pardee J. D., Spudich J. A. (1982) Purification of muscle actin. Methods Enzymol. 85(Pt B), 164–181 [DOI] [PubMed] [Google Scholar]
- 28.Kron S. J., Spudich J. A. (1986) Fluorescent actin filaments move on myosin fixed to a glass surface. Proc. Natl. Acad. Sci. USA 83, 6272–6276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meijering E., Dzyubachyk O., Smal I. (2012) Methods for cell and particle tracking. Methods Enzymol. 504, 183–200 [DOI] [PubMed] [Google Scholar]
- 30.Sweeney H. L., Rosenfeld S. S., Brown F., Faust L., Smith J., Xing J., Stein L. A., Sellers J. R. (1998) Kinetic tuning of myosin via a flexible loop adjacent to the nucleotide binding pocket. J. Biol. Chem. 273, 6262–6270 [DOI] [PubMed] [Google Scholar]
- 31.Coureux P. D., Sweeney H. L., Houdusse A. (2004) Three myosin V structures delineate essential features of chemo-mechanical transduction. EMBO J. 23, 4527–4537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cammarato A., Dambacher C. M., Knowles A. F., Kronert W. A., Bodmer R., Ocorr K., Bernstein S. I. (2008) Myosin transducer mutations differentially affect motor function, myofibril structure, and the performance of skeletal and cardiac muscles. Mol. Biol. Cell 19, 553–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holmes K. C., Angert I., Kull F. J., Jahn W., Schröder R. R. (2003) Electron cryo-microscopy shows how strong binding of myosin to actin releases nucleotide. Nature 425, 423–427 [DOI] [PubMed] [Google Scholar]
- 34.Greenberg M. J., Moore J. R. (2010) The molecular basis of frictional loads in the in vitro motility assay with applications to the study of the loaded mechanochemistry of molecular motors. Cytoskeleton (Hoboken) 67, 273–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gorga J. A., Fishbaugher D. E., VanBuren P. (2003) Activation of the calcium-regulated thin filament by myosin strong binding. Biophys. J. 85, 2484–2491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dweck D., Reynaldo D. P., Pinto J. R., Potter J. D. (2010) A dilated cardiomyopathy troponin C mutation lowers contractile force by reducing strong myosin-actin binding. J. Biol. Chem. 285, 17371–17379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Homsher E., Kim B., Bobkova A., Tobacman L. S. (1996) Calcium regulation of thin filament movement in an in vitro motility assay. Biophys. J. 70, 1881–1892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li X. E., Tobacman L. S., Mun J. Y., Craig R., Fischer S., Lehman W. (2011) Tropomyosin position on F-actin revealed by EM reconstruction and computational chemistry. Biophys. J. 100, 1005–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ly S., Lehrer S. S. (2012) Long-range effects of familial hypertrophic cardiomyopathy mutations E180G and D175N on the properties of tropomyosin. Biochemistry 51, 6413–6420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chang A. N., Greenfield N. J., Singh A., Potter J. D., Pinto J. R. (2014) Structural and protein interaction effects of hypertrophic and dilated cardiomyopathic mutations in alpha-tropomyosin. Front. Physiol. 5, 460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Orzechowski M., Moore J. R., Fischer S., Lehman W. (2014) Tropomyosin movement on F-actin during muscle activation explained by energy landscapes. Arch. Biochem. Biophys. 545, 63–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Viswanathan M. C., Schmidt W., Rynkiewicz M. J., Agarwal K., Gao J., Katz J., Lehman W., Cammarato A. (2017) Distortion of the Actin A-triad results in contractile disinhibition and cardiomyopathy. Cell Reports 20, 2612–2625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Davis J., Davis L. C., Correll R. N., Makarewich C. A., Schwanekamp J. A., Moussavi-Harami F., Wang D., York A. J., Wu H., Houser S. R., Seidman C. E., Seidman J. G., Regnier M., Metzger J. M., Wu J. C., Molkentin J. D. (2016) A tension-based model distinguishes hypertrophic versus dilated cardiomyopathy. Cell 165, 1147–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Green E. M., Wakimoto H., Anderson R. L., Evanchik M. J., Gorham J. M., Harrison B. C., Henze M., Kawas R., Oslob J. D., Rodriguez H. M., Song Y., Wan W., Leinwand L. A., Spudich J. A., McDowell R. S., Seidman J. G., Seidman C. E. (2016) A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science 351, 617–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tardiff J. C., Carrier L., Bers D. M., Poggesi C., Ferrantini C., Coppini R., Maier L. S., Ashrafian H., Huke S., van der Velden J. (2015) Targets for therapy in sarcomeric cardiomyopathies. Cardiovasc. Res. 105, 457–470 [DOI] [PMC free article] [PubMed] [Google Scholar]