

Abstract
Case series
Patient: Female, 29 • Female, 69 • Female, 52 • Female, 71 • Male, 62 • Female, 67
Final Diagnosis: Neuroendocrine carcinoma
Symptoms: Abdominal pain
Medication: —
Clinical Procedure: —
Specialty: Oncology
Objective:
Unusual clinical course
Background:
Neuroendocrine tumors (NETs) encompass a diverse group of varying clinicopathological entities arising from cells of the endocrine and nervous systems. The presentation of these unique tumors can range from occult disease discovered incidentally to hyperactive, metastatic secretory tumors. NETs most commonly originate in the gastrointestinal and respiratory tract, although they may occur at any site in the body due to the wide distribution of neuroendocrine cells. Their classification system is complex and continues to evolve, and the current system uses histological grade in defining these subtypes. Neuroendocrine carcinomas (NECs), or high-grade, poorly-differentiated NETs, are the most aggressive subtype. Surgical resection remains the primary treatment modality and may be curative, thus early diagnosis is paramount. Management of advanced NETs remains both a diagnostic and therapeutic challenge; however, advances in our understanding of these unique neoplasms as well as an evolving classification system has led to the development of adjunctive therapeutic approaches aimed to minimize morbidity and improve patient outcomes.
Case Report:
We present 6 cases of unusual sites of high-grade neuroendocrine carcinomas involving the cervix, gallbladder, oesophagus, ovary, prostate, and urinary bladder.
Conclusions:
Our case series highlights the heterogenous and aggressive nature of this subtype of NETs as well as their diagnostic and therapeutic difficulties. We also review the evolution of the NET classification system and its impact on the management of these malignancies.
MeSH Keywords: Carcinoma, Neuroendocrine; Carcinoma, Small Cell; Receptors, Somatostatin
Background
Neuroendocrine tumors (NETs) represent a diverse group of rare neoplasms arising from cells of the neuroendocrine system. NETs most commonly originate in the gastrointestinal tract, although they may originate anywhere in the body due to the wide distribution of the neuroendocrine system [1–3]. Despite their malignant histopathological appearance, the majority of NETs are slow-growing with an indolent nature. Thus, the term ‘carcinoid’ was originally applied to describe a subset of these neoplasms, referring to their behavior as ‘cancer-like’ as opposed to the more aggressive behavior observed in other carcinomas [4–6]. However, advances in our understanding of the endocrine-related properties of these tumors resulted in progressive abandonment of this sobriquet and subsequent reclassification as neuroendocrine neoplasms [7,8].
NETs share a unifying feature in their ability to produce and secrete different peptides and neuroamines [9]. This hormone hyperactivity can therefore produce a wide array of clinical signs and symptoms. Nonfunctional, or non-hormone-secreting NETs, tend be more indolent, and symptoms from this subset of malignancies are usually caused by the mass effect of the tumor itself, often resulting in delayed presentation and diagnosis at a more advanced stage.
Attempts to achieve a unifying classification system of NETs to guide management has been frustrating, and current international grading systems incorporate histological characteristics such as tumor differentiation and tumor grade. Under the 2010 World Health Organization classification scheme, NETs may be classified as grade (G) 1 NETs, G2 NETs, and G3 neuroendocrine carcinomas, which have small-cell and large-cell subtypes. These histologic grades are assigned based on mitotic counts and the Ki-67 labeling (proliferation) index. Neuroendocrine carcinomas (NECs) are poorly-differentiated NETS and resemble small-cell neuroendocrine carcinomas of the lung. They are characterized by a high mitotic count and Ki-67 index, and are the most aggressive NET subtype, and thus have the worst prognosis [10].
We present 6 patients with unusual sites of high-grade, poorly-differentiated neuroendocrine carcinomas which highlight the heterogenous nature of this aggressive subtype and difficulties with regards to management. We also conducted a literature review and discuss the ever-changing classification system of these unique malignancies.
Case Report
Patient #1: Small-cell neuroendocrine carcinoma of the cervix
A 29-year-old woman presented with a lesion on her cervix which was observed during childbirth. She was asymptomatic prior to giving birth. An MRI pelvis demonstrated a large exophytic lesion arising from the lower cervix and distending and distorting the endocervical canal close to the external os (Figure 1). This frond-like exophytic mass lesion measured at least 72×43 mm. The lesion expanded to the lower cervix and extended into the upper vagina. There was also a suspicious small lymph node identified in the left obturator station, measuring 10 mm. A staging CT revealed no distant metastasis. Six weeks postpartum the cervical lesion was biopsied. Histopathology revealed fragments of mostly necrotic tumor which focally expressed keratin (CAM 5.2 [BD Biosciences, CA, USA, 1: 10] in a dot-like staining pattern) and neuroendocrine markers (focal positivity for synaptophysin [Dakocytomation, Hamburg, Germany, 1: 100] and chromogranin A [Dakocytomation, Hamburg, Germany,1: 500] with strong positivity for neural cell adhesion molecule [CD56] [ThermoScientific, CA, USA, 1: 100]) (Figure 2). These features were consistent with a diagnosis of high-grade, poorly-differentiated neuroendocrine carcinoma of the cervix, FIGO Stage IB, or limited stage.
Figure 1.

MRI pelvis. Sagittal (A) and coronal (B) section demonstrating a large exophytic lesion (red arrow) arising from the lower cervix and distending and distorting the endocervical canal, which is in intimate contact with the external os. The lesion measures at least 72×43 mm, extending to the lower cervix, and on the sagittal images extending into the upper vagina.
Figure 2.

Small-Cell neuroendocrine carcinoma of the cervix. The tumor cells consist of diffuse sheets of small malignant cells (A) with extensive necrosis (B) and frequent mitotic activity (C). Immunohistochemistry shows positivity for neuroendocrine marker CD56 (D).
She commenced chemotherapy and received 4 cycles of cisplatin and etoposide, which was well tolerated. She then received consolidative radiation to the pelvis and a dose of 45 Gray (Gy) in 25 fractions (Fr) followed by 2 brachytherapy sessions. A restaging MRI pelvis reported a complete response, with no residual tumor seen. She proceeded to prophylactic cranial irradiation with 30 Gy in 10 fractions. She remains in remission 9 years after diagnosis.
Patient #2: Small-cell neuroendocrine carcinoma of the gallbladder
A 69-year-old woman presented with a 4-month history of upper-abdominal discomfort.
She attended her General Practitioner who requested an ultrasound, which revealed gallstones. The patient elected to proceed to surgery and underwent a cholecystectomy. Histopathological examination of the gallbladder specimen revealed small-cell undifferentiated carcinoma with focal tumor necrosis and invasion into the lymphovascular space and liver parenchyma. The tumor was focally positive for CD56, and Ki-67 labeling index showed a high proliferation rate with almost 100% staining (Figure 3). A staging CT revealed liver metastases and necrotic regional lymph nodes (Figure 4).
Figure 3.

Small-cell neuroendocrine carcinoma of the gallbladder. Histopathological examination of the gallbladder specimen reveals involvement of all layers of the gallbladder wall and focal tumor necrosis (A, B). On Immunohistochemistry, CD 56 shows diffuse positive membranous staining (C) and Ki 67 shows a high proliferation rate with almost 100% staining (D).
Figure 4.
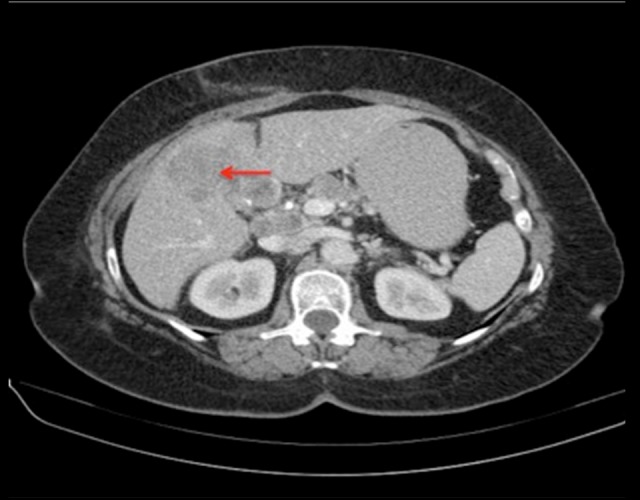
Transaxial CT image demonstrates a large hypoenhancing area in the medial aspect of the left hepatic lobe predominantly involving segment 4B (red arrow). Lymphadenopathy involving the nodes in the porta hepatis is also noted.
She received cisplatin and etoposide and tolerated treatment well. A restaging CT after 3 cycles revealed a reduction in size of the coeliac axis and porta hepatis lymph nodes. However, there were new multiple liver metastases within the right lobe of the liver. She remained clinically well and proceeded to second-line FOLFIRI. Her treatment course was complicated by 2 admissions due to nausea and severe hyponatremia caused by paraneoplastic syndrome. After 4 cycles, she was admitted with increasing shortness of breath and right sided pleuritic chest pain. A CT pulmonary angiogram revealed no evidence of a pulmonary embolism but reported progressive liver disease. She deteriorated clinically over the next few days and died 6 months after her initial diagnosis.
Patient #3: High-grade neuroendocrine carcinoma of the oesophagus
A 52-year-old woman presented with a 3-month history of epigastric discomfort, dysphagia to solids, and weight loss. An esophagoduodenostomy (OGD) was performed and revealed a mid-oesophageal ulcer. Histopathological analysis of the biopsies was consistent with a high-grade neuroendocrine carcinoma (Figure 5). Immunohistochemistry revealed positivity for neuroendocrine markers CD56 and thyroid transcription factor 1 (TTF-1) (Leica Microsystems, Newcastle Upon Tyne, UK, 1: 100). A staging CT revealed thickening of the wall of the mid-oesophagus corresponding to the site of the ulcer seen on esophago-gastroduodenoscopy (OGD) (Figure 6). There was also mediastinal lymphadenopathy and multiple bilobar liver metastases reported on imaging.
Figure 5.

High-grade neuroendocrine carcinoma of the oesophagus with liver metastases. Histopathological analysis of the biopsied liver lesion reveals appearances consistent with metastatic high-grade neuroendocrine carcinoma (A). On immunohistochemistry there is positivity for epithelial marker CK 7 (B) and neuroendocrine marker CD 56 (C). Ki-67 shows a high proliferative index (D).
Figure 6.
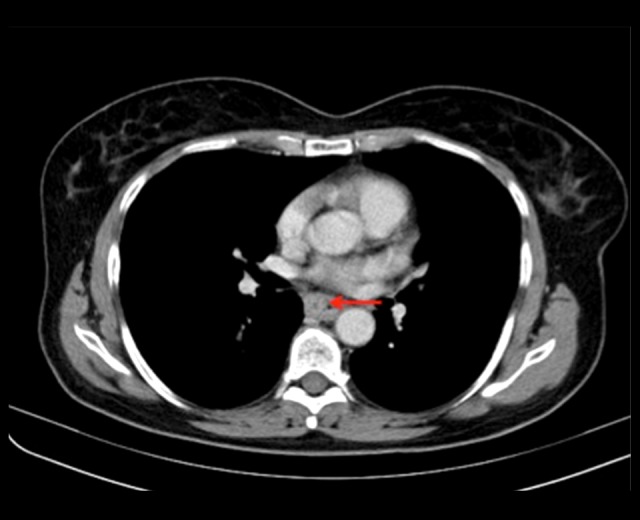
Transaxial section demonstrates circumferential thickening of the wall of the mid-oesophagus (red arrow), corresponding to the site of the ulcer discovered on endoscopy. There are also enlarged lymph nodes in the aorto-pulmonary window and paraaortic regions.
She proceeded to 3 cycles of carboplatin and etoposide, which she tolerated well. A restaging CT to assess response revealed significant reduction in size of the mediastinal adenopathy, oesophageal thickening, and focal liver lesions. She completed a further 3 cycles of chemotherapy and remained clinically well. However, a restaging CT revealed progression of disease in the liver as well as interval enlargement of the mediastinal lymph nodes.
She commenced second-line treatment with cyclophosphamide, doxorubicin, and vincristine (CAV) and tolerated 6 cycles without complications. Unfortunately, a CT reported further progression of disease in the liver. A liver biopsy was performed and again revealed metastatic neuroendocrine carcinoma. Staining was positive for CD56, synaptophysin and neuron-specific enolase (NSE) (Dakocytomation, Hamburg, Germany, 1: 1500). Ki-67 labeling index showed a proliferation index of approximately 90%.
Despite her disease appearing to be refractory to treatment, she remained well both clinically and biochemically, with normal liver function. She started third-line chemotherapy with single-agent irinotecan. During treatment she suffered from increased nausea, vomiting, and diarrhea.
Approximately 20 months after her initial diagnosis, after 6 cycles of third-line chemotherapy, she presented to the Emergency Department with seizures. A CT brain revealed multiple enhancing lesions identified at scattered sites bilaterally in the cerebellum and cerebrum, consistent with brain metastases (Figure 7). A CT revealed progressive metastatic disease in the liver and several small retroperitoneal lymph nodes. She received palliative whole-brain radiation therapy (30 Gy in 10 treatments). She was discharged home with palliative care support but continued to deteriorate and died 6 weeks later.
Figure 7.
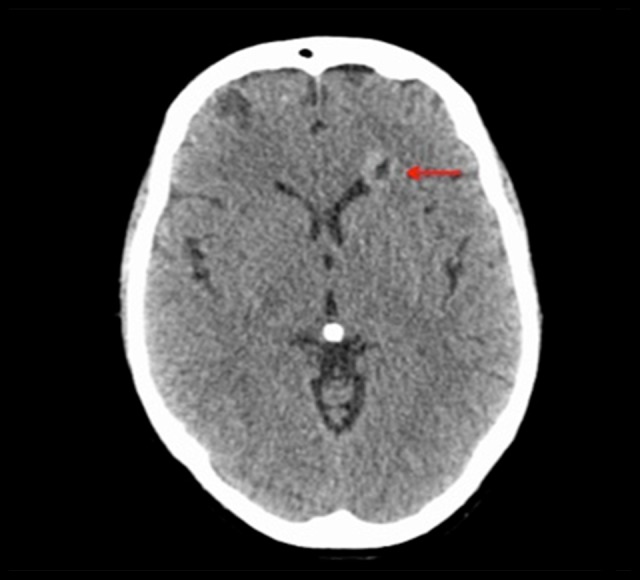
CT Brain with contrast demonstrates one of many bilateral enhancing lesions in the cerebrum (red arrow), compatible with multiple metastases.
Patient #4: Small-cell neuroendocrine carcinoma of the ovary
A 71-year-old woman presented with a 2-month history of abdominal swelling and increasing discomfort, nausea, reduced appetite, and fatigue. Physical examination revealed a palpable pelvic mass. A routine blood panel was within normal limits. However, CA-125 was noted to be elevated at 1170 U/ml.
A CT revealed a 17-cm pelvis mass, abdominal ascites, and intra-peritoneal deposits, as well as a small left pleural effusion (Figure 8). She underwent ultrasound-guided drainage of the ascites, with symptom relief and percutaneous biopsy of the pelvis mass. Histopathological analysis showed a poorly-differentiated carcinoma with morphological features of small-cell carcinoma (Figure 9). Immunohistochemistry revealed tumor cells expressing cytokeratin (CK) AE1/AE3 (Dakocytomation, Hamburg, Germany 1: 70) and Wilms tumor protein (WT-1) (Dakocytomation, Hamburg, Germany, 1: 150). There was weak expression of chromogranin A and very focal expression of CD56, confirming a diagnosis of stage IIIC high-grade small-cell neuroendocrine carcinoma of the ovary.
Figure 8.

CT Coronal (A) and sagittal (B) sections. There is a large soft-tissue mass measuring 17×91 mm in the pelvis, involving the serosa of the sigmoid colon and rectum and obscuring the uterus and ovaries (red arrows), as well as mass effect on the bladder. There is also large-volume intra-abdominal ascites with peritoneal nodularity consistent with intra-abdominal spread of malignancy.
Figure 9.

Histopathological analysis of the pelvis mass biopsy reveals dense sheets of small cells with scanty cytoplasm, hyperchromatic nuclei, frequent mitoses, and inconspicuous nucleoli, consistent with small-cell carcinoma (A, B). On immunohistochemistry, CK AE1/AE3 shows positive staining (C) and WT-1 is also positive (D).
She completed 6 cycles of palliative cisplatin and etoposide and tolerated treatment well. A restaging scan revealed a good partial response, with a reduction in size of the pelvic mass. The peritoneal nodularity and ascites had resolved. Her images were discussed at the oncology MDM and, in view of the residual disease, consolidative irradiation was recommended. She received external beam radiation therapy to the residual pelvic mass, receiving a total dose of 50 Gy in 25 treatments.
She remained on surveillance, but a restaging CT 3 months later showed significant recurrent disease throughout the abdomen and pelvis. She elected to proceed with second-line chemotherapy in the form of CAV. Treatment is ongoing.
Patient #5: High-grade neuroendocrine carcinoma of the prostate
A 62-year-old man was diagnosed with an adenocarcinoma of the prostate, Gleason score 6 (3+3), in 2010. He was treated with radical radiotherapy and brachytherapy as well as hormonal therapy for 6 months. Six years later, although asymptomatic, a gradually rising PSA led to a repeat prostatic biopsy. Histopathological analysis revealed the presence of prostatic carcinoma in almost all biopsy specimens. In addition, immunohistochemistry revealed strong positivity for synaptophysin, and less intensive but definite positivity for CD56 and chromogranin A, supporting a diagnosis of high-grade neuroendocrine carcinoma (Figure 10).
Figure 10.

High-grade neuroendocrine carcinoma of the prostate. Histopathological analysis reveals the presence of prostatic neuroendocrine carcinoma in practically all biopsy specimens (A, B). On immunohistochemistry, synaptophysin shows positive cytoplasmic staining (C) and CD56 shows positive membranous staining (D).
An MRI pelvis revealed a prostatic adenocarcinoma with left para-aortic, right internal iliac, and left external iliac lymphadenopathy, stage T3bN2Mx (Figure 11). The right seminal vesicle and right neurovascular bundle were also involved by disease. A staging CT revealed numerous bilateral lung nodules however these were felt to be related to his history of sarcoidosis. To date he has received 5 cycles of cisplatin and etoposide. Treatment is ongoing.
Figure 11.
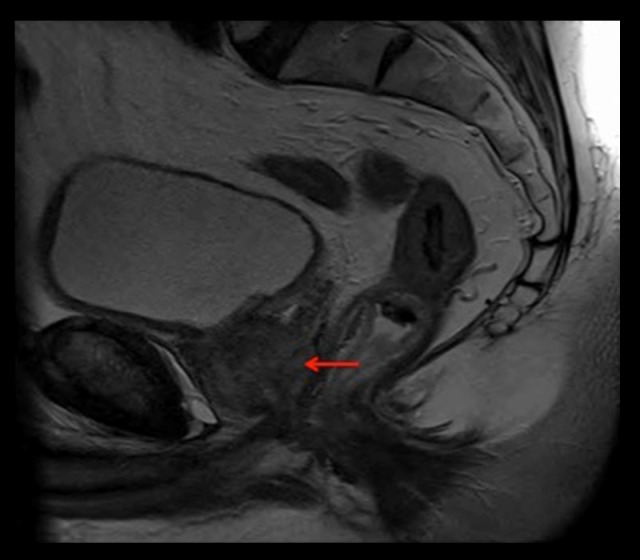
MRI pelvis demonstrates a stage T3b N2 Mx lesion of the prostate, gland volume 37 cc (red arrow). The right seminal vesicle and right neurovascular bundle are involved by disease. Left para-aortic, right internal iliac, and left external iliac lymphadenopathy are also shown.
Patient #6: High-grade neuroendocrine carcinoma of the urinary bladder
A 67-year-old woman presented with a 3-week history of painless hematuria. Cystoscopy demonstrated a sizeable bladder mass for which she underwent a transurethral resection of a bladder tumor. Histology revealed a high-grade poorly-differentiated neuroendocrine carcinoma of the bladder, large-cell type, with deep submucosal and detrusor invasion. Ki-67 labeling index showed a very high proliferation fraction of virtually 100%. The tumor stained positive for CD 56 and synaptophysin. Staging investigations revealed very mild diffuse thickening along the central and right lateral aspect of the urinary bladder fundus, consistent with the macroscopic location of the bladder tumor. There was no evidence of metastatic disease or regional lymphadenopathy (Figure 12). She had indeterminate pulmonary nodules possibly related to a 30-pack-year history of smoking and was not felt to be suspicious for meta-static disease. Thus, final staging of the tumor was pT3N0M0.
Figure 12.
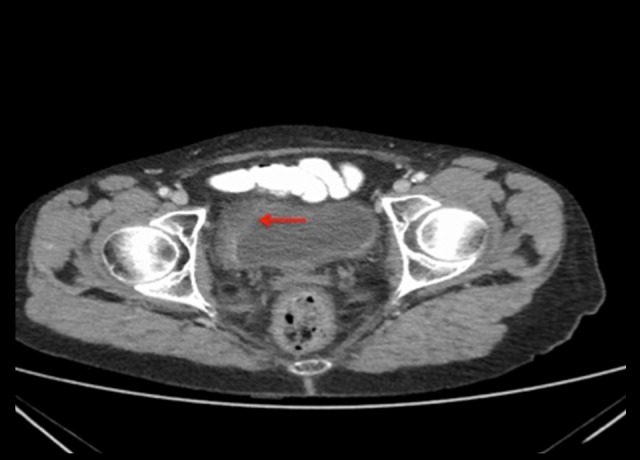
CT Pelvis demonstrates thickening along the central and right lateral aspect of the urinary bladder fundus, consistent with the previous location of the bladder tumor (red arrow), with induration of surrounding fat.
She received a radical treatment of radiotherapy (50 Gy in 25 fractions) with concurrent systemic chemotherapy (4 cycles of cisplatin and etoposide), which was well tolerated. She continued on surveillance with regular cystoscopies and remains well with no evidence of disease recurrence at 2.5 years since diagnosis.
Discussion
Neuroendocrine tumors (NETs) are a heterogenous group of unique neoplasms that originate from cells of the diffuse neuroendocrine system, and may be found throughout the body [9,11]. NETs are most commonly observed in the gastrointestinal tract and lung, where a rich supply of these cells can be found. The cells of the neuroendocrine system are characterized by their ability to produce polypeptide hormones and biogenic amines, such as catecholamines and serotonin, which can also be found in neurons [12,13]. This hormone hypersecretion can result in dramatic clinical symptoms and management can be challenging.
The most recent data from Ireland reports an incidence of 4.6 cases of invasive NETS (excluding those of the lung) per 100 000 population per year [14]. Since 1994, there has been a fairly steady increase in cases, thought to be due to more sensitive imaging techniques. Previously, the prognosis of these patients was poor, with a 5-year relative survival (RS) for all invasive NETs of 35–50%. However, this has dramatically improved with more radical surgical approaches and the development of systemic treatment options, with 5-year survival rates now at 75–95% [14–16].
The majority of NETs are sporadic and risk factors for tumorigenesis are poorly understood. A genetic component has also been recognized. Mutations in genes promoting neuroendocrine carcinogenesis have been observed in inherited genetic syndromes, including MEN types 1 and 2 and von Hippel-Lindau syndrome (VHL) [1,5]. Approximately two-thirds of sporadic NETs are found in the diffuse endocrine system of the gastrointestinal tract, which functions to regulate secretion, absorption, and motility of the gastrointestinal tract [17]. Pancreatic NETs include insulinomas, gastrinomas, VIPomas, and glucagonomas. Another one-quarter occur in the lungs and thymus. Catecholamine-secreting tumors (e.g., pheochromocytomas and paragangliomas), medullary carcinoma of the thyroid, chromophobe pituitary tumors, small-cell lung cancer, and Merkel cell tumors are also included.
NETs are typically slow-growing, and the term ‘carcinoid’ was originally coined to describe their cancer-like morphology [6]. The histology of carcinoid tumors was initially described by Langhans in 1867 [18], and Lubarsch was the first to report 2 cases of ileal carcinoid tumors at autopsy in 1888 [19,20]. The german pathologist Obendorfer was credited with applying the term carcinoid to describe their unique characteristic in exhibiting carcinoma-like morphology under the microscope despite their relatively benign clinical behavior. Advances in microscopic and immunohistochemistry techniques led to further comprehension of their endocrine-related properties, subsequently leading to their reclassification as neuroendocrine neoplasms.
NETs have the unique potential of secreting hormones and vasoactive peptides, and can thus be further classified as functional, or hormone secreting, or nonfunctional. This characteristic gave them another sobriquet of APUDomas, assigned due to their high amine precursor uptake (L-DOPA and 5-hydroxytryptophan) and their high content of the enzyme amino acid decarboxylase [4,9,11]. The release of the monoamine neurotransmitter serotonin and its ‘flushing’ effect was first described by Kulchitsky in 1953, but fewer than 10% of patients suffer from the ‘carcinoid syndrome’ of flushing, hypotension, diarrhea, wheezing, and heart disease caused by hormone overproduction [4]. While nonfunctional tumors may also secrete peptides, which are as yet undetectable, these tumors tend to be more aggressive and present at a more advanced stage, with metastases observed in approximately 40% of patients at diagnosis [21]. Symptoms are largely due to the mass effect caused by tumor bulk and can cause symptoms such as abdominal pain or jaundice caused by biliary obstruction [1,22]. Weight loss, nausea, and vomiting are also frequently observed [1,23].
Attempts to classify NETs to facilitate prognostic indicators have been frustrating, and to date there is no universal classification system for NETs of all tumor sites. The nomenclature of NETs has evolved considerably over the last few decades. Originally, the 1980 WHO classification focused on staining techniques to classify NETs into enterochromaffin cell, gastrin cell, and other carcinoids [7]. However, in 1995 the term carcinoid was replaced by the term neuroendocrine tumor (NET) [7,8]. Initially NETs were classified into 4 groups (I to IV) based upon size and angioinvasion: benign, benign or low-grade malignant, low-grade malignant, and high-grade malignant. The WHO 2000 classification then used degree of differentiation to divide NETs from the GI and pancreatobiliary tracts into well-differentiated endocrine tumors, well-differentiated endocrine carcinomas, and poorly-differentiated endocrine carcinomas [24]. Well-differentiated endocrine tumors were further classified into benign tumors and low-grade malignant tumors, based on the tumor size, mitotic rate, Ki-67 labeling index, lymphovascular invasion, and sites of metastases [24]. Poorly-differentiated endocrine carcinomas encompass small-cell and large-cell carcinomas and are considered high-grade malignant tumors. However, both well-differentiated and poorly-differentiated endocrine carcinomas are invasive cancers with the ability to metastasize to distant organs [24].
The most recent histological classification system of the European Neuroendocrine Tumor Society (ENETs) and the World Health Organization (WHO) incorporate tumor differentiation and grade, which often correlate with mitotic count and Ki-67 proliferation index [10]. In general, the majority of NETs fall into one of 3 broad histological categories (Table 1):
Well-differentiated, low-grade (grade 1);
Well-differentiated, intermediate grade (grade 2);
Poorly-differentiated, high-grade (grade 3).
Table 1.
2010 ENETS/WHO Nomenclature and Classification for Neuroendocrine neoplasms arising in the gastrointestinal tract [10].
| Differentiation | Grade | Mitotic count | Ki-67 Index | Traditional | ENETS/WHO |
|---|---|---|---|---|---|
| Well-differentiated | Low-grade (Grade 1) | <2 per 10 HPF | < 3% | Carcinoid, Islet cell, pancreatic (neuro) endocrine tumor | Neuroendocrine Tumor (NET) Grade 1 |
| Intermediate-grade (Grade 2) | 2–20 per 10 HPF | 3–20% | Carcinoid, atypical carcinoid, Islet cell, pancreatic (neuro) endocrine tumor | Neuroendocrine Tumor (NET) Grade 2 | |
| Poorly-differentiated | High-grade (Grade 3) >20 per 10 HPF >20% Large-cell neuroendocrine carcinoma Neuroendocrine Tumor (NET) Grade 3, large-cell |
Small-cell carcinoma | Neuroendocrine Tumor (NET) Grade 3, small-cell |
ENETS – European Neuroendocrine Tumor Society; WHO – World Health Organization; HPF – high-power fields.
Increased mitotic rate and high Ki-67 labeling index are associated with a more aggressive clinical course and worse prognosis, and are most often observed in the poorly-differentiated, grade 3 subtypes, which may be further divided into small-cell and large-cell variants [25,26]. The proliferative rate in most cases of these grade 3 NECs is well in excess of the cutoffs proposed to distinguish them from well-differentiated NETs (>20 percent Ki-67 labeling index, >20 per 10 high-power fields [HPF] mitotic rate). Furthermore, recent studies have suggested this new classification system better reflects behavior and can predict metastatic potential and risk of recurrence, but some studies suggest anatomical site should also be included in this system [27,28].
While NECs are included in many published studies of NETs of the digestive system, they represent a distinct subgroup, with aggressive clinical behavior resembling that of small-cell carcinoma or large-cell NEC of the lung. In the largest series of poorly-differentiated pancreatic NEC, 88% of the patients had lymph node or distant metastatic disease at presentation, reflecting their high growth fraction and propensity for early dissemination [29]. The median survival was 11 months (range 0 to 104 months), and the 2- and 5-year survival rates were 22.5% and 16.1%, respectively, markedly worse than their grade 1 and 2 counterparts [29].
This heterogeneity associated with NETs creates a diagnostic dilemma. Limitations in diagnostic imaging modalities led to the publication of the Delphi consensus, which aimed to standardize diagnostic imaging of neuroendocrine tumors [30]. Anatomic imaging in the form of CT, MRI, PET, transabdominal ultrasonography, GI endoscopy, and endoscopic ultrasonography may be used to evaluate staging and achieve a tissue diagnosis. Functional imaging, in the form of somatostatin receptor scintigraphy (SRS) using radiolabeled octreotide, also provides a useful diagnostic adjunct in well-differentiated sub-types as they may target receptors, uptake pathways, or metabolism unique to NETs. Furthermore, combined use of anatomic and functional imaging has been proven to increase the accuracy of staging in the diagnosis of NETs [31].
Serology is also important in the identification of various circulating biochemical markers and can be used as a diagnostic adjunct as well as an indicator of tumor burden and response to therapy. Immunohistochemistry (IHC) markers commonly used to demonstrate neuroendocrine differentiation include chromogranin A, a secretory granule in large dense-core vesicles of neuronal and neuroendocrine cells, and their levels appear to be highest in carcinoid tumors, MTCs, and pheochromocytomas [32]. Chromogranin B, neuron-specific enolase (NSE), and urinary 5-HIAA quantification may also be useful [9].
Our first patient was diagnosed with small-cell neuroendocrine carcinoma of the cervix, based on focal positivity for synaptophysin and chromogranin A, and strongly positive when stained for CD56 and other IHC markers associated with NECs. NECs of the cervix are a rare clinical entity, observed in only 1–3% of all cervical tumors [33,34]. These tumors may be associated with local aggression and involvement of papillomavirus, typically observed in cervical cancer, as well as early dissemination of the disease, which is observed in small-cell NECs of any site.
Our second patient was diagnosed with a neuroendocrine carcinoma of the gallbladder (GB), which is extremely rare, representing only 0.2% of all NETs [35,36]. Most cases are detected incidentally during histological examination after cholecystectomy. It is postulated that GB NETs originate from either a multipotent stem cell or neuroendocrine cells in intestinal or gastric metaplasia of the gallbladder epithelium, which occurs following cholelithiasis and or chronic inflammation [35].
Large-cell NECs of the oesophagus, as described in our third patient, have only been described in isolated case reports, with a reported incidence between 0.05% and 2.4% [25,37–41].
Primary ovarian carcinoma of small-cell subtype, diagnosed in our fourth patient, is also a rare clinical entity, with an incidence of less than 1%. We know of only 1 case series of 11 patients with this type of histopathology published in the literature [42]. In two-thirds of the patients, small-cell ovarian neuroendocrine carcinomas are associated with paraneoplastic hypercalcemia, but the calcium levels in our patient were in the normal range. This tumor tends to occur in younger women and is associated with poor prognosis. The 1-year survival rate is 50%, with an overall 5-year survival rate of approximately 10% [43].
Small-cell NEC of the prostate is another rare origin of high-grade NETs that is responsible for less than 0.5–1% of all prostate cancers and is classically associated with a more aggressive clinical course and poor prognosis [44,45]. This tumor can occur concomitantly with adenocarcinomas or as isolated disease, and approximately one-half have mixed tumors [46]. Furthermore, PSA levels do not correlate with burden of disease, as was the case in our patient [47].
Finally, NECs of the urinary bladder are rare, as over 90% of cancers of the bladder are urothelial carcinomas, and the 2 most common non-urothelial epithelial malignancies of the bladder are squamous cell carcinomas and adenocarcinomas. NECs of the bladder are thought to represent only 0.5–1% of primary bladder malignancies [48,49].
The primary treatment for NECs is surgical resection and should be attempted even if the tumor size is small or slow-growing, as it may relieve symptoms of excessive hormone production. However, surgery as a single-treatment strategy is rarely curative. Rates of relapse after radical surgery are high, thus guidelines recommend platinum-based adjuvant therapy in this setting [10]. In patients who are not surgical candidates due to comorbidities or tumor localization, a definitive course of radiotherapy and chemotherapy may be a reasonable alternative approach.
A multimodality approach is recommended in advanced disease, and systemic chemotherapy plays an important role. While overall survival of patients with metastatic NECs treated with chemotherapy varies widely (from 7 to 19 months), it still represents a considerable improvement over those opting for best supportive care (1 month) [10].
Despite a lack of large prospective trials, platinum-based chemotherapy is considered the standard treatment based on extrapolation of data from small-cell lung cancer, with response rates in the largest, most recent series of approximately 30% and median survival of around 1 year [10,50–55]. Of note, greater response rates have been reported in those patients with high Ki-67 values (greater than 55%), observed in the more aggressive NECs (42% vs. 15%), although a poorer survival (10 vs. 14 months) was also reported when compared to those patients with lower Ki-67 values (less than 55%) [56]. Evidence for salvage therapy in patients progressing on first-line platinum-based regimens is limited; however, oxaliplatin-based (FOLFOX) or irinotecan-based (FOLFIRI) regimens have produced response rates of 23–40% in small series [10,56–59].
Of interest, several studies, predominantly based on pancreatic NETs, have raised questions that challenge the assumption that poorly-differentiated histology and high tumor grade are equivalent [60–64]. These studies report a small subset of patients with NETs that appear histologically well-differentiated, with fewer than 20 mitoses per 10 HPF (G2 by mitotic count) but are associated with high Ki-67 proliferation indices (>20%) that fall into the G3 range. The clinical behavior of this intermediate group appears more aggressive than in well-differentiated G2 tumors but less aggressive than the poorly-differentiated NECs [60,65]. Optimal management of this select subgroup is unclear, but clinical data suggest the G3 well-differentiated NETs are less likely to respond to platinum-based chemotherapy than are poorly-differentiated NECs, thus alternative therapeutic strategies may be needed [66].
With regards to well-differentiated NETs, management with long- and intermediate-acting somatostatin analogues (lantreo-tide and octreotide, respectively) has become an important component of therapy for carcinoid tumors. These agents are useful in controlling symptoms due to the frequent tumor expression of somatostatin receptors such as somatostatin receptor 2 (SSTR-2) [66]. Use of long-acting somatostatin analogues has led to symptomatic response rates of up to 75% in functional NET and temporary tumor shrinkage in 9% [67,68]. Unfortunately, the use of these agents is rarely associated with tumor regression [5]. Recent advances, however, in our understanding of the molecular pathogenesis of these neoplasms has led to the development of molecular therapies targeting the mammalian target of rapamycin (mTOR) and tyrosine kinase receptors, such as everolimus and sunitinib, respectively, and has resulted in improved survival even in some patients with advanced disease [5].
Finally, the role of prophylactic cranial irradiation (PCI) in high-grade, poorly-differentiated neuroendocrine carcinomas is unclear. While extrapulmonary small-cell neuroendocrine cancer may share certain features with pulmonary NECs, as mentioned above, previous studies have shown differences in etiology, clinical trajectory, and survival [69,70]. Furthermore, the incidence of brain metastases appears to be lower than in pulmonary SCLC; therefore, PCI is not routinely recommended [69–72]. Research with larger series is needed to further evaluate the potential role for PCI [73].
Conclusions
The prognosis for patients with NETs has improved considerably with the evolution of their classification system, coupled with more aggressive surgical intervention. Therapeutic advances for well-differentiated NETs have also contributed with the development of long-acting somatostatin agonists and targeted second-line therapy. Recent studies have demonstrated that advanced disease may have 5-year survival rates as high as 77% to 95% when treated aggressively with resection of primary tumor and adjunctive therapy, representing a marked improvement when compared with older studies reporting a much lower survival rate of 36% at 5 years [56].
NECs, however, constitute a more aggressive subtype with a rapid clinical course, and survival and prognosis are markedly worse in these patients. This case series highlights 6 unusual presentations of this aggressive subtype, showing its heterogeneity and difficulties in management. While similar cases have been described in isolated case reports, to our knowledge this is the largest case series describing the broad and diverse origins of NECs. There is also evidence to suggest that the classification of grade 3 poorly-differentiated NECs may continue to evolve, improving our therapeutic approach to optimize patient outcomes.
Footnotes
Conflicts of interest
None.
References:
- 1.Mougey AM, Adler DG. Neuroendocrine tumors: Review and clinical update. Hospital Physician. 2007;43(11):12–20. [Google Scholar]
- 2.Taal BG, Visser O. Epidemiology of neuroendocrine tumors. Neuroendocrinology. 2004;80(Suppl. 1):3–7. doi: 10.1159/000080731. [DOI] [PubMed] [Google Scholar]
- 3.Modlin IM, Lye KD, Kidd M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer. 2003;97:934–59. doi: 10.1002/cncr.11105. [DOI] [PubMed] [Google Scholar]
- 4.Arnold R, Göke R, Wied M, Behr T. Neuroendocrine gastroentero-pancreatic (GEP) tumors (Chapter 15) In: Scheppach W, Bresalier RS, Tytgat GNJ, editors. Gastrointestinal and liver tumors. Berlin Heidelberg: Springer-Verlag; 2003. pp. 195–233. [Google Scholar]
- 5.Kapoor R, Bhattacharyya T, Gupta R, et al. A systematic review of management of neuroendocrine tumors: An experience from a tertiary care centre from India. Clin Cancer Investig J. 2014;3:363–72. [Google Scholar]
- 6.Oberndorfer S. [Carcinoid tumors of the small intestine] Frankf Z Pathol. 1907;1:425–29. [in German] [Google Scholar]
- 7.Capella C, Heitz PU, Höfler H, et al. Revised classification of neuroendocrine tumors of the lung, pancreas and gut. Virchows Arch. 1995;425:547. doi: 10.1007/BF00199342. [DOI] [PubMed] [Google Scholar]
- 8.Modlin IM, Shapiro MD, Kidd M. Siegfried oberndorfer: Origins and perspectives of carcinoid tumors. Hum Pathol. 2004;35(12):1440–51. doi: 10.1016/j.humpath.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 9.Barakat MT, Meeran K, Bloom SR. Neuroendocrine tumors. Endocr Relat Cancer. 2004;11:1–18. doi: 10.1677/erc.0.0110001. [DOI] [PubMed] [Google Scholar]
- 10.Garcia-Carbonero R, Sorbye H, Baudin E, et al. ENETS consensus guidelines for high-grade gastroenteropancreatic neuroendocrine tumors and neuroendocrine carcinomas. Neuroendocrinology. 2016;103(2):186–94. doi: 10.1159/000443172. [DOI] [PubMed] [Google Scholar]
- 11.Polak JM, Bloom SR. The diffuse neuroendocrine system. Studies of this newly discovered controlling system in health and disease. J Histochem Cytochem. 1979;27:1398–400. doi: 10.1177/27.10.512327. [DOI] [PubMed] [Google Scholar]
- 12.Pearse AG. Common cytochemical and ultrastructural characteristics of cells producing polypeptide hormones (the APUD series) and their relevance to thyroid andultimobranchial C cells and calcitonin. Proc R Soc Lond B Biol Sci. 1968;170(1018):71–80. doi: 10.1098/rspb.1968.0025. [DOI] [PubMed] [Google Scholar]
- 13.Pearse AG. The cytochemistry and ultrastructure of polypeptide hormone-producing cells of the APUD series and the embryologic, physiologic and pathologic implications of the concept. J Histochem Cytochem. 1979;17:303–13. doi: 10.1177/17.5.303. [DOI] [PubMed] [Google Scholar]
- 14.National Cancer Registry of Ireland (NCRI) Cancer trends No 18. – Neuroendocrine cancers. 2013. Available from: https://www.ncri.ie/sites/ncri/files/pubs/CancerTrendsNo.18-NeuroendocrineCancers.pdf.
- 15.Hausman MS, Thompson NW, Gauger PG, Doherty GM. The surgical management of MEN-1 pancreatoduodenal neuroendocrine disease. Surgery. 2004;136:1205–11. doi: 10.1016/j.surg.2004.06.049. [DOI] [PubMed] [Google Scholar]
- 16.Norton JA, Kivlen M, Li M, et al. Morbidity and mortality of aggressive resection in patients with advanced neuroendocrine tumors. Arch Surg. 2003;138:859–66. doi: 10.1001/archsurg.138.8.859. [DOI] [PubMed] [Google Scholar]
- 17.Kurirya H, Swiedb AM. Large-cell neuroendocrine carcinoma of the esophagus: A case from Saudi Arabia. Case Rep Gastroenterol. 2015;9(3):327–34. doi: 10.1159/000441381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Langhans T. [About a gland polyp in the ileum.] Virchows Arch Pathol Anat. 1867;38:550–60. [in German] [Google Scholar]
- 19.Lubarsch O. [About the primary cancer of the ileum, along with remarks about the coexistence of cancer and tuberculosis.] Virchows Arch Pathol Anat. 1888;111:280–317. [in German] [Google Scholar]
- 20.Kaplan EL. The carcinoid syndromes. In: Friesen SR, editor. Surgical endocrinology: Clinical syndromes. 1st ed. Philadelphia, PA: J.B. Lippincott; 1978. pp. 120–44. [Google Scholar]
- 21.Matthews BD, Heniford BT, Reardon PR, et al. Surgical experience with nonfunctioning neuroendocrine tumors of the pancreas. Am Surg. 2000;66:1116–22. [PubMed] [Google Scholar]
- 22.Legaspi A, Brennan MF. Management of islet cell carcinoma. Surgery. 1988;104:1018–23. [PubMed] [Google Scholar]
- 23.Plockinger U, Wiedenmann B. Diagnosis of non-functioning neuro-endocrine gastro-enteropancreatic tumors. Neuroendocrinology. 2004;80(Suppl. 1):35–38. doi: 10.1159/000080739. [DOI] [PubMed] [Google Scholar]
- 24.Kloppel G, Perren A, Heitz PU. The gastroenteropancreatic neuroendocrine cell system and its tumors: The WHO classification. Ann NY Acad Sci. 2004;1014:13–27. doi: 10.1196/annals.1294.002. [DOI] [PubMed] [Google Scholar]
- 25.Panzuto F, Boninsegna L, Fazio N, et al. Metastatic and locally advanced pancreatic endocrine carcinomas: analysis of factors associated with disease progression. J Clin Oncol. 2011;29(17):2372–77. doi: 10.1200/JCO.2010.33.0688. [DOI] [PubMed] [Google Scholar]
- 26.Hochwald SN, Zee S, Conlon KC, et al. Prognostic factors in pancreatic endocrine neoplasms: An analysis of 136 cases with a proposal for low-grade and intermediate-grade groups. J Clin Oncol. 2002;1(11):20. 2633–42. doi: 10.1200/JCO.2002.10.030. [DOI] [PubMed] [Google Scholar]
- 27.Jernman J, Välimäki MJ, Louhimo J, et al. The novel WHO 2010 classification for gastrointestinal neuroendocrine tumors correlates well with the metastatic potential of rectal neuroendocrinetumors. Neuroendocrinology. 2012;95:317–24. doi: 10.1159/000333035. [DOI] [PubMed] [Google Scholar]
- 28.Yamaguchi T, Fujimori T, Tomita S, et al. Clinical validation of the gastrointestinal NET grading system: Ki67 index criteria of the WHO 2010 classification is appropriate to predict metastasis or recurrence. Diagn Pathol. 2013;8:65. doi: 10.1186/1746-1596-8-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Basturk O, Tang L, Hruban RH, et al. Poorly differentiated neuroendocrine carcinomas of the pancreas: A clinicopathologic analysis of 44 cases. Am J Surg Pathol. 2014;38:437–47. doi: 10.1097/PAS.0000000000000169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ricke J, Klose KJ, Mignon M, et al. Standardisation of imaging in neuroendocrine tumors: Results of a European Delphi process. Eur J Radiol. 2001;37:8–17. doi: 10.1016/s0720-048x(00)00187-x. [DOI] [PubMed] [Google Scholar]
- 31.Gotthardt M, Dirkmorfeld LM, Wied MU, et al. Influence of somatostatin receptor scintigraphy and CT/MRI on the clinical management of patients with gastrointestinal neuroendocrine tumors: An analysis in 188 patients. Digestion. 2003;68:80–85. doi: 10.1159/000074519. [DOI] [PubMed] [Google Scholar]
- 32.Nobels FR, Kwekkeboom DJ, Coopmans W, et al. Chromogranin A as serum marker for neuroendocrine neoplasia: Comparison with neuron-specific enolase and the alpha-subunit of glycoprotein hormones. J Clin Endocrinol Metab. 1997;82:2622–28. doi: 10.1210/jcem.82.8.4145. [DOI] [PubMed] [Google Scholar]
- 33.Van Nagell JR, Jr, Powell DE, Gallion HH, et al. Small cell carcinoma of the uterine cervix. Cancer. 1988;62(8):1586–93. doi: 10.1002/1097-0142(19881015)62:8<1586::aid-cncr2820620822>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 34.Bifulco G, Mandato VD, Giampaolino P, et al. Small cell neuroendocrine cervical carcinoma with 1-year follow-up: Case report and review. Anticancer Res. 2009;29(2):477–84. [PubMed] [Google Scholar]
- 35.Monier A, Saloum N, Szmigielski W, et al. Neuroendocrine tumor of the gallbladder. Pol J Radiol. 2015;80:228–31. doi: 10.12659/PJR.893705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ozawa K, Kinoshita M. A case of double carcinoid tumors of the gallbladder. Dig Dis Sci. 2003;48(9):1760–61. doi: 10.1023/a:1025499112957. [DOI] [PubMed] [Google Scholar]
- 37.Hoang MP, Hobbs CM, Sobin LH, Albores-Saavedra J. Carcinoid tumor of the esophagus: A clinicopathologic study of four cases. Am J Surg Pathol. 2002;26:517–22. doi: 10.1097/00000478-200204000-00016. [DOI] [PubMed] [Google Scholar]
- 38.Huang Q, Wu H, Nie L, et al. Primary high-grade neuroendocrine carcinoma of the esophagus: A clinicopathologic and immunohistochemical study of 42 resection cases. Am J Surg Pathol. 2013;37:467–83. doi: 10.1097/PAS.0b013e31826d2639. [DOI] [PubMed] [Google Scholar]
- 39.Okamoto H, Fujishima F, Kasajima A, et al. A large cell neuroendocrine carcinoma of the esophagus. Jpn J Gastroenterol Surg. 2012;45:914–22. [Google Scholar]
- 40.Lee CG, Lim YJ, Park SJ, et al. The clinical features and treatment modality of esophageal neuroendocrine tumors: A multicenter study in Korea. BMC Cancer. 2014;14:569. doi: 10.1186/1471-2407-14-569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Estrozi B, Bacchi CE. Neuroendocrine tumors involving the gastroenteropancreatic tract: A clinicopathological evaluation of 773 cases. Clinics (Sao Paulo) 2011;66:1671–75. doi: 10.1590/S1807-59322011001000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eichhorn JH, Young RH, Scully RE. Primary ovarian small cell carcinoma of pulmonary type. A clinicopathologic, immunohistologic, and flow cytometric analysis of 11 cases. Am J Surg Pathol. 1992;16(10):926–38. doi: 10.1097/00000478-199210000-00002. [DOI] [PubMed] [Google Scholar]
- 43.Dykgraaf RH, de Jong D, van Veen M, et al. Clinical management of ovarian small-cell carcinoma of the hypercalcemic type: A proposal for conservative surgery in an advanced stage of disease. Int J Gynecol Cancer. 2009;19(3):348–53. doi: 10.1111/IGC.0b013e3181a1a116. [DOI] [PubMed] [Google Scholar]
- 44.Hoof P, Tsai-Nguyen G, Paulson S, et al. Neuroendocrine carcinoma of the prostate gland. Proc (Bayl Univ Med Cent) 2016;29(1):68–69. doi: 10.1080/08998280.2016.11929365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abbas F, Civantos F, Benedetto P, Soloway MS. Small cell carcinoma of the bladder and prostate. Urology. 1995;46(5):617–30. doi: 10.1016/S0090-4295(99)80290-8. [DOI] [PubMed] [Google Scholar]
- 46.Têtu B, Ro JY, Ayala AG, et al. Small cell carcinoma of the prostate Part I. A clinicopathologic study of 20 cases. Cancer. 1987;59(10):1803–9. doi: 10.1002/1097-0142(19870515)59:10<1803::aid-cncr2820591019>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 47.Oesterling JE, Hauzeur CG, Farrow GM. Small cell anaplastic carcinoma of the prostate: A clinical, pathological and immunohistological study of 27 patients. J Urol. 1992;147(3 Pt 2):804–7. doi: 10.1016/s0022-5347(17)37390-1. [DOI] [PubMed] [Google Scholar]
- 48.Sehgal SS, Wein AJ, Bing Z, et al. Neuroendocrine tumor of the bladder. Rev Urol. 2010;12(4):e197–201. [PMC free article] [PubMed] [Google Scholar]
- 49.Mazzucchelli R, Morichetti D, Lopez-Beltran A, et al. Neuroendocrine tumors of the urinary system and male genital organs: clinical significance. BJU Int. 2009;103:1464–70. doi: 10.1111/j.1464-410X.2009.08451.x. [DOI] [PubMed] [Google Scholar]
- 50.Hager S, Ackermann CJ, Joerger M, et al. Anti-tumour activity of platinum compounds in advanced prostate cancer – a systematic literature review. Ann Oncol. 2016;27(6):975–84. doi: 10.1093/annonc/mdw156. [DOI] [PubMed] [Google Scholar]
- 51.Steineck G, Reuter V, Kelly WK, et al. Cytotoxic treatment of aggressive prostate tumors with or without neuroendocrine elements. Acta Oncol. 2002;41:668–74. doi: 10.1080/028418602321028292. [DOI] [PubMed] [Google Scholar]
- 52.Amato RJ, Logothetis CJ, Hallinan R, et al. Chemotherapy for small cell carcinoma of prostatic origin. J Urol. 1992;147:935–37. doi: 10.1016/s0022-5347(17)37427-x. [DOI] [PubMed] [Google Scholar]
- 53.Papandreou CN, Daliani DD, Thall PF, et al. Results of a phase II study with doxorubicin, etoposide, and cisplatin in patients with fully characterized small-cell carcinoma of the prostate. J Clin Oncol. 2002;20:3072–80. doi: 10.1200/JCO.2002.12.065. [DOI] [PubMed] [Google Scholar]
- 54.Moore SR, Reinberg Y, Zhang G. Small cell carcinoma of prostate: Effectiveness of hormonal versus chemotherapy. Urology. 1992;39:411–16. doi: 10.1016/0090-4295(92)90235-o. [DOI] [PubMed] [Google Scholar]
- 55.Tetu B, Ro JY, Ayala AG, et al. Small cell carcinoma of the prostate. Part I. A clinicopathologic study of 20 cases. Cancer. 1987;59:1803–9. doi: 10.1002/1097-0142(19870515)59:10<1803::aid-cncr2820591019>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 56.Sørbye H, Welin S, Langer SW, et al. Predictive and prognostic factors for treatment and survival in 305 patients with advanced gastrointestinal neuroendocrine carcinoma (WHO G3): The NORDIC NEC study. Ann Oncol. 2013;24:152–60. doi: 10.1093/annonc/mds276. [DOI] [PubMed] [Google Scholar]
- 57.Yamaguchi T, Machida N, Morizane C, et al. Multicenter retrospective analysis of systemic chemotherapy for advanced neuroendocrine carcinoma of the digestive system. Cancer Sci. 2014;105:1176–81. doi: 10.1111/cas.12473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hentic O, Hammel P, Couvelard A, et al. FOLFIRI regimen: An effective second-line chemotherapy after failure of etoposide-platinum combination in patients with neuroendocrine carcinomas grade 3. Endocr Relat Cancer. 2012;19:751–57. doi: 10.1530/ERC-12-0002. [DOI] [PubMed] [Google Scholar]
- 59.Hadoux J, Malka D, Planchard D, et al. Post-first-line FOLFOX chemotherapy for grade 3 neuroendocrine carcinoma. Endocr Relat Cancer. 2015;22:289–98. doi: 10.1530/ERC-15-0075. [DOI] [PubMed] [Google Scholar]
- 60.Coriat R, Walter T, Terris B, et al. Gastroenteropancreatic well-differentiated grade 3 neuroendocrine tumors: Review and position statement. Oncologist. 2016;21:1191–99. doi: 10.1634/theoncologist.2015-0476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Panzuto F, Boninsegna L, Fazio N, et al. Metastatic and locally advanced pancreatic endocrine carcinomas: Analysis of factors associated with disease progression. J Clin Oncol. 2011;29:2372–77. doi: 10.1200/JCO.2010.33.0688. [DOI] [PubMed] [Google Scholar]
- 62.Vélayoudom-Céphise FL, Duvillard P, Foucan L, et al. Are G3 ENETS neuroendocrine neoplasms heterogeneous? Endocr Relat Cancer. 2013;20:649–57. doi: 10.1530/ERC-13-0027. [DOI] [PubMed] [Google Scholar]
- 63.Scoazec JY, Couvelard A, Monges G, et al. Well-differentiated grade 3 digestive neuroendocrine tumors: myth or reality? The PRONET Study Group. J Clin Oncol. 2012;30(15 Suppl.):4129. [Google Scholar]
- 64.Sorbye H, Strosberg J, Baudin E, et al. Gastroenteropancreatic high-grade neuroendocrine carcinoma. Cancer. 2014;120(18):2814–23. doi: 10.1002/cncr.28721. [DOI] [PubMed] [Google Scholar]
- 65.Basturk O, Yang Z, Tang LH, et al. The high-grade (WHO G3) pancreatic neuroendocrine tumor category is morphologically and biologically heterogenous and includes both well differentiated and poorly differentiated neoplasms. Am J Surg Pathol. 2015;39(5):683–90. doi: 10.1097/PAS.0000000000000408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Heetfeld M, Chougnet CN, Olsen IH, et al. Characteristics and treatment of patients with G3 gastroenteropancreatic neuroendocrine neoplasms. Endocr Relat Cancer. 2015;22(4):657–64. doi: 10.1530/ERC-15-0119. [DOI] [PubMed] [Google Scholar]
- 67.de Herder WW, Hofland LJ, van der Lely AJ, Lamberts SW. Somatostatin receptors in gastroentero-pancreatic neuroendocrine tumors. Endocr Relat Cancer. 2003;10:451–58. doi: 10.1677/erc.0.0100451. [DOI] [PubMed] [Google Scholar]
- 68.Nikou GC, Lygidakis NJ, Toubanakis C, et al. Current diagnosis and treatment of gastrointestinal carcinoids in a series of 101 patients: the significance of serum chromogranin-A, somatostatin receptor scintigraphy and somatostatin analogues. Hepatogastroenterology. 2005;52:731–41. [PubMed] [Google Scholar]
- 69.Brennan SM, Gregory DL, Stillie A, et al. Should extrapulmonary small cell cancer be managed like small cell lung cancer? Cancer. 2010;116(4):888–95. doi: 10.1002/cncr.24858. [DOI] [PubMed] [Google Scholar]
- 70.Cicin I, Karagol H, Uzunoglu S, et al. Extrapulmonary small cell carcinoma compared with small-cell lung carcinoma. Cancer. 2007;110:1068–75. doi: 10.1002/cncr.22887. [DOI] [PubMed] [Google Scholar]
- 71.Soto DE, Eisbruch A. Limited-stage extrapulmonary small cell carcinoma: Outcomes after modern chemotherapy and radiotherapy. Cancer J. 2007;13:243–46. doi: 10.1097/PPO.0b013e31813ffe7c. [DOI] [PubMed] [Google Scholar]
- 72.Rieber J, Schmitt J, Warth A, et al. Outcome and prognostic factors of multimodal therapy for pulmonary large-cell neuroendocrine carcinomas. Eur J Med Res. 2015;20:64. doi: 10.1186/s40001-015-0158-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chu QD, Hill HC, Douglass HO, Jr, et al. Predictive factors associated with long-term survival in patients with neuroendocrine tumors of the pancreas. Ann Surg Oncol. 2002;9:855–62. doi: 10.1007/BF02557521. [DOI] [PubMed] [Google Scholar]