

Abstract
Herein we summarize important discoveries made over many years about Leydig cell function and regulation. Fetal Leydig cells produce the high levels of androgen (testosterone or androstenedione, depending upon the species) required for differentiation of male genitalia and brain masculinization. Androgen production declines with loss of these cells, reaching a nadir at postpartum. Testosterone then gradually increases to high levels with adult Leydig cell development from stem cells. In the adult, luteinizing hormone (LH) binding to Leydig cell LH receptors stimulates cAMP production, increasing the rate of cholesterol translocation into the mitochondria. Cholesterol is metabolized to pregnenolone by the CYP11A1 enzyme at the inner mitochondrial membrane, and pregnenolone to testosterone by mitochondria and smooth endoplasmic reticulum enzymes. Cholesterol translocation to the inner mitochondrial membrane is mediated by a protein complex formed at mitochondrial contact sites that consists of the cholesterol binding translocator protein, voltage dependent anion channel, and other mitochondrial and cytosolic proteins. Steroidogenic acute regulatory protein acts at this complex to enhance cholesterol movement across the membranes and thus increase testosterone formation. The 14-3-3γ and ε adaptor proteins serve as negative regulators of steroidogenesis, controlling the maximal amount of steroid formed. Decline in testosterone production occurs in many aging and young men, resulting in metabolic and quality-of-life changes. Testosterone replacement therapy is widely used to elevate serum testosterone levels in hypogonadal men. With knowledge gained of the mechanisms involved in testosterone formation, it is also conceivable to use pharmacological means to increase serum testosterone by Leydig cell stimulation.
Keywords: androgen, testosterone, steroid hormones, hormone action, mitochondria, hypogonadism
A summary of important discoveries made over the course of many years about Leydig cell function and regulation, and discussion of important issues that remain to be understood.
Introduction
We are pleased, indeed honored, to write this review article entitled “Leydig Cells: Formation, Function and Regulation” for the Special Issue of Biology of Reproduction commemorating the 50th Anniversary of the founding of the Society for the Study of Reproduction (SSR). There have been numerous major figures in the SSR among the many investigators who have contributed to our current knowledge of Leydig cells. In this review, our intent is to summarize important discoveries made over the years that have contributed to current understanding of Leydig cell function and regulation, and how such understanding is contributing to the treatment of conditions resulting in reduced serum levels of testosterone. We regret that we have been able to reference the contributions of only some of the many outstanding investigators whose work has contributed significantly to this important field. In this introduction, we outline the content of our review. Specifics (and most references) are presented under sections below.
Studies of Leydig cells began with the German zoologist and anatomist Franz von Leydig who, in 1850, described the presence of interstitial cells in the testes of several mammals [1, 2]. Fifty years later, Bouin and Ancel first suggested that androgens are produced by the interstitial Leydig cells [3]. About 60 years after that, Baillie demonstrated that these cells contain the enzyme 3β-hydroxysteroid dehydrogenase [4]. At about the same time, Hall and Eik-Nes [5] and Ewing and Eik-Nes [6] showed that pituitary gonadotropic hormones stimulate androgen formation by testes in vitro and ex vivo. In 1969, Hall and colleagues demonstrated that the conversion of cholesterol to testosterone occurred in the interstitial Leydig cells [7]. Thus, from the time of the discovery of interstitial cells in 1850, it took over a century to appreciate the major steps involved in Leydig cell function.
We now know that there are two distinct periods of androgen production in males, the fetal and adult periods; and there are two distinct populations of androgen-producing Leydig cells: the fetal and adult Leydig cells (Figure 1). Fetal Leydig cells produce the high levels of androgen (testosterone or androstenedione, depending upon the species) that are required for the differentiation of the male genitalia and for brain masculinization. Testosterone production declines with the postnatal decline in numbers of the fetal Leydig cells, reaching a nadir early in the postpartum period. Thereafter, testosterone gradually increases to high levels with the development of the adult Leydig cells from stem cells of the neonatal testis. Once formed, the adult Leydig cells rarely turn over or die.
Figure 1.

Fetal and adult periods of testosterone production. Fetal Leydig cells produce the high levels of testosterone that are required for the differentiation of the male genitalia and for brain masculinization. Testosterone production declines with the postnatal decline in numbers of the fetal Leydig cells, reaching a nadir early in the postpartum period. Thereafter, testosterone gradually increases to high levels with the development of the adult Leydig cells from stem cells of the neonatal testis. LH is not required either for the development of fetal Leydig cells or for their initial testosterone production. Later, however, the fetal Leydig cells express LH receptor and respond to LH stimulation.
Current understanding of Leydig cell function and its regulation has been made possible by the development of experimental approaches by which Leydig cells can be examined both in vivo and in vitro, as well as through the use of cell lines. Luteinizing hormone (LH) secreted by the pituitary gland in response to gonadotropin-releasing hormone (GnRH) from the hypothalamus, initiates steroid formation by binding to the Leydig cell LH receptor (LHR) which, through coupling to G protein, stimulates Leydig cell cyclic adenosine 3’,5’-monophosphate (cAMP) production. Cyclic AMP, in turn, stimulates cholesterol translocation from intracellular sources into the mitochondria, the rate-determining step in steroid formation. This is followed by pregnenolone formation from cholesterol in the mitochondria, and then its conversion to testosterone by enzymes of the smooth endoplasmic reticulum (Figure 2). Declines in testosterone production occur with aging and other conditions, resulting in reduced serum testosterone levels (hypogonadism) and accompanying metabolic and quality-of-life changes. Testosterone replacement therapy (TRT) is available and widely used to elevate serum testosterone levels in hypogonadal men. Knowledge of the mechanisms involved in testosterone formation has also made it conceivable to use pharmacological means to increase serum (and intratesticular) testosterone by stimulating the Leydig cells themselves.
Figure 2.

The steroidogenic pathway. In Leydig cells, androgens are derived from cholesterol. Cholesterol is from de novo synthesis, lipoproteins, lipid droplets, or plasma membrane. Lipoproteins, LDL (through the LDL-receptor pathway), HDL (through the SR-BI pathway), and lipid droplets contain esterified cholesterol (yellow) that can be used in steroidogenesis after de-esterification (green). Cholesterol is imported into mitochondria through a large protein complex, the transduceosome, composed of mitochondrial and cytosolic proteins. Cholesterol reaches CYP11A1 in the inner mitochondrial membrane where it is metabolized to pregnenolone. Pregnenolone is converted by 3β-HSD located at the mitochondria and endoplasmic reticulum. Subsequent metabolism to androgen and its metabolites is by Leydig cell species-specific expression of CYPs and HSD.
Fetal leydig cells
The male phenotype depends in part upon the expression of the fetal hormones anti-Müllerian hormone (AMH), secreted by fetal Sertoli cells, and androgen and INSL3, produced by fetal Leydig cells [8]. AMH induces regression of the Müllerian ducts, and androgen induces differentiation of the Wolffian ducts into male reproductive organs [8]. In the mouse, cells expressing steroidogenic factor 1 (SF-1; NR5A1) give rise to the fetal Leydig cells [8, 9]. The differentiation of the SF-1 expressing cells to fetal Leydig cells is promoted by platelet-derived growth factor A and desert hedgehog [9]. Although the fetal Leydig cells are themselves mitotically inactive, their numbers increase considerably during embryonic development, suggesting that the new cells must arise from the differentiation of progenitor cells rather than from the division of existing fetal Leydig cells [8, 10].
In the rat, the fetal Leydig cells begin to produce testosterone by gestational day 15.5, with peak production just prior to birth. In the mouse, the fetal Leydig cells produce androstenedione which is then converted into testosterone by the fetal Sertoli cells [11, 12]. Initially, LH is not required either for the development of fetal Leydig cells or for their androgen production [8, 13]. Later, however, the fetal Leydig cells express LHR and respond to LH stimulation [13, 14]. Although plasma LH levels remain high at the end of gestation, regression of fetal Leydig cells begins during late fetal life and continues thereafter [15–18]. Some fetal Leydig cells do persist in adult life [8, 19, 20]. However, it is unlikely that these cells contribute significantly to testosterone production in the adult [9, 21]. The observation that fetal and adult Leydig cells express different genes suggests that the two cell populations arise and function distinctly [9].
Development of adult leydig cells from stem cells
During the first 2–3 postnatal weeks in rats, fetal Leydig cells are gradually replaced by adult Leydig cells [8, 21]. It had been suggested that adult Leydig cell development is independent of the fetal Leydig cells, but rather that the cells arose from stem cells [18, 22, 23]. The work of Matthew Hardy and his colleagues showed that adult rat Leydig cells do indeed develop from stem cells of the early neonatal (postnatal day 7) testis [24]. These investigators demonstrated that there are cells in the neonatal rat testis that are 3βHSD-negative, LHR-negative, and platelet-derived growth factor receptor α (PDGFRα)-positive. Depending upon the in vitro conditions to which they are exposed, these cells were shown to have the ability to proliferate indefinitely (self-renew) or differentiate to become 3β-HSD positive and ultimately produce testosterone. Additionally, when transplanted into the interstitial compartment of host adult rat testes, the cells differentiated in vivo to become 3β-HSD positive.
Stem cells indistinguishable from those in the neonatal testis have also been shown to surround the seminiferous tubules and blood vessels of adult testes [25]. These cells were isolated and, as with the stem cells of the neonatal testis, were found able to undergo self-renewal indefinitely or to differentiate and ultimately produce testosterone [25]. In a recent study, we reported that stem Leydig cells obtained from the adult rat can transdifferentiate into uterine and prostatic epithelium, but not into epidermis [26]. Leydig cell ablation studies, using either ethane dimethane sulfonate or genetic approaches to eliminate the adult cells, have also contributed to our understanding of the origin of adult Leydig cells [27, 28]. These studies demonstrated that although adult stem cells are normally quiescent, they will regenerate new Leydig cells after the experimentally induced loss the adult cells. However, the role normally played by the stem cells of the adult testis is not known.
Adult leydig cell function and regulation
Groundbreaking early findings
As indicated in the introduction, studies of Leydig cell function and regulation have had a long history that began in 1850 when Franz Von Leydig first described the interstitial cells of the testis. In the early 1960s, Hall and Eik-Nes reported that stimulating rabbit testis slices with LH in vitro induced testosterone production [5]. At that time, however, it was uncertain which cells produced testosterone. Studies conducted in the late 1950s and 1960s showed that interstitial cells of the testis metabolize cholesterol to pregnenolone and are responsible for most, if not all, androgen synthesis in the testis. Christensen and Mason, in 1965, described methods used to separate the seminiferous tubules from the interstitial tissue of rat testes [29]. When incubated separately, both tubules and interstitial tissue transformed progesterone to testosterone, though the interstitial tissue did so far more effectively. These results confirmed previous evidence that the interstitial tissue is the principal source of testicular androgens. Later, Hall and his colleagues [7] reported that testosterone and androstenedione were produced when interstitial cells, but not seminiferous tubules, were incubated with cholesterol. These results confirmed previous evidence suggesting that interstitial cells represent the major, and probably the only, source of testicular androgens. The finding by Hall et al [7] that Leydig cells metabolize cholesterol to testosterone was of particular importance because it is this quality that defines steroidogenic cells.
The introduction of important methodologic approaches by Larry Ewing made it possible to greatly advance understanding of Leydig cell function and regulation. Ewing was among the founders of SSR and later the Society's President. He also served as Editor-In-Chief of Biology of Reproduction. While a graduate student, Ewing developed in vitro testicular perfusion by which Leydig cell function could be studied while the cells remained within the uncompromised three-dimensional architecture of the testis. He and his colleagues showed that in response to LH infused into the testis, testosterone could be synthesized at high levels for several hours [30]. Species-specific pathways for the biosynthesis of testosterone from pregnenolone were identified by measuring the secretion products of in vitro perfused testes infused with specific intermediates and inhibitors [31]. Significant differences were noted in testosterone secretion between species [32]. A linear, positive correlation was seen between the ability of Leydig cells to produce testosterone in response to LH and the Leydig cell content of smooth endoplasmic reticulum as measured by stereology [33]. These results suggested that LH regulates not only Leydig cell steroidogenesis but also Leydig cell ultrastructure. To test this further, it was shown that the experimental inhibition of Leydig cell testosterone secretion in vivo by LH suppression was reflected in quantitative changes in Leydig cell smooth endoplasmic reticulum, and that there was also a significant linear relationship between the recovery of steroidogenic capacity and the restoration of LH secretion and Leydig cell morphology [34, 35]. These studies indicated that LH regulates not only Leydig cell steroidogenesis but also membrane biogenesis and/or turnover within the Leydig cells. Later studies indicated that though LH is required to maintain Leydig cell structure, it is not required to maintain Leydig cell number [36]. Additionally, it was shown that the treatment of rats with LH at the time of hypophysectomy, but not with follicle-stimulating hormone, prolactin, thyroidstimulating hormone, or growth hormone, maintained the capacity of the testes to produce testosterone [32], providing further evidence of trophic effect elicited by LH on Leydig cell testosterone production.
The development in 1981 by Mario Ascoli of the hormone-dependent steroid producing mouse tumor Leydig cell line, MA-10, was an important step that, over the years, has allowed the undertaking of numerous studies designed to understand the mechanism regulating Leydig cell steroidogenesis [37]. However, MA-10 cells do not express CYP17A1, and thus the major steroid product made is progesterone. Attempts to generate androgen-producing Leydig cell lines have been met with limited success. In 1968, Sato and colleagues developed the R2C rat tumor Leydig cell line that produced progesterone in a constitutive, hormone-independent, manner [38]. In 1980, Mather developed the TM3 mouse cell line believed to be derived from immature Leydig cells [39]. Although these cells produced testosterone, there was only limited if any response to LH. In 1982, Rebois established the MLTC-1 mouse Leydig tumor cell line [40]. These cells are similar to MA-10 cells in that they were generated from the same tumor as MA-10 cells and the main steroid product is progesterone. In 1987, Finaz and colleagues developed the K9 Leydig cell line, cells that were able to produce testosterone in response to hormone treatment and had characteristics of normal Leydig cells [41]. In 2012, the BLTK-1 Leydig cell line was developed from a testicular tumor [42]. These cells retain functional LHCGR-mediated steroidogenesis, producing progesterone, testosterone, and estradiol.
MA-10 cells remain the most widely used of the cell lines, and have been instrumental in advancing our knowledge of Leydig cell steroid formation and regulation. However, despite the benefits provided by MA-10 cells, there are significant differences between these cells and normal Leydig cells. The most important is that the cells produce progesterone rather than testosterone. Less well appreciated is that there are major differences in their responsiveness to gonadotropin treatment. The response of these cells to gonadotropin is reminiscent of adrenal cortical cells rather than Leydig cells, which respond more slowly and produce 20–30 times less steroid than adrenal cells.
Though important results had been obtained from studies of testis slices, perfused testes, and cell lines, Ewing and others recognized the need for better methods by which to isolate highly purified primary Leydig cells, and for culture methods by which the isolated cells would retain the ability to respond to LH and to produce testosterone at levels comparable to those produced by cells of the perfused testis. Before the important studies of Ewing and Klinefelter of 1987–1989 [43–45], Leydig cells could be isolated, but when cultured they rapidly lost the capacity to produce LH-stimulated testosterone. In 1987, Klinefelter and Ewing developed an isolation procedure that achieved >95% pure Leydig cells and a culture system in which these cells responded to maximally stimulating LH with 10-fold increase in testosterone production over 3 hours [43]. This was many-fold higher than had been achieved previously. Subsequent modifications of the culture system resulted in the maintenance of high levels of testosterone production by purified adult rat Leydig cells for up to 3 days [44, 45]. The development of these in vitro methods for isolating, purifying, and culturing Leydig cells were instrumental in subsequent detailed studies of Leydig cell function and regulation.
Current understanding of Leydig cell steroidogenic function
Since the groundbreaking studies described above a great deal has been discovered about the hormones and enzymes involved in testosterone biosynthesis, the how testosterone production is regulated.
Hypothalamic/pituitary/gonadal axis
The hypothalamic–pituitary axis is integrally involved in the regulation of testicular testosterone production. GnRH binds to a membrane receptor on pituitary gonadotrophs and stimulates the biosynthesis and secretion of LH (reviewed in 46, 47). LH binds LHR on the surface of Leydig cells and stimulates intracellular signaling cascades. Dufau and Catt [48] demonstrated that in response to LH, cAMP is produced. The cAMP pathway, through protein kinase A (PKA), is of critical importance in steroid formation by virtue of its acute stimulation of the translocation of cholesterol to the outer mitochondrial membrane. Cholesterol is then transferred from the outer to the inner mitochondrial membrane where it is converted to pregnenolone by CYP11A1 [49]. In addition, the chronic stimulation of Leydig cells by LH and cAMP is essential for regulating the expression levels of the proteins and enzymes involved in steroidogenesis, and thus for the trophic regulation of steroidogenesis, responsible for the sustainable production of steroids over a long period of time.
Steroidogenic enzymes
The two classes of enzymes involved in testosterone biosynthesis, the cytochrome P450 proteins of the mitochondria and the hydroxysteroid dehydrogenases of the smooth endoplasmic reticulum, catalyze the conversion of cholesterol to testosterone (Figure 2). Four steroidogenic enzymes are involved in testosterone biosynthesis from cholesterol: Cytochrome P450 Family 11 Subfamily A1 (CYP11A1), 3β-hydroxysteroid dehydrogenase/Δ5 → Δ4 isomerase (3β-HSD), CYP17A1, and 17β-HSD3 [50, 51]. CYP11A1 is located in mitochondria at the inner mitochondrial membrane; 3β-HSD is present in mitochondria but predominantly in smooth endoplasmic reticulum; and CYP17A1 and 17β-HSD3 are found only in smooth endoplasmic reticulum. CYP11A1 (aka P450scc) catalyzes the conversion of cholesterol to pregnenolone and, in this way, determines the biosynthetic capacity of the Leydig cells. The observation that both 17α-hydroxylase and 17,20 lyase activities reside in a single protein, CYP17A1 [52], represented a major contribution to understanding the enzymes involved in steroid biosynthesis in men; 17α-hydroxylase activity distinguishes between the synthesis of mineralocorticoids (aldosterone) and glucocorticoids (cortisol), and 17,20 lyase activity distinguishes between the synthesis of glucocorticoids and sex steroids. In rodents, CYP17A1 is exclusively found in the gonads. The proximity of 3β-HSD to CYP17A1 and 17β-HSD3 in the smooth endoplasmic reticulum makes it possible for each intermediate involved in testosterone synthesis to move easily and directly to the next enzyme.
Cholesterol translocation
The first catalytic reaction in androgen biosynthesis by CYP11A1 occurs in mitochondria. A number of studies demonstrated that this reaction is not rate limiting [49, 52, 53]. Before this reaction, cholesterol must be translocated from its various locations in the cell (primarily the plasma membrane and intracellular stores) to the outer mitochondrial membrane. The translocation of cholesterol to the inner mitochondrial membrane is the rate-limiting and -determining step in steroidogenesis.
Cholesterol can be synthesized de novo from acetate [54, 55] and stored in lipid droplets, or it can come from Leydig cell membranes including the plasma membrane [56]. As indicated above, the acute regulation of steroidogenesis is by LH (or human chorionic gonadotropin, hCG), which binds to a G protein-coupled receptor and activates adenylate cyclase, resulting in cAMP production and then cAMP-dependent protein kinase activation. This leads to the initiation of cholesterol release from lipid droplets or from the plasma membrane. Testosterone biosynthesis begins with the conversion of cholesterol to pregnenolone by the mitochondrial enzyme CYP11A1. In 1975, Cooke and colleagues showed that inhibition of protein synthesis affected LH-induced steroid production by Leydig cells [57]. Four years later, the Cooke lab proposed that LH affects the stability of a regulatory protein involved steroid formation [58], the identification of which was not known. Finally, in 1991, Orme-Johnson and colleagues described an LH-induced, 30 kDa phosphoprotein in steroidogenic cells, including mouse Leydig cells [59, 60], the levels of which increased in parallel with steroid production. The protein was found to be localized to mitochondria and later was shown to result from the cleavage of a 37-kDa precursor protein. The protein was cloned and named steroidogenic acute regulatory protein (STAR) [61]. Detailed studies [49, 62, 63] demonstrated that STAR acts at the mitochondria to trigger cholesterol movement across the membranes. PKA-dependent phosphorylation of STAR was shown to be critical for its function [62]. The rapid induction of STAR formation by LH and its targeting and processing at the outer mitochondrial membrane increase testosterone formation [49, 64]. The importance of STAR was made evident by the observation that mutations of the STAR gene resulted in a severe deficiency in mineralocorticoids and, consistent with this, that there were severe defects in adrenal steroids seen in STAR knockout mice, mimicking features of lipoid congenital adrenal hyperplasia in patients [65]. The STAR transgene was found to restore steroidogenic function to STAR–/– mice [65]. Gonadal hormones in the knockout mice did not differ significantly from levels in wild-type littermates, suggesting that although adrenal steroid production was dramatically reduced in the STAR knockout mice, the mice retained their capacity for androgen biosynthesis [66]. However, Star-deficient mice were reported to exhibit female external genitalia, suggesting possible effects of STAR knockout on androgen production by the fetal testis [67].
Once targeted to the outer mitochondrial membrane, cholesterol must be translocated to the inner mitochondrial membrane, and there converted to pregnenolone by CYP11A1. It has been suggested that actin, microtubules, and microfilaments may play roles in regulating cholesterol transport into mitochondria [68–70], possibly by direct effects on elements of the steroidogenic pathway and/or by effects on interactions among endoplasmic reticulum, lipid droplets, and mitochondria [71]. Numerous studies have suggested that cholesterol translocation is mediated by the formation of a mitochondrial scaffold, the transduceosome, created by protein–protein interactions of cytosolic and outer mitochondrial membrane proteins [64]. This complex contains proteins that mediate the import of cholesterol from cytosolic sources into mitochondria, including the hormone-induced STAR, translocator protein (TSPO), and voltage dependent anion channel 1 (VDAC1). Aghazadeh et al. reported that the protein 14-3-3γ and ε adaptor proteins are also part of the transduceosome and that they both serve as negative regulators of steroidogenesis [72, 73]. The proteins were found to be hormonally induced and to function at the initiation of steroidogenesis by delaying maximal steroidogenesis in MA-10 mouse tumor Leydig cells. Treatment of MA-10 cells with a cAMP analog was found to trigger the interaction of 14-3-3γ with STAR in the cytosol, limiting STAR activity. Over time, STAR suppression ceased, allowing for induction of STAR activity and increased rate of steroid formation. Once 14-3-3γ releases STAR, a second regulatory mechanism for cholesterol import to mitochondria is activated that involves 14-3-3ε. The regulatory role of 14-3-3ε has a later onset but is maintained long term. 14-3-3ε anchors to mitochondria through interacting with VDAC1-TSPO complex, thereby regulating cholesterol import and steroidogenesis. When steroidogenesis is highly induced, 14-3-3ε interactions with VDAC1 are increased. Therefore, 14-3-3-ε intercalation between TSPO and VDAC1 blocks their efficient interactions, thus affecting the rate of cholesterol import into mitochondria [73].
TSPO (18 kDa) is an outer mitochondrial membrane protein that is abundant in steroid synthesizing cells and has high affinity for cholesterol [74]. Cholesterol binds at a specific binding site of TSPO, the cholesterol recognition/interaction amino acid consensus (CRAC) motif [75–77]. There is strong evidence indicating that cholesterol targeting to CYP11A1 is achieved through the formation of a multiprotein complex of outer and inner mitochondrial proteins that includes TSPO, VDAC, ATPase family AAA domain-containing protein 3 (ATAD3), and CYP11A1 (Figure 3). Data from a number of independent laboratories, published over the course of many years, have indicated an important role of TSPO in steroidogenesis. In particular, knockdown of Tspo expression using antisense oligonucleotides reduced the ability of cultured cells to form steroids. Additionally, several TSPO-specific ligands were shown to stimulate cholesterol import into mitochondria and thus steroid formation by MA-10 and primary Leydig cells in vitro, and to result in elevated testosterone production when administered in vivo [78–82]. Consistent with this, blocking the CRAC domain of TSPO was shown to block hormone-induced steroid formation in cells both in vitro and in vivo [83–87]. These studies strongly support the contention that TSPO plays an important role in cholesterol import into mitochondria and thus in steroidogenesis [88–90]. It should be noted, however, that the specific mechanism by which it does so was not determined. Additionally, it is technically challenging to be certain as to whether the effects seen on steroidogenesis in such studies were affected by TSPO knockdown alone or reduced cell viability [80].
Figure 3.
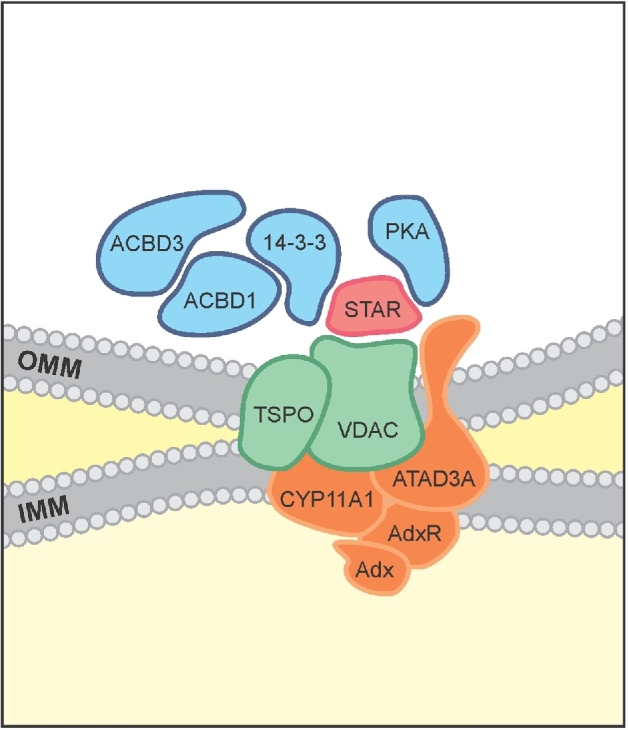
Protein–protein interactions driving cholesterol import into mitochondria. Cholesterol import into mitochondria is the result of series of protein–protein interactions. VDAC and TSPO are proteins found in most mitochondria, and ATAD3A is found in many cells. The presence of CYP11A1, adrenodoxin reductase and adenodoxin as well as the extremely high levels of expression of the cholesterol binding protein TSPO are characteristics of steroidogenic cell mitochondria. ACBD1 is a TSPO endogenous ligand. In response to hormone treatment, the outer mitochondrial membrane (OMM) TSPO and VDAC complex recruits ACBD3 which brings PKA to mitochondria. The hormone-induced STAR protein contains a mitochondrial signal sequence and is targeted to the OMM, where it interacts with VDAC and is locally phosphorylated by PKA for maximal activity. 14-3-3 adaptor proteins, binding to either STAR (14-3-3γ) or VDAC1 (14-3-3ɛ), provide negative control of maximally produced steroid formation, thus allowing for sustainable steroid formation. This complex is termed the transduceosome because it transduces the cAMP signal directly at the OMM. The OMM proteins TSPO and VDAC, together with the IMM proteins ATAD3 and CYP11A1, are part of the larger 800-kDa metabolon composed of proteins that bring cholesterol directly to CYP11A1 for metabolism.
Although studies conducted over the course of many years and by many labs concluded that TSPO plays a significant role in steroid biosynthesis, this conclusion recently has been called into question [91–94]. In one study, no effect on TSPO expression was seen after Tspo deletion in MA-10 cells [94]. This was in contrast to previous reports showing significant reduction of steroid production in the same cell line after TSPO knockdown using antisense oligodeoxynucleotides [95] or antisense knockdown [80]. As yet, the explanation for the difference in results is uncertain. In the same study, Selvaraj and his colleagues reported that a TSPO drug ligand PK 11195 stimulated progesterone production in Tspo knockout MA-10 cell lines generated using CRISPR/Cas9 technology, and suggested from this that the ligand's ability to stimulate steroid formation was unrelated to its binding to TSPO [94]. It should be pointed out, however, that whereas TSPO drug ligands at nanomolar and low micromolar concentrations have specificity for TSPO, at high micromolar concentrations they do not [88]. Moreover, in a recent study, we generated Tspo knockout MA-10 cell lines via CRISPR/Cas9 technology, and found that the mitochondrial membrane potential (ΔΨM) was significantly reduced compared to control cells, and that the cells made little to no steroids [96]. Additionally, whereas mice with Leydig cell-targeted Tspo conditional knockout were reported to show lack of effect on androgen production [92], the Papadopoulos lab generated steroidogenic cell-targeted Tspo knockout mice that showed a lack of ability to produce steroid (corticosterone) in response to adrenocorticotropic hormone (ACTH) [97]. Notably, increased accumulation of lipid droplets was seen in Leydig cells of the knockouts, suggesting an effect on lipid homeostasis in the testis. In agreement with these findings, Barron and colleagues generated TSPO KO mice that showed reduced total steroidogenic output and age-dependent androgen deficiency [98]. Recently, using zinc finger nuclease technology to perform Tspo-targeted genome editing, a null mutant rat line was generated lacking TSPO expression as was a line expressing a truncated TSPO protein which lacks the fifth transmembrane domain, containing the cholesterol recognition amino acid consensus motif [99]. The Tspo mutations in both rat models resulted in accumulation of esterified cholesterol in all steroidogenic cells examined, and with loss of corticosteroid formation in response to ACTH. Basal testosterone production was also reduced in the Tspo homozygous mutant rats [99].
It should be noted that even if the studies reporting no effect of Tspo knockout on gonadal steroid formation were found to be correct, such studies would not disprove the conclusion of many investigations that TSPO plays a critical role in steroidogenesis. Rather, it might be the case that the role of TSPO is not indispensable. For example, potential compensatory mechanisms may become involved in steroid formation when TSPO is knocked out in cells or in animals, and particularly so in the latter case when the knockouts are in vivo. This might be the case considering that TSPO is an evolutionarily conserved protein and that no humans have been identified lacking TSPO or with TSPO mutations. It also should be noted that the role of TSPO in different steroid-producing organs might differ. For example, the effect of knocking out STAR in the adrenal and testis has been reported to differ significantly, with an earlier and far more dramatic phenotype seen in the adrenal than the testis [66, 100]. Effects seen in MA-10 cells also seem likely to differ from both the adrenal and testis. Such differences might result from differences in the amounts of the transduceosome components and cell-specific protein–protein interactions. For example, it has been reported that there is 10 times more 14-3-3ɛ in Leydig than in adrenalcortical cells [73], perhaps explaining why the response of Leydig cells to LH or hCG is reduced compared to corticosteroids formed in response to ACTH. Clearly, further investigation is needed to clarify and extend our understanding of the relationships among TSPO, STAR, 14-3-3γ and ɛ, and other proteins of the transduceosome in relation to steroidogenesis in Leydig and adrenal cells, as well as in other steroidogenic cells.
Regulatory pathways
Steroid hormone synthesis must be a precisely regulated process because insufficient or excess production is detrimental. In addition to the well-established regulation of steroid formation by PKA, several regulators (signaling molecules, kinases, transcription factors) of Leydig cell differentiation and function were identified in the last two decades. These include the signaling molecules PDGF and DHH; the kinases MAPK, PKG, CAMKI, and AMPK; and the transcription factors NUR77, MEF2, and GATA4 [101]. Moreover, a cell-autonomous AMPK-dependent mechanism actively represses steroidogenesis, thus preventing excessive production of steroid hormones [102]. Several nuclear receptors have also been shown to have either direct or indirect roles in Leydig cell function (e.g. NR5A1) [103]. In addition to phosphorylation and protein kinases, protein phosphatases were also shown to be critical in the regulation of steroid hormone production by Leydig cells [104].
Reduced serum levels of testosterone and its treatment
Reduced serum levels of testosterone (hypogonadism) can occur in both young and aging men. Indeed, a significant decline in serum testosterone levels affects about 5 million American men [105, 106] including 20%–50% of men over age 60 and approximately 15% of men who are among the 15% of couples who seek infertility-related medical appointments [107–109]. There are many other men who also present with what is referred to as “low T,” including men with sickle cell disease and spinal cord injury [110]. In some men, reduced serum testosterone results from reduced serum LH (hypogonadotropic hypogonadism) [109]. In most, however, serum LH either does not change or increases, indicative of primary testicular deficiency (primary hypogonadism) [107–108]. Although changes in GnRH gene expression and LH pulse amplitude often occur with aging, LH administration typically results in lesser stimulation of testosterone in aged than young men [109], indicating reduced LH responsiveness of old Leydig cells. Whether in aging or young men, reduced serum testosterone is associated with a number of metabolic and quality-of-life changes, including decreased lean body mass, bone mineral density, muscle mass, libido and sexual function, increased adiposity, osteoporosis and cardiovascular disorders, and altered mood [108, 110].
In hypogonadal men in whom there are deficiencies in central stimulation (hypogonadotropic hypogonadism, Kallman's syndrome), serum testosterone can be elevated directly by administering LH or hCG, or indirectly with clomiphene or aromatase inhibitors. However, hypogonadism in most patients is not the result of central deficiencies, but rather results from the decreased responsiveness of the Leydig cells to LH. In such men, attempts to increase Leydig cell testosterone production and thus serum testosterone levels by LH administration typically are not effective.
Administering exogenous testosterone, known as testosterone replacement therapy, reverses many of the symptoms of low testosterone. The availability of new forms of testosterone supplementation and consumer marketing have contributed to prescription testosterone sales having increased 500% since 1993, with particularly dramatic increases in the USA since 2000 [111]. The primary objective of TRT is to raise serum testosterone levels into the eugonadal range. The testosterone preparations in use are injections; scrotal and nonscrotal transdermal patches; and oral, buccal, and gel preparations [112–114]. With injections, serum testosterone levels initially are supraphysiologic and then reduced, requiring testosterone levels to be monitored and sometimes adjusted between injections. Testosterone administered by gels and other transdermal methods are easier to use and produce more constant testosterone concentrations. However, recent studies suggest that there may be increased risk of cardiovascular disease in older men after TRT [115–117], resulting in the FDA cautioning (September 2014) that men who take exogenous testosterone may face increased risk of stroke and heart attack. There are also reports suggesting that exogenous testosterone treatment might increase the risk of prostate cancer [118]. Although there is agreement that testosterone replacement in young hypogonadal men is relatively safe and has beneficial effects, exogenous testosterone typically will suppress LH, resulting in reduced Leydig cell testosterone production and therefore in the suppression of spermatogenesis. The recovery of spermatogenesis after cessation of treatment often requires 6–15 months or more [111]. Thus, exogenous testosterone administration is inappropriate for men who wish to father children [119–120]. Although there are methods by which to increase serum testosterone without TRT, including hCG or aromatase inhibitors for men with secondary hypogonadism, these approaches typically are ineffective in men with primary hypogonadism [109].
In rodents as in humans, serum testosterone levels decline progressively with aging [121, 122]. In both, these decreases result from reduced testosterone production by aging Leydig cells, not from a reduction in cell numbers. As in men, aging in Brown Norway rats is characterized by reduced serum testosterone and unchanged or increased LH levels, and by the reduced ability of the Leydig cells to produce testosterone in response to LH [123, 124]. Defects in aging Brown Norway rat Leydig cells include reductions in each of LH-stimulated cAMP production, STAR, TSPO, CYP11A1, and downstream steroidogenic enzymes [125–127]. Although steroidogenic enzyme levels are reduced in aged cells, high levels of testosterone are produced if enough cholesterol is available to the inner mitochondrial membrane steroidogenic enzyme CYP11A1 [127].
Knowledge of the steps and mechanisms in testosterone formation has made it possible to consider the use of pharmacological means to increase serum (and intratesticular) testosterone by stimulating the Leydig cells themselves. Activation of TSPO by specific drug ligands was found to result in increased testosterone production by aged Leydig cells in vitro, and treating old rats with TSPO drug ligands resulted in elevated serum testosterone levels [82]. Whether or not such increase would be specific to Leydig cells remains uncertain. It should be noted however that in aging rats, Leydig cell TSPO levels are reduced by 50% [127]. Thus, ligand activated TSPO might reestablish normal TSPO activity and be responsible for the recovery of testosterone formation perhaps in a testis-specific manner. Similarly, TSPO drug ligands have been shown to induce neurosteroid formation in the human brain, but only in cases of neurological and psychiatric disease symptoms conditions where TSPO levels were found to be reduced [128, 129]. These findings, and the very high levels of TSPO in Leydig cells, hold promise that an appropriate dose of administered TSPO drug ligand might elevate testosterone production by Leydig cells specifically, with minor if any effects on the adrenal and/or brain in normal in vivo settings.
Another promising possible approach to increasing serum testosterone in hypogonadal men might be to target 14-3-3γ and/or ɛ, negative regulators of testosterone production. If the negative regulation of the proteins could be removed, there would be increased testosterone production by the Leydig cells and therefore increased testosterone levels in the serum and intratesticular fluid. The primary interaction sites for 14-3-3γ and ɛ were identified to be STAR S194 [72] and VDAC1 S167 [73], respectively. STAR S194 is the site where PKA acts to phosphorylate STAR and induce its steroidogenic activity [130]. VDAC S167 is located on the lateral side of the protein accessible for interaction with outer mitochondrial membrane partners such as TSPO [73]. In subsequent studies, cell penetrating peptide sequences conjugated to a short sequence of VDAC1 containing S167, and of STAR containing S194, were shown to successfully compete out 14-3-3ε and 14-3-3γ interactions in MA-10 cells [72, 73]. As a result, the negative regulatory roles of 14-3-3γ and ε were ablated and therefore the cells produced more steroids both acutely at the initiation of steroidogenesis, or long-term, respectively. Furthermore, peptides containing VDAC1 S167, when administered directly to the testes of adult male Sprague-Dawley rats, induced increased intratesticular and plasma testosterone levels in a manner independent of LH in vivo [73]. Such an approach, or targeting TSPO with specific drug ligands, hold promise for providing new means by which to increase serum testosterone levels without administering LH-suppressive testosterone. In addition to providing potential benefit to aging men, the design of new therapies that increase intratesticular bioactive androgen levels without affecting the hypothalamic–pituitary axis could be of importance for subfertile and infertile young men, including most men diagnosed with idiopathic infertility and present with reduced circulating testosterone levels, and men with orchitis and following trauma (injury to genitalia, spinal cord injury), torsion, surgery, chemotherapy, irradiation, and in response to some medications (acquired hypogonadism).
Footnotes
Grant support: This work was supported by grants from the National Institutes of Health [R01 AG021092 and R21 AG051259] and the John Stauffer Dean's Chair in Pharmaceutical Sciences (University of Southern California).
References
- 1. Leydig F. Zur Anatomie der männlichen Geschlechtsorgane und Analdrüsen der Säugethiere. Zeitschrift f Wiss Zool 1850;2:1–57. [Google Scholar]
- 2. Le Minor JM, Sick H. About the 350th anniversary of the foundation of the chair of anatomy of the faculty of medicine at Strasbourg (1652-2002). Hist Sci Med 2003;37:31–40. [PubMed] [Google Scholar]
- 3. Bouin P, Ancel P. Recherches sur les cellules interstitielles du testicle des mammifères. Arch Zool Exp Gen 1903;1:437–523. [Google Scholar]
- 4. Baille AH. Further observations on the growth and histochemistry of Leydig tissue in the postnatal prepubertal mouse testis. J Anat 1964;98:403–418. [PMC free article] [PubMed] [Google Scholar]
- 5. Hall PF, Eik-Nes KB. The action of gonadotropic hormones upon rabbit testis in vitro. Biochim Biophys Acta 1962;63(3):411–422. [DOI] [PubMed] [Google Scholar]
- 6. Ewing LL, Eik-Nes KB. On the formation of testosterone by the perfused rabbit testis. Can J Biochem 1966;44(10):1327–1344. [DOI] [PubMed] [Google Scholar]
- 7. Hall PF, Irby DC, De Kretser DM. Conversion of cholesterol to androgens by rat testes: comparison of interstitial cells and seminiferous tubules. Endocrinology 1969;84(3):488–496. [DOI] [PubMed] [Google Scholar]
- 8. Habert R, Lejeune H, Saez JM. Origin, differentiation and regulation of fetal and adult Leydig cells. Mol Cell Endocrinol 2001;179(1-2):47–74. [DOI] [PubMed] [Google Scholar]
- 9. Barsoum IB, Kaur J, Ge RS, Cooke PS, Yao HH. Dynamic changes in fetal Leydig cell populations influence adult Leydig cell populations in mice. FASEB J 2013;27(7):2657–2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Orth JM. Proliferation of Sertoli cells in fetal and postnatal rats: a quantitative autoradiographic study. Anat Rec 1982;203(4):485–492. [DOI] [PubMed] [Google Scholar]
- 11. O'Shaughnessy PJ, Baker PJ, Vainio HM, McMahon AP. Localization of 17beta-hydroxysteroid dehydrogenase/17-ketosteroid reductase isoform expression in the developing mouse testis—androstenedione is the major androgen secreted by fetal/neonatal leydig cells. Endocrinology 2000;141(7):2631–2637. [DOI] [PubMed] [Google Scholar]
- 12. Shima Y, Morohashi KI. Leydig progenitor cells in fetal testis. Mol Cell Endocrinol 2017;445:55–64. [DOI] [PubMed] [Google Scholar]
- 13. O'Shaughnessy PJ, Baker P, Sohnius U, Haavisto AM, Charlton HM, Huhtaniemi I. Fetal development of Leydig cell activity in the mouse is independent of pituitary gonadotroph function. Endocrinology 1998;139(3):1141–1146. [DOI] [PubMed] [Google Scholar]
- 14. Migrenne S, Pairault C, Racine C, Livera G, Géloso A, Habert R. Luteinizing hormone-dependent activity and luteinizing hormone-independent differentiation of rat fetal Leydig cells. Mol Cell Endocrinol 2001;172(1-2):193–202. [DOI] [PubMed] [Google Scholar]
- 15. Habert R, Picon R. Control of testicular steroidogenesis in foetal rat: effect of decapitation on testosterone and plasma luteinizing hormone-like activity. Acta Endocrinol 1982;99:466–473. [DOI] [PubMed] [Google Scholar]
- 16. Habert R, Rouiller-Fabre V, Lecerf L, Levacher C, Saez JM. Developmental changes in testosterone production by the rat testis in vitro during late fetal life. Arch Androl 1992;29(2):191–197. [DOI] [PubMed] [Google Scholar]
- 17. Huhtaniemi I, Pelliniemi LJ. Fetal Leydig cells. Cellular origin, morphology, life span, and special functional features. Exp Biol Med 1992;201(2):125–140. [DOI] [PubMed] [Google Scholar]
- 18. Habert R. In vivo acute testicular testosterone response to injection of luteinizing hormone in the rat fetus. Acta Endocrinol 1993;128:268–273. [DOI] [PubMed] [Google Scholar]
- 19. Kerr JB, Knell CM. The fate of fetal Leydig cells during the development of the fetal and postnatal rat testis. Development 1988;103:535–544. [DOI] [PubMed] [Google Scholar]
- 20. Mendis-Handagama SM, Ariyaratne HB. Differentiation of the adult Leydig cell population in the postnatal testis. Biol Reprod 2001;653:660–671. [DOI] [PubMed] [Google Scholar]
- 21. Haider SG. Cell biology of Leydig cells in the testis. Int Rev Cytol 2004;233:181–241. [DOI] [PubMed] [Google Scholar]
- 22. Ge RS, Shan LX, Hardy MP. Pubertal development of Leydig cells. In: Payne AH, Hardy MP, Russell LD (eds.), The Leydig Cell. Vienna, IL: Cache River Press; 1996:159. [Google Scholar]
- 23. Lo KC, Lei Z, Rao ChV, Beck J, Lamb DJ. De novo testosterone production in luteinizing hormone receptor knockout mice after transplantation of leydig stem cells. Endocrinology 2004;145(9):4011–4015. [DOI] [PubMed] [Google Scholar]
- 24. Ge RS, Dong Q, Sottas CM, Papadopoulos V, Zirkin BR, Hardy MP. In search of rat stem Leydig cells: identification, isolation, and lineage-specific development. Proc Natl Acad Sci USA 2006;103(8):2719–2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen H, Wang Y, Ge R, Zirkin BR. Leydig cell stem cells: Identification, proliferation and differentiation. Mol Cell Endocrinol 2017;445:65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nanjappa MK, Medrano TI, Prins GS, Chen H, Zirkin BR, Cooke PS. Transdifferentiation of adult rat stem Leydig cells into prostatic and uterine epithelium, but not epidermis. Andrology 2017;5(6):1165–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen H, Huhtaniemi I, Zirkin BR. Depletion and repopulation of Leydig cells in the testes of aging brown Norway rats. Endocrinology 1996;137(8):3447–3452. [DOI] [PubMed] [Google Scholar]
- 28. Smith LB, O'Shaughnessy PJ, Rebourcet D. Cell-specific ablation in the testis: what have we learned? Andrology 2015;3(6):1035–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Christensen AK, Mason NR. Comparative ability of seminiferous tubules and interstitial tissue of rat tastes to synthesize androgen from progesterone-4-14C in vitro. Endocrinology 1965;76(4):646–656. [DOI] [PubMed] [Google Scholar]
- 30. Vandermark NL, Ewing LL. Factors affecting testicular metabolism and function. Reproduction 1963;6(1):1–8. [DOI] [PubMed] [Google Scholar]
- 31. Chubb C, Ewing LL. Steroid secretion by in vitro perfused testes: Inhibitors of testosterone biosynthesis. Am J Physiol 1979;237:E239–E246. [DOI] [PubMed] [Google Scholar]
- 32. Zirkin BR, Ewing LL, Kromann N, Cochran RC. Testosterone secretion by rat, rabbit, guinea pig, dog, and hamster testes perfused in vitro: correlation with Leydig cell ultrastructure. Endocrinology 1980;1076:1867–1874. [DOI] [PubMed] [Google Scholar]
- 33. Ewing LL, Zirkin BR. Leydig cell structure and steroidogenic function. Rec Prog Horm Res 1983;39:599–635. [DOI] [PubMed] [Google Scholar]
- 34. Wing T-Y, Ewing LL, Zirkin BR. Effect of luteinizing hormone withdrawal on Leydig cell smooth endoplasmic reticulum and steroidogenic reactions which convert pregnenolone to testosterone. Endocrinology 1984;1156:2290–2296. [DOI] [PubMed] [Google Scholar]
- 35. Ewing LL, Wing T-Y, Cochran RC, Kromann N, Zirkin BR. Effect of luteinizing hormone on Leydig cell structure and testosterone secretion. Endocrinology 1983;112(5):1763–1769. [DOI] [PubMed] [Google Scholar]
- 36. Keeney DS, Mendi-Handagama ML, Zirkin BR, Ewing LL. Effect of long term deprivation of luteinizing hormone on Leydig cell volume, Leydig cell number, and steroidogenic capacity of the rat testis. Endocrinology 1988;123(6):2906–2915. [DOI] [PubMed] [Google Scholar]
- 37. Ascoli M. Characterization of several clonal lines of cultured Leydig tumor cells: gonadotropin receptors and steroidogenic responses. Endocrinology 1981;108(1):88–95. [DOI] [PubMed] [Google Scholar]
- 38. Shin SI, Yasumura Y, Sato GH. Studies on interstitial cells in tissue culture. II. Steroid biosynthesis by a clonal line of rat testicular interstitial cells. Endocrinology 1968;82(3):614–616. [DOI] [PubMed] [Google Scholar]
- 39. Mather JP. Establishment and characterization of two distinct mouse testicular epithelial cell lines. Biol Reprod 1980;23(1):243–252. [DOI] [PubMed] [Google Scholar]
- 40. Rebois RV. Establishment of gonadotropin-responsive murine Leydig tumor cell line. J Cell Biol 1982;941:70–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Finaz C, Lefèvre A, Dampfhoffer D. Construction of a Leydig cell line synthesizing testosterone under gonadotropin stimulation: a complex endocrine function immortalized by cell hybridization. Proc Natl Acad Sci USA 1987;84(16):5750–5753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Forgacs AL, Ding Q, Jaremba RG, Huhtaniemi IT, Rahman NA, Zacharewski TR. BLTK1 murine Leydig cells: a novel steroidogenic model for evaluating the effects of reproductive and developmental toxicants. Toxicol Sci 2012;127(2):391–402. [DOI] [PubMed] [Google Scholar]
- 43. Klinefelter GR, Hall PF, Ewing LL. Effect of luteinizing hormone deprivation in situ on steroidogenesis of rat Leydig cells purified by a multistep procedure. Biol Reprod 1987;363: 769–783. [DOI] [PubMed] [Google Scholar]
- 44. Klinefelter GR, Ewing LL. Optimizing testosterone production by purified adult rat Leydig cells in vitro. In Vitro Cell Dev Biol 1988;24(6):545–549. [DOI] [PubMed] [Google Scholar]
- 45. Klinefelter GR, Ewing LL. Maintenance of testosterone production by purified adult rat Leydig cells for 3 days in vitro. In Vitro Cell Dev Biol 1989;25(3):283–288. [DOI] [PubMed] [Google Scholar]
- 46. Plant TM. 60 Years of Neuroendocrinology: The hypothalamo-pituitary-gonadal axis. J Endocrinol 2015;226(2):T41–T54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kaprara A, Huhtaniemi IT. The hypothalamus-pituitary-gonad axis: tales of mice and men. Metabolism 2017; in press. [DOI] [PubMed] [Google Scholar]
- 48. Dufau ML, Catt KJ. Gonadotropin receptors and regulation of steroidogenesis in the testis and ovary. Vitam Horm 1978;36:461–592. [DOI] [PubMed] [Google Scholar]
- 49. Papadopoulos V, Miller WL. Role of mitochondria in steroidogenesis. Best Practice Res Clin Endocrinol Metab 2012;26(6):771–790. [DOI] [PubMed] [Google Scholar]
- 50. Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev 2011;32(1):81–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Payne AH, Hales DB. Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocr Rev 2004;25(6):947–970. [DOI] [PubMed] [Google Scholar]
- 52. Hall PF. Cytochromes P-450 and the regulation of steroid synthesis. Steroids 1986;48(3-4):131–196. [DOI] [PubMed] [Google Scholar]
- 53. Jefcoate C. High-flux mitochondrial cholesterol trafficking, a specialized function of the adrenal cortex. J Clin Invest 2002;110(7):881–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hu J, Zhang Z, Shen WJ, Azhar S. Cellular cholesterol delivery, intracellular processing and utilization for biosynthesis of steroid hormones. Nutr Metab (Lond) 2010;7(1):47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Payne AH, Hardy MP. The Leydig Cell in Health and Disease. Totoawa, NJ: Humana Press, 2007. [Google Scholar]
- 56. Venugopal S, Martinez-Arguelles DB, Chebbi S, Hullin-Matsuda F, Kobayashi T, Papadopoulos V. Plasma membrane origin of the steroidogenic pool of cholesterol used in hormone-induced acute steroid formation in Leydig cells. J Biol Chem 2016;291(50):26109–26125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cooke BA, Janszen FH, Clotscher WF, van der Molen HJ. Effect of protein-synthesis inhibitors on testosterone production in rat testis interstitial tissue and Leydig-cell preparations. Biochem J 1975;150(3):413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cooke BA, Lindh LM, van der Molen HJ. The mechanism of action of lutropin on regulator protein(s) involved in Leydig-cell steroidogenesis. Biochem J 1979;184(1):33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Epstein LF, Orme-Johnson NR. Acute action of luteinizing hormone on mouse Leydig cells: accumulation of mitochondrial phosphoproteins and stimulation of testosterone synthesis. Mol Cell Endocrinol 1991;81(1-3):113–126. [DOI] [PubMed] [Google Scholar]
- 60. Epstein LF, Orme-Johnson NR. Regulation of steroid hormone biosynthesis. Identification of precursors of a phosphoprotein targeted to the mitochondrion in stimulated rat adrenal cortex cells. J Biol Chem 1991;266:19739–19745. [PubMed] [Google Scholar]
- 61. Clark BJ, Wells J, King SR, Stocco DM. The purification, cloning, and expression of a novel luteinizing hormone-induced mitochondrial protein in MA-10 mouse Leydig tumor cells. Characterization of the steroidogenic acute regulatory protein (StAR). J Biol Chem 1994;269:28314–28322. [PubMed] [Google Scholar]
- 62. Clark BJ. ACTH action on StAR biology. Front Neurosci 2016;10:547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Miller WL, Bose HS. Early steps in steroidogenesis: intracellular cholesterol trafficking. J Lipid Res 2011;52(12):2111–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rone MB, Midzak AS, Issop L, Rammouz G, Jagannathan S, Fan J, Ye X, Blonder J, Veenstra T, Papadopoulos V. Identification of a dynamic mitochondrial protein complex driving cholesterol import, trafficking, and metabolism to steroid hormones. Mol Endocrinol 2012;26(11):1868–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Caron KM, Ikeda Y, Soo SC, Stocco DM, Parker KL, Clark BJ. Characterization of the promoter region of the mouse gene encoding the steroidogenic acute regulatory protein. Mol Endocrinol 1997;11(2):138–147. [DOI] [PubMed] [Google Scholar]
- 66. Hasegawa T, Zhao L, Caron KM, Majdic G, Suzuki T, Shizawa S, Sasano H, Parker KL. Developmental roles of the steroidogenic acute regulatory protein (StAR) as revealed by StAR knockout mice. Mol Endocrinol 2000;14(9):1462–1471. [DOI] [PubMed] [Google Scholar]
- 67. Caron KM, Soo SC, Wetsel WC, Stocco DM, Clark BJ, Parker KL. Targeted disruption of the mouse gene encoding steroidogenic acute regulatory protein provides insights into congenital lipoid adrenal hyperplasia. Proc Natl Acad Sci USA 1997;94(21):11540–11545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hall PF, Almahbobi G. The role of the cytoskeleton in the regulation of steroidogenesis. J Steroid Biochem Mol Biol 1992;43(8):769–777. [DOI] [PubMed] [Google Scholar]
- 69. Sewer MB, Li D. Regulation of steroid hormone biosynthesis by the cytoskeleton. Lipids 2008;43(12):1109–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Shen WJ, Zaidi SK, Patel S, Cortez Y, Ueno M, Azhar R, Azhar S, Kraemer FB. Ablation of vimentin results in defective steroidogenesis. Endocrinology 2012;153(7):3249–3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Issop L, Rone MB, Papadopoulos V. Organelle plasticity and interactions in cholesterol transport and steroid biosynthesis. Mol Cell Endocrinol 2013;371(1-2):34–46. [DOI] [PubMed] [Google Scholar]
- 72. Aghazadeh Y, Rone M, Hales DB, Blonder J, Veenstra T, Hales DB, Culty M, Papadopoulos V. Hormone-induced 14-3-3γ adaptor protein regulates steroidogenic acute regulatory protein activity and steroid biosynthesis in MA-10 Leydig cells. J Biol Chem 2012;28719:15380–15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Aghazadeh Y, Martinez-Arguelles DB, Fan J, Culty M, Papadopoulos V. Induction of androgen formation in the male by a TAT-VDAC1 fusion peptide blocking 14-3-3ɛ protein adaptor and mitochondrial VDAC1 interactions. Mol Ther 2014;22(10):1779–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Rone MB, Fan J, Papadopoulos V. Cholesterol transport in steroid biosynthesis: Role of protein–protein interactions and implications in disease states. Biochim Biophys Acta 2009;1791(7):646–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Li H, Papadopoulos V. Peripheral-type benzodiazepine receptor function in cholesterol transport. Identification of a putative cholesterol recognition/interaction amino acid sequence and consensus pattern. Endocrinology 1998;139(12):4991–4997. [DOI] [PubMed] [Google Scholar]
- 76. Jamin N, Neumann J, Ostuni M, Vu T, Yao Z, Murail S, Robert J, Giatzakis C, Papadopoulos V, Lacapère J. Characterization of the cholesterol recognition amino acid consensus sequence of the peripheral-type benzodiazepine receptor. Mol Endocrinol 2005;19(3):588–594. [DOI] [PubMed] [Google Scholar]
- 77. Li F, Liu J, Zheng Y, Garavito RM, Ferguson-Miller S. Crystal structures of translocator protein (TSPO) and mutant mimic of a human polymorphism. Science 2015;347(6221):555–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Papadopoulos V, Fan J, Zirkin B. Translocator protein (18 kDa): an update on its function in steroidogenesis. J Neuroendocrinol 2017, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Gavish M, Weizman R. Role of peripheral-type benzodiazepine receptors in steroidogenesis. Clin Neuropharmacol 1997;20(6):473–481. [DOI] [PubMed] [Google Scholar]
- 80. Kelly-Hershkovitz E, Weizman R, Spanier I, Leschiner S, Lahav M, Weisinger G, Gavish M. Effects of peripheral-type benzodiazepine receptor antisense knockout on MA-10 Leydig cell proliferation and steroidogenesis. J Biol Chem 1998;273(10):5478–5483. [DOI] [PubMed] [Google Scholar]
- 81. Papadopoulos V, Baraldi M, Guilarte TR, Knudsen TB, Lacapere JJ, Lindemann P, Norenberg MD, Nutt D, Weizman A, Zhang MR, Gavish M. Translocator protein (18kDa): new nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol Sci 2006;27(8):402–409. [DOI] [PubMed] [Google Scholar]
- 82. Chung JY, Chen H, Midzak A, Burnett AL, Papadopoulos V, Zirkin BR. Drug ligand-induced activation of translocator protein (TSPO) stimulates steroid production by aged brown Norway rat Leydig cells. Endocrinology 2013;154(6):2156–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Gazouli M, Han Z, Papadopoulos V. Identification of a peptide antagonist to the peripheral-type benzodiazepine receptor that inhibits hormone-stimulated leydig cell steroid formation. J Pharmacol Exp Ther 2002;303(2):627–632. [DOI] [PubMed] [Google Scholar]
- 84. Midzak A, Akula N, Lecanu L, Papadopoulos V. Novel androstenetriol interacts with the mitochondrial translocator protein and controls steroidogenesis. J Biol Chem 2011;286(11):9875–9887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Midzak A, Denora N, Laquintana V, Cutrignelli A, Lopedota A, Franco M, Altomare CD, Papadopoulos V. 2-Phenylimidazo[1,2-a]pyridine-containing ligands of the 18-kDa translocator protein (TSPO) behave as agonists and antagonists of steroidogenesis in a mouse leydig tumor cell line. Eur J Pharm Sci 2015;76:231–237. [DOI] [PubMed] [Google Scholar]
- 86. Midzak AS, Akula N, Rone MB, Papadopoulos V. Computational modeling and biological validation of novel non-steroidal ligands for the cholesterol recognition/interaction amino acid consensus (CRAC) motif of the mitochondrial translocator protein (TSPO). Pharmacol Res 2015;99:393–403. [DOI] [PubMed] [Google Scholar]
- 87. Papadopoulos V, Nowzari FB, Krueger KE. Hormone-stimulated steroidogenesis is coupled to mitochondrial benzodiazepine receptors. Tropic hormone action on steroid biosynthesis is inhibited by flunitrazepam. J Biol Chem 1991;266:3682–3687. [PubMed] [Google Scholar]
- 88. Papadopoulos V, Aghazadeh Y, Fan J, Campioli E, Zirkin B, Midzak A. Translocator protein-mediated pharmacology of cholesterol transport and steroidogenesis. Mol Cell Endocrinol 2015;408:90–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Aghazadeh Y, Zirkin BR, Papadopoulos V. Pharmacological regulation of the cholesterol transport machinery in steroidogenic cells of the testis. Vitam Horm 2015;98:189–227. [DOI] [PubMed] [Google Scholar]
- 90. Midzak A, Zirkin B, Papadopoulos V. Translocator protein: pharmacology and steroidogenesis. Biochm Soc Trans 2015;43(4):572–578. [DOI] [PubMed] [Google Scholar]
- 91. Stocco DM, Zhao AH, Tu LN, Morohaku K, Selvaraj V. A brief history of the search for the protein(s) involved in the acute regulation of steroidogenesis. Mol Cell Endocrinol 2017;441:7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Morohaku K, Pelton SH, Daugherty DJ, Butler WR, Deng W, Selvaraj V. Translocator protein/peripheral benzodiazepine receptor is not required for steroid hormone biosynthesis. Endocrinology 2014;155(1):89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Tu LN, Morohaku K, Manna PR, Pelton SH, Butler WR, Stocco DM, Selvaraj V. Peripheral benzodiazepine receptor/translocator protein global knock-out mice are viable with no effects on steroid hormone biosynthesis. J Biol Chem 2014;289(40):27444–27454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Tu LN, Zhao AH, Stocco DM, Selvaraj V. PK11195 effect on steroidogenesis is not mediated through the translocator protein (TSPO). Endocrinology 2015;156(3):1033–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Hauet T, Yao ZX, Bose HS, Wall CT, Han Z, Li W, Hales DB, Miller WL, Culty M, Papadopoulos V. Peripheral-type benzodiazepine receptor-mediated action of steroidogenic acute regulatory protein on cholesterol entry into leydig cell mitochondria. Mol Endocrinol 2005;19(2):540–554. [DOI] [PubMed] [Google Scholar]
- 96. Fan J, Wang K, Zirkin BR, Papadopoulos V. CRISPR/CAS9-mediated Tspo gene mutations lead to reduced mitochondrial membrane potential and steroid formation in MA-10 mouse tumor Leydig cells. Endocrinology 2018;159:1130–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Fan J, Campioli E, Midzak A, Culty M, Papadopoulos V. Conditional steroidogenic cell-targeted deletion of TSPO unveils a crucial role in viability and hormone-dependent steroid formation. Proc Natl Acad Sci USA 2015;112(23):7261–7266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Barron AM, Ji B, Kito S, Suhara T, Higuchi M. Steroidogenic abnormalities in translocator protein knockout mice and significance in the aging male. Biochem J 2018;475:75–85. [DOI] [PubMed] [Google Scholar]
- 99. Owen DR, Fan J, Campioli E, Venugopal S, Midzak A, Daly E, Harlay A, Issop L, Libri V, Kalogiannopoulou D, Oliver E, Gallego-Colon E et al. TSPO mutations in rats and a human polymorphism impair the rate of steroid synthesis. Biochem J 2017;474(23):3985–3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Miller WL. Steroidogenic acute regulatory protein (StAR), a novel mitochondrial cholesterol transporter. Biochim Biophys Acta 2007;1771:663–676. [DOI] [PubMed] [Google Scholar]
- 101. Tremblay JJ. Molecular regulation of steroidogenesis in endocrine Leydig cells. Steroids 2015;103:3–10. [DOI] [PubMed] [Google Scholar]
- 102. Abdou HS, Bergeron F, Tremblay JJ. A cell-autonomous molecular cascade initiated by AMP-activated protein kinase represses steroidogenesis. Mol Cell Biol 2014;34:4257–4271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Martin LJ, Tremblay JJ. Nuclear receptors in leydig cell gene expression and function. Biol Reprod 2010;83:3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Gorostizaga A, Cornejo Maciel F, Brion L, Maloberti P, Podestá EJ, Paz C. Tyrosine phosphatases in steroidogenic cells: regulation and function. Mol Cell Endocrinol 2007;265–266:131–137. [DOI] [PubMed] [Google Scholar]
- 105. Surampudi PN, Wang C, Swerdloff R. Hypogonadism in the aging male diagnosis, potential benefits, and risks of testosterone replacement therapy. Int J Endocrinol 2012;2012:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Bhasin S, Huang G, Travison TG, Basaria S. Age-related changes in the male reproductive axis. In: De Groot LJ, et al. (eds.), Endotext. South Dartmouth, MA: MDText.Com, Inc.; 2014:2000–2014. [Google Scholar]
- 107. Kim HH, Schlegel PN. Endocrine manipulation in male infertility. Urol Clin North Am 2008;35:303–318. [DOI] [PubMed] [Google Scholar]
- 108. Hwang K, Walters RC, Lipshultz LI. Contemporary concepts in the evaluation and management of male infertility. Nat Rev Urol 2011;8:86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Sigman M, Howards SS. 2012 male infertility. In: Walsh PC, Retik AB, Vaughan ED Jr (eds.), Campbell's Textbook of Urology, 7th ed Philadelphia: W B Saunders Co; 1998: 1287. [Google Scholar]
- 110. Bhasin S, Basaria S. Diagnosis and treatment of hypogonadism in men. Best Prac Res Clin Endocrinol Metab 2011;25:251–270. [DOI] [PubMed] [Google Scholar]
- 111. Kolettis PN, Purcell ML, Parker W, Poston T, Nangia A. Medical testosterone: an iatrogenic cause of male infertility and a growing problem. Urology 2015;85:1068–1073. [DOI] [PubMed] [Google Scholar]
- 112. Abadilla KA, Dobs AS. Topical testosterone supplementation for the treatment of male hypogonadism. Drugs 2012;72:1591–1603. [DOI] [PubMed] [Google Scholar]
- 113. Gooren LJ, Bunck MCM. Transdermal testosterone delivery: testosterone patch and gel. World J Urol 2003;21:316–319. [DOI] [PubMed] [Google Scholar]
- 114. Wang C, Ilani N, Arvert S, McLachlan RI, Soulis T, Watkinson A. Efficacy and safety of the 2% formulation of testosterone topical solution applied to the axillae in androgen-deficient men. Clin Endocrinol 2011;75:836–843. [DOI] [PubMed] [Google Scholar]
- 115. Finkle WD, Greenland S, Ridgeway GK, Adams JL, Frasco MA, Cook MB, Fraumeni JF Jr, Hoover RN. Increased risk of non-fatal myocardial infarction following testosterone therapy prescription in men. PLoS One 2014;9:e85805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Vigen R, O’Donnell CI, Barón AE, Grunwald GK, Maddox TM, Bradley SM, Barqawi A, Woning G, Wierman ME, Plomondon ME, Rumsfeld JS, Ho PM. Association of testosterone therapy with mortality, myocardial infarction, and stroke in men with low testosterone levels. JAMA 2013;310:1829–1836. [DOI] [PubMed] [Google Scholar]
- 117. Xu L, Freeman G, Cowling BJ, Schooling CM. Testosterone therapy and cardiovascular events among men: a systematic review and meta-analysis of placebo-controlled randomized trials. BMC Med 2013;11:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Bosland MC. Testosterone treatment is a potent tumor promoter for the rat prostate. Endocrinology 2014;155:4629–4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Pavlovich CP, King P, Goldstein M, Schlegel PN. Evidence of a treatable endocrinopathy in infertile men. J Urol 2001;165:837–841. [PubMed] [Google Scholar]
- 120. Ramasamy R, Stahl PJ, Schlegel PN. Medical therapy for spermatogenic failure. Asian J Androl 2012;14:57–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Chen H, Hardy MP, Huhtaniemi I, Zirkin BR. Age-related decreased Leydig cell testosterone production in the Brown Norway rat. J Androl 1994;15:551–557. [PubMed] [Google Scholar]
- 122. Wang C, Leung A, Sinha-Hikim AP. Reproductive aging in the male brown-Norway rat: a model for the human. Endocrinology 1993;133:2773–2781. [DOI] [PubMed] [Google Scholar]
- 123. Grzywacz FW, Chen H, Allegretti J, Zirkin BR. Does age-associated reduced Leydig cell testosterone production in brown Norway rats result from under-stimulation by luteinizing hormone? J Androl 1998;19:625–630. [PubMed] [Google Scholar]
- 124. Bonavera JJ, Swerdloff RS, Leung A, Lue YH, Baravarian S, Superiano L, Sinha-Hikim AP, Wang C. In the male brown-Norway (BN) male rat, reproductive aging is associated with decreased LH-pulse amplitude and area. J Androl 1997;18:359–365. [PubMed] [Google Scholar]
- 125. Luo L, Chen H, Zirkin BR. Leydig cell aging: steroidogenic acute regulatory protein (StAR) and cholesterol side-chain cleavage enzyme. J Androl 2001;22:149–156. [PubMed] [Google Scholar]
- 126. Luo L, Chen H, Zirkin BR. Temporal relationships among testosterone production, steroidogenic acute regulatory protein (StAR), and P450 side-chain cleavage enzyme (P450scc) during Leydig cell aging. J Androl 2005;26:25–31. [PubMed] [Google Scholar]
- 127. Culty M, Luo L, Yao ZX, Chen H, Papadopoulos V, Zirkin BR. Cholesterol transport, peripheral benzodiazepine receptor, and steroidogenesis in aging Leydig cells. J Androl 2002;23:439–447. [PubMed] [Google Scholar]
- 128. Nothdurfter C, Baghai TC, Schüle C, Rupprecht R. Translocator protein (18 kDa) (TSPO) as a therapeutic target for anxiety and neurologic disorders. Eur Arch Psychiatry Clin Neurosci 2012;262 Suppl 2:107–112. [DOI] [PubMed] [Google Scholar]
- 129. Da Pozzo E, Giacomelli C, Costa B, Cavallini C, Taliani S, Barresi E, Da Settimo F, Martini C. TSPO PIGA ligands promote neurosteroidogenesis and human astrocyte well-being. Int J Mol Sci 2016;17:1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Arakane F, Kallen CB, Watari H, Foster JA, Sepuri NB, Pain D, Stayrook SE, Lewis M, Gerton GL, Strauss JF 3rd. The mechanism of action of steroidogenic acute regulatory protein (StAR). J Biol Chem 1998;273:16339–16345. [DOI] [PubMed] [Google Scholar]