

Abstract
Mitochondrial diseases (MDs) result from alteration of the mitochondrial respiratory chain (MRC) function. Despite the prevalence of MDs in the population, the paucity of animal models available limits the understanding of these disorders. Mutations in SDHA, a gene that codes for the alpha subunit of succinate dehydrogenase (SDH), can cause some forms of MD. SDHA is a crucial contributor to MRC function. In order to expand the range of MD animal models available, we attempted to generate a Sdha knockout rat. Since homozygous Sdha-/- rats could neither be identified in newborn litters, nor as early as embryonic day 14, we evaluated wild-type (WT) and heterozygous Sdha+/- genotypes. No differences in behavioral, biochemical, or molecular evaluations were observed between WT and Sdha+/- rats at 6 weeks or 6 months of age. However, 30% of Sdha+/- rats displayed mild muscle fiber atrophy with rare fibers negative for cytochrome oxidase and SDH on histochemical staining. Collectively, our data provide additional evidence that modeling SDH mutations in rodents may be challenging due to animal viability, and heterozygous rats are insufficiently symptomatic at a phenotypic and molecular level to be of significant use in the study of SDH deficiency.
Keywords: Complex II, Electron paramagnetic resonance, Electron transport chain, Leigh syndrome, Mitochondrial disease, Neuropathology
INTRODUCTION
Mitochondrial diseases (MDs) are a group of rare diseases in both children and adults with a prevalence between 5 in 100 000 and 1 in 5000 that lead to altered mitochondrial energy metabolism (1–3). MDs can be caused by errors in mitochondrial DNA (mtDNA), nuclear DNA (nDNA), or a combination (4), as both mtDNA and nDNA code for proteins that participate in the mitochondrial respiratory chain (MRC). Such alterations may decrease the activity of any of the MRC complexes, which can result in physiological consequences including reduced metabolic capacity, reduced ATP synthesis, and increased oxidative and nitrosative stress (5, 6). Symptoms of MD can be related to dysfunction of any organ, resulting in abnormalities of the motor, sensory, gastrointestinal, endocrine, and cardiovascular systems, as well as intolerance of some general anesthetics and anti-epileptic drugs, increased susceptibility to infection, and pregnancy loss (5–7). The combination of clinical and pathological heterogeneity provides extensive diagnostic challenges (5, 8). Diagnostic approaches for MD are currently nonstandard and better diagnostic tools and treatments for MD are needed (1, 2, 5, 8).
Currently only 5 murine models and 2 rat models are available for study. Murine models provide limited information, as they have an extremely low proportion of oxidative myofibers (9, 10) and may not properly recapitulate human muscular disease. Additionally, neither of the available MD rat models incorporate a MRC deficiency in Complex II. The paucity of animal models available for the study of MD limits the understanding of these disorders, thereby preventing the development of improved diagnosis and treatment approaches.
Mutations in SDHA, a nuclear gene that codes for the alpha subunit of succinate dehydrogenase (SDH), can cause some forms of MD including Leigh syndrome and leukodystrophy (11–13). SDH, also known as mitochondrial complex II, is a critical contributor to the MRC, and the inhibition of SDH reduces the number of electrons that are available for downstream complexes and thus decreases ATP production (14, 15). Mutations in SDHA have been associated with Leigh syndrome, a neurometabolic disorder presenting with symptoms of ataxia, muscle hypotonia, spasticity, and necrotizing lesions of the basal ganglia, diencephalon, cerebellum, or brainstem (11, 12). Other individuals with deficient SDHA present with neuroendocrine tumors (pheochromocytoma or paraganglioma), gastrointestinal stromal tumor (GIST), and abnormal energy metabolism, such as hypoglycemia and lactic academia (12, 16, 17).
In order to expand the variety of MD states available for study, we used CRISPR-Cas9 gene editing in an attempt to generate a Complex II-deficient Sdha KO rat. Homozygous Sdha KO rats could not be identified in newborn litters and could not be identified as early as embryonic day 14, which is consistent with a lethal phenotype early in embryonic development. As a result, we evaluated wild-type (WT) and heterozygous Sdha+/- rats with the expectation that Sdha+/- rats would demonstrate some features associated with human SDHA deficiency. Behavioral, pathological, biochemical, and molecular evaluations were performed on WT and Sdha+/- rats at 6 weeks and 6 months of life, and the vast majority of these assays yielded entirely normal results. However, 30% of Sdha+/- rats displayed mild muscle fiber atrophy and rare fibers that were negative for cytochrome oxidase (COX) and SDH on enzyme histochemistry. There was no evidence of neoplasia upon gross and histological evaluation of the rats.
MATERIALS AND METHODS
Live Animal Studies
All studies were performed with approval from the IACUC at The Medical College of Wisconsin.
Generation of Rats
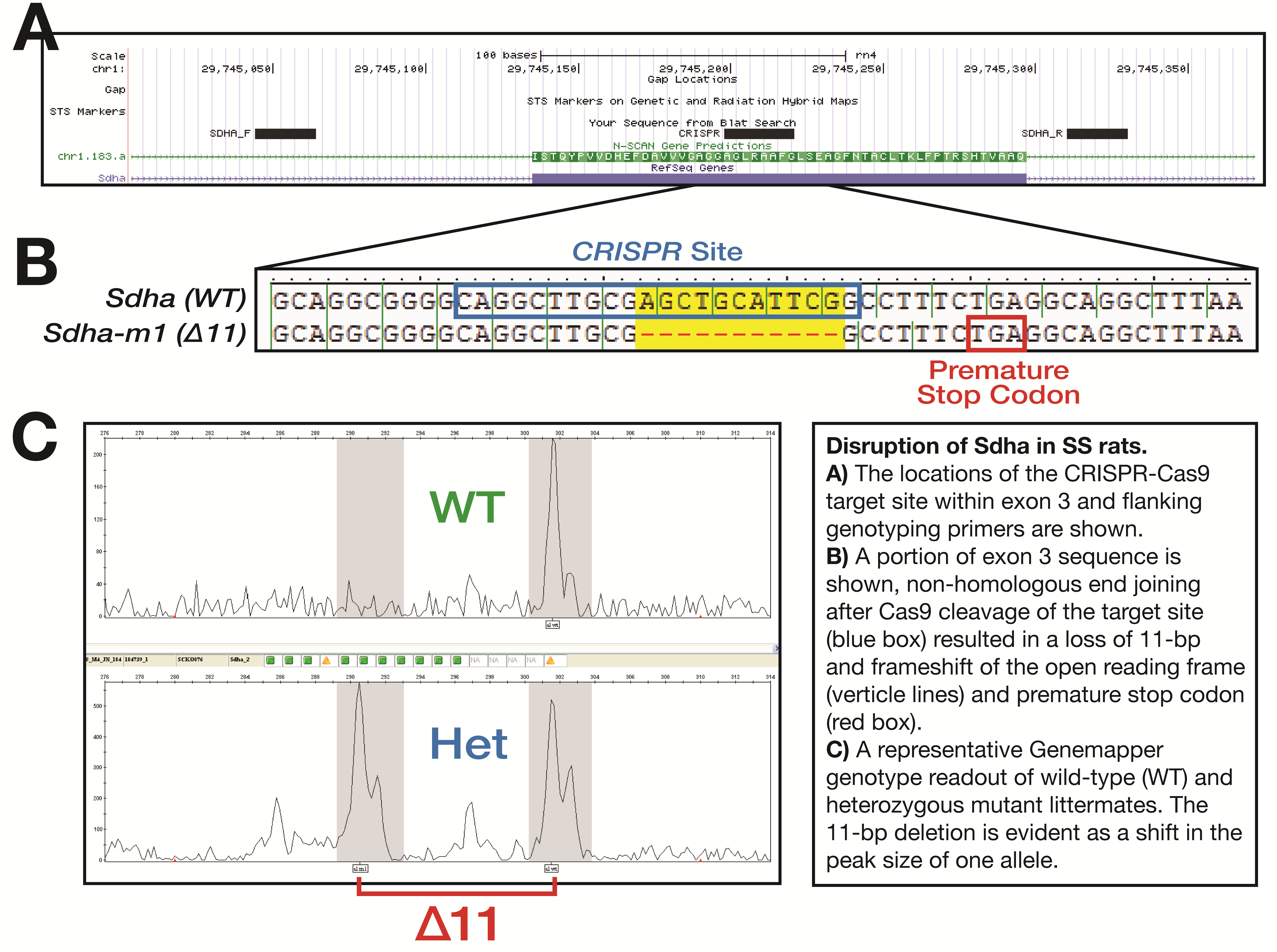
Sdha + /- rats (SS-Sdhaem1Mcwi) were produced by injecting a CRISPR targeting the Sdha exon 3 sequence GCAGGCTTGCGAGCTGCATTCGG (protospacer adjacent motif [PAM] underlined; Supplementary Data Fig. S1A) into SS (SS/JrHsdMcwi) rat embryos. Briefly, the pX330 plasmid (18) was modified with oligos to direct SpCas9 cleavage to the above target sequence. Founder animals were screened as previously described (19) and a founder animal harboring an 11-bp deletion (AGCTGCATTCG) in exon 3 was identified by Sanger sequencing (Supplementary Data Fig. S1B). This genetic change was expected to produce a severe truncation of the gene product, as it corresponded to a unique CRISPR site in an early exon that was shared by all known transcriptional forms. The founders were then backcrossed to the parental strain and subsequent litters were genotyped by fragment analysis (20) using fluorescently labeled M13-tagged primers Sdha_F: 5′-M13-GCAGCTTTTCTTTCCCATGA-3′ and Sdha_R: 5′-TGGGTAGGCATCAAAAGAGG-3′ (Supplementary Data Fig. S1C).
Behavior
At 6 months of age, a grip strength (Columbus Instruments, Columbus, OH) meter was used to assess the animals’ forelimb grip strength by placing the animals’ front paws on a horizontal wire grid and allowing the animal to try to pull itself away from the experimenter. Antigravity hanging performance was assessed by placing the animals on a rigid wire mesh and lifting them ∼40 cm above a cage filled with extra bedding. The time it took for the animals to fall back into the cage was recorded or the test was stopped after 60 seconds. Animals were placed on a rotarod (IITC Life Sciences, Woodland Hills, CA) and time to fall off the rod, distance traveled, and the RPM of the rod at the conclusion of the test were recorded. For grip strength, inverted screen and rotarod tests, 3 independent measurements were taken, and the maximum value was used for statistical analysis. The animals were allowed a minimum of 1-minute (grip strength, antigravity hanging) or 5-minute recovery time (rotarod) between trials and a maximum of 2 independent tests per day. Open field activity was performed in a square (90 cm × 90 cm) Plexiglas apparatus for 10 minutes in which the animals freely roam. Total distance traveled, maximum speed and average speed were assessed using ANY-maze software (Stoelting, Wood Dale, IL).
Gross Evaluation and Tissue Collection
At 6 weeks and 6 months of age, rats were killed for tissue collection. Internal organs and brain were placed in zinc-buffered formalin after being externally inspected for evidence of tumor nodules or discoloration. Quadriceps, gastrocnemius, triceps and diaphragm muscles were removed and frozen in liquid nitrogen-cooled isopentane. Initial assays were divided between quadriceps muscle (mtDNA quantification, electron paramagnetic resonance spectroscopy [EPR]) and gastrocnemius muscle (histology, Western blot) to allow conservation of muscle and maximize flexibility in future assays if abnormalities were detected.
MtDNA Content Quantification
Real time mtDNA quantification was performed as previously described (21). Briefly, DNA from quadriceps muscle was extracted using a Qiagen Blood Core Kit (#158389), diluted, combined with forward/reverse primer, and iTAQ SYBR Green Supermix with ROX (BioRad, Hercules, CA). Triplicate assays were measured and analyzed on a 7900HT Fast Real-Time PCR system (Applied Biosystems, Foster City, CA) using SDS V2.3 software. The relative mtDNA copy number was determined from the threshold difference between the nuclear and mitochondrial targeted primer sets.
Muscle Histology
Hematoxylin and eosin (H&E), Gomori trichrome, NADH, SDH, and COX stains were performed on gastrocnemius samples using standard techniques, which were cross sectioned at 8 µm and mounted on slides. Images were taken using an Olympus BX53 microscope with an Olympus DP72 camera and cell Sens Standard software (Olympus, Center Valley, PA).
Organ Histology
Formalin-fixed major organs (brain, heart, lung, kidney, spleen, intestines, colon, and gonads) from 5 animals per treatment group were sectioned and grossly examined for evidence of tumor nodule formation, discoloration, and other focal lesions, and representative samples were submitted to the Children’s Hospital of Wisconsin Research Institute Histology Core for routine processing and staining with H&E (22). Organ and muscle histology was reviewed by a board certified anatomic pathologist and neuropathologist.
Immunohistochemistry
Fiber type was evaluated by double-staining for Type 2b myosin (DSHB antibody BF-F3, 1:50 dilution) and dystrophin (Abcam ab15277, 1:100 dilution) on 8-µm sections of frozen gastrocnemius muscle. Images were taken using a Zeiss Axio Imager Z1 microscope.
Western Blots
Muscle tissues from the gastrocnemius muscle was frozen at the time of necropsy and stored at −80°C until analysis. Frozen muscles were sliced into 8-μm sections and homogenized with lysing buffer (EMD Millipore, Temecula, CA) containing cOmplete mini protease inhibitor cocktail tablet (Roche, Basel, Switzerland) and PhosSTOP phosphatase inhibitor (Roche). Western blot procedures were performed as previously described (22, 23). Transferred proteins were probed with Total OXPHOS Blue Native WB Antibody Cocktail (Abcam ab110412). Quantification of protein levels normalized to GAPDH was performed with Image Lab Software (BioRad). Three to eight animals per treatment group were used for analysis.
EPR
Quadriceps muscle samples were extruded by syringe into 3-mm internal-diameter quartz tubes and frozen in liquid nitrogen within 30 seconds of harvesting. Samples were stored either at −80°C or in liquid nitrogen for up to 60 days, over which time EPR signals of representative samples were observed not to change. Samples were of sufficient depth to completely fill the active length of the EPR resonator. EPR spectra were recorded at a temperature of 12 K on an upgraded Bruker EMX-TDU/L/E-532LX spectrometer system equipped with an ER4112-SHQ resonator operating at 9.49 GHz (precise frequencies were measured with an HP 5350B counter). Temperature was maintained with a cryogen-free ColdEdge/Bruker Stinger RDK-408 4K cryocooler and Sumitomo F-70L compressor, an Oxford ESR900 cryostat fed by a Stinger recirculating compressor, and an Oxford Mercury iTC temperature controller. Spectra were recorded with 10 G (1.0 mT) magnetic field modulation at 100 kHz, and at 5 mW microwave power. Other acquisition parameters were chosen such that the spectral resolution (∼3 G; ∼0.3 mT) was limited by the modulation amplitude. Signals were analyzed using the intensities of 4 signals: (1) the composite “g = 1.92” signal due to reduced Complex I & II [2Fe2S]+ and [4Fe4S]+ clusters, with a sharp trough at 3528 G; (2) the doublet signal due to the low potential Complex I N4b and N3 clusters centered around 3600 G; (3) the aconitase [3Fe4S]+ cluster derivative-shaped signal at 3352 to 3390 G; and (4) the Complex III reduced Rieske [2Fe2S]+g1 signal with a peak at 3300 G. Relative intensities were measured as either peak-to-trough magnitudes (g = 1.92 FeS from 3509 to 3528 G, and aconitase from 3352 to 3390 G), or else were baseline-adjusted, windowed (Rieske from 2287 to 3315 G, and N4b from 3560 to 3679 G), and integrated (Bruker Xenon).
Statistical Analysis
Prism 7.0 software (GraphPad, Inc., La Jolla, CA) was used to perform the Student t-test on animal weight, forelimb grip strength, open field, individual muscle weights. mtDNA content and Western blot data were analyzed using two-way ANOVA. Student t-tests and standard deviation (SD) calculations for EPR were carried out using Wavemetrics IGORPro v.7.05.
RESULTS
Assessment of Embryonic Lethality in Sdha-/- Rats
Homozygous SDHA mutations or compound heterozygous SDHA mutations in patients often results in early death (12). We found that 100% of Sdha-/- rats die prior to birth. To determine when embryonic lethality occurs in Sdha-/- rats, we collected embryos at day E14 (n = 11). Each embryo was genotyped and we found that no Sdha-/- embryos were viable at day E14. Embryos collected prior to day E14 would not have been suitable for our level of evaluation; thus, we focused our efforts on the assessment of heterozygous rats as a model of possible SDH deficiency.
Whole Animal Studies
General observation of animals did not show differential behavior between WT and Sdha+/- rats from 0 to 6 months of age; thus, behavioral testing suitable for the evaluation of mild behavioral phenotypes was performed in an effort to detect subtle abnormalities in 6-month-old animals. No significant difference in weight was observed between WT and Sdha+/- rats at 6 months of life. Forelimb grip strength, rotarod, and open field activity were assessed to evaluate muscle weakness and stamina. No significant differences in grip force (WT: 924.60 ± 71.72, Sdha+/-: 992.00 ± 30.82 g force), open field activity (WT: 30.14 ± 1.93, Sdha+/-: 29.08 ± 2.04 m traveled; WT: 0.05 ± 0.00, Sdha+/-: 0.05 ± 0.00 average meters per second; WT: 0.48 ± 0.03, Sdha+/-: 0.54 ± 0.03 maximum meters per second) or rotarod (WT: 32.87 ± 1.67, Sdha+/-: 35.38 ± 3.47 RPM; WT: 106.00 ± 23.51, Sdha+/-: 139.00 ± 16.66 seconds; WT: 8.42 ± 0.89, Sdha+/-: 10.40 ± 2.22 m) assays were observed in 6-month-old Sdha+/- rats (Fig. 1).
FIGURE 1.

Weight and behavioral analysis of Sdha+/- rats. Wild-type (WT, n = 7) and Sdha+/- (Het, n = 7) rats did not display any significant differences in total bodyweight at 6 months of age. Wild type (n = 5) and Sdha+/- (n = 5) rats did not produce significant differences upon testing of motor and coordination abilities, using rotarod, grip strength, and open field behavioral assays at 6 months of age. Data were analyzed using the Student t-test.
MtDNA Content
No significant differences in mtDNA content of the quad muscle were observed between WT and Sdha+/- rats at 6 weeks or 6 months of life. The average relative mtDNA content of the WT (n = 6) rats was 1546 ± 478 copies compared with 1761 ± 436 copies in heterozygous (n = 5) animals at 6 weeks (p > 0.05). At 6 months, WT (n = 10) rats had an average relative mtDNA content of 1282 ± 116 and heterozygous (n = 10) rats had 1781 ± 238 (p > 0.05; Fig. 2).
FIGURE 2.
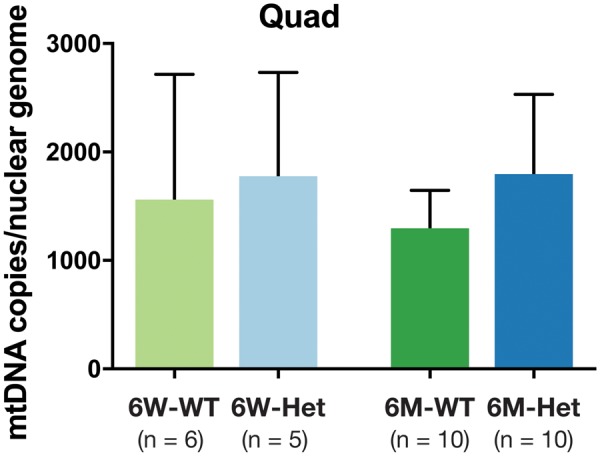
MtDNA content in Sdha+/- rats. The mtDNA copy numbers for 6-week (6W) and 6-month-old (6M) wild-type and Sdha+/- rats. There was no significant mtDNA copy number differences at 6 weeks of age between the wild type (n = 6) and Sdha+/- (n = 5) rats. There were also no significant differences between the 6-month-old wild type (n = 10) and Sdha+/- (n = 10) rats. Data were analyzed using two-way ANOVA. Error bars indicate SEM.
MRC Studies
No significant differences in MRC protein expression were detected in the gastrocnemius of 6-week-old (Complex I: WT = 0.77 ± 0.09, Sdha+/- = 0.77 ± 0.15; Complex II-70: WT = 0.34 ± 0.05, Sdha+/- = 0.22 ± 0.04; Complex III-Core 2: WT = 1.40 ± 0.22, Sdha+/- = 1.42 ± 0.28; Complex IV Subunit IV: WT = 0.80 ± 0.12, Sdha+/- = 0.73 ± 0.10; Complex V Alpha: WT = 1.24 ± 0.23, Sdha+/- = 1.35 ± 0.28) or 6-month-old Sdha+/- rats (Complex I: WT = 0.76 ± 0.08, Sdha+/- = 0.56 ± 0.05; Complex II-70: WT = 0.37 ± 0.05, Sdha+/- = 0.25 ± 0.03; Complex III-Core 2: WT = 0.89 ± 0.13, Sdha+/- = 0.70 ± 0.08; Complex IV Subunit IV: WT = 0.58 ± 0.07, Sdha+/- = 0.58 ± 0.07; Complex V Alpha: WT = 0.91 ± 0.13, Sdha+/- = 0.86 ± 0.11; Fig. 3).
FIGURE 3.
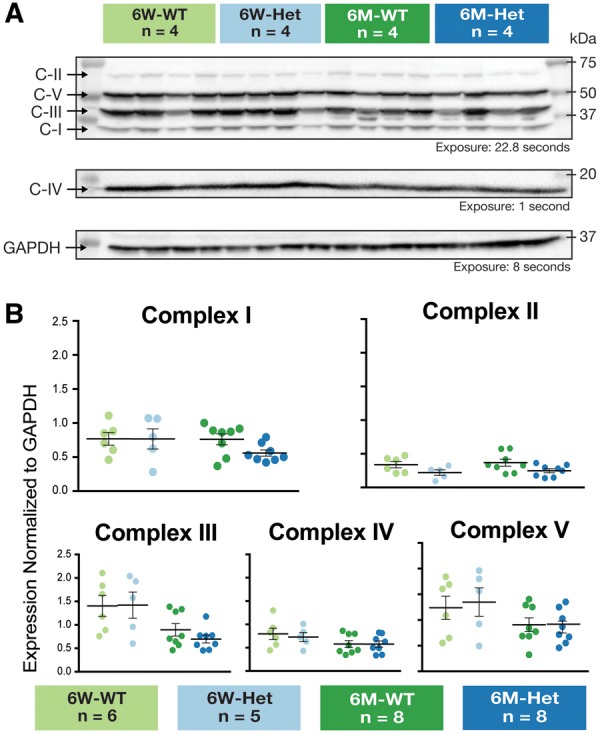
Expression of mitochondrial respiratory chain proteins in Sdha+/- rats. Western blot analysis of gastrocnemius muscle did not yield any significant differences in mitochondrial respiratory chain proteins (C-I, C-II-70, C-III-Core 2, C-IV Subunit IV, and C-V Alpha) between 6-week wild-type (n = 4) and 6 week Sdha+/- (n = 4) or 6-month wild-type (n = 4) and 6-month Sdha+/- (n = 4) rats. C-IV is shown on a different panel because its optimal exposure time was much lower. GAPDH is shown in a different panel as it required stripping/reprobing to evaluate. (A) Western blots of 6-week wild-type, 6-week Sdha+/-, 6-month wild-type, and 6-month Sdha+/- rats (n = 4) as representative sample of gastrocnemius muscle that was quantified. (B) Quantified mitochondrial respiratory chain protein expression for 6-week-old wild type (n = 6), 6-week Sdha+/- (n = 5), 6-month-wild type (n = 8), and 6-month Sdha+/- (n = 8) rats were normalized to GAPDH and graphed. Data were analyzed using two-way ANOVA.
Additionally, no differences were observed in MRC protein expression in the brain of 6-month-old rats (Complex I: WT = 0.68 ± 0.09, Sdha+/- = 0.61 ± 0.18; Complex II: WT = 0.25 ± 0.05, Sdha+/- = 0.27 ± 0.01; Complex III: WT = 0.60 ± 0.07, Sdha+/- = 0.50 ± 0.10; Complex IV: WT = 0.66 ± 0.12, Sdha+/- = 0.84 ± 0.21; Complex V: WT = 0.93 ± 0.21, Sdha+/- = 0.73 ± 0.19) or the liver (Complex I: WT = 0.47 ± 0.16, Sdha+/- = 0.26 ± 0.17; Complex II: WT = 0.98 ± 0.11, Sdha+/- = 0.95 ± 0.04; Complex III: WT = 1.64 ± 0.33, Sdha+/- = 1.50 ± 0.47; Complex IV: WT = 1.17 ± 0.19, Sdha+/- = 1.22 ± 0.27; Complex V: WT = 2.39 ± 0.36, Sdha+/- = 1.66 ± 0.17; data not shown). Notably, complex II protein expression was not significantly decreased in Sdha+/- animals, despite the allelic loss in the major catalytic subunit, Sdha (11, 24).
EPR Studies
EPR was performed to assess abnormalities of electron placement and flow through the muscle mitochondria. EPR signal abnormalities that are particularly useful when considering mitochondrial dysfunction include (1) those that report on reactive oxygen species (ROS)-mediated oxidative stress, and (2) those that report on changes in redox potential. In the spectra reported herein, the signal that reports on oxidative stress is that due to the [3Fe4S]+ cluster of aconitase, which arises due to ROS-mediated loss of the noncovalently bound Fea iron atom from the EPR-silent intact [4Fe4S]2+ cluster of active aconitase. This signal exhibits a crossover at 3372 G at the microwave frequency employed in this study. A number of signals report on redox potential; those at 3528 G (a composite signal, the “g = 1.92” signal, due to multiple clusters in Complexes I & II), 3598 G (Complex I N4b), 3630 G (Complex I N3), and 3302 G (Complex III Rieske cluster), are all due to reduced iron sulfur clusters. The intensity of these signals decreases as the redox potential across Complexes I & II increases. The N4b and N3 clusters exhibit very low redox potentials, close to the NADH/NAD+ couple midpoint, and are thus very sensitive markers for elevated redox potential. The low field signals at 1130 G and 1570 G are due to ferriheme and transferrin, respectively; these signals may be highly enhanced in conditions of extreme stress such as hemorrhage. The 4 traces shown in Figure 4 for female WT, female Sdha+/-, male WT, and male Sdha+/- represent averaged data from 5, 7, 4, and 2 animals, respectively. Analysis of the intensities of individual signals indicated that the variation of EPR signals within the 4 separate populations (for aconitase, g = 1.92, and N4b/N3 signals, the SD was 20% of the intensity, and for the Rieske cluster the SD = 30%) was greater than the variation among them, that is, that there were no significant differences between the EPR signals from the 4 populations female WT, female Sdha+/-, male WT, and male combined WT & Sdha+/- (male Sdha+/- only consisted of 2 samples and was not statistically analyzed independently; however, the individual male Sdha+/- signals were within the SDs for the other groups). The consistency of the intensities of the N4b and N3 signals, in particular, indicates that the global redox potential is likely dictated by the NADH/NAD+ couple, as expected for normally functioning mitochondria. The aconitase signals are not only invariant across the populations but are small and provide no evidence for oxidative stress (21). The Rieske signal is the only resolved signal solely due to Complex III, whose redox state may have been expected to be altered in the Sdha+/- animals. However, there was no evidence for any change in the extent of reduction of the Complex III Rieske cluster from EPR. Therefore, the EPR data strongly argue against any bulk mitochondrial dysfunction in any of the 4 populations.
FIGURE 4.

Electron paramagnetic resonance spectra from muscle tissue. The traces show EPR spectra from quadriceps muscle samples from female wild-type (n = 5), female Sdha+/- (n = 7), male wild-type (n = 4), and male Sdha+/- (n = 2) rats. The EPR signals in the 4 groups do not display any statistically significant differences. Data were analyzed using the Student t-test.
Pathological Studies
Histologic evaluation of rat gastrocnemius muscle revealed normal looking fibers in both WT and Sdha+/- animals. A mild histological phenotype, defined by rare, mildly atrophic were observed following H&E staining in 3 of 10 (30%) of 6-month-old Sdha+/- animals. Some abnormal fibers showed slight basophilia and some showed internal nucleation, but these features were not uniform and the degree of basophilia and internal nucleation was not suggestive of active or recent myofiber regeneration (Fig. 5). Additionally, rare occurrence of enzymatically deficient fibers was detected on COX and SDH staining in 6-month-old Sdha+/- animals. These abnormal fibers were not present throughout the muscle, and they were not detected in the majority of 6-month-old Sdha+/- rats. While atrophic fibers were deficient in COX and SDH, not all COX or SDH-deficient fibers were atrophic. No notable findings were seen upon Gomori trichrome staining.
FIGURE 5.
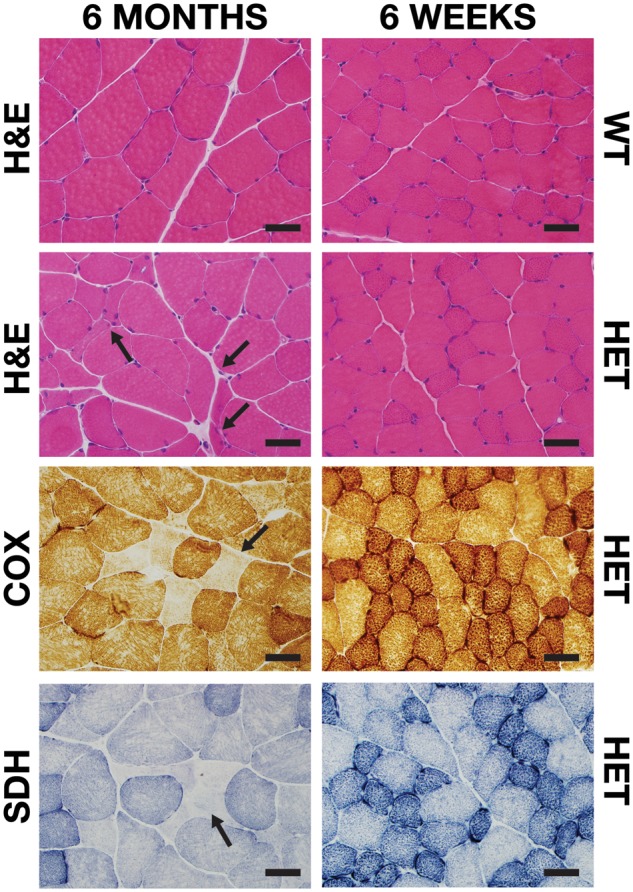
Histological staining of Sdha+/- muscle. Representative areas of transversely sectioned gastrocnemius muscle after sectioning and staining. Upon H&E staining, mild fiber atrophy (arrows), basophilic fibers, and internal nucleation were observed in 6-month-old Sdha+/- (HET) animals. COX and SDH staining revealed rare, atrophic, COX and SDH-deficient fibers (arrows) in 6-month-old Sdha+/- animals. Scale bar = 40 µm.
No change in Type 2b myosin and dystrophin composition was present on immunofluorescence staining of muscle from 6-month-old Sdha+/- rats, which labeled ∼60% of myofibers in 3 WT and 3 Sdha+/- rats (data not shown). This is consistent with our expectation that a rat model would have a greater proportion of oxidative fibers in comparison to a mouse model (10), and additional evaluations of fiber type were not pursued because of the lack of apparent phenotypic difference on this first evaluation. This finding indicates that muscle fiber composition was not different between WT and Sdha+/- animals, while also demonstrating that fiber type grouping (which can be seen in neuropathic disease) was not present.
Gross observation of brain, heart, lung, kidney, spleen, intestines, colon, and gonads revealed no evidence of abnormalities. Further histological evaluation of organs revealed no structural abnormalities.
DISCUSSION
In this report, we evaluated Sdha+/- rats to determine if they could serve as a potential model of Leigh syndrome, a type of MD. We found that Sdha+/- rats displayed a mild histopathological phenotype of muscle cells with the rare occurrence of atrophic fibers in a subset of animals. However, no disease phenotype was present behavior, MRC protein expression, or mtDNA content. Overall, our findings indicate that Sdha+/- rats do not exhibit the necessary phenotype to serve as a model for Leigh Syndrome. Furthermore, it appears that the muscle tissue from these rats retains adequate SDH activity and mitochondrial ETC complex II levels, suggesting that even in vitro cell lines established from these animals are unlikely to model a deficiency of SDH.
Given the embryonic lethality of homozygous Sdha-/- rats, this study focused on identifying whether heterozygous Sdha+/- rats would be sufficient to model an incomplete degree of SDH deficiency. While it is not surprising that Sdha+/- rats do not display the full Leigh syndrome phenotype that is associated with biallelic mutations (13), the lack of a discernible disease phenotype on the vast majority of our assays defied our expectations. Despite extensive and diverse testing methods, the sole abnormality detected in Sdha+/- rats was the presence of rare, atrophic fibers that were negative on SDH and COX histochemical staining. Despite the observance of mild histopathological changes, no differences were observed in MRC protein expression between Sdha+/- and WT rats at 6 weeks or 6 months of age. Additionally, our EPR data also indicated no significant differences in mitochondrial function, oxidative stress burden, or Complex II levels between Sdha+/- and WT rats. These findings indicate true SDH deficiency in this genetic context is only present focally and in rare fibers, and that assays using tissue homogenates (Western blot, EPR) were insufficiently discriminative to detect these changes.
As SDHA is thought to act as a tumor suppressor gene (25–27), gross and histological evaluations for neoplasia were also performed across a range of tissues. While no tumors were found, this is less surprising in a heterozygous model due to the absence of a “second hit” to the WT allele to completely remove tumor suppression. While it is possible that additional aging or environmental exposure to carcinogens would stimulate neuroendocrine tumor formation in this model, such studies were beyond the scope of the initial investigation of this model and are somewhat less likely to succeed given the relatively intact SDH function that is observed in these rats.
Interestingly, despite the lack of viable homozygous Sdha subunit knockout animals, there are patients who have presented with homozygous SDHA mutations or compound heterozygous SDHA mutations and are associated with a severe clinical phenotype (death at <2 years of age) (12). It has been suggested that individuals with homozygous SDHA mutations can survive because the missense mutations they carry only have a mild effect on protein function and do not result in a total loss of protein function (12). Our finding of nonviability in Sdha-/- embryos was also observed in Sdhd-/- mice (28). Thus, we corroborate that functional SDH is very important to embryo survival, but the effects of a total loss of a subunit of the protein are not yet able to be studied in live animals. Further studies may be conducted to selectively knock-in the mutations seen in homozygous patients, as these alterations in SDH may not be as destructive to the embryo and thus enable survival.
In summary, Sdha+/- rats did not demonstrate the symptoms associated with human SDHA deficiency, such as neurodegeneration, altered energy metabolism, or neuroendocrine tumors. Collectively, our data provide additional evidence that modeling SDH mutations in rodents may be challenging on the grounds of animal viability, and heterozygous rats are insufficiently symptomatic at a phenotypic and molecular level to be of significant use in the study of SDH deficiency.
Supplementary Material
{kind=link}
ACKNOWLEDGMENTS
Behavioral testing was performed at the Behavioral Core Facility provided by the Neuroscience Research Center at the Medical College of Wisconsin. Organ histology was performed at the Children’s Hospital of Wisconsin Research Institute’s Histology Core Facility. Immunofluorescence microscopy was performed at the Children’s Hospital of Wisconsin Research Institute’s Imaging Core Facility. The Myosin Heavy Chain Type IIB monoclonal antibody developed by S. Schiaffino, Universita degli Studi di Padova, was obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology, Iowa City, Iowa.
FUNDING
This project was supported by the Children’s Hospital of Wisconsin Research Institute, The Clinical and Translational Science Institute (CTSI) at the Medical College of Wisconsin (as a subaward of funding from the National Center for Advancing Translational Sciences, National Institutes of Health, Award Number UL1TR001436), the W.M. Keck Foundation, and by an Alpha Omega Alpha Carolyn L. Kuckein Student Research Fellowship. Electron paramagnetic resonance spectroscopy instrumentation was funded in part by an NSF Major Research Instrumentation award (CHE-1532168 to B.B. and Richard C. Holz) and Bruker Biospin. A.M.G. and L.G.D.D.M. were supported by the Todd Wehr Foundation.
MWL is a member of advisory boards for Audentes Therapeutics, Solid Biosciences, and Ichorion Therapeutics and is a consultant for Wave Life Sciences. The remaining authors have no duality or conflicts of interest to declare.
Supplementary Data can be found at http://www.jnen.oxfordjournals.org.
REFERENCES
- 1. Thorburn DR. Mitochondrial disorders: Prevalence, myths and advances. J Inherit Metab Dis 2004;27:349–62 [DOI] [PubMed] [Google Scholar]
- 2. Thorburn DR, Sugiana C, Salemi R et al. , . Biochemical and molecular diagnosis of mitochondrial respiratory chain disorders. Biochim Biophys Acta 2004;1659:121–8 [DOI] [PubMed] [Google Scholar]
- 3. Dimmock DP, Lawlor MW.. Presentation and diagnostic evaluation of mitochondrial disease. Pediatr Clin North Am 2017;64:161–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wallace DC. Mitochondrial diseases in man and mouse. Science 1999;283:1482–8 [DOI] [PubMed] [Google Scholar]
- 5. Parikh S, Goldstein A, Koenig MK et al. , . Practice patterns of mitochondrial disease physicians in North America. Part 1: Diagnostic and clinical challenges. Mitochondrion 2014;14:26–33 [DOI] [PubMed] [Google Scholar]
- 6. Chinnery PF, Mitochondrial disorders overview In: Pagon RA, Adam MP, Ardinger HH et al., , eds. GeneReviews(R). Seattle, WA: 1993. [PubMed] [Google Scholar]
- 7. Scaglia F, Towbin JA, Craigen WJ et al. , . Clinical spectrum, morbidity, and mortality in 113 pediatric patients with mitochondrial disease. Pediatrics 2004;114:925–31 [DOI] [PubMed] [Google Scholar]
- 8. Haas RH, Parikh S, Falk MJ et al. , . Mitochondrial disease: A practical approach for primary care physicians. Pediatrics 2007;120:1326–33 [DOI] [PubMed] [Google Scholar]
- 9. Johnson MA, Polgar J, Weightman D, Appleton D.. Data on the distribution of fibre types in thirty-six human muscles. An autopsy study. J Neurol Sci 1973;18:111–29 [DOI] [PubMed] [Google Scholar]
- 10. Lawlor MW, Read BP, Edelstein R et al. , . Inhibition of activin receptor type IIB increases strength and lifespan in myotubularin-deficient mice. Am J Pathol 2011;178:784–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Burnichon N, Briere JJ, Libe R et al. , . SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet 2010;19:3011–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hoekstra AS, Bayley JP.. The role of complex II in disease. Biochim Biophys Acta 2013;1827:543–51 [DOI] [PubMed] [Google Scholar]
- 13. Renkema GH, Wortmann SB, Smeets RJ et al. , . SDHA mutations causing a multisystem mitochondrial disease: Novel mutations and genetic overlap with hereditary tumors. Eur J Hum Genet 2015;23:202–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Drose S. Differential effects of complex II on mitochondrial ROS production and their relation to cardioprotective pre- and postconditioning. Biochim Biophys Acta 2013;1827:578–87 [DOI] [PubMed] [Google Scholar]
- 15. Raimundo N, Baysal BE, Shadel GS.. Revisiting the TCA cycle: Signaling to tumor formation. Trends Mol Med 2011;17:641–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Casey RT, Ascher DB, Rattenberry E et al. , . SDHA related tumorigenesis: A new case series and literature review for variant interpretation and pathogenicity. Mol Genet Genomic Med 2017;5:237–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Oudijk L, Gaal J, Korpershoek E et al. , . SDHA mutations in adult and pediatric wild-type gastrointestinal stromal tumors. Mod Pathol 2013;26:456–63 [DOI] [PubMed] [Google Scholar]
- 18. Cong L, Ran FA, Cox D et al. , . Multiplex genome engineering using CRISPR/Cas systems. Science 2013;339:819–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Geurts AM, Cost GJ, Remy S et al. , . Generation of gene-specific mutated rats using zinc-finger nucleases. Methods Mol Biol 2010;597:211–25 [DOI] [PubMed] [Google Scholar]
- 20. Moreno C, Kennedy K, Andrae JW, Jacob HJ.. Genome-wide scanning with SSLPs in the rat. Methods Mol Med 2005;108:131–8 [DOI] [PubMed] [Google Scholar]
- 21. Bennett B, Helbling D, Meng H et al. , . Potentially diagnostic electron paramagnetic resonance spectra elucidate the underlying mechanism of mitochondrial dysfunction in the deoxyguanosine kinase deficient rat model of a genetic mitochondrial DNA depletion syndrome. Free Radic Biol Med 2016;92:141–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tinklenberg J, Meng H, Yang L et al. , . Treatment with ActRIIB-mFc produces myofiber growth and improves lifespan in the acta1 H40Y murine model of nemaline myopathy. Am J Pathol 2016;186:1568–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lawlor MW, Viola MG, Meng H et al. , . Differential muscle hypertrophy is associated with satellite cell numbers and Akt pathway activation following activin type IIB receptor inhibition in Mtm1 p.R69C mice. Am J Pathol 2014;184:1831–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Baysal BE, Lawrence EC, Ferrell RE.. Sequence variation in human succinate dehydrogenase genes: Evidence for long-term balancing selection on SDHA. BMC Biol 2007;5:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. King A, Selak MA, Gottlieb E.. Succinate dehydrogenase and fumarate hydratase: Linking mitochondrial dysfunction and cancer. Oncogene 2006;25:4675–82 [DOI] [PubMed] [Google Scholar]
- 26. Pollard PJ, Briere JJ, Alam NA et al. , . Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet 2005;14:2231–9 [DOI] [PubMed] [Google Scholar]
- 27. Briere JJ, Favier J, Benit P et al. , . Mitochondrial succinate is instrumental for HIF1alpha nuclear translocation in SDHA-mutant fibroblasts under normoxic conditions. Hum Mol Genet 2005;14:3263–9 [DOI] [PubMed] [Google Scholar]
- 28. Bayley JP, Devilee P, Taschner PE.. The SDH mutation database: An online resource for succinate dehydrogenase sequence variants involved in pheochromocytoma, paraganglioma and mitochondrial complex II deficiency. BMC Med Genet 2005;6:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.