

SUMMARY
Topoisomerase IIβ binding protein 1 (TopBP1) is a critical protein-protein interaction hub in DNA replication checkpoint control. It was proposed that TopBP1 BRCT5 interacts with Bloom syndrome helicase (BLM) to regulate genome stability through either phospho-Ser304 or phospho-Ser338 of BLM. Here we show that TopBP1 BRCT5 specifically interacts with the BLM region surrounding pSer304, not pSer338. Our crystal structure of TopBP1 BRCT4/5 bound to BLM reveals recognition of pSer304 by a conserved pSer-binding pocket, and interactions between a FVPP motif N-terminal to pSer304 and a hydrophobic groove on BRCT5. This interaction utilizes the same surface of BRCT5 that recognizes the DNA damage mediator, MDC1, however the binding orientations of MDC1 and BLM are reversed. While the MDC1 interactions are largely electrostatic, the interaction with BLM has higher affinity and relies on a mix of electrostatics and hydrophobicity. We suggest similar evolutionarily conserved interactions may govern interactions between TopBP1 and 53BP1.
Keywords: TopBP1, BLM, Bloom syndrome, BRCT, phospho-peptide interactions, X-ray crystallography, fluorescence polarization
eTOC BLURB
TopBP1 is a hub for DNA damage signaling interactions. Sun et al. determine the structure of TopBP1 BRCT5 bound to its phosphorylated partner, Bloom syndrome helicase (BLM). The structure reveals a strikingly different binding mode than that observed for another TopBP1 BRCT5 interaction partner, MDC1.
INTRODUCTION
Topoisomerase IIβ Binding Protein 1(TopBP1) is a key protein interaction hub that regulates DNA replication, checkpoint activation and damage response (Garcia et al., 2005; Wardlaw et al., 2014). TopBP1 protein interactions are mediated by its nine BRCA1 associated C-terminus (BRCT) repeats, as well as its ATR activation domain (AAD) that is particularly critical for its role in DNA replication stress signaling which initiates with ATR activation. Although dozens of protein–protein interactions (PPIs) involving the TopBP1 BRCT domains have been reported in the literature, how many of these distinct domains collaborate with different protein partners remains unclear. It has been shown that the N terminal three BRCTs (BRCT0/1/2) of TopBP1 can interact with the phosphorylated Rad9 tail of the Rad9-Hus1-Rad1 (9-1-1) complex to assist ATR-mediated activation of CHK1 in mammalian cells (Delacroix et al., 2007; Greer et al., 2003; Lee et al., 2007). The C-terminal BRCT7/8 next to the AAD can bind to phosphorylated ATR and has been suggested to help TopBP1 to facilitate ATR kinase activity and substrate binding (Liu et al., 2011). This BRCT7/8 also interacts with BRCA1 associated C-terminal helicase/Fanconi Anemia J group proteins (BRIP1/FANCJ). This subsequently extends single stranded DNA regions and enhances replication protein A (RPA) loading at stalled replication forks (Gong et al., 2010).
The internal BRCT5 has been implicated in TopBP1 recruitment to sites of DNA damage under certain circumstances (Cescutti et al., 2010; Yamane et al., 2002) and several DNA damage-associated proteins have been suggested to interact with this domain in a phosphorylation-dependent manner. The first potential partner identified for BRCT5 was 53BP1, whose interaction was suggested to mediate recruitment of TopBP1 to sites of DNA double strand breaks (DSBs) during G1 (Cescutti et al., 2010; Yamane et al., 2002). Another DNA double strand break mediator, MDC1, has also been shown to interact with BRCT5, via phosphorylated Ser-Asp-Thr (SDT) repeats in MDC1 (Leung et al., 2013; Wang et al., 2011). However, more recent studies have suggested that, in cells, TopBP1 instead binds to MDC1 via BRCT1 (Blackford et al., 2015; Choi and Yoo, 2016).
Recently, two reports have shown that BRCT5 may also interact with Bloom syndrome, RecQ-like helicase (BLM) in a phosphorylation and cell-cycle dependent manner (Blackford et al., 2015; Wang et al., 2013). In humans, mutations in BLM cause Bloom syndrome, a disease characterized by growth retardation, immunodeficiency, genomic instability, and cancer predisposition (German, 1993). Disruption of BLM-TopBP1 interaction in cells leads to elevated sister chromatid exchanges (SCEs) and chromosomal aberrations, features that are commonly found in Bloom syndrome patients (Blackford et al., 2015; Chaganti et al., 1974; German et al., 1965; Wang et al., 2013). While some evidence suggests that TopBP1 interacts with BLM pSer338 to stabilize BLM during S phase (Wang et al., 2013), other evidence has suggested that pSer304 of BLM is more crucial for this interaction, and that TopBP1 has no effect on BLM stability (Bass et al., 2016; Blackford et al., 2015). Here we use fluorescence polarization to demonstrate that TopBP1 BRCT5 specifically binds phosphopeptides corresponding to the pSer304 region of BLM but not the pSer338 region. The X-ray crystal structure of mammalian TopBP1 in complex with a BLM peptide reveals specific recognition of pSer304 by the phosphate binding pocket of BRCT5 and recognition of the residues N-terminal to pSer304 by a hydrophobic groove and positively charged loop in BRCT5. The same surface is used by BRCT5 to bind a phosphorylated SDT repeat in the DNA damage checkpoint mediator, MDC1, however the orientation of BLM binding is reversed compared to MDC1. We suggest that TopBP1 BRCT5 can engage alternative protein partners to regulate DNA replication checkpoints.
RESULTS
TopBP1 BRCT5 interacts with BLM mainly through pSer304 and not pSer338
To assess the likelihood that either Ser304 or Ser338 could serve as targets for TopBP1 BRCT5, we probed the conservation of sequences in this region in a number of vertebrate organisms (Figure 1A). Both Ser304 and Ser338 belong to the BLM N-terminal domain (1-636) that is largely unstructured and poorly conserved. The alignment shows striking conservation of residues N-terminal to the phosphorylation site of Ser304, with acidic residues conserved at the −7 and −5 positions, and a hydrophobic F(V/I)PP motif conserved from −4 to −1 (Figure 1A). In contrast, there is much poorer sequence conservation around Ser338. To directly investigate TopBP1 BRCT5 interactions with peptide targets in vitro, we synthesized FITC labeled BLM peptides corresponding to both sites (297-DTDFVPPpSPEEII-309, 331-KEDVLSTpSKDL-341) and tested their ability to interact with TopBP1 BRCT5 using fluorescence polarization (FP) spectroscopy. TopBP1 interacts tightly with pSer304 BLM peptide (KD = 3.9 ± 0.3 μM), but weakly with the pSer338 BLM peptide (KD ≥ 300 μM) (Figure 1B). To test whether the interaction between TopBP1 BRCT5 and pSer304 BLM peptide is phosphorylation dependent, we dephosphorylated the pSer304 BLM peptide using λ phosphatase (STAR Methods) and examined its interaction with TopBP1 BRCT5 in vitro by FP. The λ phosphatase treated peptide binds TopBP1 BRCT5 much more weakly (KD ≥ 60 μM) than the phosphorylated peptide (Figure 1C). In addition, control peptides treated under same condition without phosphatase (KD = 2.6 ± 0.4 μM) or with heat inactivated phosphatase both bind BRCT5 at similar affinity compared to untreated BLM pSer304 peptide (KD= 3.7 ± 0.8 μM), indicating the treatment itself did not affect peptide affinity to TopBP1 BRCT5. These results show that the phosphorylation of Ser304 is essential for its interaction with TopBP1 BRCT5.
Figure 1. BLM interacts with TopBP1 via pSer304, not pSer338.
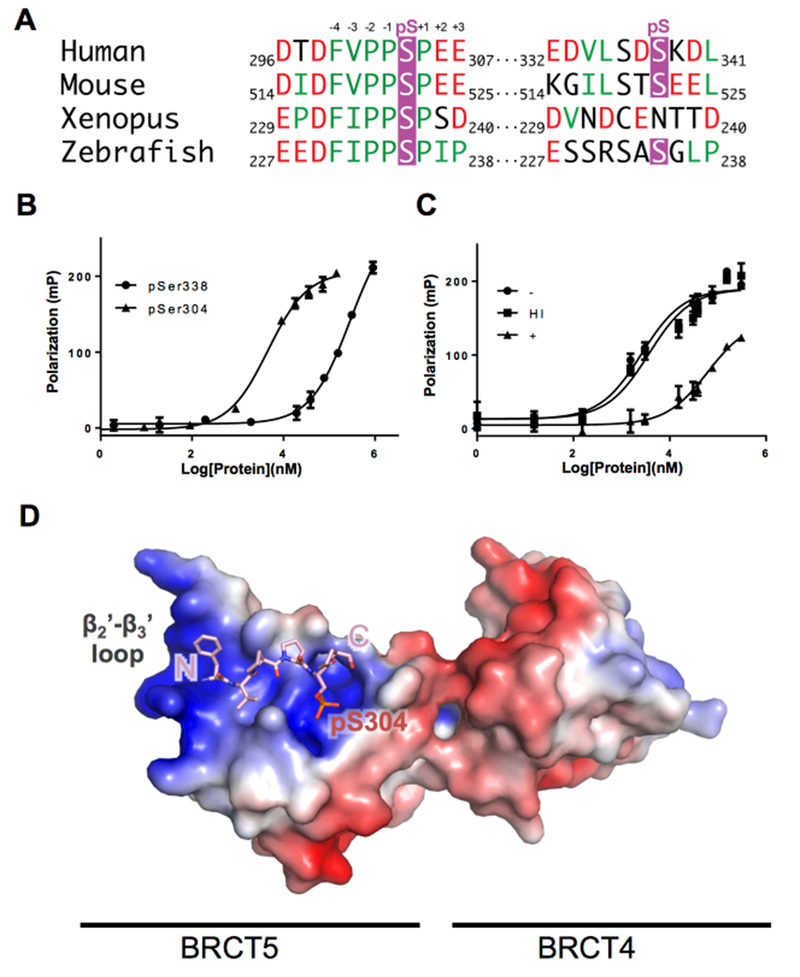
A. Sequence alignment of BLM orthologs around pSer304 and pSer338. Phosphorserine residues are colored in purple, hydrophobic residues are colored in green, and negatively charged residues are colored in red.
B. TopBP1 BRCT4/5 binds more tightly to BLM pSer304 than BLM pSer338. GST-hTopBP1 BRCT5 protein was titrated against BLM phosphopeptides corresponding to either the pSer304 or pSer338 regions and binding was monitored by fluorescence polarization (FP) spectroscopy.
C. TopBP1 BRCT4/5 binds BLM pSer304 in a phosphorylation dependent manner. BLM pSer304 peptide was dephosphorylated by treatment with λ phosphatase (+) and its interaction with GST-hTopBP1 BRCT5 protein was measure by FP. BLM pSer304 peptide mock-treated without λ phosphatase (−) or with heat inactivated phosphatase (HI) were also included as controls.
D. Structural overview of TopBP1 BRCT4/5 in complex with pSer304 BLM peptide. An electrostatic charge surface is displayed for TopBP1 BRCT4/5 while the modeled BLM phosphopeptide is shown in pink sticks. See also Figure S1–3 and Table S1.
Crystal structure of TopBP1 BRCT4/5 bound to phosphorylated BLM
To gain molecular insight into the interactions involved in pSer304-dependent BLM recognition by TopBP1, we crystallized and determined the structure of TopBP1 BRCT4/5 bound to a pSer304-containing BLM peptide. Crystallization trials using human TopBP1 BRCT4/5 were unsuccessful, however we were able to crystallize and determine the structure of the murine TopBP1 BRCT4/5 – BLM complex at 2.6 Å resolution (STAR Methods). The murine TopBP1 BRCT4/5 and murine BLM Ser304 regions are highly conserved with the human counterparts and interact with similar affinity (Figure 1A; Supplementary Figure 1). The murine complex crystallizes in P21 space group with 8 copies of TopBP1 BRCT4/5 per asymmetric unit. There exists a translational crystallographic symmetry between protomers ACBE and HDGF, with BLM bound to only half of these protomers (Figure S2). Comparisons of the unbound (EFGH) and bound (ABCD) structures of BRCT4/5 suggest that the TopBP1 structure is largely unchanged upon peptide binding (averaged root-mean-square deviation [rmsd] Cα = 0.147 Å2 within each set, Cα =0.469 Å2 between the peptide-bound and unbound sets). The structural differences are largely limited to the α1-β2 and β2’-β3’ loops (Supplementary Figure 2B). The β2’-β3’ loop directly contacts the BLM peptide, which restrains the loop conformation compared to the TopBP1 protomers with no bound peptide. The differences in the BRCT4 α1-β2 loops are likely caused by differences in crystal packing. The overall structure of murine TopBP1 BRCT4/5 adopts a head-to-head packing that is identical to human TopBP1 BRCT4/5, and nearly all residues involved in the BRCT-BRCT interface are conserved (Supplementary Figure 1A). As suggested in previous BLM-TopBP1 interaction studies (Blackford et al., 2015; Wang et al., 2013), our structure shows that TopBP1 interacts with BLM exclusively through its BRCT5 domain (Figure 1D, 2, Figure S2C) and indeed FP measurements indicate that BRCT5 and BRCT4/5 bind the BLM peptide with nearly identical affinities (Figure S3A).
Figure 2. Comparison of TopBP1 recognition of BLM and MDC1.
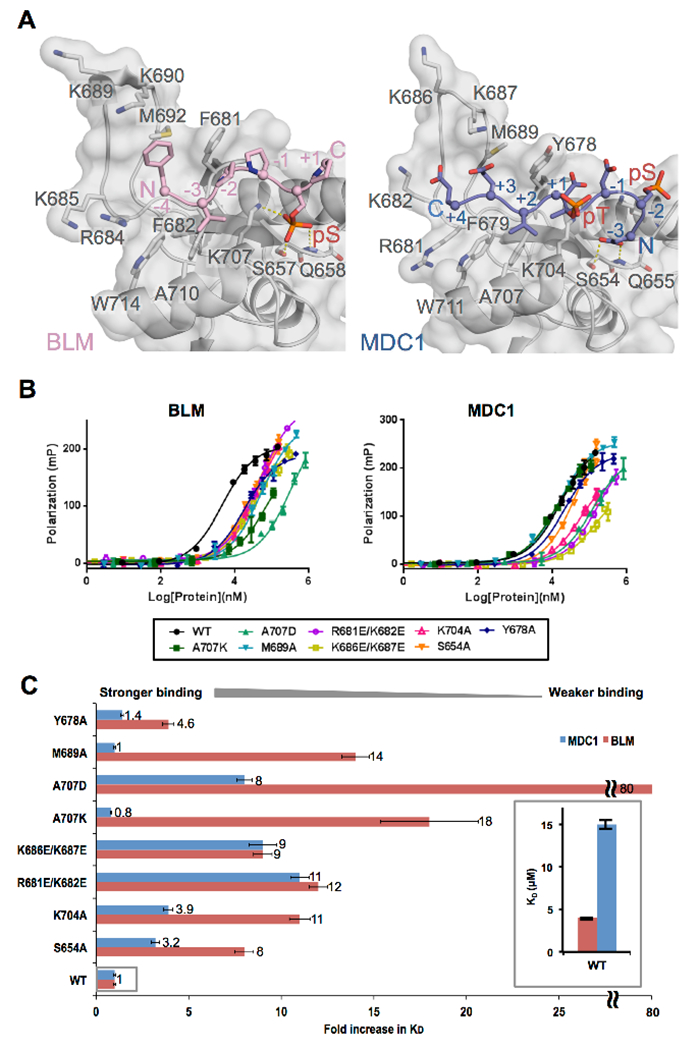
A. Structural comparison of TopBP1-BLM and TopBP1-MDC1 complexes. Mouse TopBP1-BLM complex (left) and human TopBP1-MDC1 (right) are aligned by their BRCT5 main-chain Cα positions. Both TopBP1 BRCT structures are displayed with semi-transparent surface over a grey cartoon, while the BLM peptide is displayed as a pink cartoon and the MDC1 peptide is displayed as a blue cartoon. Key interacting residues are displayed as sticks and hydrogen bonds are indicated by yellow dash-lines. Note that the region in mouse TopBP1 N-terminal to BRCT4/5 is three residues larger than the human protein. As a result, the numbers for homologous residues are three larger for mouse compared to human.
B. Comparison of the binding affinities of BLM and MDC1 phosphopeptides for a panel of human TopBP1 BRCT5 variants using FP. WT TopBP1 BRCT5 as well as a panel of eight missense variants were titrated against either BLM phosphopeptide (left) or the MDC1 phosphopeptide (right) and their binding affinities were assessed by FP.
C. Effects of human TopBP1 BRCT5 missense mutations on the binding of either MDC1 or BLM. The fold increase in KD is plotted for each BRCT5 variant normalized against the binding affinity for the WT. Results for BRCT5-MDC1 interactions are shown in blue, while the results for BRCT5-BLM interactions are shown in pink. The inset shows the relative difference in KD between WT BRCT5-MDC1 and WT BRCT5-BLM.
Modeling of TopBP1-BLM binding interaction
The electron density enabled us to model the core of the BLM phosphopeptide sequence 300Phe-Val-Pro-Pro-pSer-Pro305 for each of the peptide-bound TopBP1 BRCT4/5 complexes (Table S1, Figure 1D). All peptide protomers adopt similar interactions with the TopBP1 BRCT5 except peptide O that interacts with two protomers (D and E) due to crystal packing (RMSD of Cα = 0.084 Å2 between L, M, N, and Cα = 0.201 Å2 with O) (Figure S3B). The BLM peptide adopts a typical left-handed type II polyproline (PPII) helical structure. The phosphate group of BLM pSer304 is bound in the phosphate-binding pocket of TopBP1 BRCT5 through a set of hydrogen-bonding interactions with side chains of Lys707, Ser657 and main chain NH of Q658 that are conserved in other BRCT – phosphopeptide structures (Leung and Glover, 2011) (Figure 2A). The conserved FVPP motif N-terminal to Ser304 contours through a hydrophobic groove on the surface of BRCT5 that leads to the β2’-β3’ loop. The tandem prolines at positions −1 and −2 pack against Phe681, the valine at −3 is buried within a hydrophobic pocket formed by residues Phe682, Ala710 and Trp714, and the phenylalanine at −4 packs against Met692 at the center of the β2’-β3’ loop. The −3 valine and phosphoserine are aligned on the same side of the PPII helix for interactions with the BRCT5 binding groove. Although we were unable to model the conserved Asp-Thr-Asp motif N-terminal to the core sequence due lack of electron density, this acidic motif could potentially interact with basic residues including Arg684, Lys685, Lys689 and Lys690 from the β2’-β3’ loop, which, together with the phosphate binding pocket, render the peptide-binding surface highly electropositive (Figure 1D). C-terminal to pSer304, the peptide tracks away from BRCT5 and in 3 of the 4 complexes, there is no density observable for the semi-conserved Glu-Glu-Ile-Ile motif. In one of the complexes however, this motif is visible and packs against another BRCT5 in the asymmetric unit (Figures S2C, 3B, D). We conclude that the primary TopBP1 binding determinants within the BLM target peptide is the core 300Phe-Val-Pro-Pro-pSer 304 region, while the acidic N-terminal motif (297Asp-Thr-Asp299) may play a secondary role to enhance this interaction.
Figure 3. TopBP1 BRCT5 may bind BLM and 53BP1 through similar mechanisms.
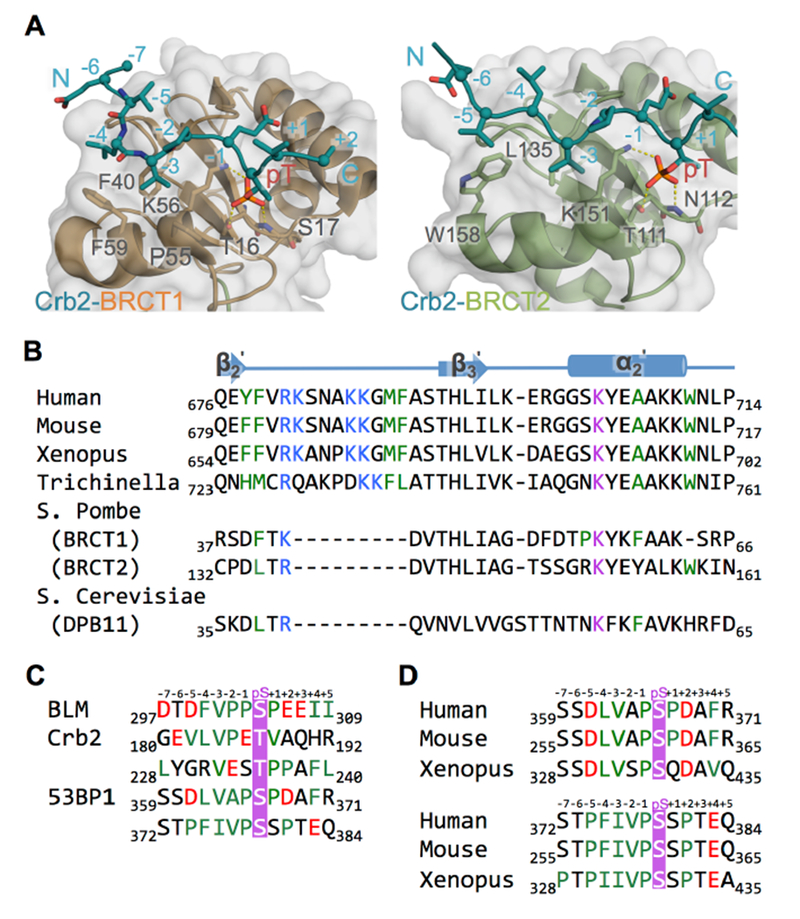
A. Structures of Rad4TopBP1 BRCT repeats bound to Crb253BP1. Rad4 BRCT1/Crb2 (left) and Rad4 BRCT2/Crb2 (right) structures (PDB code: 4BU0) are aligned with the TopBP1/BLM complex as in Figure 2A. Both structures have surface and cartoon displayed for Rad4TopBP1 (BRCT1 in orange, BRCT2 in green) and only cartoon displayed for Crb253BP1 (blue). Interacting residues are displayed as sticks. Hydrogen bonds are indicated by yellow dash-lines.
B. Sequence alignment of the peptide binding region TopBP1 BRCT5 homologues. Residues from the positively charged β2’-β3’ loop are colored blue, residues from phosphate binding pocket are colored purple, and residues lining hydrophobic groove are colored green.
C. Sequence alignment of BLM, Crb2 and 53BP1 phosphopeptide partners for TopBP1 BRCT5. Phosphorylated residues are colored purple, negatively charged residues are colored red and hydrophobic residues are colored green.
D. Sequence alignment of potential TopBP1 BRCT5 binding regions in 53BP1 homologues. Coloring is as in Figure 3C.
Comparison of BLM and MDC1 recognition by TopBP1
MDC1 has also been identified as a potential binding partner of TopBP1 BRCT4/5 and the structure of a consensus MDC1 SDT repeat region (GFIpSDpTDVEEE) bound to TopBP1 BRCT4/5 was solved by X-ray crystallography (Leung et al., 2013). MDC1 interacts with TopBP1 in a manner not observed in other BRCT-peptide structures, with one MDC1 peptide sandwiched between two BRCT4/5 domains. There is no direct interface between the two BRCT4/5 protomers, and most of the MDC1 interaction involves just one of the protomers. Evidence that a TopBP1 dimer binds MDC1 more tightly than a monomer in solution comes from FP binding studies that show the untagged monomeric TopBP1 BRCT4/5 binds MDC1 significantly weaker than dimeric GST-BRCT4/5 or GST-BRCT5 (Leung et al., 2013) (Table 1). In contrast, both the untagged and GST-tagged forms interact with BLM with similar dissociation constants in the 3-6 μM range that is significantly higher than MDC1 (Figure S3A, Table 1). This suggests that TopBP1 BRCT4/5 interacts with BLM as a monomer and with much higher affinity than MDC1.
Table 1: Summary of Fluorescence Polarization Results.
| Protein | TopBP1-peptide KD (µM) | |||
|---|---|---|---|---|
| TopBP1 | MDC1 | BLM | ||
| GST-hBRCT5 | WT | 15 ± 1 | 285 ± 10 | pSer338 |
| 60 ± 10 | Ser304 | |||
| 3.9 ± 0.3 | pSer304 | |||
| S654A | 48 ± 6 | 33 ± 3 | ||
| K704A | 59 ± 6 | 42 ± 3 | ||
| R681E/K682E | 170 ± 10 | 45 ± 2 | ||
| K686E/K687E | 130 ± 20 | 36 ± 3 | ||
| A707K | 12 ± 0.9 | 70 ± 20 | ||
| A707D | 120 ± 10 | 320 ± 40 | ||
| M689A | 15 ± 1 | 53 ± 4 | ||
| Y678A | 21 ± 2 | 18 ± 2 | ||
| hBRCT4/5 | 100 ± 10 | 3.2± 0.8 | ||
| GST-hBRCT4/5 | 30 ± 4 | 6 ± 1 | ||
| mBRCT4/5 | 170± 20 | 6 ± 2 | ||
In order to understand how TopBP1 BRCT4/5 interacts with two different partners, we first compared these interactions by superimposition of the structures of the TopBP1-BLM and TopBP1-MDC1 complexes (Figure 2A). The TopBP1 BRCT4/5 structures are quite similar between the two complexes (RMSD of Cα= 0.497). The same BRCT5 groove is used to engage the two extended phosphopeptide partners, however, the orientation of the two peptides relative to the BRCT5 binding surface is reversed. To further probe the interactions of the two peptides with TopBP1, we compared the impact on peptide binding of a panel of human TopBP1 BRCT5 mutations using the FP assay (Figure 2B). Mutation of either Ser654 or Lys704 in the phosphate binding pocket (equivalent of S657A and K707A in mouse) causes an 8- to 11-fold reduction in BLM binding affinity, consistent with the role of these residues in hydrogen bonding with the pSer304 (Figure 2C). These same mutations only result in a 3.2- to 3.9-fold reduction of affinity in MDC1 for TopBP1. The reduced importance of this pocket for MDC1 binding is consistent with the structure. In the TopBP1 protomer that makes the most extensive contacts with MDC1, the phosphate binding pocket does not bind a phosphate and instead binds the −3 Asp of MDC1. The −3 Asp only partially mimics a pSer, preserving hydrogen bonding interactions with Ser657 and the main chain NH of Gln658. In the other TopBP1 protomer, the MDC1 pThr is partially docked into the phosphate binding pocket. Two different pairs of charge reversal mutations within the basic β2’-β3’ loop (R681E/K682E and K686E/K687E, equivalent of R684E/K685E or K689E/K690E in mouse) reduced TopBP1 affinity ~9- to 12-fold suggesting charged interactions involving this loop are important to stabilize either complex. In the MDC1 complex, the acidic residues at +3 and +4 are in proximity to the β2’-β3’ loop, while in the BLM complex, we propose it is the DTD motif at positions −5 to −7 that contacts this loop. Both MDC1 and BLM present a valine (position −3 in BLM, position +2 in MDC1) that docks into the TopBP1 hydrophobic pocket (Figure 2A). The floor of this pocket is formed by a conserved alanine (Ala707 in human, Ala710 in mouse). Mutation of this alanine shows strikingly different effects on binding of the two peptides. Replacement of the alanine with either a positive (A707K) or negative (A707D) charge dramatically reduces BLM binding, however only the A707D reduces MDC1 peptide binding. This result suggests that the hydrophobic nature of this pocket is critical for the BLM interaction, but is much less important for the MDC1 interaction. Additional hydrophobic contacts are observed in the BLM complex that is not found in the MDC1 complex (Figure 2A). The BLM proline at −2 docks against TopBP1 Phe681 and the BLM phenylalanine at −4 packs into a shallow hydrophobic depression in the surface of the β2’-β3’ loop formed by Met692, Val683 and Phe681. Mutations of residues that constitute these surfaces in the human protein (M689A or Y678A) have no appreciable impact on MDC1 binding, however these mutations result in significant reductions in BLM binding (Figure 2B,C).
Taken together, this data indicates that BLM binds TopBP1 BRCT5 in a way that utilizes the electrostatic complementarity between the phosphopeptide and BRCT5, as well as hydrophobic contacts from the PPII helix that impart additional specificity and binding affinity. In contrast, the MDC1-TopBP1 complex appears to be largely electrostatically driven and of much lower affinity than the BLM complex.
DISCUSSION
TopBP1 is distinguished by its range of protein partners that interact with the diverse BRCT domains of TopBP1. TopBP1 BRCT5 has been particularly interesting as it has been proposed to bind multiple phosphoprotein targets: BLM, MDC1 and 53BP1 (although the relevant phosphorylated residue in the latter has not yet been identified). Our work shows that the recognition of BLM is highly specific for the region surrounding pSer304 and likely does not involve pSer338, which was also proposed as a possible binding target (Wang et al., 2013). While MDC1 recognition involves the same surface on BRCT5, the MDC1 peptide binds in an opposite orientation to that observed for BLM, and is of lower affinity and specificity, relying primarily on electrostatic interactions between the highly negatively charged MDC1 phosphopeptide and the positively charged surface of BRCT5. Our results raise further doubts as to whether MDC1 is a physiological binding partner for BRCT5 of TopBP1 (Blackford et al., 2015; Choi and Yoo, 2016), although a similar electrostatic interaction may also explain reports of interactions between BRCT5 and single stranded DNA, which have been suggested to play a role in the recognition of stalled DNA replication forks (Acevedo et al., 2016).
TopBP1 BRCT4/5 has not only been shown to interact with BLM and MDC1, but has also been implicated in binding the key DNA damage signaling factor, 53BP1, in a phosphorylation-dependent interaction (Cescutti et al., 2010; Yamane et al., 2002). Insight into this interaction has been provided by structural studies of the S. pombe orthologs of these proteins, Rad4TopBP1 and Crb253BP1 (Qu et al., 2013). Rad4TopBP1 contains a pair of BRCTs (BRCT1 and BRCT2) which can both bind either of two Crb253BP1 phosphopeptides containing a VxxpT motif in a manner that is similar to the binding of BLM by TopBP1 BRCT5, both in terms of the left-handed helical tracking of the peptide across the BRCT surface and docking of the −3 Val into the BRCT hydrophobic pocket (Figure 3A). The major difference between TopBP1 BRCT5 and either BRCT1 or BRCT2 of Rad4TopBP1 is the lack of the positively charged β2’-β3’ loop in the Rad4TopBP1 BRCTs (Figure 3A,B) and the Crb2 phosphopeptide binding partner does not contain the conserved negatively charged residues at positions −4 to −7 observed in BLM (Figure 3C).
To probe the possibility that similar interactions might be responsible for TopBP1-53BP1 interactions in the mammalian homologs, we scanned the known 53BP1 phosphorylation sites for potential TopBP1 binding sites that contain VxxpS/T motifs. Several sites match this motif with the best matches centering on pSer366 and pSer379/pSer380 (Figure 3C,D). While other 53BP1 phosphosites conform to the VxxpS/T motif, the pSer366 and pSer379/pSer380 sites are flanked by a conserved proline-containing hydrophobic region from −1 to −4 and additional acidic or potentially phosphorylated residues that could provide electrostatic interactions with the β2’-β3’ loop (Figure 3D).
Agents that increase the replication stress load in cancer cells are particularly effective chemotherapeutics. Cells in which the BLM-TopBP1 interaction is disrupted show increased replication stress, as evidenced by the increased DNA replication origin firing, chromosomal aberrations and SCEs (Blackford et al., 2015). Therefore, the structure of the BRCT domains of TopBP1 in complex with a BLM peptide lays the foundation for targeting of the BLM-TopBP1 interaction with small molecules as potential chemotherapeutic agents.
STAR METHODS
Contact for reagent and resource sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact: Mark Glover (mark.glover@ualberta.ca)
Cloning, expression and purification
Human TopBP1 BRCT5 (641-746) and BRCT4/5 (549-746), and mouse TopBP1 BRCT4/5 (553-749) were cloned into pGEX-6P-1 (GE Healthcare). Mutants (Y678A, A707D, A707K, M689A) of human TopBP1 BRCT5 were created from the WT template using QuickChange Lightning site-directed mutagenesis kit (Stratagene) (Braman et al., 1996; Kunkel, 1985; Nelson and McClelland, 1992; Sugimoto et al., 1989; Taylor et al., 1985; Vandeyar et al., 1988). All the GST fusion proteins were expressed in Escherichia coli BL21-Gold cells and purified using glutathione affinity chromatography with glutathione sepharose 4B beads (GE Healthcare) and eluted in elution buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 20 mM reduced glutathione and 0.1% βME). GST-fusion protein of BRCT5 was purified by Superdex 200 16/60 column in storage buffer (10 mM Tris-HCl pH 8.0, 150 mM NaCl, 1 mM DTT). GST-fusion protein of TopBP1 BRCT4/5 was cleaved with PreScission protease overnight at 4 °C. BRCT4/5 was purified by anion exchange chromatography (buffer A: 50 mM HEPES pH 7.0, 0.1% βME; buffer B: 50 mM HEPES pH 7.0, 1M NaCl, 0.1% βME). Residual GST was removed by incubation with glutathione sepharose 4B beads (GE Healthcare) prior to a final purification step on a Superdex 75 26/60 column in storage buffer (10 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM DTT).
Crystallization
Mouse TopBP1 BRCT4/5 concentrated to 10 mg/mL was incubated with 3-fold molar excess of BLM peptide (Ac-DTDFVPPpSPEEII-NH2, Genosphere Biotechnologies) for 2 hours on ice. Crystals of the complex were grown at room temperature using hanging drop vapor diffusion by mixing 1 μL of protein:peptide complex with 1 μL of reservoir solution (0.1 M sodium citrate pH 5.4 and 24% PEG 8000). Co-crystals were flash-cooled in reservoir solution supplemented with 20% glycerol.
Data collection and structure determination
Data for crystals of the BRCT4/5-pSer304 BLM peptide complex were collected at the CMCF 08ID-1 beamline (Canadian Light Source, Saskatoon). Intensity data were processed by DENZO, scaled and reduced using SCALEPACK (Adams et al., 2010) to the space group P21 with unit cell dimensions: a = 98.62Å, b = 97.0 Å, c = 127.3Å, α = 90.0 °, β =94.3 °, γ =90.0 ° (Otwinowski and Minor, 1997). The human TopBP1 BRCT4/5 structure (PDB ID: 3UEN) was used in PHASER 25.6 to successfully find 8 copies in the asymmetric unit (McCoy et al., 2007). Model building was carried out in COOT and refined using TLS refinement (peptide bound molecule ABCD and unbound molecule EFGH are grouped separately) in PHENIX (Adams et al., 2010; Emsley and Cowtan, 2004). The BRCT4/5 molecules are related by translational symmetry (Supplementary Figure 2A). BRCT4/5 molecules A and C lack the N-terminal 549–550 residues, C-terminal 742–746 residues, and α1-β2 loop residues 584–588. Molecules B and D lack the N-terminal 549–550 residues, C-terminal 743–746 residues, and α1-β2 loop residues 584–589. The BRCT4/5 molecules were fully refined before building of the four peptides. Peptides were positioned by first docking in the key pSer304 residues using Ligandfit in Phenix, and the rest of peptide chain subsequently built manually in COOT (Koska et al., 2008). Peptide chains L and M lack the N-terminal 7 to 5 residues and C-terminal +1 to +5 residues, peptide N lacks the N-terminal 7 to 5 residues and C-terminal +1 to +5 residue, and peptide O only lacks the N-terminal 7 to 5 residues. The final model was refined in Phenix at 2.6 Å resolution to Rwork and Rfree of 0.219 and 0.256, respectively. The Ramachandran plot contained 96.7% of all residues in the favored region. All the 7 Ramachandran outliers come from the two flexible loop regions (β2’-β3’ loop and α1-β2 loop) of TopBP1 BRCT4/5. Data collection and refinement statistics for the structures are listed in Table 1. Models were validated with MolProbity (Chen et al., 2010). Alignments with structure information were done by PROMAL3D. Bias reduced omit maps were generated by Phenix (Terwilliger et al., 2008). All structure figures were prepared with PyMOL (Version 1.8, www.pymol.org). Please see supplemental materials for PDB Validation Report.
Fluorescence polarization assay
FP measurements were carried out using an Envision multi-label plate reader (Perkin Elmer) using 384-well OptiPlates (Perkin Elmer). All pSer304 related BLM peptides (FITC-DTDFVPPpSPEEII-NH2, Ac-DTDFVPPpSPEEII-NH2, Ac-DTDFVPPpSPEEIIKK-NH2, Ac-DFVPPpSPEEII-NH2) were synthesized and purified by Genosphere Biotechnologies. The rest of peptides (FITC-KEDVLSTpSKDL-NH2, FITC-GFIDpSDpTDVEEE-NH2) were synthesized and purified by Biomatik. FP assays were performed by mixing 10 nM FITC-labelled phospho-peptide with freshly concentrated TopBP1 in FP assay buffer (10 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM DTT, 0.05 % Tween-20) and incubated for 15 minutes at room temperature. FP measurements were carried out at an excitation wavelength of 485 nm and emission wavelength of 538 nm. Curve fitting and KD calculations were obtained using PRISM software (GraphPad Prism). KD values summarized in Table 1 are the average of at least three individual measurements.
Lambda (λ) protein phosphatase treatment
Unphosphoylated Ser304 BLM peptide (FITC-DTDFVPPSPEEII-NH2) was made by treating pSer304 BLM peptide (FITC-DTDFVPPpSPEEII-NH2) with λ protein phosphatase (New England Biolabs) for 1 hour at 30 °C in reaction buffer (50 mM Tris-HCl, pH 7.5, 0.1 mM Na2EDTA, 5 mM DTT, and 0.01% Brij 35, 2 mM MnCl2). Mock-treated control reaction was carried out in the same reaction buffer lacking λ phosphatase. As an additional control, λ phosphatase was inactivated by heating 1 hour at 65 °C in the presence of 50 mM EDTA. The binding affinities of the peptides for GST-TopBP1 BRCT5 were determined by FP.
Data and software availability
Coordinates and scattering data for the BLM-TopBP1 BRCT4/5 complex have been deposited in the Protein Data Bank (RCSB accession: 5U6K). Intensity data were processed by DENZO, scaled and reduced using SCALEPACK. Initial molecular replacement of our data was done using TopBP1 BRCT4/5 structure (PDB ID: 3UEN) in PHASER 25.6. Model building was carried out in COOT and PHENIX. All structure figures were prepared with PyMOL. FP data were analyzed using GraphPad PRISM. All software resources are listed in the Key Resource Table, and the use of each package in the data analysis is described in the sub-headings of the STAR Methods Table.
Supplementary Material
HIGHLIGHTS.
TopBP1 BRCT4/5 interacts with BLM through pSer304, not pSer338
BLM binds via the BRCT5 phosphate binding pocket and hydrophobic cleft
Structural and mutagenesis results indicate TopBP1 uses the same BRCT5 surface to recognize MDC1 and BLM peptides in opposite orientations
ACKNOWLEDGEMENTS
We thank Dr. Pawel Grochulski and the staff at the Canadian Light Source CMCF beamline 08ID-1 for assistance with synchrotron data collection. This work was funded by NCI program project grant PO1CA092584 to J.N.M.G, CIHR grant 114975 to J.N.M.G and NSERC Discovery grant 2016-05163 to J.N.M.G. A.N.B. is supported by a Cancer Research UK Career Development Fellowship (C29215/A20772).
Footnotes
SUPPLEMENTAL INFORMATION
Figure S1-3 and Table S1 can be found with this article online
REFERENCES
- Acevedo J, Yan S, and Michael WM (2016). Direct Binding to Replication Protein A (RPA)-coated Single-stranded DNA Allows Recruitment of the ATR Activator TopBP1 to Sites of DNA Damage. J Biol Chem 291, 13124–13131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW , et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass TE, Luzwick JW, Kavanaugh G, Carroll C, Dungrawala H, Glick GG, Feldkamp MD, Putney R, Chazin WJ, and Cortez D (2016). ETAA1 acts at stalled replication forks to maintain genome integrity. Nat Cell Biol 18, 1185–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackford AN, Nieminuszczy J, Schwab RA, Galanty Y, Jackson SP, and Niedzwiedz W (2015). TopBP1 interacts with BLM to maintain genome stability but is dispensable for preventing BLM degradation. Mol Cell 57, 1133–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braman J, Papworth C, and Greener A (1996). Site-directed mutagenesis using double-stranded plasmid DNA templates. Methods Mol Biol 57, 31–44. [DOI] [PubMed] [Google Scholar]
- Cescutti R, Negrini S, Kohzaki M, and Halazonetis TD (2010). TopBP1 functions with 53BP1 in the G1 DNA damage checkpoint. EMBO J 29, 3723–3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaganti RS, Schonberg S, and German J (1974). A manyfold increase in sister chromatid exchanges in Bloom’s syndrome lymphocytes. Proc Natl Acad Sci U S A 71, 4508–4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SH, and Yoo HY (2016). Mdc1 modulates the interaction between TopBP1 and the MRN complex during DNA damage checkpoint responses. Biochem Biophys Res Commun 479, 5–11. [DOI] [PubMed] [Google Scholar]
- Delacroix S, Wagner JM, Kobayashi M, Yamamoto K, and Karnitz LM (2007). The Rad9-Hus1-Rad1 (9-1-1) clamp activates checkpoint signaling via TopBP1. Genes Dev 21, 1472–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Garcia V, Furuya K, and Carr AM (2005). Identification and functional analysis of TopBP1 and its homologs. DNA Repair 4, 1227–1239. [DOI] [PubMed] [Google Scholar]
- German J (1993). Bloom-Syndrome - a Mendelian Prototype of Somatic Mutational Disease. Medicine 72, 393–406. [PubMed] [Google Scholar]
- German J, Archibald R, and Bloom D (1965). Chromosomal Breakage in a Rare and Probably Genetically Determined Syndrome of Man. Science 148, 506–507. [DOI] [PubMed] [Google Scholar]
- Gong Z, Kim JE, Leung CC, Glover JN, and Chen J (2010). BACH1/FANCJ acts with TopBP1 and participates early in DNA replication checkpoint control. Mol Cell 37, 438–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer DA, Besley BD, Kennedy KB, and Davey S (2003). hRad9 rapidly binds DNA containing double-strand breaks and is required for damage-dependent topoisomerase II beta binding protein 1 focus formation. Cancer Res 63, 4829–4835. [PubMed] [Google Scholar]
- Koska J, Spassov VZ, Maynard AJ, Yan L, Austin N, Flook PK, and Venkatachalam CM (2008). Fully automated molecular mechanics based induced fit protein-ligand docking method. J Chem Inf Model 48, 1965–1973. [DOI] [PubMed] [Google Scholar]
- Kunkel TA (1985). Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc Natl Acad Sci U S A 82, 488–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kumagai A, and Dunphy WG (2007). The Rad9-Hus1-Rad1 checkpoint clamp regulates interaction of TopBP1 with ATR. J Biol Chem 282, 28036–28044. [DOI] [PubMed] [Google Scholar]
- Leung CC, and Glover JN (2011). BRCT domains: easy as one, two, three. Cell Cycle 10, 2461–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung CC, Sun L, Gong Z, Burkat M, Edwards R, Assmus M, Chen J, and Glover JN (2013). Structural insights into recognition of MDC1 by TopBP1 in DNA replication checkpoint control. Structure 21, 1450–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Shiotani B, Lahiri M, Marechal A, Tse A, Leung CC, Glover JN, Yang XH, and Zou L (2011). ATR autophosphorylation as a molecular switch for checkpoint activation. Mol Cell 43, 192–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J Appl Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson M, and McClelland M (1992). Methods Enzymol 216, 24. [DOI] [PubMed] [Google Scholar]
- Qu M, Rappas M, Wardlaw CP, Garcia V, Ren JY, Day M, Carr AM, Oliver AW, Du LL, and Pearl LH (2013). Phosphorylation-dependent assembly and coordination of the DNA damage checkpoint apparatus by Rad4(TopBP1). Mol Cell 51, 723–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto M, Esaki N, Tanaka H, and Soda K (1989). A simple and efficient method for the oligonucleotide-directed mutagenesis using plasmid DNA template and phosphorothioate-modified nucleotide. Anal Biochem 179, 309–311. [DOI] [PubMed] [Google Scholar]
- Taylor JW, Ott J, and Eckstein F (1985). The rapid generation of oligonucleotide-directed mutations at high frequency using phosphorothioate-modified DNA. Nucleic Acids Res 13, 8765–8785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC, Grosse-Kunstleve RW, Afonine PV, Moriarty NW, Adams PD, Read RJ, Zwart PH, and Hung LW (2008). Iterative-build OMIT maps: map improvement by iterative model building and refinement without model bias. Acta Crystallogr D Biol Crystallogr 64, 515–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandeyar MA, Weiner MP, Hutton CJ, and Batt CA (1988). A simple and rapid method for the selection of oligodeoxynucleotide-directed mutants. Gene 65, 129–133. [DOI] [PubMed] [Google Scholar]
- Wang J, Chen J, and Gong Z (2013). TopBP1 controls BLM protein level to maintain genome stability. Mol Cell 52, 667–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Gong Z, and Chen J (2011). MDC1 collaborates with TopBP1 in DNA replication checkpoint control. J Cell Biol 193, 267–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardlaw CP, Carr AM, and Oliver AW (2014). TopBP1: A BRCT-scaffold protein functioning in multiple cellular pathways. DNA Repair 22, 165–174. [DOI] [PubMed] [Google Scholar]
- Yamane K, Wu X, and Chen J (2002). A DNA damage-regulated BRCT-containing protein, TopBP1, is required for cell survival. Mol. Cell. Biol. 22, 555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.