

Abstract
The design, synthesis, and biological evaluation of a new class of HIV-1 protease inhibitors containing stereochemically defined fused tricyclic polyethers as the P2 ligands and a variety of sulfonamide derivatives as the P2′ ligands, are described. A number of ring sizes and various substituent effects were investigated to enhance the ligand-backbone interactions in the protease active site. Inhibitors 5c and 5d containing this unprecedented fused 6-5-5 ring system as the P2 ligand, an aminobenzothiazole as the P2′ ligand and a difluorophenylmethyl as the P1 ligand exhibited exceptional enzyme inhibitory potency and maintained excellent antiviral activity against a panel of highly multidrug-resistant HIV-1 variants. The umbrella-like P2 ligand for these inhibitors has been synthesized efficiently in an optically active form using a Pauson-Khand cyclization reaction as the key step. The racemic alcohols were resolved efficiently using a lipase catalyzed enzymatic resolution. Two high resolution X-ray structures of inhibitor-bound HIV-1 protease revealed extensive interactions with the backbone atoms of HIV-1 protease and provided molecular insight into the binding properties of these new inhibitors.
Keywords: HIV-1 protease, inhibitors, antiviral, multidrug-resistant, synthesis, X-ray crystal structure, Umb-THF, backbone binding
Graphical abstract
INTRODUCTION
Protein X-ray structure-based molecular design has emerged as one of the most powerful strategies in modern drug design.1–3 Its utility is particularly notable in the area of design and development of HIV-1 protease inhibitors.4,5 The development of protease inhibitors (PIs) and their combination with reverse transcriptase inhibitors marked the beginning of a new era of management of HIV infection and AIDS in the late 1990s.6,7 HIV-1 PIs are a critical component of current combination antiretroviral therapies (ART) which significantly improved life expectancy and mortality rates of HIV/AIDS patients in developing countries.8,9 HIV-1 PIs inhibit viral enzymes, block viral replication and generate morphologically immature and noninfectious virions. However, selective drug pressure led to the emergence of drug-resistant HIV-1 variants, many of which are widely cross-resistant.10,11 This has raised serious concerns with regards to the future management of patients with HIV-1 infection and AIDS. A recent report documented that a growing number of HIV/AIDS patients are harboring multidrug-resistant HIV-1 variants that are difficult to treat and these viruses can be transmitted.12,13 Therefore, the development of new PIs that exhibit broad-spectrum activity against multidrug-resistant HIV-1 strains and show improved pharmacological properties is an urgent priority.
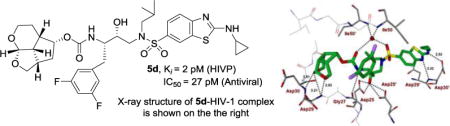
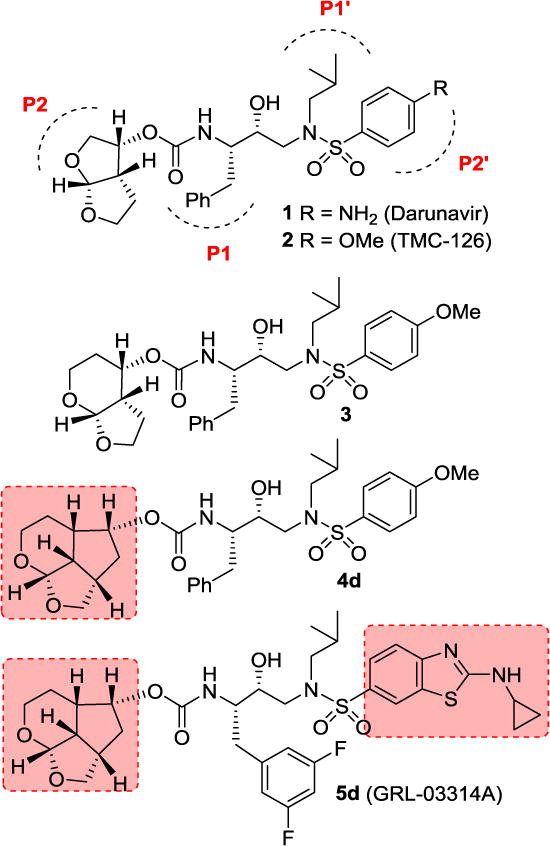
In an effort to design and develop conceptually novel PIs to combat drug-resistance, we designed and synthesized a wide range of exceptionally potent and structurally intriguing PIs that exhibited potent antiviral activity against multidrug-resistant HIV-1 variants and showed favorable drug-like properties.14,15 One of our key strategies of molecular design involves incorporation of cyclic ether and polyether-like templates where the ether oxygens are positioned to make specific hydrogen bonds with the backbone atoms of active site residues.16,17 Our molecular design strategy is based upon the evidence that a protease cannot alter its overall backbone conformation without compromising its catalytic fitness for viral replication.18,19 Therefore, our inhibitor design strategy involved maximizing active site interactions, particularly by promoting a network of strong hydrogen bonding interactions with the HIV-1 protease backbone.18,20 We incorporated a stereochemically defined bis-tetrahydrofuran (bis-THF) ligand in Darunavir (DRV 1, Figure 1) to promote hydrogen bonding interactions with Asp29 and Asp30 backbone NHs in the S2-subsite.16,21–24 Our structure-based design strategies also created other intriguing PIs with ligands such as cyclopentyl-tetrahydrofuran (Cp-THF), tetrahydropyrano-tetrahydrofuran (Tp-THF), tris-tetrahydrofuran (Tris-THF), and crown-tetrahydrofuran (Crn-THF).18,25,26 These inhibitors have been shown to bind extensively with the backbone atoms of active site of HIV-1 protease and maintain robust antiviral activity against a wide range of multidrug-resistant HIV-1 variants. Furthermore, our detailed X-ray structural analysis of inhibitor-bound HIV-1 protease provided molecular insights into the ligand-binding site interactions responsible for their impressive potency against multidrug-resistant HIV-1 variants.18,27,28 Based upon our comparison of the X-ray structures of DRV-bound HIV-1 protease and inhibitor 3-bound HIV-1 protease, we have investigated structural templates that would enhance the backbone-binding interactions, as well as further improve van der Waals interactions within the S2 subsite of the HIV-1 protease active site. Herein, we report the design, synthesis, and X-ray structural studies of structurally novel PIs which incorporate an unprecedented 6-5-5 ring-fused octahydrocyclopentylpyranofuran as the P2 ligand with the (R)-hydroxyethylsulfonamide isostere containing a substituted benzenesulfonamide or an aminobenzothiazole sulfonamide as the P2′ ligands.
Figure 1.
Structures of HIV-1 protease inhibitors 1-5.
The design and synthesis of novel PIs continue to be an active area of research. A number of novel HIV-1 PIs have been reported that incorporated a variety of heterocyclic P2 ligands for improving interactions at the S2 subsite.29–32 Also, other designed efforts were focused on optimizing both P1 and P2’ moieties to improve potency.29,30,33 More recently, bicyclic piperazine sulfonamide-based new PIs have been reported in which the piperazine NH forms the key interaction with the catalytic aspartates of the HIV-1 protease.34,35
Results and Discussion
Our examination of the X-ray crystal structure of DRV (1)-bound HIV-1 protease (PDB ID: 2IEN)27 and its methoxy derivative 2-bound HIV-1 protease suggested that a stereochemically defined hexahydrofuropyranol-derived urethane as the P2-ligand in inhibitor 3 would further improve backbone binding interactions with the protease active site. We also speculated that the extra methylene group on the tetrahydropyran would increase van der Waals interaction in the active site. The resulting inhibitor 3 indeed, showed enzyme inhibitory Ki value of 2.7 pM and antiviral EC50 value of 0.5 nM. Inhibitor 3 also maintained excellent antiviral activity against multidrug-resistant HIV-1 variants similar to DRV and inhibitor 2.36 To promote further stability of the acetal functionality and improve hydrogen bonding interactions with the Asp29 and Asp30 NHs, we speculated that a fused tricyclic ring could increase the dihedral angle of the acetal template and a cyclopentyl ring spacer could make stronger hydrogen bonding with Asp29 and Asp30 backbone amide NHs. A preliminary model of this inhibitor showed a more optimal alignment of the acetal oxygens with the backbone amide NHs of Asp29 and Asp30 with respect to DRV. This highly preorganized and conformationally constrained octahydro-2H-1,7-dioxacyclopenta[cd]indene would incorporate three extra methylene groups over bis-THF ligand in DRV. These additional methylene groups would be optimally located to provide more favorable van der Waals interactions in the hydrophobic space surrounding the Ile47, Val32, Leu76, and Ile50′ residues.
Based upon these possible ligand-binding site interactions, we have designed a 6-5-5 ring-fused octahydrocyclopentylpyranofuran derivative as the novel P2 ligand shown in inhibitor 4d. This umbrella-like tetrahydropyranofuran (Umb-THF) with some degree of flexibility may show better adaptability to protease mutation. With this new structural scaffold, we planned to investigate the preference for ligand stereochemistry. Also, in order to promote stronger hydrogen bonding interactions as well as to improve hydrophobic contacts, we have planned to investigate inhibitors with an Umb-THF P2 ligand in combination with a fluorinated phenylmethyl P1 ligand and a benzothiazole derivative to interact with the Asp30′ residue in the S2′-subsite as represented in inhibitor 5d. Many benzothiazole structural features are embedded in medicinally important compounds for either improving drug-like properties or interaction with the biological target.37,38 Recently, we reported PIs incorporating a cyclopropylaminobenzothiazole-based P2′ ligand in combination with a crown-THF P2 ligand.25 Interestingly, the replacement of a bis-THF with a crown-THF P2 ligand led to enhanced van der Waals interactions in the S2 site. These increased hydrophobic interactions, in addition to the robust hydrogen bond interaction pattern of a cyclopropylaminobenzothiazole-based P2′ ligand, led to PIs with unprecedented potency against multidrug-resistant HIV-1 strains.
For our preliminary investigation, we planned to utilize a deoxy sugar such as tri-O-acetyl-D-glucal (6) to synthesize the tricyclic scaffold in a stereodefined manner. Also, for rapid assembly of the tricyclic scaffold, we planned to carry out a cobalt-mediated intramolecular Pauson-Khand reaction to provide tricyclic enone.39–41 As shown in Scheme 1, reaction of the commercially available tri-O-acetyl-D-glucal (6) with propargyl alcohol in the presence of iodine in THF at 23 °C for 1 h afforded propargyl ether derivative 7 in 95% yield.42 For Pauson-Khand cyclization, we planned to utilize the N-methylmorpholine-N-oxide (NMO) promoted reaction protocol reported by Schreiber and co-workers.43,44 Thus, reaction of compound 7 with dicobalt octacarbonyl [Co2(CO)8] in hexane at 23 °C for 5 h provided the corresponding cobalt complex. Subsequent treatment of this complex with NMO in CH2Cl2 at 23 °C for 48 h afforded the tricyclic enone 8 as a single isomer in 32% yield. As part of our initial investigation, we planned to convert the 6-acetoxymethyl group into a methyl ether as well as a methyl group. Towards this goal, enone 8 was subjected to transfer hydrogenation using a substoichiometric amount of 10% Pd-C in the presence of ammonium formate in MeOH at reflux for 15 min.45 Transesterification of the resulting acetate derivative with triethylamine in MeOH at 23 °C for 3 h provided saturated keto alcohol 9 as a single isomer in 60% isolated yield for the two-steps. Keto alcohol 9 was converted to ligand alcohol 10 by treatment with Meerwein’s salt (Me3O+BF4−) and proton-sponge in CH2Cl2 at 0 °C to 23 °C for 48 h, followed by reduction of the keto group with NaBH4 in MeOH at 0 °C for 1 h. Alcohol derivative 10 was isolated as a single isomer in 55% yield over two-steps. Keto alcohol 9 was converted to ligand alcohol 11 as follows. Keto alcohol 9 was reacted with mesyl chloride and triethylamine in CH2Cl2 at -20 °C to 23 °C for 48 h. The resulting mesylate was treated with LAH in THF at 0 °C to 23 °C for 36 h to furnish alcohol 11 as a single isomer in 35% yield over two-steps.
Scheme 1.
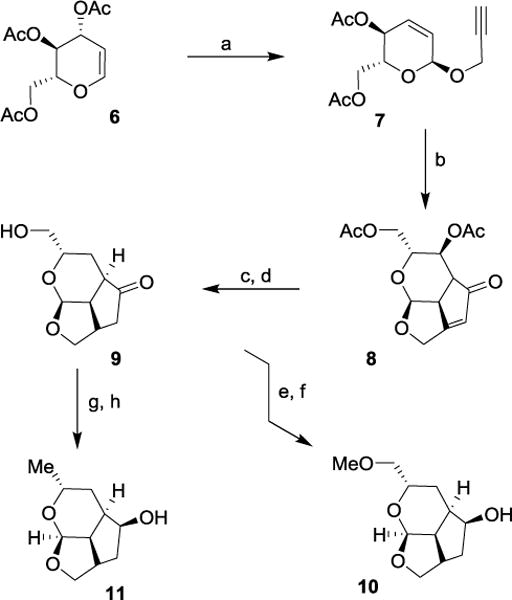
Synthesis of substituted tricyclic P2 ligands 10 and 11. Reagents and conditions. (a) Propargyl alcohol, I2, THF, 23 °C, 1 h, (95%); (b) Co2(CO)8, hexane, 23 °C, 5 h then NMO, CH2Cl2, 23 °C, 48 h, (32%); (c) HCO2NH4, 10% Pd/C, MeOH, reflux, 15 min, (70%); (d) Et3N, MeOH, H2O, 23 °C, 3 h, (86%); (e) Me3O+BF4−, proton-sponge, CH2Cl2, 0 °C to 23 °C, 48 h, (63%); (f) NaBH4, MeOH, 0 °C, 1 h, (88%); (g) MsCl, Et3N, CH2Cl2, -20 °C, 1.5 h (70%); (h) LAH, THF, 0 °C to 23 °C, 36 h, (50%).
The Pauson-Khand route provided a rapid access to our designed ligands. To establish structure-activity relationships for these tricyclic ligands, we then devised a more general route to provide convenient access to both enantiomers of the ligand alcohols. As shown in Scheme 2, reaction of dihydropyran 12 with propargyl alcohol in the presence of N-bromosuccinimide (NBS) at -20 °C for 2 h and then at 23 °C for 15 h furnished bromo ether 13 in 98% yield.46 Bromo ether 13 was treated with DBU and the resulting mixture was heated at 110 °C for 5 h to provide the dehydrobromination product 14 in 85% yield. Enyne 14 was subjected to Pauson-Khand reaction with Co2(CO)8 and NMO as described above to furnish tricyclic enone 15 in 20% yield. Transfer hydrogenation of enone 15 gave the corresponding saturated ketone which was reduced with NaBH4 in MeOH at 0 °C for 1 h to provide racemic endo alcohol 16 in 75% yield over two steps. Racemic alcohol 16 was subjected to lipase (PS-30 on celite) catalyzed enzymatic resolution47,48 in vinyl acetate as the solvent at 23 °C for 6 h to provide optically active alcohol (+)-16 in 53% yield and optically active acetate derivative 17 in 47% yield. Saponification of acetate 17 with K2CO3 in MeOH at 23 °C for 1 h furnished optically pure ligand alcohol (−)-16 in 99% yield. Enantiomeric ligands were converted to the corresponding p-nitrophenyl carbonate derivative. Enantiopurity of these ligand alcohols was 94% ee as determined by HPLC analysis using a chiral column. The optically active alcohol (−)-16 was converted to its 4-nitrophenyl carbonate derivative 18. The absolute stereochemistry of alcohols was determined based upon X-ray analysis of carbonate derivative 18 derived as shown in Figure 2.49
Scheme 2.
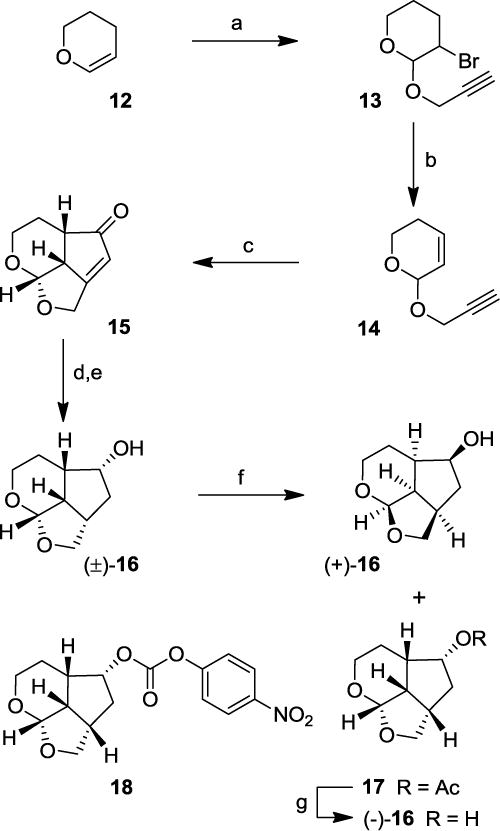
Synthesis of optically active ligand alcohols (−)-16 and (+)-16. Reagents and conditions. (a) NBS, propargyl alcohol, CH2Cl2, -20 °C to 23 °C, 17 h, (98%); (b) DBU, heat 110 °C, 5 h, (85%); (c) Co2(CO)8, CH2Cl2, 23 °C, 1 h, then NMO, CH2Cl2, 0 °C to 23 °C, 3 h (20%); (d) 10% Pd/C, HCO2NH4, MeOH, reflux, 15 min then (e) NaBH4, MeOH, 0 °C, 1 h (75% for 2-steps) NaBH4, MeOH (75%); (f) Lipase (PS-30), vinyl acetate, THF, 23 °C, 6 h, (+)-16 (53%), 17 (47%); (g) K2CO3; MeOH, 23 °C, 1 h, (99%).
Figure 2.
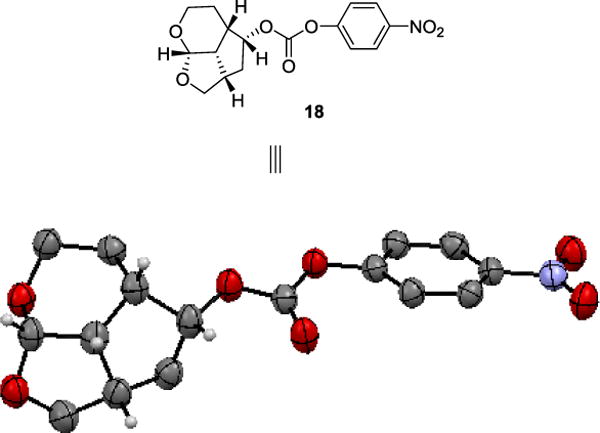
X-Ray Crystal Structure of Activated Carbonate 18.
For the synthesis of octahydro-2,H-4,5-dioxacyclopenta[cd]inden-2-ol derivatives, we utilized commercially available 2,3-dihydrofuran as the starting material. As shown in Scheme 3, dihydrofuran 19 was converted to tricyclic enone 20 in three steps involving (i) bromoetherification of 19 with NBS and homopropargyl alcohol; (ii) dehydrobromination with DBU and (iii) Pauson-Khand reaction of the resulting enyne to provide 20 in 16% yield over three steps. Catalytic transfer hydrogenation of enone 20 followed by reduction with NaBH4 provided racemic alcohol 21. This alcohol was subjected to enzymatic resolution to provide alcohol (−)-21 and acetate derivative 22. Saponification of acetate 22 afforded alcohol (+)-21 in excellent yield.
Scheme 3.
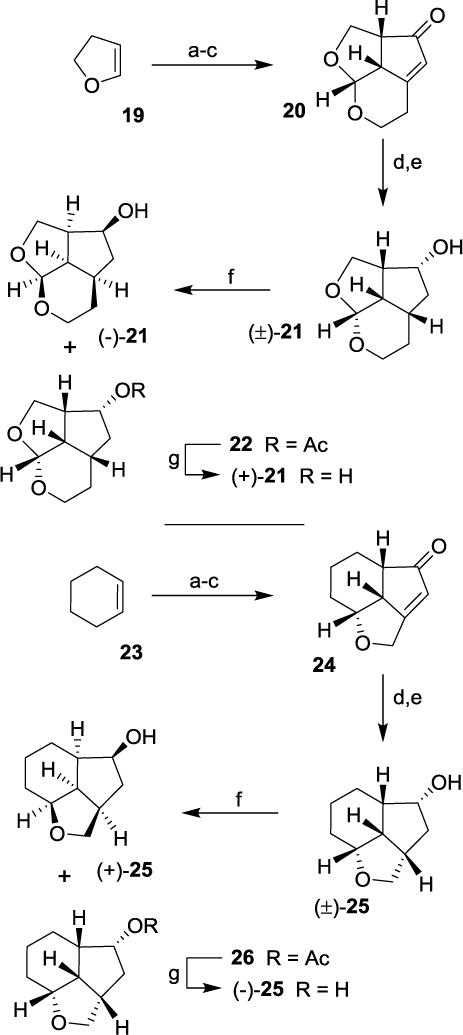
Synthesis of optically active ligand alcohols 21 and 25. Reagents and conditions. (a) NBS, homopropargyl alcohol (for 20)/propargyl alcohol (for 24), CH2Cl2, -20 °C to 23 °C, 17 h (63-93%); (b) DBU, heat 110 °C, 5 h, (84-92%); (c) Co2(CO)8, CH2Cl2, -23 °C, 1 h, then NMO, CH2Cl2, 0 °C to 23 °C, 3 h (20- 58%); (d) 10% Pd/C, HCO2NH4, MeOH, reflux (e) NaBH4, MeOH (63% over 2-steps); (f) Lipase (PS-30), vinyl acetate, THF, 23 °C, (−)-21 (47%), 22 (53%), (+)-25 (48%), 26 (52%); (g) K2CO3; MeOH, 23 °C, 1 h, (90-95%).
For SAR studies, we also prepared decahydroindeno[7,1-bc]furan-6-ol using commercially available cyclohexene. As shown in Scheme 3, cyclohexene 23 was converted to enone 24 as described above. Transfer hydrogenation followed by NaBH4 reduction provided racemic alcohol 25. Enzymatic resolution of alcohol 25 provided optically active alcohol (+)-25 and acetate 26. Saponification of acetate 26 provided optically active alcohol (−)-25 in excellent yield. The stereochemical assignment of the depicted stereochemistry is based upon correlation with sign of optical rotation of optically active alcohol (−)-16.
The synthesis of the designed PIs was carried out in a two-step sequence involving synthesis of activated carbonates followed by reaction of these carbonates with appropriate hydroxyethylaminesulfonamide isosteres. The syntheses of various activated carbonates are shown in Scheme 4. All optically active ligand alcohols synthesized above were converted to their respective activated carbonates. As shown, reaction of ligand alcohols (−)-16 and 10 with 4-nitrophenylchloroformate in the presence of pyridine in CH2Cl2 at 0 °C to 23 °C for 12 h provided activated carbonates 18 and 27a in 88% and 79% yields, respectively. Accordingly, other activated carbonates 27b-g were readily prepared in good yields as shown.36
Scheme 4.
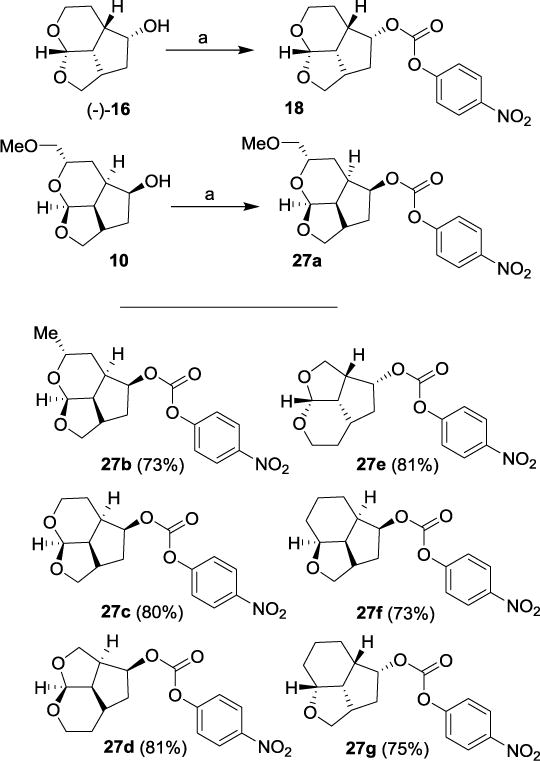
Synthesis of activated carbonates 18, 27a-g. Reagents and conditions. (a) 4-(NO2)PhOCOCl, pyridine, CH2Cl2, 0 °C to 23 °C, 12 h, 18 (88%) and 27a (79%).
The synthesis of various inhibitors containing Umb-THF as the P2 ligands on the hydroxysulfonamide isostere is shown in Scheme 5. Reactions of activated carbonates 27a and 27b with known 4-methoxybenzenesulfonamide isostere 28 in the presence of N,N-diisopropylethylamine (DIPEA) in CH3CN at 23 °C for 72 h provided inhibitors 4a and 4b in 90% and 97% yields, respectively. Other activated carbonates 18, 27c-g were reacted with amine 28 under similar conditions to afford inhibitors 4c-h in very good yields (81-99%). The full structures of these inhibitors are shown in Table 1. The synthesis of inhibitors with benzothiazole sulfonamides 29 and 30 is shown in Scheme 6. Reaction of activated carbonates 27c and 18 with known cyclopropylaminobenzothiazole sulfonamide isostere 29 in CH3CN at 23 °C for 96 h furnished inhibitors 5a and 5b in excellent yields (78-90%). Similarly, reactions of carbonates 27c and 18 with cyclopropylaminobenzothiazole isostere 30 with a fluorinated P1-ligand provided inhibitors 5c and 5d in excellent yields.
Scheme 5.

Synthesis of PIs 4a-4h. Reagents and conditions. (a) 27a and 27b, DIPEA, CH3CN, 23 °C, (4a, 90%; 4b, 97%).
Table 1.
HIV-1 protease inhibitory and antiviral activity of PIs 4a-h
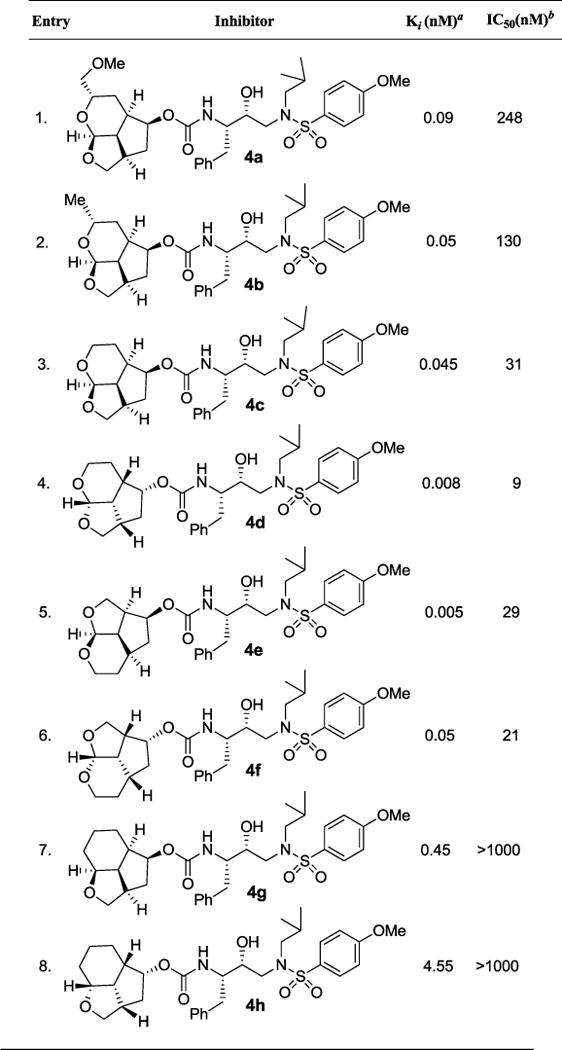
|
Ki values represents at least 5 data points. Standard error in all cases was less than 7%. Darunavir exhibited Ki= 16 pM.
Values are means of at least three experiments. Standard error in all cases was less than 5%. Darunavir exhibited antiviral IC50 = 3.2 nM, saquinavir IC50 = 21 nM.
Scheme 6.
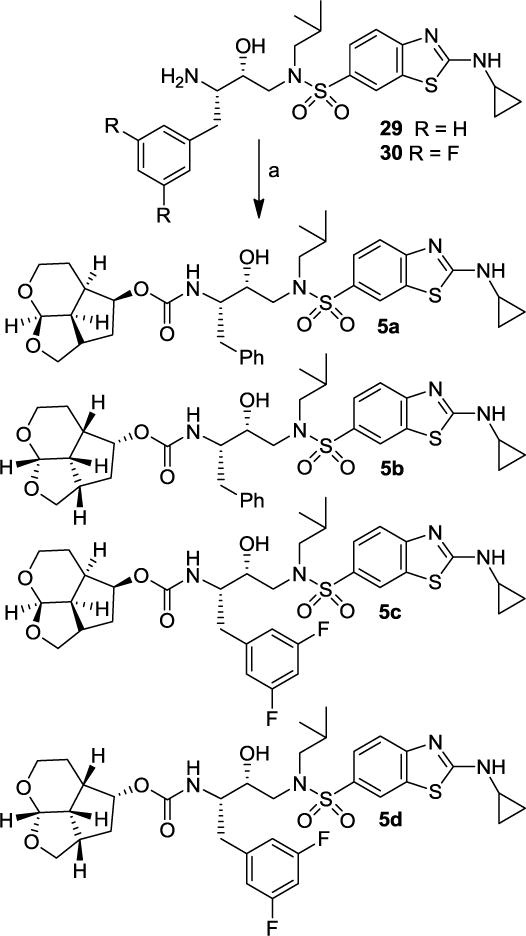
Synthesis of PIs 5a-d. Reagents and conditions. (a) 18 or 27c, DIPEA, CH3CN, 23 °C, (78-90%).
Our examination of the preliminary model of the tricyclic ligand derived from D-glucal for compound 4a indicated that the side chain methoxy group may not form hydrogen bond with any residues in the S2-subsite. The results of HIV-1 protease inhibitory and antiviral assays are shown in Table 1. The protocol for enzyme assays is similar to the reported procedure of Toth and Marshall.50 The antiviral activity was determined as described previously using MT-2 human T-lymphoid cells exposed to HIV-1LAI.51 For our preliminary assessment of ligand substituent stereochemistry, we planned to utilize a hydroxyethyamine sulfonamide isostere with a 4-methoxy benzene sulfonamide ligand as the P2′-ligand.36 Inhibitor 4a with a methoxymethyl side chain on the tricyclic P2 ligand 10, showed potent enzyme inhibitory activity with a Ki of 0.09 nM. It showed antiviral activity IC50 value of 248 nM. The removal of methoxy group resulted in inhibitor 4b which showed further improvement of both enzyme inhibitory as well as antiviral activity (entry 2). Interestingly, the removal of the methyl group from the ligand, as shown in inhibitor 4c, while not leading to a relevant change in HIV-1 protease inhibitory activity, resulted in a 4-fold improvement in antiviral activity compared to inhibitor 4b. Our initial model of tricyclic ligands suggested that the ring stereochemistry in ligand 10 would be preferred, although the enantiomeric ligand also appears to form hydrogen bonding interaction with aspartate residue in S2 subsite. Interestingly, enantiomeric tricyclic ligand in inhibitor 4d exhibited significant enhancement of both enzyme inhibitory and antiviral activity. Inhibitor 4d showed an enzyme Ki of 8 pM and antiviral IC50 value of 9 nM (entry 4). We then investigated the effect of each ring size for the tricyclic ligands. Inhibitor 4e with a tetrahydrofuro-tetrahydropyran fused ligand showed very potent enzyme activity, however, antiviral potency was not improved over the isomeric ligand in inhibitor 4c. Similarly, enantiomeric ligand in inhibitor 4f, showed very good enzyme Ki and antiviral IC50 value, but less potent than the isomeric ligand in inhibitor 4d.
To ascertain the effect of the tetrahydropyran ring oxygen of the tricyclic ligand, we have synthesized the corresponding deoxygenated ligands. As shown, both inhibitors 4g and 4h containing the cyclohexyl derivatives, showed significant reduction of HIV-1 protease inhibitory activity. Also, both inhibitors did not show any appreciable antiviral activity (IC50 > 1 μM, entries 7, 8). This result indicated the importance of the ring oxygen and our preliminary model indicates possible involvement in strong hydrogen bonding interactions with both Asp29 and Asp30 backbone NHs in the S2-subsite.
We then explored further optimization of PIs containing tricyclic Umb-THF ligands as the P2-ligands in combination with other benzothiazole ligands as the P2′-ligand. The results are shown in Table 2. Interestingly, both compounds 5a and 5b containing enantiomeric P2-ligands in combination with cyclopropylaminobenzothiazole as the P2′-ligand displayed comparable enzyme inhibitory and antiviral activity (entries 1, 2). Further modification of ligand-binding site interactions by incorporation of a lipophilic 3,5-difluoro phenylmethyl group as the P1-ligand resulted in very potent PIs. Both inhibitors 5c and 5d exhibited exceptionally potent activity, particularly antiviral activity in MT-2 cells (entries 3 and 4), showing antiviral IC50 values of 0.023 nM and 0.027 nM, respectively. In comparison, DRV and SQV displayed antiviral IC50 values of 3.2 and 21 nM, respectively.
Table 2.
HIV-1 protease inhibitory and antiviral activity of PIs 5a-d

|
Ki values represents at least 5 data points. Standard error in all cases was less than 7%. Darunavir exhibited Ki= 16 pM.
Values are means of at least three experiments. Standard error in all cases was less than 5%. Darunavir exhibited antiviral IC50 = 3.2 nM, saquinavir IC50 = 21 nM.
The current antiretroviral therapy and treatment guidelines are updated regularly. However, PIs continue to be an important component of current ART regimens. Particularly, PIs are extensively used for the treatment of naïve and experienced HIV/AIDS patients. Unfortunately, heavily-ART regimen-experienced HIV/AIDS patients tend to have drug-failure with the currently available PIs including DRV.51,52 Consequently, design and discovery of new classes of potent PIs that exhibit a high genetic barrier are critical to effective long-term treatment options. Towards this goal, our design objectives include the design of highly potent PIs that maintain potency against a variety of existing multi-PI-resistant HIV-1 variants with better selectivity index and safety profiles. In preliminary studies, we therefore examined the activity of both potent Umb-THF containing PIs (5c and 5d) against a panel of HIV-1 variants that had been selected in vitro with three widely used FDA-approved PIs, ATV, LPV, and APV. Each of these HIV-1 variants were selected in vitro by propagating HIV-1NL4-3 in the presence of increasing concentrations of each PI (up to 5 μM) in MT-4 cells as described previously.51,53
Each variant was highly resistant to the PI drug, with which the variant was selected. The results are shown in Table 3. As can be seen, among two current clinically used PIs, LPV and DRV, LPV lost significant activity against the three HIV-1 variants. DRV displayed relatively better results, however, it too failed to block replication of each of these three variants very effectively. DRV exhibited an IC50 value fold-change ranging from 8- to 67-fold. Interestingly, both inhibitors 5c and 5d maintained superior activity against all three HIV-1 variants showing no loss of antiviral activity compared to wild-type HIVNL4-3. Both PIs exerted exceptionally potent antiviral activity with EC50 values ranging from 0.0003 to 0.002 μM. These PIs contain structurally novel fused tricylic enantiomeric ligands as the P2 ligand, a cyclopropylaminobenzothiazole derivative as the P2′-ligand, and a 3,5-difluorophenylmethyl as the P1 ligand. Our detailed X-ray crystallographic studies of inhibitors 5c- and 5d-bound HIV-1 protease provided molecular insight into the binding features responsible for the superior properties of inhibitors 5c and 5d.
Table 3.
Comparison of the antiviral activity of 5c and 5d and other PIs against highly PI-resistant HIV-1 variants.
| Virusa | EC50 (μM)b | |||||
|---|---|---|---|---|---|---|
| LPV | APV | ATV | DRV | 5c | 5d | |
| HIV-1NL4-3 | 0.032 ± 0.001 | 0.087 ± 0.003 | 0.0033 ± 0.0001 | 0.003 ± 0.002 | 0.002 ± 0.0007 | 0.0012 ± 0.0007 |
| HIV-1ATVR5μM | >1 (>31) | >1 (> 11) | > 1 (> 303) | 0.024 ± 0.004 (8) | 0.0015 ± 0.0001 (1) | 0.0006 ± 0.0001 (0.3) |
| HIV-1LPVR5μM | >1 (>31) | 0.19 ± 0.06 (2) | 0.029 ± 0.004 (9) | 0.026 ± 0.004 (9) | 0.002 ± 0.001 (2) | 0.002 ± 0.001 (1) |
| HIV-1APVR5μM | 0.39 ± 0.02 (12) | >1 (> 11) | 0.07 ± 0.04 (21) | 0.2 ± 0.1 (67) | 0.00032 ± 0.00001 (0.3) | 0.0003 ± 0.0001 (0.2) |
The amino acid substitutions identified in the protease-encoding region compared to the wild-type HIV-1NL4-3 were L23I, E34Q, K43I, M46I, I50L, G51A, L63P, A71V, V82A, T91A in HIV-1ATVR5μM; L10F, M46I, I54V, V82A in HIV-1LPVR5μM; or L10F, M46I, I50V, I85V in HIV-1APVR5μM.
The EC50 (50% effective concentration) values were determined by using MT-4 cells as target cells. MT-4 cells (105/mL) were exposed to 100 TCID50s of each HIV-1, and the inhibition of p24 Gag protein production by each drug was used as an endpoint. All assays were conducted in duplicate, and the data shown represent mean values (± S.D.) derived from the results of two independent experiments.
The X-ray structures of the wild-type HIV-1 protease co-crystallized independently with inhibitors 5c and 5d were refined at a resolution of 1.25 Å and 1.13 Å, respectively.54 Both structures contain one HIV-1 protease dimer and each inhibitor (5c and 5d) binds in two orientations related by 180° rotation with relative occupancy of 50/50% and 55/45%, respectively. The overall dimer structure is comparable to the X-ray structure of HIV-1 protease-bound DRV with root mean square difference of 0.15 Å for 5c complex and 0.18 Å for 5d for 198 equivalent Cα atoms.27 In both these X-ray structures, the large difluorophenylmethyl P1-group results in larger disparity of about 0.4-0.6 Å between the corresponding Cα atoms in the flap regions and residues of 80’s loop compared to DRV-bound HIV-1 protease structure.27 The key interactions of inhibitor 5d with HIV-1 protease are highlighted in Figure 3. In the major conformation, inhibitor 5d forms a strong hydrogen bond through its urethane NH with the carbonyl oxygen of Gly27. The inhibitor 5d also forms a water-mediated tetracoordinated hydrogen bonding interaction involving the inhibitor 5d carbonyl oxygen and one of the sulfonamide oxygens with amide NHs of Ile50 and Ile50′ located in the flaps. Inhibitor 5d also forms extensive interactions through its P2 and P2′-ligands in both S2 and S2′-subsites of HIV-1 protease. These inhibitor-HIV-1 protease interactions are similar in inhibitor 5c-bound HIV-1 protease complex shown in Figure 4.
Figure 3.
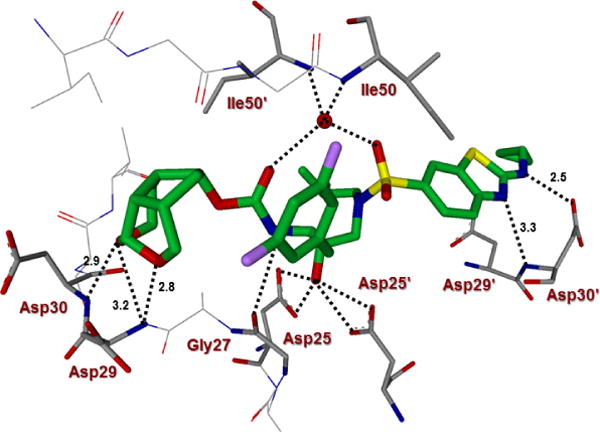
Inhibitor 5d-bound HIV-1 protease X-ray structure is shown (pdb code: 6CDJ). The inhibitor carbon atoms are shown in green and hydrogen bonds are shown by black dotted lines.
Figure 4.
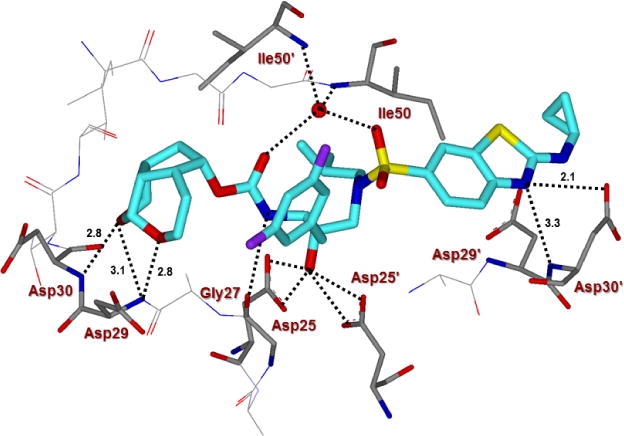
Inhibitor 5c-bound HIV-1 protease X-ray structure is shown (pdb code: 6CDL). The inhibitor carbon atoms are shown in cyan and hydrogen bonds are shown by black dotted lines.
The new P2-tricyclic ligand scaffold is involved in extensive interactions in the S2-subsite of HIV-1 protease. The new ligand basically retains all key hydrogen bonding interactions similar to the bis-THF oxygens of DRV-bound HIV-1 protease structures. The tricyclic umbrella-like scaffold containing the acetal oxygens is suitably positioned to interact with backbone NHs of Asp29 and Asp30. Furthermore, the Umb-THF makes significantly better hydrophobic contacts than the bis-THF ligand in DRV. In particular, as shown in the X-ray structure of 5c and 5d-bound HIV-1 protease in Figures 3 and 4, the Umb-THF, tetrahydropyranyl oxygen of 5d ligand forms a pair of tight hydrogen bonds with Asp30 and Asp29 backbone NHs, while the tetrahydrofuranyl oxygen on the other hand forms a strong hydrogen bond with Asp29 backbone NH. In addition, both enantiomeric Umb-THF ligands form significant van der Waals interactions with the side chain atoms of Ile50, Ile47, Val32, and Ile84 in the S2 subsite as shown in Figure 5.
Figure 5.
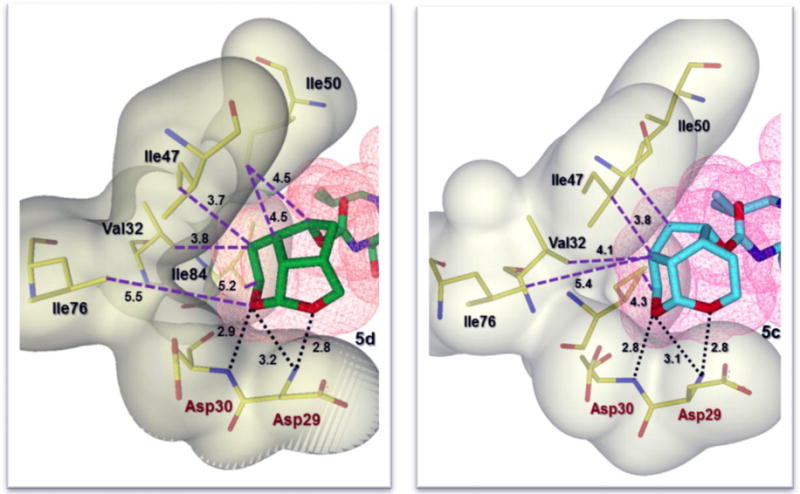
Side by side comparison of the new Umb-THF moiety of inhibitor 5d (left, green carbon chain) with the enantiomeric Umb-THF moiety of inhibitor 5c (right, cyan carbon chain) inside the S2 subpocket. Both ligands form extensive van der Waals interactions (Val32, Ile47, and Ile50 for 5d and Val32, Ile47, Ile50 and Ile84 for 5c) in the S2 subsite. Also, they are located close to the periphery of the protease active site and form three strong hydrogen bonds in a similar fashion (black dotted lines).
Interestingly, the bulging P2 ligand in inhibitor 5d is nicely accommodated by shifting the Gly48 carbonyl group in flap region into an alternate conformation, on the other hand, the enantiomeric Umb-THF ligand in inhibitor 5c rotated towards the CD1 atom of Ile50 to form a van der Waals contact. The new tricyclic P2 ligand is conformationally constrained and bigger in size than the bis-THF ligand in DRV. The new ligand not only forms stronger hydrogen bonds with the backbone atoms in the S2-site, but also makes enhanced van der Waals interactions in S2-site compared to bis-THF ligand of DRV. The P1-ligand for inhibitors 5c and 5d shows similar halogen bond interactions as the Crn-THF derived fluorinated inhibitor reported by us recently.26,55 One of the fluorine atoms of the P1-ligand forms strong polar interactions with the backbone NH group of Ile50 (C-F•••H-N) at a distance of 3.2 Å for inhibitor 5c and 2.9 Å for inhibitor 5d and 3.0 Å long interactions with the backbone carbonyl group of Gly49.55,56 Furthermore, one fluorine atom forms hydrophobic interactions with Gly49, Ile50, and Pro81 side chains. The second fluorine atom interacts with the guanidinium group of Arg8′, which is involved in a critical intersubunit ion pair with Asp29. This fluorine atom also forms van der Waals interactions with the side chain of Val82′.
The binding properties of inhibitors 5c and 5d in the S2′-subsite are slightly different. As can be seen in the overlay structures of 5c and 5d in Figure 6, the aminobenzothiazole group sits nicely in the S2′-subpocket, where the thiazole nitrogen and the cyclopropyl amine moiety form hydrogen bonds with the backbone amide NH of Asp30′. In the X-ray structure of inhibitor 5c-bound HIV-1 protease, the cyclopropyl group exists in two different conformations. In one conformation, it forms van der Waals contacts with Ile47′, and Lys45′. These interactions are absent in the other conformation. For inhibitors 5c and 5d, the thiazole nitrogen specifically formed a strong hydrogen bond with the backbone amide NH of Asp30′ at a distance of 3.3 Å for both inhibitors 5c and 5d. The P2’-amine NH also formed strong hydrogen bond with Asp30′ carboxylate group at a distance of 2.8 Å and 2.5 Å in the respective X-ray structures for PIs 5c and 5d. This P2′ ligand also makes more hydrophobic contacts with the protease than the 4-aminobenzenesulfonamide ligand of DRV. These interactions are extensive and may account for the high potency of these new PIs.
Figure 6.

Stereoview of the overlay of X-ray crystal structures of inhibitors 5c (magenta) and 5d (green) into the active site of HIV-1 protease (PDB codes: 6CDL and 6CDJ). Both P2 ligands make van der Waals interactions with Val32, Ile47, and Ile50’ in the S2 subsite. All key hydrogen bonds are shown as black dotted lines.
Conclusions
In summary, we have designed and developed a new class of highly potent HIV-1 PIs incorporating stereochemically defined tricyclic umbrella-like scaffolds as the P2-ligands. The ligands have been specifically designed to interact with the backbone atoms of HIV-1 protease in the S2-subsite, in particular, with the Asp29 and Asp30 backbone amide NHs. The new ligands also make favorable van der Waals interactions in the active site. Both enantiomeric P2 ligands provided very potent inhibitors, namely compounds 5c and 5d; however, inhibitor 5d with the (2aS,2a1S,4R,4aS,7aS)-octahydro-2H-1,7-dioxacyclopenta[cd]inden-4-ol-derived ligand showed slightly better antiviral activity. Both PIs with a difluorophenylmethyl as the P1-ligand and an aminobenzothiazole as the P2′-ligand showed exceptionally potent activity against a panel of highly resistant multidrug-resistant HIV-1 variants. The data show that the PIs are superior to other clinically approved PIs. The ligands were synthesized in racemic form using a Pauson-Khand cyclization as the key step. The racemic ligands were efficiently resolved using lipase PS-30 catalyzed acylation in high optical purity. Our structure-activity studies revealed that the tetrahydropyranyl oxygen is critical to inhibitors’ potency. Our X-ray structural studies of inhibitor 5c and 5d-bound HIV-1 protease provided molecular insight into their ligand-binding site interactions. As it turned out, both oxygens on the tricyclic ligand form very strong hydrogen bonding interactions with the backbone amide NHs of Asp29 and Asp30. Furthermore, the new ligand fills in the hydrophobic pocket in the S2 site and makes good van der Waals interactions with several key amino acid residues. The combinations of ligands in inhibitors 5c and 5d led to exceptional HIV-1 protease inhibitory potency and antiviral activity. Also, these inhibitors maintained nearly full potency against a panel of highly resistant multidrug-resistant HIV-1 strains. Further design and optimization of inhibitors are currently underway in our laboratories.
Experimental Section
General Methods
All chemicals and reagents were purchased from commercial suppliers and used without further purification unless otherwise noted. The following reaction solvents were distilled prior to use: dichloromethane from calcium hydride, diethyl ether and tetrahydrofuran from Na/benzophenone, methanol and ethanol from activated magnesium under argon. All reactions were carried out under an argon atmosphere in either flame or oven-dried (120 °C) glassware. TLC analysis was conducted using glass-backed Thin-Layer Silica Gel Chromatography Plates (60 Å, 250 μm thickness, F-254 indicator). Column chromatography was performed using 230-400 mesh, 60 Å pore diameter silica gel. 1H and 13C NMR spectra were recorded at room temperature on a Bruker AV800, DRX-500 and ARX-400. Chemical shifts (δ values) are reported in parts per million, and are referenced to the deuterated residual solvent peak. NMR data is reported as: δ value (chemical shift, J-value (Hz), integration, where s = singlet, d = doublet, t = triplet, q = quartet, brs = broad singlet). Optical rotations were recorded on a Perkin Elmer 341 polarimeter. HRMS and LRMS spectra were recorded at the Purdue University Department of Chemistry Mass Spectrometry Center. HPLC analysis and purification was done an on Agilent 1100 series instrument using a YMC Pack ODS-A column of 4.6 mm ID for analysis and either 10 mm ID or 20 mm ID for purification. The purity of all test compounds was determined by HPLC analysis to be ≥95% pure.
(2aR,2a1R,4S,4aR,6S,7aS)-6-(Methoxymethyl)octahydro-2H-1,7-dioxacyclopenta[cd]inden-4-yl ((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)carbamate (4a)
To a stirred solution of activated alcohol 27a (14 mg, 0.04 mmol) and isostere 28 (18 mg, 0.044 mmol) in acetonitrile (2 mL) was added DIPEA (32 μL, 0.18 mmol) at 23 °C under argon atmosphere. The reaction mixture was stirred at 23 °C until completion. Upon completion, solvents were removed under reduced pressure and crude product was purified by silica gel column chromatography (65% EtOAc in hexane) to give 4a (21.5 mg, 90%). 1H NMR (800 MHz, CDCl3) δ 7.72 (d, J = 8.4 Hz, 2H), 7.30 – 7.27 (m, 2H), 7.22 (dd, J = 14.2, 7.4 Hz, 3H), 7.00 – 6.96 (m, 2H), 5.34 (d, J = 5.5 Hz, 1H), 5.09 – 5.05 (m, 1H), 4.99 (d, J = 8.0 Hz, 1H), 4.11 – 4.07 (m, 1H), 3.93 – 3.86 (m, 4H), 3.86 – 3.82 (m, 3H), 3.80 (t, J = 8.6 Hz, 1H), 3.64 (d, J = 7.6 Hz, 1H), 3.43 – 3.36 (m, 4H), 3.36 – 3.32 (m, 1H), 3.29 (dd, J = 9.9, 5.1 Hz, 1H), 3.13 (dd, J = 14.8, 7.9 Hz, 1H), 3.04 (t, J = 12.3 Hz, 2H), 2.95 (dd, J = 13.3, 8.3 Hz, 1H), 2.87 (dd, J = 13.9, 8.2 Hz, 1H), 2.81 (dd, J = 13.2, 6.6 Hz, 1H), 2.77 – 2.71 (m, 1H), 2.49 (q, J = 9.3 Hz, 1H), 2.39 – 2.32 (m, 1H), 2.01 (dt, J = 12.3, 5.7 Hz, 1H), 1.84 (dd, J = 13.4, 6.7 Hz, 1H), 1.76 (d, J = 14.1 Hz, 1H), 0.91 (d, J = 6.5 Hz, 3H), 0.87 (d, J = 6.5 Hz, 3H); 13C NMR (200 MHz, CDCl3) δ 163.2, 156.4, 137.9, 130.1, 129.7, 129.5, 128.7, 126.7, 114.5, 101.6, 79.7, 75.7, 72.8, 71.1, 66.2, 59.4, 58.8, 55.8, 55.1, 53.8, 41.1, 40.0, 38.8, 36.0, 35.6, 27.4, 23.0, 20.3, 20.1; LRMS-ESI (m/z): 647.3 [M+H]+; HRMS-ESI (m/z): [M+Na]+ calcd for C33H46N2O9SNa, 669.2822; found 669.2818.
(2aR,2a1R,4S,4aR,6R,7aS)-6-Methyloctahydro-2H-1,7-dioxacyclopenta[cd]inden-4-yl ((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)carbamate (4b)
Activated alcohol 27b (10 mg, 0.029 mmol) was treated with isostere amine 28 (14 mg, 0.034 mmol) by following the procedure outlined for inhibitor 4a to give inhibitor 4b (17 mg, 97%). 1H NMR (400 MHz, CDCl3) δ 7.75 – 7.69 (m, 2H), 7.31 – 7.19 (m, 5H), 6.97 (d, J = 8.9 Hz, 2H), 5.29 (d, J = 4.7 Hz, 1H), 5.09 – 5.03 (m, 2H), 4.05 – 3.96 (m, 1H), 3.90 – 3.76 (m, 7H), 3.62 (dd, J = 8.8, 2.9 Hz, 1H), 3.13 (dd, J = 15.1, 7.5 Hz, 1H), 3.04 (d, J = 14.3 Hz, 2H), 2.95 (dd, J = 13.4, 8.3 Hz, 1H), 2.88 – 2.79 (m, 2H), 2.77 – 2.70 (m, 1H), 2.49 – 2.41 (m, 1H), 2.36 – 2.27 (m, 1H), 1.98 (dt, J = 14.3, 7.5 Hz, 1H), 1.84 (dt, J = 14.2, 7.3 Hz, 2H), 1.74 (d, J = 14.1 Hz, 1H), 1.50 (d, J = 14.2 Hz, 1H), 1.07 (d, J = 6.2 Hz, 3H), 0.91 (d, J = 6.6 Hz, 3H), 0.86 (d, J = 6.6 Hz, 3H); 13C NMR (200 MHz, CDCl3) δ 163.1, 156.5, 137.9, 130.1, 129.6, 129.5, 128.6, 126.7, 114.5, 101.7, 79.6, 72.9, 70.9, 63.1, 58.8, 55.8, 55.1, 53.7, 41.2, 39.6, 38.8, 36.5, 35.6, 29.8, 28.7, 27.4, 21.9, 20.3, 20.0; LRMS-ESI (m/z): 617.3 [M+H]+; HRMS-ESI (m/z): [M+Na]+ calcd for C32H44N2O8SNa, 639.2717; found 639.2722.
(2aR,2a1R,4S,4aR,7aR)-Octahydro-2H-1,7-dioxacyclopenta[cd]inden-4-yl ((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)carbamate (4c)
Activated alcohol 27c (9.0 mg, 0.027 mmol) was treated with isostere amine 28 (13 mg, 0.03 mmol) by following the procedure outlined for inhibitor 4a to give inhibitor 4c (13 mg, 81%). 1H NMR (400 MHz, CDCl3) δ 7.74 – 7.67 (m, 2H), 7.29 – 7.16 (m, 5H), 7.01 – 6.95 (m, 2H), 5.19 (d, J = 5.3 Hz, 1H), 4.98 – 4.86 (m, 2H), 3.96 – 3.90 (m, 1H), 3.88 – 3.80 (m, 5H), 3.76 (td, J = 11.8, 11.4, 6.1 Hz, 3H), 3.67 – 3.61 (m, 1H), 3.27 (qd, J = 11.7, 3.5 Hz, 1H), 3.15 (dd, J = 15.1, 8.5 Hz, 1H), 3.11 – 2.91 (m, 3H), 2.80 (dd, J = 13.4, 6.9 Hz, 2H), 2.61 (dt, J = 14.9, 8.0 Hz, 1H), 2.50 – 2.35 (m, 2H), 2.06 – 1.94 (m, 1H), 1.91 – 1.67 (m, 3H), 0.92 (d, J = 6.6 Hz, 3H), 0.87 (d, J = 6.6 Hz, 3H); 13C NMR (200 MHz, CDCl3) δ 163.2, 156.1, 137.9, 130.0, 129.6, 129.5, 128.6, 126.6, 114.5, 100.7, 79.2, 73.1, 72.0, 60.4, 58.9, 55.8, 55.0, 53.9, 42.0, 39.5, 36.0, 35.8, 35.2, 29.8, 27.4, 21.5, 20.3, 20.0; LRMS-ESI (m/z): 603.2 [M+H]+; HRMS-ESI (m/z): [M+Na]+ calcd for C31H42N2O8SNa, 625.2560; found 625.2556.
(2aS,2a1S,4R,4aS,7aS)-Octahydro-2H-1,7-dioxacyclopenta[cd]inden-4-yl ((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)carbamate (4d)
Activated alcohol 18 (22 mg, 0.07 mmol) was treated with isostere amine 28 (32 mg, 0.08 mmol) by following the procedure outlined for inhibitor 4a to give inhibitor 4d (39 mg, 99%). 1H NMR (400 MHz, CDCl3) δ 7.71 (d, J = 8.9 Hz, 2H), 7.31 –7.18 (m, 5H), 6.97 (d, J = 8.9 Hz, 2H), 5.21 (d, J = 4.9 Hz, 1H), 5.04 (d, J = 8.2 Hz, 1H), 4.95 (q, J = 6.0 Hz, 1H), 3.93 – 3.83 (m, 7H), 3.55 (dd, J = 8.4, 5.9 Hz, 1H), 3.38 (ddd, J = 13.0, 8.5, 4.6 Hz, 1H), 3.15 – 3.00 (m, 4H), 2.94 (dd, J = 13.3, 8.3 Hz, 1H), 2.82 (td, J = 15.6, 13.5, 7.8 Hz, 2H), 2.67 – 2.57 (m, 1H), 2.52 – 2.38 (m, 2H), 1.95 (dt, J = 13.4, 7.0 Hz, 1H), 1.83 (tt, J = 13.5, 6.7 Hz, 2H), 1.55 – 1.48 (m, 1H), 0.90 (d, J = 6.6 Hz, 3H), 0.86 (d, J = 6.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 163.1, 156.2, 137.9, 130.0, 129.6, 128.6, 126.6, 114.5, 100.7, 79.4, 72.8, 71.9, 60.0, 58.8, 55.8, 55.1, 53.7, 53.6, 41.7, 39.7, 35.9, 35.7, 35.5, 30.4, 27.3, 21.9, 20.3, 20.0; LRMS-ESI (m/z): 603.3 [M+H]+; HRMS-ESI (m/z): [M+Na]+ calcd for C31H42N2O8SNa, 625.2560; found 625.2557.
(2aS,2a1R,3S,4aR,7aS)-Octahydro-2H-1,7-dioxacyclopenta[cd]inden-3-yl ((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)carbamate (4e)
Activated alcohol 27d (12 mg, 0.036 mmol) was treated with isostere amine 28 (17 mg, 0.04 mmol) by following the procedure outlined for inhibitor 4a to give inhibitor 4e (18 mg, 84%). 1H NMR (400 MHz, CDCl3) δ 7.74 – 7.69 (m, 2H), 7.32 – 7.26 (m, 2H), 7.24 – 7.20 (m, 3H), 6.98 (d, J = 8.9 Hz, 2H), 5.09 (d, J = 5.5 Hz, 1H), 4.93 (d, J = 8.1 Hz, 1H), 4.86 – 4.76 (m, 1H), 3.99 – 3.91 (m, 1H), 3.88 – 3.82 (m, 5H), 3.81 – 3.74 (m, 2H), 3.54 (dd, J = 11.5, 3.4 Hz, 1H), 3.34 – 3.27 (m, 1H), 3.14 (dd, J = 15.2, 8.4 Hz, 1H), 3.07 – 2.94 (m, 3H), 2.86 – 2.72 (m, 3H), 2.34 (td, J = 8.6, 5.6 Hz, 1H), 2.16 – 2.06 (m, 1H), 1.91 – 1.66 (m, 4H), 1.45 (d, J = 12.8 Hz, 1H), 0.92 (dd, J = 7.0, 2.5 Hz, 3H), 0.87 (d, J = 6.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 163.2, 156.0, 137.7, 129.6, 129.5, 128.7, 126.7, 114.5, 101.4, 76.0, 72.9, 63.1, 58.9, 55.8, 55.4, 55.1, 53.9, 44.1, 38.6, 35.7, 33.7, 28.7, 27.4, 24.1, 20.3, 20.0; LRMS-ESI (m/z): 603.2 [M+H]+; HRMS-ESI (m/z): [M+Na]+ calcd for C31H42N2O8SNa, 625.2560; found 625.2553.
(2aR,2a1S,3R,4aS,7aR)-Octahydro-2H-1,7-dioxacyclopenta[cd]inden-3-yl ((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)carbamate (4f)
Activated alcohol 27e (12 mg, 0.036 mmol) was treated with isostere amine 28 (17 mg, 0.04 mmol) by following the procedure outlined for inhibitor 4a to give inhibitor 4f (19.4 mg, 90%). 1H NMR (400 MHz, CDCl3) δ 7.70 (d, J = 8.8 Hz, 2H), 7.32 – 7.27 (m, 2H), 7.23 (d, J = 7.7 Hz, 3H), 7.01 – 6.96 (m, 2H), 5.12 (d, J = 5.4 Hz, 1H), 4.92 (d, J = 8.4 Hz, 1H), 4.83 (q, J = 8.2 Hz, 1H), 4.06 – 3.91 (m, 2H), 3.88 – 3.82 (m, 5H), 3.59 – 3.52 (m, 1H), 3.49 – 3.41 (m, 1H), 3.10 (dd, J = 14.9, 8.1 Hz, 1H), 3.05 – 2.89 (m, 5H), 2.81 (td, J = 15.1, 13.4, 7.0 Hz, 2H), 2.42 – 2.31 (m, 1H), 2.17 – 2.07 (m, 1H), 1.92 – 1.71 (m, 4H), 1.46 (d, J = 14.1 Hz, 1H), 0.90 (d, J = 6.5 Hz, 3H), 0.87 (d, J = 6.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 163.2, 156.2, 137.7, 130.0, 129.6, 128.7, 126.7, 114.5, 101.4, 76.1, 72.6, 63.3, 60.5, 58.9, 55.8, 55.5, 55.3, 53.8, 44.2, 38.6, 35.4, 33.8, 28.8, 27.4, 24.1, 20.3, 20.0; LRMS-ESI (m/z): 603.3 [M+H]+; HRMS-ESI (m/z): [M+Na]+ calcd for C31H42N2O8SNa, 625.2560; found 625.2550.
(2aR,2a1R,4S,4aR,7aS)-Decahydroindeno[7,1-bc]furan-4-yl ((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)carbamate (4g)
Activated alcohol 27f (18 mg, 0.054 mmol) was treated with isostere amine 28 (26 mg, 0.06 mmol) by following the procedure outlined for inhibitor 4a to give inhibitor 4g (27 mg, 84%). 1H NMR (400 MHz, CDCl3) δ 7.72 (d, J = 8.8 Hz, 2H), 7.25 – 7.17 (m, 5H), 6.97 (d, J = 8.9 Hz, 2H), 5.16 (d, J = 7.7 Hz, 1H), 4.96 (q, J = 5.3 Hz, 1H), 3.86 (s, 3H), 3.83 – 3.77 (m, 4H), 3.72 – 3.66 (m, 1H), 3.59 (d, J = 8.1 Hz, 1H), 3.09 (t, J = 12.7 Hz, 3H), 2.95 (dd, J = 13.3, 8.4 Hz, 1H), 2.86 – 2.78 (m, 2H), 2.74 – 2.68 (m, 1H), 2.45 (q, J = 9.2 Hz, 1H), 2.04 (tt, J = 10.5, 6.1 Hz, 2H), 1.98 – 1.88 (m, 2H), 1.84 (dd, J = 12.8, 6.0 Hz, 1H), 1.65 – 1.58 (m, 1H), 1.52 – 1.45 (m, 2H), 1.35 – 1.28 (m, 1H), 1.16 (dd, J = 11.9, 5.0 Hz, 1H), 0.91 (d, J = 6.6 Hz, 3H), 0.86 (d, J = 6.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 163.1, 156.8, 138.1, 130.2, 129.6, 128.5, 126.5, 114.4, 79.8, 77.4, 77.0, 74.7, 58.7, 55.7, 55.2, 53.7, 43.4, 43.0, 38.6, 38.5, 35.7, 29.8, 27.3, 26.1, 22.0, 20.3, 20.0, 16.9; LRMS-ESI (m/z): 601.3 [M+H]+; HRMS-ESI (m/z): [M+Na]+ calcd for C32H44N2O7SNa, 623.2767; found 623.2761.
(2aS,2a1S,4R,4aS,7aR)-Decahydroindeno[7,1-bc]furan-4-yl ((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)carbamate (4h)
Activated alcohol 27g (18 mg, 0.05 mmol) was treated with isostere amine 28 (26 mg, 0.06 mmol) by following the procedure outlined for inhibitor 4a to give inhibitor 4h (32 mg, 99%). 1H NMR (400 MHz, CDCl3) δ 7.72 (d, J = 8.8 Hz, 2H), 7.29 – 7.22 (m, 4H), 7.21 – 7.16 (m, 1H), 6.99 – 6.94 (m, 2H), 5.20 (d, J = 8.7 Hz, 1H), 4.92 (q, J = 5.1 Hz, 1H), 3.88 – 3.83 (m, 4H), 3.82 – 3.76 (m, 2H), 3.66 (t, J = 8.6 Hz, 1H), 3.45 (d, J = 8.3 Hz, 1H), 3.11 (d, J = 6.4 Hz, 3H), 2.95 (dd, J = 13.4, 8.4 Hz, 1H), 2.85 – 2.75 (m, 2H), 2.74 – 2.65 (m, 1H), 2.45 (td, J = 9.4, 6.1 Hz, 1H), 2.09 – 2.01 (m, 1H), 1.99 – 1.91 (m, 1H), 1.84 (dq, J = 13.3, 6.4 Hz, 2H), 1.71 – 1.60 (m, 2H), 1.55 – 1.42 (m, 3H), 1.23 – 1.16 (m, 1H), 0.90 (d, J = 6.6 Hz, 3H), 0.85 (d, J = 6.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 163.1, 156.9, 138.2, 130.2, 129.6, 128.5, 126.5, 114.4, 80.2, 77.4, 77.0, 74.8, 73.1, 58.7, 55.7, 55.0, 53.7, 43.5, 43.2, 38.7, 38.5, 35.9, 27.3, 26.4, 22.6, 20.3, 20.0, 16.8; LRMS-ESI (m/z): 601.3 [M+H]+; HRMS-ESI (m/z): [M+Na]+ calcd for C32H44N2O7SNa, 623.2767; found 623.2761.
(2aR,2a1R,4S,4aR,7aR)-Octahydro-2H-1,7-dioxacyclopenta[cd]inden-4-yl ((2S,3R)-4-((2-(cyclopropylamino)-N-isobutylbenzo[d]thiazole)-6-sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (5a)
Activated alcohol 27c (9 mg, 0.027 mmol) was treated with isostere amine 29 (15.7 mg, 0.032 mmol) by following the procedure outlined for inhibitor 4a to give inhibitor 5a (14.4 mg, 78%). 1H NMR (400 MHz, CDCl3) δ 8.08 (d, J = 1.7 Hz, 1H), 7.68 (dd, J = 8.5, 1.8 Hz, 1H), 7.56 (d, J = 8.5 Hz, 1H), 7.29 – 7.18 (m, 5H), 7.14 – 7.06 (m, 1H), 5.19 (d, J = 5.3 Hz, 1H), 4.97 (dt, J = 20.3, 7.4 Hz, 2H), 3.89 (dd, J = 20.2, 8.2 Hz, 4H), 3.76 (dq, J = 8.7, 4.2, 3.7 Hz, 1H), 3.68 – 3.61 (m, 1H), 3.28 (td, J = 10.1, 8.4, 3.6 Hz, 1H), 3.18 (dd, J = 15.0, 8.3 Hz, 1H), 3.06 (t, J = 11.5 Hz, 2H), 3.02 – 2.97 (m, 1H), 2.83 (dd, J = 13.3, 6.7 Hz, 2H), 2.75 (dq, J = 6.7, 3.5 Hz, 1H), 2.60 (dt, J = 14.5, 7.5 Hz, 1H), 2.43 (ddd, J = 22.4, 12.3, 6.3 Hz, 2H), 2.02 – 1.97 (m, 1H), 1.91 – 1.78 (m, 2H), 1.58 (dd, J = 14.2, 6.8 Hz, 2H), 0.95 – 0.90 (m, 5H), 0.88 (d, J = 6.5 Hz, 3H), 0.80 – 0.77 (m, 2H); 13C NMR (200 MHz, CDCl3) δ 172.9, 156.1, 155.9, 137.9, 131.5, 130.6, 129.5, 128.6, 126.6, 125.5, 121.1, 118.9, 100.7, 79.2, 73.1, 72.0, 60.4, 59.0, 55.1, 53.9, 42.0, 39.5, 36.0, 35.8, 35.2, 31.7, 27.4, 26.8, 22.8, 21.5, 20.3, 20.1, 8.2; LRMS-ESI (m/z): 685.3 [M+H]+; HRMS-ESI (m/z): [M+H]+ calcd for C34H45N4O7S2, 685.2730; found 685.2735.
(2aS,2a1S,4R,4aS,7aS)-Octahydro-2H-1,7-dioxacyclopenta[cd]inden-4-yl ((2S,3R)-4-((2-(cyclopropylamino)-N-isobutylbenzo[d]thiazole)-6-sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (5b)
Activated alcohol 18 (10 mg, 0.03 mmol) was treated with isostere amine 29 (17.5 mg, 0.036 mmol) by following the procedure outlined for inhibitor 4a to give inhibitor 5b (18 mg, 90%). 1H NMR (400 MHz, CDCl3) δ 8.07 (s, 1H), 7.67 (dd, J = 8.5, 1.9 Hz, 1H), 7.56 (d, J = 8.5 Hz, 1H), 7.31 – 7.18 (m, 5H), 7.06 (brs, 1H), 5.21 (d, J = 4.6 Hz, 1H), 5.03 (d, J = 8.7 Hz, 1H), 4.98 – 4.90 (m, 1H), 3.96 – 3.83 (m, 5H), 3.60 – 3.52 (m, 1H), 3.41 – 3.33 (m, 1H), 3.16 (dd, J = 15.1, 8.3 Hz, 1H), 3.10 – 2.96 (m, 3H), 2.91 – 2.73 (m, 3H), 2.62 (dq, J = 14.1, 7.8, 7.3 Hz, 1H), 2.46 (tt, J = 16.6, 9.2 Hz, 2H), 2.00 – 1.92 (m, 1H), 1.85 (dq, J = 14.4, 6.7 Hz, 1H), 1.54 – 1.45 (m, 3H), 0.93 (dd, J = 13.5, 5.9 Hz, 5H), 0.87 (d, J = 6.6 Hz, 3H), 0.79 (p, J = 5.3, 4.9 Hz, 2H); 13C NMR (200 MHz, CDCl3) δ 173.1, 156.3, 155.8, 137.9, 131.4, 130.6, 129.6, 128.6, 126.7, 125.5, 121.0, 118.7, 100.7, 79.4, 72.9, 72.0, 60.1, 58.9, 55.2, 53.8, 41.8, 39.6, 35.9, 35.6, 31.7, 27.4, 26.8, 22.8, 21.9, 20.3, 20.1, 8.1; LRMS-ESI (m/z): 685.3 [M+H]+; HRMS-ESI (m/z): [M+H]+ calcd for C34H45N4O7S2, 685.2730; found 685.2723.
(2aR,2a1R,4S,4aR,7aR)-Octahydro-2H-1,7-dioxacyclopenta[cd]inden-4-yl ((2S,3R)-4-((2-(cyclopropylamino)-N-isobutylbenzo[d]thiazole)-6-sulfonamido)-1-(3,5-difluorophenyl)-3-hydroxybutan-2-yl)carbamate (5c)
Activated alcohol 27c (6 mg, 0.018 mmol) was treated with isostere amine 30 (11 mg, 0.02 mmol) by following the procedure outlined for inhibitor 4a to give inhibitor 5c (12 mg, 92%). 1H NMR (400 MHz, CDCl3) δ: 8.10 (d, J = 1.6 Hz, 1H), 7.70 (dd, J = 8.5, 1.8 Hz, 1H), 7.57 (d, J = 8.6 Hz, 1H), 6.92 (s, 1H), 6.78 (dd, J = 16.1, 7.2 Hz, 2H), 6.68 – 6.61 (m, 1H), 5.21 (d, J = 5.1 Hz, 1H), 5.13 (d, J = 9.7 Hz, 1H), 4.99 (q, J = 6.3 Hz, 1H), 3.94 (t, J = 8.5 Hz, 2H), 3.89 – 3.79 (m, 3H), 3.64 (dt, J = 9.0, 4.4 Hz, 1H), 3.34 (q, J = 6.6, 6.2 Hz, 1H), 3.06 (dtd, J = 33.4, 14.4, 13.4, 8.1 Hz, 4H), 2.89 – 2.82 (m, 1H), 2.77 (ddq, J = 10.2, 6.8, 3.9 Hz, 2H), 2.68 – 2.60 (m, 1H), 2.52 – 2.39 (m, 2H), 2.07 – 1.97 (m, 1H), 1.85 (dt, J = 13.7, 6.6 Hz, 1H), 1.62 (dt, J = 13.1, 7.3 Hz, 1H), 1.38 (q, J = 5.8 Hz, 2H), 0.93 (d, J = 6.5 Hz, 4H), 0.89 (d, J = 6.6 Hz, 4H), 0.80 – 0.77 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 173.1, 164.3 (d, J = 12.9 Hz), 161.8 (d, J = 12.9 Hz), 156.1, 156.0, 142.3 (t, J = 8.9 Hz), 131.6, 130.4, 125.5, 121.1, 118.9, 112.4 (d, J = 18.7 Hz), 102.1, 100.7, 79.4, 73.1, 72.0, 60.1, 59.1, 54.9, 53.8, 41.9, 39.6, 36.0, 35.5, 29.9, 27.5, 26.9, 21.7, 20.3, 20.1, 8.2; LRMS-ESI (m/z): 721.2 [M+H]+; HRMS-ESI (m/z): [M+H]+ calcd for C34H43F2N4O7S2, 721.2542; found 721.2536.
(2aS,2a1S,4R,4aS,7aS)-Octahydro-2H-1,7-dioxacyclopenta[cd]inden-4-yl ((2S,3R)-4-((2-(cyclopropylamino)-N-isobutylbenzo[d]thiazole)-6-sulfonamido)-1-(3,5-difluorophenyl)-3-hydroxybutan-2-yl)carbamate (5d)
Activated alcohol 18 (10 mg, 0.029 mmol) was treated with isostere amine 30 (19 mg, 0.036 mmol) by following the procedure outlined for inhibitor 4a to give inhibitor 5d (17 mg, 80%). 1H NMR (400 MHz, CDCl3) δ: 8.09 (s, 1H), 7.69 (dd, J = 8.5, 1.8 Hz, 1H), 7.57 (d, J = 8.5 Hz, 1H), 7.11 (s, 1H), 6.79 (d, J = 6.2 Hz, 2H), 6.68 – 6.61 (m, 1H), 5.22 (t, J = 7.4 Hz, 2H), 4.99 (q, J = 6.0 Hz, 1H), 4.08 (s, 1H), 3.96 – 3.78 (m, 5H), 3.60 (dd, J = 8.7, 5.6 Hz, 1H), 3.42 – 3.33 (m, 1H), 3.10 (dd, J = 11.0, 4.9 Hz, 2H), 3.05 (d, J = 3.8 Hz, 1H), 2.98 (dd, J = 13.3, 8.3 Hz, 1H), 2.88 (t, J = 6.6 Hz, 1H), 2.75 (dq, J = 6.7, 3.4 Hz, 1H), 2.69 – 2.61 (m, 1H), 2.51 – 2.42 (m, 2H), 1.98 (dt, J = 13.5, 6.8 Hz, 1H), 1.84 (dd, J = 14.0, 6.9 Hz, 2H), 1.54 (dd, J = 13.2, 6.6 Hz, 2H), 0.92 (d, J = 6.5 Hz, 4H), 0.88 (d, J = 6.6 Hz, 4H), 0.80 – 0.78 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 173.1, 164.3 (d, J = 13.0 Hz), 161.8 (d, J = 13.0 Hz), 156.1 (d, J = 31.9 Hz), 142.4 (t, J = 8.7 Hz), 131.5, 130.5, 125.5, 121.1, 118.8, 112.5 (d, J = 24.5 Hz), 102.3, 100.8, 79.6, 72.9, 71.9, 59.9, 59.1, 55.0, 53.8, 41.6, 39.8, 36.0 (d, J = 7.5 Hz), 35.2, 29.8, 27.5, 26.8, 21.9, 20.3, 20.1, 8.1; LRMS-ESI (m/z): 721.3 [M+H]+; HRMS-ESI (m/z): [M+H]+ calcd for C34H43F2N4O7S2, 721.2542; found 721.2537.
((2R,3S,6S)-3-Acetoxy-6-(prop-2-yn-1-yloxy)-3,6-dihydro-2H-pyran-2-yl)methyl acetate (7)
To a stirred solution of tri-O-acetyl-D-glucal 6 (10 g, 36.73 mmol) in THF (170 mL) were added propargyl alcohol (2.16 mL, 37.1 mmol) and iodine (1.86 g, 7.35 mmol) at 23 °C under argon atmosphere. The reaction mixture was stirred at 23 °C for 1 h. After this period, the reaction mixture was diluted with ether, quenched by the addition of saturated aqueous Na2S2O3 solution and the layers were separated. The aqueous layer was extracted with ether, combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (20% EtOAc in hexane) to give 7 (9.35 g, 95%). 1H NMR (400 MHz, CDCl3) δ 5.92 (d, J = 10.2 Hz, 1H), 5.84 (dt, J = 10.2, 2.3 Hz, 1H), 5.34 (dq, J = 9.6, 1.6 Hz, 1H), 5.26 – 5.23 (m, 1H), 4.31 (d, J = 2.4 Hz, 2H), 4.26 (dd, J = 12.1, 5.2 Hz, 1H), 4.19 (dd, J = 12.1, 2.4 Hz, 1H), 4.09 (ddd, J = 9.5, 5.2, 2.5 Hz, 1H), 2.46 (t, J = 2.4 Hz, 1H), 2.10 (s, 3H), 2.08 (s, 3H).
((2a1S,4aS,5S,6R,7aS)-5-Acetoxy-4-oxo-2a1,4,4a,5,6,7a-hexahydro-2H-1,7-dioxacyclopenta[cd]inden-6-yl)methyl acetate (8)
To a stirred solution of 7 (500 mg, 1.86 mmol) in hexanes (6 mL) was added Co2(CO)8 (700 mg, 2.05 mmol) at 23 °C under argon atmosphere. The reaction mixture was stirred at 23 °C for 5 h. After this period, the mixture was filtered through a plug of Celite, washed with dichloromethane and concentrated under reduced pressure. To the above crude mixture in dichloromethane (30 mL) was added NMO (1.31 g, 11.19 mmol) at 0 °C and the reaction mixture was stirred for 48 h at 23 °C. After this period, solvent was removed under reduced pressure and the crude product was purified by silica gel column chromatography (70% EtOAc in hexane) to afford 8 (176 mg, 32%). 1H NMR (400 MHz, CDCl3) δ 6.17 (s, 1H), 5.53 (d, J = 5.7 Hz, 1H), 4.94 – 4.85 (m, 1H), 4.80 (d, J = 15.2 Hz, 1H), 4.59 (d, J = 15.3 Hz, 1H), 4.09 – 4.05 (m, 2H), 3.74 (dt, J = 10.2, 3.0 Hz, 1H), 3.46 – 3.38 (m, 2H), 1.98 (d, J = 7.2 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 205.2, 180.8, 170.5, 169.8, 126.2, 96.3, 66.7, 66.0, 62.8, 47.6, 46.0, 20.7; LRMS-ESI (m/z): 297.0 [M+H]+.
(2aR,2a1R,4aR,6S,7aS)-6-(Hydroxymethyl)hexahydro-2H-1,7-dioxacyclopenta[cd]inden-4(4aH)-one (9)
To a stirred solution of 8 (176 mg, 0.59 mmol) in MeOH (5 mL) were added HCOONH4 (375 mg, 5.9 mmol) and 10% Pd/C (35 mg) at 23 °C under argon atmosphere. The reaction mixture was refluxed for 15 min. After this period, the reaction mixture was cooled to 23 °C and filtered through a plug of Celite. Methanol was removed under reduced pressure. To the crude residue was added chloroform to precipitate out of the excess HCOONH4, filtered and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (50% EtOAc in hexane) to afford saturated ketone (100 mg, 70%). [α]D20 +5.9 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.24 (d, J = 5.4 Hz, 1H), 4.10 – 3.99 (m, 2H), 3.87 (dd, J = 9.5, 6.1 Hz, 1H), 3.79 (ddd, J = 11.9, 6.3, 3.2 Hz, 1H), 3.69 (dd, J = 9.5, 1.7 Hz, 1H), 2.98 – 2.84 (m, 2H), 2.74 (ddd, J = 18.7, 10.5, 2.0 Hz, 1H), 2.61 – 2.54 (m, 1H), 2.17 (dd, J = 18.7, 3.0 Hz, 1H), 2.03 (s, 3H), 1.54 (ddd, J = 13.6, 12.0, 6.2 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 216.1, 170.9, 100.4, 72.2, 66.2, 65.6, 44.2, 42.3, 38.5, 35.4, 22.8, 20.9; LRMS-ESI (m/z): 263.0 [M+Na]+.
To a stirred solution of above ketone (87 mg, 0.36 mmol) in MeOH (3 mL) were added H2O (2 mL) and Et3N (1 mL) at 23 °C under argon atmosphere. The reaction mixture was stirred at 23 °C for 3 h. After this period, methanol was removed under reduced pressure. The reaction mixture was diluted with dichloromethane, dried over Na2SO4 and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (70% EtOAc in hexane) to give 9 (86 mg, 86%). [α]D20 +6.4 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.26 (d, J = 5.5 Hz, 1H), 3.86 (dd, J = 9.5, 6.1 Hz, 1H), 3.69 (dd, J = 9.5, 1.7 Hz, 1H), 3.66 – 3.58 (m, 2H), 3.51 (dd, J = 11.8, 6.3 Hz, 1H), 2.98 – 2.85 (m, 2H), 2.79 – 2.68 (m, 1H), 2.62 – 2.54 (m, 1H), 2.39 (brs, 1H), 2.18 (dd, J = 18.7, 3.0 Hz, 1H), 2.02 – 1.94 (m, 1H), 1.58 (ddd, J = 13.7, 11.8, 6.3 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 216.7, 100.5, 72.0, 68.2, 65.2, 44.2, 42.4, 38.4, 35.6, 22.4; LRMS-ESI (m/z): 221.1 [M+Na]+.
(2aR,2a1R,4S,4aR,6S,7aS)-6-(Methoxymethyl)octahydro-2H-1,7-dioxacyclopenta[cd]inden-4-ol (10)
To a stirred solution of 9 (30 mg, 0.15 mmol) in dichloromethane (3 mL) were added proton-sponge (130 mg, 0.6 mmol) and trimethyloxonium tetrafluoroborate (90 mg, 0.6 mmol) at 0 °C under argon atmosphere. The reaction mixture was warmed to 23 °C and stirred for 48 h. After this period, the reaction mixture was quenched by the addition of H2O and the layers were separated. The aqueous layer was extracted with dichloromethane and combined organic extracts were washed with HCl (4N) and NaHCO3. The reaction mixture was dried over Na2SO4 and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (35% EtOAc in hexane) to give ketone (20 mg, 63%). [α]D20 +2.8 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.31 (d, J = 5.3 Hz, 1H), 3.89 (dd, J = 9.5, 6.1 Hz, 1H), 3.77 – 3.68 (m, 2H), 3.45 – 3.40 (m, 2H), 3.36 (s, 3H), 2.98 – 2.86 (m, 2H), 2.75 (ddd, J = 18.7, 10.6, 2.0 Hz, 1H), 2.59 (ddt, J = 8.4, 4.0, 1.9 Hz, 1H), 2.20 (dd, J = 18.7, 2.9 Hz, 1H), 2.03 (dt, J = 13.7, 2.4 Hz, 1H), 1.74 – 1.66 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 216.8, 100.9, 75.0, 72.2, 66.7, 59.4, 44.4, 42.7, 38.5, 35.6, 22.8; LRMS-ESI (m/z): 235.0 [M+Na]+.
To a stirred solution of above ketone (17 mg, 0.08 mmol) in MeOH (3 mL) was added NaBH4 (3.65 mg, 0.096 mmol) at 0 °C under argon atmosphere. The reaction mixture was stirred at 0 °C for 1 h. After this period, the reaction mixture was quenched by the addition of saturated aqueous NH4Cl and the layers were separated. The aqueous layer was extracted with EtOAc, combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (70% EtOAc in hexane) to afford 10 (15 mg, 88%). [α]D20 +42.5 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.18 (d, J = 5.3 Hz, 1H), 4.34 (dq, J = 11.9, 3.6 Hz, 1H), 4.11 (brs, 1H), 3.77 – 3.73 (m, 2H), 3.49 – 3.42 (m, 2H), 3.40 (d, J = 5.2 Hz, 1H), 3.38 (s, 3H), 2.80 – 2.71 (m, 1H), 2.47 (td, J = 9.2, 8.6, 5.4 Hz, 1H), 2.20 – 2.12 (m, 1H), 1.99 (ddd, J = 13.5, 8.5, 4.7 Hz, 1H), 1.89 – 1.81 (m, 2H), 1.80 – 1.76 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 101.5, 77.4, 75.9, 70.7, 66.5, 59.4, 43.0, 39.1, 38.5, 23.6; LRMS-ESI (m/z): 237.1 [M+Na]+.
(2aR,2a1R,4S,4aR,6R,7aS)-6-Methyloctahydro-2H-1,7-dioxacyclopenta[cd]inden-4-ol (11)
To a stirred solution of alcohol 9 (50 mg, 0.25 mmol) in dichloromethane (3 mL) were added Et3N (70.3 μL, 0.5 mmol) and methanesulfonyl chloride (39 μL, 0.5 mmol) at -20 °C under argon atmosphere. The reaction mixture was stirred at -20 °C for 1.5 h. After this period the reaction mixture was quenched by the addition of H2O and the layers were separated. The aqueous layer was extracted with dichloromethane, combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (70% EtOAc in hexane) to afford ketone (49 mg, 70%). 1H NMR (400 MHz, CDCl3) δ 5.24 (d, J = 5.4 Hz, 1H), 4.20 (d, J = 4.6 Hz, 2H), 3.89 – 3.79 (m, 2H), 3.69 (dd, J = 9.6, 1.7 Hz, 1H), 3.03 (s, 3H), 2.98 – 2.86 (m, 2H), 2.76 (ddd, J = 18.7, 10.5, 2.0 Hz, 1H), 2.65 – 2.58 (m, 1H), 2.17 (dd, J = 18.6, 2.7 Hz, 1H), 2.05 (dt, J = 13.6, 2.5 Hz, 1H), 1.61 (ddd, J = 13.6, 12.1, 6.2 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 215.7, 100.5, 72.2, 71.5, 65.4, 44.1, 42.0, 38.0, 37.8, 35.3, 22.1.
To a stirred solution of above ketone (49 mg, 0.18 mmol) in THF (3 mL) was added LAH (20 mg, 0.53 mmol) 0 °C under argon atmosphere. The reaction mixture was warmed to 23 °C and stirred for 36 h. After this period, the reaction mixture was quenched by the addition of NaOH (2N), filtered and washed with EtOAc. The reaction mixture was dried over Na2SO4 and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (50% EtOAc in hexane) to afford 11 (16.5 mg, 50%). 1H NMR (400 MHz, CDCl3) δ 5.13 (d, J = 5.3 Hz, 1H), 4.32 – 4.23 (m, 1H), 4.09 (dt, J = 11.6, 4.8 Hz, 1H), 3.75 (d, J = 7.1 Hz, 2H), 3.47 (d, J = 12.1 Hz, 1H), 2.80 – 2.72 (m, 1H), 2.43 (td, J = 9.2, 8.5, 5.3 Hz, 1H), 2.16 – 2.09 (m, 1H), 2.04 – 1.97 (m, 1H), 1.96 – 1.90 (m, 1H), 1.80 (d, J = 14.1 Hz, 1H), 1.54 (ddd, J = 14.1, 11.8, 7.0 Hz, 1H), 1.18 (d, J = 6.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 101.7, 77.3, 70.7, 63.7, 43.1, 41.1, 39.2, 38.9, 29.9, 22.4.
trans-3-Bromo-2-(prop-2-yn-1-yloxy)tetrahydro-2H-pyran (13)
To a stirred solution of 3,4-dihydro-2H-pyran 12 (5.39 mL, 59.44 mmol) and propargyl alcohol (10.4 mL, 178.32 mmol) in dichloromethane (80 mL) was added NBS (11.63 g, 65.37 mmol) in small portions over 0.5 h at -20 °C under argon atmosphere. The reaction mixture was stirred at -20 °C for 2 h and further 15 h at 23 °C. After this period, the reaction mixture was quenched by the addition of water and extracted with dichloromethane. The extracts were washed with saturated aqueous NaHSO3 solution, aqueous K2CO3 solution, water, dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (10% Et2O in hexane) to afford 13 (12.7 g, 98%). 1H NMR (400 MHz, CDCl3) δ 4.85 (d, J = 3.9 Hz, 1H), 4.30 (dd, J = 4.8, 2.4 Hz, 2H), 4.01 (dt, J = 5.7, 4.0 Hz, 1H), 3.89 (ddd, J = 11.7, 8.6, 3.5 Hz, 1H), 3.65 – 3.58 (m, 1H), 2.46 (t, J = 2.4 Hz, 1H), 2.44 – 2.34 (m, 1H), 1.96 (dddd, J = 18.6, 10.3, 7.6, 4.4 Hz, 2H), 1.52 (dtd, J = 15.1, 6.0, 3.1 Hz, 1H).
6-(Prop-2-yn-1-yloxy)-3,6-dihydro-2H-pyran (14)
A mixture of 13 (10.0 g, 45.65 mmol) and DBU (34.0 mL, 228.25 mmol) was stirred at 110 °C for 5 h under argon atmosphere. After this period, the reaction mixture was cooled, 90 mL of anhydrous ether was added and stirred for 1 h. The mixture was filtered through a plug of Celite, washed with ether and concentrated under reduced pressure by using cold bath. The crude product was purified by silica gel column chromatography (15% Et2O in pentane) to afford 14 (5.36 g, 85%) as a volatile liquid. 1H NMR (400 MHz, CDCl3) δ 6.09 – 6.03 (m, 1H), 5.73 (dtd, J = 10.1, 2.8, 1.3 Hz, 1H), 5.09 (s, 1H), 4.27 (d, J = 2.4 Hz, 2H), 3.88 (td, J = 11.4, 3.6 Hz, 1H), 3.72 (ddt, J = 11.1, 6.1, 1.1 Hz, 1H), 2.41 (t, J = 2.4 Hz, 1H), 2.37-2.25 (m, 1H), 1.94 – 1.85 (m, 1H).
2a1,5,6,7a-Tetrahydro-2H-1,7-dioxacyclopenta[cd]inden-4(4aH)-one (15)
To a stirred solution of 14 (1.57 g, 11.4 mmol) in dichloromethane (80 mL) was added Co2(CO)8 (4.3 g, 12.50 mmol) at 23 °C under argon atmosphere. The reaction mixture was stirred at 23 °C for 1 h. After this period, the above mixture was diluted by the addition of dichloromethane (500 mL), NMO (8.0 g, 68.40 mmol) was added at 0 °C, and the reaction mixture was stirred for 3 h at 23 °C. The mixture was filtered through a plug of Celite, washed with dichloromethane and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (40% EtOAc in hexane) to afford 15 (380 mg, 20%). 1H NMR (400 MHz, CDCl3) δ 6.04 (s, 1H), 5.32 (d, J = 5.2 Hz, 1H), 4.70 (qt, J = 15.9, 1.7 Hz, 2H), 3.61 (ddd, J = 12.1, 5.3, 4.0 Hz, 1H), 3.39 (ddd, J = 12.0, 9.2, 2.8 Hz, 1H), 3.18 (dtt, J = 6.9, 4.8, 2.0 Hz, 1H), 2.92 (dt, J = 9.3, 6.5 Hz, 1H), 1.94 – 1.84 (m, 1H), 1.46 (dddd, J = 14.3, 9.2, 6.6, 3.9 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 211.8, 180.2, 123.9, 97.7, 66.2, 61.3, 47.3, 44.1, 24.4.
Octahydro-2H-1,7-dioxacyclopenta[cd]inden-4-ol ((±)-16)
To a stirred solution of 15 (165 mg, 0.99 mmol) in MeOH (10 mL) were added HCO2NH4 (626 mg, 9.93 mmol) and 10% Pd/C (25 mg) at 23 °C under argon atmosphere. The reaction mixture was refluxed for 15 min. After this period, the reaction mixture was cooled to 23 °C and filtered through a plug of Celite. MeOH was removed under reduced pressure. To the crude residue was added chloroform to precipitate out of the excess HCOONH4, filtered and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (45% EtOAc in hexane) to afford ketone (128 mg, 77%). 1H NMR (400 MHz, CDCl3) δ 5.03 (d, J = 4.8 Hz, 1H), 3.94 (dd, J = 9.2, 6.2 Hz, 1H), 3.76 – 3.71 (m, 1H), 3.62 – 3.52 (m, 2H), 2.96 – 2.82 (m, 2H), 2.67 (dd, J = 19.0, 9.8 Hz, 1H), 2.53 – 2.45 (m, 1H), 2.18 (dd, J = 18.9, 3.8 Hz, 1H), 2.10 – 2.02 (m, 1H), 1.72 (ddt, J = 13.5, 10.7, 6.5 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 217.1, 100.8, 73.1, 58.7, 43.6, 42.6, 41.2, 35.4, 21.1.
To a stirred solution of above ketone (88 mg, 0.52 mmol) in MeOH (12 mL) was added NaBH4 (24.0 mg, 0.63 mmol) at 0 °C under argon atmosphere. The reaction mixture was stirred at 0 °C for 1 h. After this period, the reaction mixture was quenched by the addition of saturated aqueous NH4Cl and the layers were separated. The aqueous layer was extracted with EtOAc, combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (50% EtOAc in hexane) to give (±)-16 (87 mg, 98%). 1H NMR (400 MHz, CDCl3) δ 5.06 (d, J = 5.4 Hz, 1H), 4.14 – 4.06 (m, 1H), 4.00 (dt, J = 11.8, 6.8 Hz, 1H), 3.89 – 3.81 (m, 1H), 3.71 (dd, J = 9.3, 4.4 Hz, 1H), 3.51 (dt, J = 11.5, 5.6 Hz, 1H), 3.37 (d, J = 9.1 Hz, 1H), 2.70 (ddq, J = 12.6, 8.1, 4.1 Hz, 1H), 2.50 (td, J = 9.6, 5.4 Hz, 1H), 2.19 (dq, J = 11.2, 5.7 Hz, 1H), 1.93 (ddd, J = 13.6, 8.4, 5.2 Hz, 1H), 1.78 (dt, J = 7.8, 4.3 Hz, 2H), 1.68 (dt, J = 13.7, 3.4 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 101.0, 77.4, 71.8, 59.9, 41.8, 40.8, 40.4, 38.0, 21.2.
(2aS,2a1S,4R,4aS,7aS)-Octahydro-2H-1,7-dioxacyclopenta[cd]inden-4-ol ((−)-16)
To a stirred solution of acetate 17 (32 mg, 0.15 mmol) in MeOH (3 mL) was added K2CO3 (31 mg, 0.23 mmol) at 23 °C under argon atmosphere. The reaction mixture was stirred at 23 °C for 1 h. After this period, the reaction mixture was quenched by the addition of saturated aqueous NH4Cl and the layers were separated. The aqueous layer was extracted with EtOAc, combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (50% EtOAc in hexane) to afford (−)-16 (26 mg, 99%). [α]D20 -43.2 (c 1.0, CHCl3).
(2aR,2a1R,4S,4aR,7aR)-Octahydro-2H-1,7-dioxacyclopenta[cd]inden-4-ol ((+)-16) and (2aS,2a1S,4R,4aS,7aS)-octahydro-2H-1,7-dioxacyclopenta[cd]inden-4-yl acetate (17)
To a solution of (±)-16 (60 mg, 0.35 mmol) in THF (4 mL) were added vinyl acetate (0.6 mL, 6.2 mmol) and Lipase PS-30 on Celite (70 mg) at 23 °C under argon atmosphere. The reaction mixture was stirred for 6 h (50:50 by 1H-NMR). After this period, the reaction mixture was filtered through a plug of Celite and solvents were removed under reduced pressure. The crude product was purified via silica gel column chromatography (30% to 50% EtOAc in hexane) to afford alcohol (+)-16 (32 mg, 53%) and acetate 17 (35 mg, 47%).
Alcohol ((+)-16)
[α]D20 +36.6 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.08 (d, J = 5.2 Hz, 1H), 4.12 (s, 1H), 4.04 (dt, J = 12.3, 7.0 Hz, 1H), 3.88 (t, J = 8.8 Hz, 1H), 3.75 (dd, J = 9.3, 4.1 Hz, 1H), 3.55 (dd, J = 11.5, 5.7 Hz, 1H), 3.31 (d, J = 8.5 Hz, 1H), 2.80 – 2.68 (m, 1H), 2.53 (td, J = 9.5, 5.5 Hz, 1H), 2.21 (dq, J = 10.1, 5.4 Hz, 1H), 1.96 (ddt, J = 13.5, 8.4, 4.8 Hz, 1H), 1.84 (t, J = 5.8 Hz, 2H), 1.73 (d, J = 13.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 101.1, 77.5, 71.9, 59.9, 41.8, 41.0, 40.8, 38.2, 21.2.
Acetate (17)
1H NMR (400 MHz, CDCl3) δ 5.22 (d, J = 5.1 Hz, 1H), 5.10 (dt, J = 8.7, 6.2 Hz, 1H), 4.01 – 3.89 (m, 2H), 3.70 (dd, J = 8.6, 6.3 Hz, 1H), 3.41 (dt, J = 11.5, 6.5 Hz, 1H), 2.65 (qt, J = 7.8, 5.1 Hz, 1H), 2.54 (ddd, J = 14.6, 11.8, 6.8 Hz, 2H), 2.15 – 2.06 (m, 1H), 2.03 (s, 3H), 1.66 (dt, J = 12.8, 8.4 Hz, 1H), 1.58 – 1.51 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 170.7, 100.6, 78.7, 72.1, 60.6, 42.2, 39.4, 35.7, 34.6, 21.9, 21.1.
4-Nitrophenyl ((2aS,2a1S,4R,4aS,7aS)-octahydro-2H-1,7-dioxacyclopenta[cd]inden-4-yl) carbonate (18)
To a stirred solution of alcohol (−)-16 (30 mg, 0.17 mmol) in dichloromethane (3 mL) was added pyridine (57 μL, 0.70 mmol) at 23 °C under argon atmosphere and the reaction mixture was cooled to 0 °C followed by addition of 4-nitrophenyl chloroformate (53 mg, 0.26 mmol). The reaction mixture was warmed to 23 °C and stirred for 12 h. Upon completion, solvents were removed under reduced pressure and crude product was purified by silica gel column chromatography (35% EtOAc in hexane) to give 18 (52 mg, 88%). [α]D20 +10.4 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.28 (d, J = 9.2 Hz, 2H), 7.38 (d, J = 9.2 Hz, 2H), 5.27 (d, J = 5.4 Hz, 1H), 5.16 (dt, J = 8.8, 6.4 Hz, 1H), 4.07 – 3.97 (m, 2H), 3.77 (dd, J = 8.9, 6.1 Hz, 1H), 3.47 (dt, J = 11.4, 6.4 Hz, 1H), 2.79 – 2.66 (m, 2H), 2.59 (ddd, J = 10.7, 9.2, 5.5 Hz, 1H), 2.29 (ddd, J = 13.7, 8.0, 6.3 Hz, 1H), 1.85 (dt, J = 13.0, 8.3 Hz, 1H), 1.73 – 1.66 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 155.4, 152.1, 145.4, 125.4, 121.9, 100.6, 83.6, 72.0, 60.5, 41.9, 39.2, 35.9, 34.7, 21.8; LRMS-ESI (m/z): 358.0 [M+Na]+.
2,2a,2a1,5,6,7a-Hexahydro-3H-1,7-dioxacyclopenta[cd]inden-3-one (20)
2,3-Dihydrofuran 19 (1.29 mL, 17.12 mmol) was treated with 3-butyn-1-ol (3.89 mL, 51.36 mmol) and NBS (3.35 g, 18.83 mmol) by following the procedure outlined for compound 13 to afford bromo ether compound (3.5 g, 93%). 1H NMR (400 MHz, CDCl3) δ 5.18 (s, 1H), 4.17 (d, J = 5.9 Hz, 1H), 4.08 (q, J = 8.0 Hz, 1H), 4.02 – 3.95 (m, 1H), 3.72 – 3.64 (m, 1H), 3.53 – 3.45 (m, 1H), 2.62 – 2.51 (m, 1H), 2.39 – 2.32 (m, 2H), 2.18 – 2.09 (m, 1H), 1.94 (td, J = 2.6, 1.2 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 108.4, 81.0, 69.5, 66.7, 65.2, 49.9, 33.7, 19.9.
Above bromo ether compound (3.25 g, 14.83 mmol) was treated with DBU (6.65 mL, 44.5 mmol) by following the procedure outlined for compound 14 to afford an alkene compound (1.73 g, 84%) as a volatile liquid. 1H NMR (400 MHz, CDCl3) δ 6.28 – 6.25 (m, 1H), 5.90 (dt, J = 4.5, 1.1 Hz, 1H), 5.83 – 5.79 (m, 1H), 4.75 – 4.67 (m, 1H), 4.56 (ddq, J = 14.0, 2.7, 1.3 Hz, 1H), 3.77 (dtd, J = 9.5, 7.0, 1.2 Hz, 1H), 3.62 (dtd, J = 9.5, 7.2, 1.2 Hz, 1H), 2.49 (tdd, J = 7.0, 2.6, 1.1 Hz, 2H), 1.97 (td, J = 2.7, 1.2 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 132.4, 125.9, 108.9, 81.4, 74.7, 69.4, 65.0, 20.3.
Above alkene compound (184 mg, 1.33 mmol) was treated with Co2(CO)8 (500 mg, 1.46 mmol) and NMO (940 mg, 7.98 mmol) by following the procedure outlined for compound 15 to afford 20 (45 mg, 20%). 1H NMR (400 MHz, CDCl3) δ 6.03 (s, 1H), 5.36 – 5.21 (m, 1H), 4.08 – 4.00 (m, 1H), 3.96 – 3.87 (m, 2H), 3.82 (t, J = 9.3 Hz, 1H), 3.20 (s, 1H), 2.94 – 2.80 (m, 1H), 2.77 – 2.61 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 208.1, 177.4, 129.4, 101.3, 65.8, 63.7, 49.1, 48.0, 31.6.
Octahydro-2H-1,7-dioxacyclopenta[cd]inden-3-ol ((±)-21)
Compound 20 (59 mg, 0.35 mmol) was treated with HCO2NH4 (224 mg, 3.55 mmol) and 10% Pd/C (20 mg) by following the procedure outlined for compound (±)-16 to afford ketone (42 mg, 70%). 1H NMR (400 MHz, CDCl3) δ 5.19 (d, J = 5.4 Hz, 1H), 4.05 – 3.98 (m, 2H), 3.80 – 3.69 (m, 2H), 2.86 – 2.74 (m, 2H), 2.68 – 2.58 (m, 1H), 2.39 – 2.34 (m, 2H), 1.96 (ddt, J = 14.3, 11.7, 5.1 Hz, 1H), 1.60 (dq, J = 14.3, 2.9 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 218.6, 101.7, 67.3, 56.0, 52.5, 42.3, 38.5, 28.3, 24.6; LRMS-ESI (m/z): 191.1 [M+Na]+.
Above ketone (20 mg, 0.12 mmol) was treated with NaBH4 (5.4 mg, 0.14 mmol) by following the procedure outlined for compound (±)-16 to give (±)-21 (18 mg, 90%). 1H NMR (400 MHz, CDCl3) δ 5.15 (d, J = 5.5 Hz, 1H), 4.34 (dd, J = 9.8, 2.4 Hz, 1H), 4.32 – 4.24 (m, 1H), 4.04 – 3.96 (m, 1H), 3.59 – 3.52 (m, 2H), 2.70 (qd, J = 7.9, 2.5 Hz, 1H), 2.39 (td, J = 8.6, 5.6 Hz, 1H), 2.21 – 2.10 (m, 1H), 2.03 (brs, 1H), 1.92 – 1.82 (m, 1H), 1.81 – 1.75 (m, 2H), 1.52 – 1.43 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 101.6, 73.8, 62.9, 55.6, 46.2, 39.2, 37.2, 29.4, 24.4; LRMS-ESI (m/z): 171.1 [M+H]+.
(2aS,2a1R,3S,4aR,7aS)-Octahydro-2H-1,7-dioxacyclopenta[cd]inden-3-ol ((−)-21) and (2aR,2a1S,3R,4aS,7aR)-octahydro-2H-1,7-dioxacyclopenta[cd]inden-3-yl acetate (22)
Compound (±)-21 (17 mg, 0.099 mmol) was treated with vinyl acetate (0.17 mL, 1.75 mmol) and Lipase PS-30 on Celite (20 mg) by following the procedure outlined for compound (+)-16 to afford alcohol (−)-21 (8 mg, 47 %) and acetate 22 (11 mg, 53%).
Alcohol ((−)-21)
[α]D20 -49.3 (c 0.8, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.15 (d, J = 5.5 Hz, 1H), 4.35 (dd, J = 9.8, 2.4 Hz, 1H), 4.32 – 4.25 (m, 1H), 4.05 – 3.97 (m, 1H), 3.59 – 3.52 (m, 2H), 2.70 (qd, J = 7.9, 2.5 Hz, 1H), 2.39 (td, J = 8.6, 5.6 Hz, 1H), 2.16 (dtd, J = 16.6, 10.5, 9.5, 5.2 Hz, 1H), 1.93 – 1.75 (m, 3H), 1.52 – 1.44 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 101.6, 73.8, 62.9, 55.6, 46.2, 39.3, 37.2, 29.5, 24.4; LRMS-ESI (m/z): 171.1 [M+H]+.
Acetate (22)
[α]D20 +59.3 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.15 (d, J = 5.5 Hz, 1H), 5.01 (dt, J = 10.5, 7.5 Hz, 1H), 4.13 (dd, J = 9.9, 2.3 Hz, 1H), 4.04 – 3.95 (m, 1H), 3.62 – 3.51 (m, 2H), 2.91 (qd, J = 7.9, 2.3 Hz, 1H), 2.40 (td, J = 8.6, 5.6 Hz, 1H), 2.25 – 2.12 (m, 1H), 2.07 (s, 3H), 1.93 – 1.84 (m, 3H), 1.54 – 1.46 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 170.9, 101.5, 75.5, 63.4, 55.4, 44.0, 38.5, 33.6, 29.0, 24.1, 21.2; LRMS-ESI (m/z): 213.1 [M+H]+.
(2aR,2a1S,3R,4aS,7aR)-Octahydro-2H-1,7-dioxacyclopenta[cd]inden-3-ol ((+)-21)
Compound 22 (11 mg, 0.15 mmol) was treated with K2CO3 (11 mg, 0.08 mmol) by following the procedure outlined for compound (−)-16 to afford (+)-21 (8 mg, 90%). [α]D20 +49.3 (c 0.8, CHCl3).
2a1,4a,5,6,7,7a-Hexahydroindeno[7,1-bc]furan-4(2H)-one (24)
Cyclohexene 23 (6.17 mL, 60.86 mmol) was treated with propargyl alcohol (10.63 mL, 182.59 mmol) and NBS (11.92 g, 66.95 mmol) by following the procedure outlined for compound 13 to afford bromo alkyne compound (8.32 g, 63%). 1H NMR (400 MHz, CDCl3) δ 4.29 (d, J = 2.5 Hz, 2H), 4.01 – 3.95 (m, 1H), 3.58 – 3.52 (m, 1H), 2.42 (t, J = 2.6, 1H), 2.36 – 2.15 (m, 2H), 1.88 – 1.62 (m, 3H), 1.40 – 1.25 (m, 3H).
Above bromo alkyne compound (8.3 g, 38.2 mmol) was treated with DBU (17.15 mL, 114.6 mmol) by following the procedure outlined for compound 14 to give compound an alkene compound (4.8 g, 92%) as a volatile liquid. 1H NMR (400 MHz, CDCl3) δ 5.90 – 5.83 (m, 1H), 5.76 (dq, J = 10.1, 2.1 Hz, 1H), 4.18 (t, J = 2.4 Hz, 2H), 4.10 – 4.03 (m, 1H), 2.41 – 2.37 (m, 1H), 2.09 – 1.88 (m, 2H), 1.84 – 1.64 (m, 3H), 1.59 – 1.50 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 131.6, 127.1, 80.5, 73.9, 71.9, 55.3, 28.1, 25.3, 19.1.
Above alkene compound (150 mg, 1.1 mmol) was treated with Co2(CO)8 (414 mg, 1.21 mmol) and NMO (770 mg, 6.6 mmol) by following the procedure outlined for compound 15 to give compound 24 (105 mg, 58%). 1H NMR (400 MHz, CDCl3) δ 5.91 (s, 1H), 4.63 – 4.52 (m, 2H), 4.36 – 4.27 (m, 1H), 3.29 (t, J = 6.6 Hz, 1H), 2.80 (q, J = 8.7 Hz, 1H), 1.99 – 1.87 (m, 1H), 1.81 (dt, J = 12.7, 6.2 Hz, 1H), 1.60 – 1.49 (m, 1H), 1.16 – 0.97 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 213.3, 182.1, 121.9, 75.3, 64.7, 47.6, 46.9, 28.8, 25.5, 21.1.
Decahydroindeno[7,1-bc]furan-4-ol ((±)-25)
Compound 24 (105 mg, 0.64 mmol) was treated with HCO2NH4 (403 mg, 6.4 mmol) and 10% Pd/C (20 mg) by following the procedure outlined for compound (±)-16 to give ketone (80 mg, 75%). 1H NMR (400 MHz, CDCl3) δ 3.72 – 3.62 (m, 3H), 2.92 – 2.83 (m, 2H), 2.73 – 2.61 (m, 1H), 2.31 (dt, J = 8.1, 4.6 Hz, 1H), 2.20 – 2.08 (m, 1H), 2.06 – 1.96 (m, 2H), 1.52 (ddt, J = 14.6, 12.8, 4.1 Hz, 1H), 1.40 – 1.17 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 217.2, 77.0, 75.9, 44.5, 44.4, 40.5, 37.1, 27.8, 22.6, 16.2; LRMS-ESI (m/z): 167.1 [M+H]+.
Above ketone (79 mg, 0.48 mmol) was treated with NaBH4 (22 mg, 0.57 mmol) by following the procedure outlined for compound (±)-16 to give compound (±)-25 (66 mg, 84%). 1H NMR (400 MHz, CDCl3) δ 3.93 (dt, J = 12.1, 4.5 Hz, 1H), 3.80 – 3.74 (m, 1H), 3.65 (td, J = 8.2, 7.4, 3.3 Hz, 3H), 2.75 (q, J = 8.0 Hz, 1H), 2.46 (td, J = 9.0, 8.5, 5.5 Hz, 1H), 2.09 – 1.95 (m, 2H), 1.92 – 1.73 (m, 3H), 1.68 (d, J = 13.9 Hz, 1H), 1.60 – 1.48 (m, 2H), 1.35 (dtt, J = 12.5, 4.8, 2.7 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 77.8, 77.6, 75.9, 43.2, 43.0, 42.5, 40.2, 26.3, 23.8, 16.9; LRMS-ESI (m/z): 169.1 [M+H]+.
(2aR,2a1R,4S,4aR,7aS)-Decahydroindeno[7,1-bc]furan-4-ol ((+)-25) and (2aS,2a1S,4R,4aS,7aR)-decahydroindeno[7,1-bc]furan-4-yl acetate (26)
Compound (±)-25 (62 mg, 0.37 mmol) was treated with vinyl acetate (0.62 mL, 6.49 mmol) and Lipase PS-30 on Celite (70 mg) by following the procedure outlined for compound (+)-16 to give alcohol (+)-25 (30 mg, 48%) and acetate 26 (40 mg, 52%).
Alcohol ((+)-25)
[α]D20 +13.8 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 3.96 (dt, J = 12.4, 4.5 Hz, 1H), 3.80 (dd, J = 9.4, 1.2 Hz, 1H), 3.70 – 3.64 (m, 3H), 2.77 (q, J = 8.1 Hz, 1H), 2.48 (td, J = 9.0, 8.4, 5.4 Hz, 1H), 2.12 – 1.99 (m, 2H), 1.95 – 1.76 (m, 3H), 1.70 (d, J = 13.9 Hz, 1H), 1.62 – 1.51 (m, 2H), 1.42 – 1.35 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 77.9, 77.7, 76.0, 43.2, 43.1, 42.5, 40.3, 26.4, 23.9, 17.0; LRMS-ESI (m/z): 169.3 [M+H]+.
Acetate (26)
1H NMR (400 MHz, CDCl3) δ 5.09 (q, J = 5.6 Hz, 1H), 3.82 (dt, J = 6.4, 3.4 Hz, 1H), 3.75 – 3.67 (m, 2H), 2.79 – 2.69 (m, 1H), 2.55 – 2.45 (m, 1H), 2.21 – 2.13 (m, 1H), 2.10 – 1.93 (m, 5H), 1.82 – 1.73 (m, 1H), 1.73 – 1.52 (m, 3H), 1.50 – 1.40 (m, 1H), 1.33 – 1.22 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 171.3, 79.2, 76.9, 74.6, 43.4, 42.8, 38.5, 37.9, 25.8, 21.8, 21.5, 16.8.
(2aS,2a1S,4R,4aS,7aR)-Decahydroindeno[7,1-bc]furan-4-ol ((−)-25)
Compound 26 (26 mg, 0.12 mmol) was treated with K2CO3 (25 mg, 0.19 mmol) by following the procedure outlined for compound (−)-16 to give compound (−)-25 (20 mg, 95%). [α]D20 -19.7 (c 1.0, CHCl3).
(2aR,2a1R,4S,4aR,6S,7aS)-6-(Methoxymethyl)octahydro-2H-1,7-dioxacyclopenta[cd]inden-4-yl (4-nitrophenyl) carbonate (27a)
Compound 10 (15 mg, 0.07 mmol) was treated with pyridine (23 μL, 0.28 mmol) and 4-nitrophenyl chloroformate (21 mg, 0.1 mmol) by following the procedure outlined for compound 18 to give 27a (21 mg, 79%). [α]D20 +23.1 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.27 (d, J = 9.2 Hz, 2H), 7.37 (d, J = 9.2 Hz, 2H), 5.38 (d, J = 5.4 Hz, 1H), 5.26 (td, J = 5.5, 3.6 Hz, 1H), 4.25 (dq, J = 8.7, 4.6 Hz, 1H), 3.89 (t, J = 8.7 Hz, 1H), 3.85 – 3.80 (m, 1H), 3.45 – 3.41 (m, 2H), 3.38 (s, 3H), 2.85 (dtd, J = 11.6, 7.8, 3.0 Hz, 1H), 2.63 – 2.54 (m, 2H), 2.26 – 2.15 (m, 1H), 2.04 (dt, J = 14.6, 3.3 Hz, 1H), 1.85 – 1.73 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 155.6, 152.5, 145.5, 125.4, 122.0, 101.5, 84.7, 75.7, 71.0, 65.7, 59.4, 41.1, 40.0, 38.4, 36.4, 23.3; LRMS-ESI (m/z): 402.1 [M+Na]+.
(2aR,2a1R,4S,4aR,6R,7aS)-6-Methyloctahydro-2H-1,7-dioxacyclopenta[cd]inden-4-yl (4-nitrophenyl) carbonate (27b)
Compound 11 (16 mg, 0.087 mmol) was treated with pyridine (28 μL, 0.34 mmol) and 4-nitrophenyl chloroformate (26 mg, 0.13 mmol) by following the procedure outlined for compound 18 to give compound 27b (22 mg, 73%). 1H NMR (400 MHz, CDCl3) δ 8.28 (dt, J = 10.2, 3.1 Hz, 2H), 7.36 (dt, J = 10.1, 3.2 Hz, 2H), 5.35 (d, J = 5.4 Hz, 1H), 5.24 (q, J = 5.5 Hz, 1H), 4.21 (dtt, J = 12.5, 6.1, 2.9 Hz, 1H), 3.88 (q, J = 7.7, 6.4 Hz, 1H), 3.81 (dd, J = 9.2, 3.5 Hz, 1H), 2.85 (qt, J = 8.0, 3.4 Hz, 1H), 2.60 – 2.50 (m, 2H), 2.21 (ddd, J = 13.7, 8.1, 5.5 Hz, 1H), 2.07 – 1.99 (m, 1H), 1.81 (dd, J = 14.2, 2.5 Hz, 1H), 1.55 (ddd, J = 14.5, 10.5, 7.2 Hz, 1H), 1.18 (t, J = 7.2 Hz, 3H); 13C NMR (200 MHz, CDCl3) δ 155.6, 152.5, 145.5, 125.5, 121.9, 101.5, 84.7, 70.9, 62.9, 41.2, 39.6, 38.4, 36.8, 28.6, 21.9. LRMS-ESI (m/z): 372.0 [M+Na]+.
4-Nitrophenyl ((2aR,2a1R,4S,4aR,7aR)-octahydro-2H-1,7-dioxacyclopenta[cd]inden-4-yl) carbonate (27c)
Alcohol (+)-16 (30 mg, 0.18 mmol) was treated with pyridine (58 μL, 0.72 mmol) and 4-nitrophenyl chloroformate (53 mg, 0.26 mmol) by following the procedure outlined for compound 18 to give compound 27c (47 mg, 80%). [α]D20 -9.5 (c 1.0, CHCl3).
4-Nitrophenyl ((2aS,2a1R,3S,4aR,7aS)-octahydro-2H-1,7-dioxacyclopenta[cd]inden-3-yl) carbonate (27d)
Alcohol (−)-21 (8 mg, 0.047 mmol) was treated with pyridine (15 μL, 0.18 mmol) and 4-nitrophenyl chloroformate (14 mg, 0.07 mmol) by following the procedure outlined for compound 18 to give compound 27d (13 mg, 81%). [α]D20 -41.2 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.31 – 8.26 (m, 2H), 7.42 – 7.36 (m, 2H), 5.19 (d, J = 5.4 Hz, 1H), 5.08 (dt, J = 10.3, 7.4 Hz, 1H), 4.25 (dd, J = 10.1, 2.1 Hz, 1H), 4.08 – 3.99 (m, 1H), 3.66 – 3.57 (m, 2H), 3.01 (qd, J = 8.0, 2.2 Hz, 1H), 2.47 (td, J = 8.5, 5.6 Hz, 1H), 2.24 (dq, J = 13.4, 6.9 Hz, 1H), 2.09 – 2.01 (m, 2H), 1.94 (ddd, J = 18.9, 9.6, 5.4 Hz, 1H), 1.54 (dd, J = 14.2, 2.2 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 155.6, 152.1, 145.6, 125.5, 121.9, 101.3, 80.4, 63.2, 55.4, 44.0, 38.5, 33.5, 28.9, 23.9; LRMS-ESI (m/z): 358.1 [M+Na]+.
4-Nitrophenyl ((2aR,2a1S,3R,4aS,7aR)-octahydro-2H-1,7-dioxacyclopenta[cd]inden-3-yl) carbonate (27e)
Alcohol (+)-21 (8 mg, 0.047 mmol) was treated with pyridine (15 μL, 0.18 mmol) and 4-nitrophenyl chloroformate (14 mg, 0.07 mmol) by following the procedure outlined for compound 18 to give compound 27e (13 mg, 81%). [α]D20 +40.8 (c 1.0, CHCl3).
(2aR,2a1R,4S,4aR,7aS)-Decahydroindeno[7,1-bc]furan-4-yl (4-nitrophenyl) carbonate (27f)
Alcohol (+)-25 (28 mg, 0.17 mmol) was treated with pyridine (55 μL, 0.68 mmol) and 4-nitrophenyl chloroformate (50 mg, 0.25 mmol) by following the procedure outlined for compound 18 to give compound 27f (40 mg, 73%). [α]D20 +3.4 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.29 – 8.24 (m, 2H), 7.39 – 7.34 (m, 2H), 5.14 (q, J = 5.5 Hz, 1H), 3.84 (dq, J = 7.9, 4.9, 4.1 Hz, 1H), 3.77 (d, J = 5.1 Hz, 2H), 2.83 (tq, J = 8.8, 4.7 Hz, 1H), 2.57 (td, J = 9.3, 6.2 Hz, 1H), 2.27 (dq, J = 10.9, 6.0 Hz, 1H), 2.17 (ddd, J = 14.1, 8.5, 5.6 Hz, 1H), 2.04 (ddd, J = 14.3, 7.7, 4.3 Hz, 1H), 1.90 (dt, J = 14.2, 4.0 Hz, 1H), 1.80 (ddt, J = 12.8, 8.4, 5.1 Hz, 2H), 1.65 – 1.52 (m, 2H), 1.35 (ddd, J = 16.5, 8.1, 4.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 155.8, 152.7, 145.4, 125.3, 122.1, 85.1, 76.8, 74.6, 43.4, 42.8, 38.9, 38.1, 25.8, 21.9, 16.7; LRMS-ESI (m/z): 356.1 [M+Na]+.
(2aS,2a1S,4R,4aS,7aR)-Decahydroindeno[7,1-bc]furan-4-yl (4-nitrophenyl) carbonate (27g)
Alcohol (−)-25 (18 mg, 0.1 mmol) was treated with pyridine (32 μL, 0.40 mmol) and 4-nitrophenyl chloroformate (32 mg, 0.16 mmol) by following the procedure outlined for compound 18 to give compound 27g (27 mg, 75%).
Determination of X-ray structures of HIV-1 protease-inhibitor complexes
HIV-1 protease was expressed and purified as described previously.55 The protease-inhibitor complex was crystallized by the hanging drop vapor diffusion method with well solutions of 1.05 M NaCl, 0.1 M sodium Cacodylate, pH 6.4 for inhibitor 5c, and 0.95 M NaCl, 0.1 M Sodium Acetate, pH 5.5 for inhibitor 5d.56–58 X-Ray diffraction data were collected on a single crystal of each complex cooled to 90 K at SER-CAT (22-ID beamline), Advanced Photon Source, Argonne National Lab (Chicago, USA) with X-ray wavelength of 1.0 Å, and processed by HKL-200059 to give an Rmerge of 8.6% and 7.8% for inhibitors 5c and 5d complexes, respectively. The crystal structures were solved by PHASER60 in CCP4i Suite61–63 using one of the previously reported isomorphous structures64 as the initial model, and refined by SHELX-201465,66 using data to resolutions of 1.25 Å and 1.13 Å for the complexes of inhibitors 5c and 5d, respectively. PRODRG-267 was used to construct the inhibitor and the restraints for refinement. COOT68,69 was used for modification of the model. Alternative conformations were modeled, and isotropic atomic displacement parameters (B factors) were applied for all atoms including solvent molecules. The final refined solvent structure comprised one Na+ ion, two Cl− ions, one acetate ion, two glycerol molecules and 207 water molecules for inhibitor 5c-protease complex, and Na+ ion, three Cl− ions, two acetate ions, one glycerol molecule and 220 waters for inhibitor 5d complex. The crystallographic statistics are listed in Table 2 SI. The coordinates and structure factors of the protease complex with PIs 5c and 5d have been deposited in the Protein Data Bank70 with code 6CDL and 6CDJ, respectively.
Supplementary Material
Acknowledgments
This research was supported by the National Institutes of Health (Grant GM53386, AKG and Grant GM 62920, ITW). X-Ray data were collected at the Southeast Regional Collaborative Access Team (SER-CAT) beamline 22ID at the Advanced Photon Source, Argonne National Laboratory. Use of the Advanced Photon Source was supported by the US Department of Energy, Basic Energy Sciences, Office of Science, under Contract No. W-31-109-Eng-38. This work was also supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health, and in part by a Grant-in-Aid for Scientific Research (Priority Areas) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (Monbu Kagakusho), a Grant for Promotion of AIDS Research from the Ministry of Health, Welfare, and Labor of Japan, and the Grant to the Cooperative Research Project on Clinical and Epidemiological Studies of Emerging and Reemerging Infectious Diseases (Renkei Jigyo) of Monbu-Kagakusho. The authors would like to thank the Purdue University Center for Cancer Research, which supports the shared NMR and mass spectrometry facilities.
ABBREVIATIONS
- APV
amprenavir
- ART
antiretroviral therapies
- ATV
atazanavir
- bis-THF
bis-tetrahydrofuran
- DIPEA
N,N-diisopropyletylamine
- DRV
darunavir
- LPV
lopinavir
- NBS
N-bromosuccinimide
- NMO
N-methylmorpholine-N-oxide
- PI
protease inhibitor
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- Umb-THF
umbrella-like-tetrahydrofuran
Footnotes
Supporting Information. The Supporting Information is available free of charge on the ACS Publication website at http://pubs.acs.org.
Full NMR spectroscopic data for all final compounds
X-ray structural data for inhibitors 5c and 5d-bound HIV-1 Protease
Molecular formula strings and some data (CSV)
PDB ID Codes. Inhibitors 5c and 5d-bound HIV-1 protease X-ray structures are: 6CDL and 6CDJ. Authors will release the atomic coordinates and experimental data upon article publication.
ORCID
Arun K. Ghosh: 0000-0003-2472-1841
References
- 1.Ghosh AK, Gemma S. Structure-Based Design of Drugs and Other Bioactive Molecules: Tools and Strategies. Wiley-VCH; Weinheim, Germany: 2014. pp. 337–354. [Google Scholar]
- 2.Hubbard RE. Structure-Based Drug Discovery An Overview. RSC Publishing; England: 2006. [Google Scholar]
- 3.Wlodawer A, Vondrasek J. Inhibitors of HIV-1 Protease a Major Success of Structure-Assisted Drug Design. Annu Rev Biophys Biomol Struct. 1998;27:249–284. doi: 10.1146/annurev.biophys.27.1.249. [DOI] [PubMed] [Google Scholar]
- 4.Ghosh AK, Osswald HL, Prato G. Recent Progress in the Development of HIV-1 Protease Inhibitors for the Treatment of HIV/AIDS. J Med Chem. 2016;59:5172–5208. doi: 10.1021/acs.jmedchem.5b01697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Edmonds A, Yotebieng M, Lusiama J, Matumona Y, Kitetele F, Napravnik S, Cole SR, Van Rie A, Behets F. The Effect of Highly Active Antiretroviral Therapy on the Survival of HIV-Infected Children in a Resource-Deprived Setting: A Cohort Study. PLoS Med. 2011;8:e1001044. doi: 10.1371/journal.pmed.1001044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Broder S. The Development of Antiretroviral Therapy and its Impact on the HIV-1/AIDS Pandemic. Antiviral Res. 2010;85:1–18. doi: 10.1016/j.antiviral.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Naggie S, Hicks C. Protease Inhibitor-Based Antiretroviral Therapy in Treatment-Naive HIV-1-Infected Patients: The Evidence Behind the Options. J Antimicrob Chemother. 2010;65:1094–1099. doi: 10.1093/jac/dkq130. [DOI] [PubMed] [Google Scholar]
- 8.Lohse N, Hansen AB, Gerstoft J, Obel N. Improved Survival in HIV-Infected Persons: Consequences and Perspectives. J Antimicrob Chemother. 2007;60:461–463. doi: 10.1093/jac/dkm241. [DOI] [PubMed] [Google Scholar]
- 9.Montaner JSG, Lima VD, Barrios R, Yip B, Wood E, Kerr T, Shannon K, Harrigan PR, Hogg RS, Daly P, Kendall P. Association of Highly Active Antiretroviral Therapy Coverage, Population Viral Load, and Yearly New HIV Diagnoses in British Columbia, Canada: a Population-Based Study. Lancet. 2010;376:532–539. doi: 10.1016/S0140-6736(10)60936-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. https://aidsinfo.nih.gov/contentfiles/lvguidelines/adultandadolescentgl.pdf, accessed on March 31, 2018.
- 11.Hue S, Gifford RJ, Dunn D, Fernhill E, Pillay D. Demonstration of Sustained Drug-Resistant Human Immunodeficiency Virus Type 1 Lineages Circulating Among Treatment-Naïve Individuals. J Virol. 2009;83:2645–2654. doi: 10.1128/JVI.01556-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Esté JA, Cihlar T. Current Status and Challenges of Antiretroviral Research and Therapy. Antiviral Res. 2010;85:25–33. doi: 10.1016/j.antiviral.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 13.Saylor D, Dickens AM, Sacktor N, Haughey N, Slusher B, Pletnikov M, Mankowski JL, Brown A, Volsky DJ, McArthur JC. HIV-Associated Neurocognitive Disorder — Pathogenesis and Prospects for Treatment. Nat Rev Neurol. 2016;12:234–248. doi: 10.1038/nrneurol.2016.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghosh AK, Chapsal BD. Design of the Anti-HIV-1 Protease Inhibitor Darunavir. In: Ganellin CR, Jefferis R, Roberts S, editors. Introduction to Biological and Small Molecule Drug Research and Development: Theory and Case Studies. Elsevier; London: 2013. pp. 355–384. [Google Scholar]
- 15.Ghosh AK, Chapsal BD. In: Aspartic Acid Proteases as Therapeutic Targets. Ghosh AK, editor. Wiley-VCH; Weinheim, Germany: 2010. pp. 169–204. [Google Scholar]
- 16.Ghosh AK, Sridhar PR, Kumaragurubaran N, Koh Y, Weber IT, Mitsuya H. Bis-Tetrahydrofuran: a Privileged Ligand for Darunavir and a New Generation of HIV Protease Inhibitors That Combat Drug Resistance. ChemMedChem. 2006;1:939–950. doi: 10.1002/cmdc.200600103. [DOI] [PubMed] [Google Scholar]
- 17.Ghosh AK, Xu CX, Rao KV, Baldridge A, Agniswamy J, Wang YF, Weber IT, Aoki M, Miguel SGP, Amano M, Mitsuya H. Probing Multidrug-Resistance and Protein–Ligand Interactions with Oxatricyclic Designed Ligands in HIV-1 Protease Inhibitors. ChemMedChem. 2010;5:1850–1854. doi: 10.1002/cmdc.201000318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghosh AK, Anderson DD, Weber IT, Mitsuya H. Enhancing Protein Backbone Binding—A Fruitful Concept for Combating Drug-Resistant HIV. Angew Chem, Int Ed. 2012;51:1778–1802. doi: 10.1002/anie.201102762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Agniswamy J, Weber IT. HIV-1 Protease: Structural Perspectives on Drug Resistance. Viruses. 2009;1:1110–1136. doi: 10.3390/v1031110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghosh AK, Chapsal BD, Weber IT, Mitsuya H. Design of HIV Protease Inhibitors Targeting Protein Backbone: An Effective Strategy for Combating Drug Resistance. Acc Chem Res. 2008;41:78–86. doi: 10.1021/ar7001232. [DOI] [PubMed] [Google Scholar]
- 21.Koh Y, Nakata H, Maeda K, Ogata H, Bilcer G, Devasamundram T, Kincaid JF, Boross P, Wang YF, Tie Y, Volarath P, Gaddis L, Harrison RW, Weber IT, Ghosh AK, Mitsuya H. Novel bis-Tetrahydrofuranylurethane-Containing Nonpeptidic Protease Inhibitor (PI) UIC-94017 (TMC114) with Potent Activity Against Multi-PI-Resistant Human Immunodeficiency Virus In Vitro. Antimicrob Agents Chemother. 2003;47:3123–3129. doi: 10.1128/AAC.47.10.3123-3129.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghosh AK, Dawson ZL, Mitsuya H. Darunavir, a Conceptually New HIV-1 Protease Inhibitor for the Treatment of Drug-Resistant HIV. Bioorg Med Chem. 2007;15:7576–7580. doi: 10.1016/j.bmc.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Meyer S, Azijn H, Surleraux D, Jochmans D, Tahri A, Pauwels R, Wigerinck P, de Béthune M. TMC114, a Novel Human Immunodeficiency Virus Type 1 Protease Inhibitor Active Against Protease Inhibitor-Resistant Viruses, Including a Broad Range of Clinical Isolates. Antimicrob Agents Chemother. 2005;49:2314–2321. doi: 10.1128/AAC.49.6.2314-2321.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Béthune MP, Sekar V, Spinosa-Guzman S, Vanstockem M, De Meyer S, Wigerinck P, Lefebvre E. Antiviral Drugs: From Basic Discovery Through Clinical Trials. John Wiley & Sons, Inc; New Jersey: 2011. Darunavir (Prezista, TMC114): From Bench to Clinic, Improving Treatment Options for HIV-Infected Patients; pp. 31–45. [Google Scholar]
- 25.Ghosh AK, Rao KV, Nyalapatla PR, Osswald HL, Martyr CD, Aoki M, Hayashi H, Agniswamy J, Wang YF, Bulut H, Das D, Weber IT, Mitsuya H. Design and Development of Highly Potent HIV-1 Protease Inhibitors with a Crown-like Oxotricyclic Core as the P2-Ligand to Combat Multidrug-Resistant HIV Variants. J Med Chem. 2017;60:4267–4278. doi: 10.1021/acs.jmedchem.7b00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aoki M, Hayashi H, Rao KV, Das D, Higashi-Kuwata N, Bulut H, Aoki-Ogata H, Takamatsu Y, Yedidi RS, Davis DA, Hattori S-i, Nishida N, Hasegawa K, Takamune N, Nyalapatla PR, Osswald HL, Jono H, Saito H, Yarchoan R, Misumi S, Ghosh AK, Mitsuya HA. Novel Central Nervous System-Penetrating Protease Inhibitor Overcomes Human Immunodeficiency Virus 1 Resistance with Unprecedented aM to pM potency. eLife. 2017;6:e28020. doi: 10.7554/eLife.28020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tie Y, Boross PI, Wang YF, Gaddis L, Hussain AK, Leshchenko S, Ghosh AK, Louis JM, Harrison RW, Weber IT. High Resolution Crystal Structures of HIV-1 Protease with a Potent Non-peptide Inhibitor (UIC-94017) Active Against Multi-Drug-resistant Clinical Strains. J Mol Biol. 2004;338:341–352. doi: 10.1016/j.jmb.2004.02.052. [DOI] [PubMed] [Google Scholar]
- 28.Kovalevsky AY, Liu F, Leshchenko S, Ghosh AK, Louis JM, Harrison RW, Weber IT. Ultra-High Resolution Crystal Structure of HIV-1 Protease Mutant Reveals Two Binding Sites for Clinical Inhibitor TMC114. J Mol Biol. 2006;363:161–173. doi: 10.1016/j.jmb.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hohlfeld K, Tomassi C, Wegner JK, Kesteleyn B, Linclau B. Disubstituted Bis-THF Moieties as New P2 Ligands in Nonpeptidal HIV-1 Protease Inhibitors. ACS Med Chem Lett. 2011;2:461–465. doi: 10.1021/ml2000356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hohlfeld K, Wegner JK, Kesteleyn B, Linclau B, Unge J. Disubstituted Bis-THF Moieties as New P2 Ligands in Nonpeptidal HIV-1 Protease Inhibitors (II) J Med Chem. 2015;58:4029–4038. doi: 10.1021/acs.jmedchem.5b00358. [DOI] [PubMed] [Google Scholar]
- 31.Bai X, Yang Z, Zhu M, Dong B, Zhou L, Zhang G, Wang J, Wang Y. Design and Synthesis of Potent HIV-1 Protease Inhibitors with (S)-Tetrahydrofuran-Tertiary Amine-Acetamide as P2−Ligand: Structure−Activity Studies and Biological Evaluation. Eur J Med Chem. 2017;137:30–44. doi: 10.1016/j.ejmech.2017.05.024. [DOI] [PubMed] [Google Scholar]
- 32.Yang ZH, Bai XG, Zhou L, Wang JX, Liu HT, Wang YC. Synthesis and Biological Evaluation of Novel HIV-1 Protease Inhibitors Using Tertiary Amine as P2-Ligands. Bioorg Med Chem Lett. 2015;25:1880–1883. doi: 10.1016/j.bmcl.2015.03.047. [DOI] [PubMed] [Google Scholar]
- 33.Yan J, Huang N, Li S, Yang LM, Xing W, Zheng YT, Hu Y. Synthesis and Biological Evaluation of Novel Amprenavir-Based P1-Substituted Bi-Aryl Derivatives as Ultra-Potent HIV-1 Protease Inhibitors. Bioorg Med Chem Lett. 2012;22:1976–1979. doi: 10.1016/j.bmcl.2012.01.037. [DOI] [PubMed] [Google Scholar]
- 34.Bungard CJ, Williams PD, Ballard JE, Bennett DJ, Beaulieu C, Bahnck-Teets C, Carroll SS, Chang RK, Dubost DC, Fay JF, Diamond TL, Greshock TJ, Hao L, Holloway MK, Felock PJ, Gesell JJ, Su HP, Manikowski JJ, McKay DJ, Miller M, Min X, Molinaro C, Moradei OM, Nantermet PG, Nadeau C, Sanchez RI, Satyanarayana T, Shipe WD, Singh SK, Truong VL, Vijayasaradhi S, Wiscount CM, Vacca JP, Crane SN, McCauley JA. Discovery of MK-8718, an HIV Protease Inhibitor Containing a Novel Morpholine Aspartate Binding Group. ACS Med Chem Lett. 2016;7:702–707. doi: 10.1021/acsmedchemlett.6b00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bungard CJ, Williams PD, Schulz J, Wiscount CM, Holloway MK, Loughran HM, Manikowski JJ, Su HP, Bennett DJ, Chang L, Chu XJ, Crespo A, Dwyer MP, Keertikar K, Morriello GJ, Stamford AW, Waddell ST, Zhong B, Hu B, Ji T, Diamond TL, Bahnck-Teets C, Carroll SS, Fay JF, Min X, Morris W, Ballard JE, Miller MD, McCauley JA. Design and Synthesis of Piperazine Sulfonamide Cores Leading to Highly Potent HIV-1 Protease Inhibitors. ACS Med Chem Lett. 2017;8:1292–1297. doi: 10.1021/acsmedchemlett.7b00386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ghosh AK, Chapsal BD, Baldridge A, Steffey MP, Walters DE, Koh Y, Amano M, Mitsuya H. Design and Synthesis of Potent HIV-1 Protease Inhibitors Incorporating Hexahydrofuropyranol-Derived High Affinity P2 Ligands: Structure – Activity Studies and Biological Evaluation. J Med Chem. 2011;54:622–634. doi: 10.1021/jm1012787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ali R, Siddiqui N. Biological Aspects of Emerging Benzothiazoles: A Short Review. J Chem. 2013:1–12. [Google Scholar]
- 38.Seth SA. Comprehensive Review on Recent Advances in Synthesis & Pharmacotherapeutic Potential of Benzothiazoles. Antiinflamm Antiallergy Agents Med Chem. 2015;14:98–112. doi: 10.2174/1871523014666150528110703. [DOI] [PubMed] [Google Scholar]
- 39.Pauson PL, Khand IU. Uses of Cobalt-Carbonyl Acetylene Complexes in Organic Synthesis. Ann N Y Acad Sci. 1977;295:2–14. [Google Scholar]
- 40.Pauson PL. The Khand reaction: A Convenient and General Route to a Wide Range of Cyclopentenone Derivatives. Tetrahedron. 1985;41:5855–5860. [Google Scholar]
- 41.Blanco-Urgoiti J, Anorbe L, Perez-Serrano L, Dominguez G, Perez-Castells J. The Pauson-Khand Reaction, A Powerful Synthetic Tool for the Synthesis of Complex Molecules. Chem Soc Rev. 2004;33:32–42. doi: 10.1039/b300976a. [DOI] [PubMed] [Google Scholar]
- 42.Saeeng R, Sirion U, Sirichan Y, Trakulsujaritchok T, Sahakitpichan P. Convertible Formation of Different Glycoside Using Molecular Iodine. Heterocycles. 2010;81:2569–2580. [Google Scholar]
- 43.Shambayati S, Crewel WE, Schreiber SL. N-Oxide Promoted Pauson-Khand Cyclizations at Room Temperature. Tetrahedron Lett. 1990;31:5289–5292. [Google Scholar]
- 44.Brummond KM, Kent JL. Recent Advances in the Pauson–Khand Reaction and Related [2+2+1] Cycloadditions. Tetrahedron. 2000;56:3263–3283. [Google Scholar]
- 45.Rao HSP, Reddy KS. Palladium Assisted Transfer Hydrogenation of Cyclic α,β-Unsaturated Ketones by Ammonium Formate. Tetrahedron Lett. 1994;35:171–174. [Google Scholar]
- 46.Dulcere JP, Crandal1 J, Faure R, Santelli M, Agati V, Mihoubi MN. Allenyl Allylic Ethers: Synthesis and Thermal Rearrangements. J Org Chem. 1993;58:5702–5708. [Google Scholar]
- 47.Bianchi D, Cesti P, Battistel E. Anhydrides as Acylating Agents in Lipase-Catalyzed Stereoselective Esterification of Racemic Alcohols. J Org Chem. 1988;53:5531–34. [Google Scholar]
- 48.Ghosh AK, Chen Y. Synthesis and Optical Resolution of High Affinity P2-Ligands for HIV-1 Protease Inhibitors. Tetrahedron Lett. 1995;36:505–508. doi: 10.1016/0040-4039(94)02296-N. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Complete crystallographic data, in CIF format, have been deposited with the Cambridge Crystallographic Data Centre. CCDC 1822740 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- 50.Toth MV, Marshall GR. A Simple, Continuous Fluorometric Assay for HIV Protease. Int J Pept Protein Res. 1990;36:544–550. doi: 10.1111/j.1399-3011.1990.tb00994.x. [DOI] [PubMed] [Google Scholar]
- 51.Koh Y, Amano M, Towata T, Danish M, Leshchenko-Yashchuk S, Das D, Nakayama M, Tojo Y, Ghosh AK, Mitsuya H. In Vitro Selection of Highly Darunavir-Resistant and Replication-Competent HIV-1 Variants by Using a Mixture of Clinical HIV-1 Isolates Resistant to Multiple Conventional Protease Inhibitors. J Virol. 2010;84:11961–11969. doi: 10.1128/JVI.00967-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hughes PJ, Cretton-Scott E, Teague A, Wensel TM. Protease Inhibitors for Patients with HIV-1 Infection: A Comparative Overview. Pharmacol Ther. 2011;36:332–345. [PMC free article] [PubMed] [Google Scholar]
- 53.Aoki M, Danish ML, Aoki-Ogata H, Amano M, Ide K, Das D, Koh Y, Mitsuya H. Loss of the Protease Dimerization Inhibition Activity of Tipranavir (TPV) and Its Association with the Acquisition of Resistance to TPV by HIV-1. J Virol. 2012;86:13384–13396. doi: 10.1128/JVI.07234-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.For details of X-ray studies, please see Supplementary Information.
- 55.Amano M, Salcedo-Gomez PM, Yedidi RS, Delino NS, Nakata H, Rao KV, Ghosh AK, Mitsuya H. GRL-09510, a Unique P2-Crown-Tetrahydrofuranylurethane-Containing HIV-1 Protease Inhibitor, Maintains Its Favorable Antiviral Activity Against Highly-Drug-Resistant HIV-1 Variants in Vitro. Scientific Reports. 2017;7:12235. doi: 10.1038/s41598-017-12052-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zürcher M, Diederich F. Structure-Based Drug Design: Exploring the Proper Filling of Apolar Pockets at Enzyme Active Sites. J Org Chem. 2008;73:4345–4361. doi: 10.1021/jo800527n. [DOI] [PubMed] [Google Scholar]
- 57.Müller K, Faeh C, Diederich F. Fluorine in Pharmaceuticals: Looking Beyond Intuition. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 58.Mahalingam B, Louis JM, Hung J, Harrison RW, Weber IT. Structural Implications of Drug-Resistant Mutants of HIV-1 Protease: High-Resolution Crystal Structures of the Mutant Protease/Substrate Analogue Complexes. Proteins. 2001;43:455–464. doi: 10.1002/prot.1057. [DOI] [PubMed] [Google Scholar]
- 59.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. In: Carter CW Jr, Sweet RM, editors. Methods in Enzymology 276: Macromolecular Crystallography, Part A. Academic Press; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 60.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser Crystallographic Software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Overview of the CCP4 Suite and Current Developments. Acta Crystallogr Sect D: Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Collaborative Computational Project, Number 4. The CCP4 Suite: Programs for Protein Crystallography. Acta Crystallogr, Sect D: Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 63.Potterton E, Briggs P, Turkenburg M, Dodson E. A Graphical User Interface to the CCP4 Program Suite. Acta Crystallogr Sect D: Biol Crystallogr. 2003;59:1131–1137. doi: 10.1107/s0907444903008126. [DOI] [PubMed] [Google Scholar]
- 64.Shen CH, Wang YF, Kovalevsky AY, Harrison RW, Weber IT. Amprenavir Complexes with HIV-1 Protease and its Drug-Resistant Mutants Altering Hydrophobic Clusters. FEBS J. 2010;277:3699–3714. doi: 10.1111/j.1742-4658.2010.07771.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sheldrick GM. A Short History of SHELX. Acta Crystallogr, Sect A: Found Crystallogr. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 66.Sheldrick GM, Schneider TR. SHELXL: High-Resolution Refinement. Meth Enzymol. 1997;277:319–343. [PubMed] [Google Scholar]
- 67.Schuettelkopf AW, van Aalten DMF. PRODRG: a Tool for High-Throughput Crystallography of Protein–Ligand Complexes. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 68.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and Development of Coot. Acta Crystallogr Sect D: Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Emsley P, Cowtan K. Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 70.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.