

Abstract
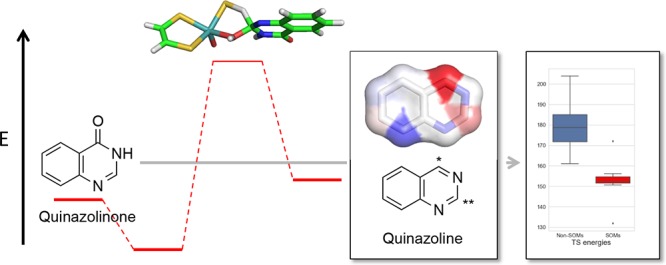
Aldehyde oxidase (AO) is a molybdenum-containing enzyme involved in the clearance of drug compounds containing aldehydes and N-containing heterocyclic fragments. AO has gained considerable interest in recent years because of examples of too fast clearance of drug compounds in development. Thus, it is important to be able to predict AO-mediated drug metabolism. Therefore, we have characterized the structural and energetic aspects of different mechanisms with density functional theory using the molybdenum cofactor as a model for the reactive part of the enzyme. For a series of 6-substituted 4-quinazolinones, the trend in activation energies is the same for three tested reaction mechanisms. Using the concerted mechanism as a model for the enzymatic reaction, the transition states (TSs) for the formation of all possible metabolites for a series of known AO substrates were determined. The lowest activation energies correspond in all cases to the experimentally observed sites of metabolism (SOMs). Various molecular properties were calculated and investigated as more easily determinable markers for reactivity. The stabilities of both intermediates and products correlate to some extent with the TS energies and may be used to predict the SOM. The electrostatic-potential-derived charges are also good markers for the prediction of the experimental SOM for this set of compounds and may pave the way for the development of fast methods for the prediction of SOM for AO substrates.
Introduction
Aldehyde oxidase (AO) is an enzyme located in the cytosol of cells and belongs to the xanthine oxidase (XO) family of molybdoenzymes that requires a molybdenum cofactor (MoCo) for the catalytic activity.1,2 The precise physiological role of AO remains unclear, but in humans, it plays an important role in drug metabolism.2,3 Although aldehydes are not often present in drug compounds, this functional group can be a result of biotransformation by other drug metabolizing enzymes, such as the cytochrome P450s, and can be subsequently oxidized to a carboxylic acid by AO. More importantly in drug discovery, AO oxidizes a range of N-containing heterocyclic aromatic rings that are often present in drug compounds. Therefore, several drug discovery projects have been discontinued after clinical evaluation owing to high clearance.2 However, AO activity can also be exploited to activate compounds, increasing the bioavailability as observed for famciclovir. In addition to the broad substrate profile of AO, it can also reduce N- and S-oxides and hydrolyze amides.4
In drug discovery projects, it is important to understand and predict the AO-mediated reactions of new compounds, preferably prior to synthesis. In several theoretical studies, different reaction mechanisms have been investigated and it may be difficult to prove which is correct. These types of investigations are typically done with density functional theory (DFT) on a model system containing a part of the MoCo and the substrate. For AO and XO, Alfaro and Jones suggest a concerted mechanism, in that the transition state (TS) has the OH of MoCo attacking the sp2-hybridized C atom while the H atom of the substrate is transferred to the S atom of the MoCo because it explains the trends in the rate of product formation for a series of 4-substituted quinazolinones and also the large observed deuterium isotope effects.5 The effect of a neighboring Glu residue, Glu1270 in human AO, on the activation energies has been estimated to enhance the stability of the TS and thus decrease the activation energy for the reaction.5,6 Other mechanisms for the related XO have been proposed in which an O instead of OH attacks the C atom of the substrate and where a proton is transferred to the neighboring O of aldehydes or N of xanthine. The next step, the hydride transfer to S of MoCo, is the rate-limiting step.7−9
The mechanistic studies have primarily been done with DFT, and the TSs are typically time-consuming to determine. Therefore, other simpler methods to estimate the most probable site of metabolism (SOM) have been applied to larger sets of compounds. For example, Torres et al. investigated the partial charges on the aromatic C atom and the stability of the hydroxylated intermediates and found that the latter energy descriptor provided a good predictor for reaction by AO.10 Models combining the stabilities of the intermediates with a steric factor have been developed to address whether the SOM is accessible to predict clearance and formation of SOMs.11,12 Xu et al.12 used these two descriptors to develop a decision-tree model for the prediction of SOM with predictivity values of 0.905 and 0.867 for the training and test set, respectively. This model predicts either the most reactive sites or medium reactive sites that are accessible as SOMs. The decision-tree model was shown to be more predictive than a model that is only based on the intermediate.
In this work, we use MoCo to study the AO reactions by DFT calculations to determine the activation energies for the possible reactions. Thus, the steric restraints from the enzyme are not included, which is a potential pitfall for using the activation energies for the prediction of SOMs. However, it has previously been observed that the electronic features of substrates play a major role in the prediction of AO metabolites.10 Several reaction mechanisms have been considered, and their reaction profiles were determined. Finally, we relate other molecular properties characterizing the reaction with the activation energies to enable fast estimates of AO oxidations on large sets of compounds.
Results and Discussion
Mechanism for 6-Substituted 4-Quinazolinones
The 6-substituted 4-quinazolinone compounds are both XO and AO substrates. Initially, activation energies were determined for the same series of 6-substituted 4-quinazolinone compounds as studied by Alfaro and Jones (see Figure 1).5 We studied both the concerted and stepwise mechanisms, as these previously have been proposed as plausible reaction mechanisms. In addition, the effect of deprotonation of the Mo-bound OH was investigated to mimic the transfer of a proton to the nearby Glu1270 residue. The different mechanisms are shown in Figure 1. All energies and distances are shown in Supporting Information, Tables S1–S3.
Figure 1.

Reaction mechanisms of 6-substituted 4-quinazolinones (R = NO2, CF3, Cl, H, CH3, OCH3, and NH2) studied in this work. (A) Reaction 1: concerted mechanism, (B) reaction 2: concerted mechanism with deprotonated MoCo, and (C) reaction 3: stepwise mechanism with an initial proton transfer to the nitrogen and subsequent hydride transfer. The H and N atoms directly involved in the mechanism are colored to ease the interpretation.
For the concerted mechanism (Figure 1A, reaction 1), the MoCo-bound OH makes a nucleophilic attack on the C atom (C2) next to the N atom while the H atom is transferred to the S atom of MoCo. The structures of the reactants, TSs, and products along with the energies are plotted in Figure 2A. As observed previously,5 4-quinazolinone compounds with electron-withdrawing substituents at position 6 have the lowest activation energies while the electron-donating substituents increase the barriers (see Table 1). In vacuum, the B3LYP-calculated activation energies range from 164 to 181 kJ/mol by comparing the TS and reactant energies, whereas the activation energies are 93–141 kJ/mol (cf. Table S1A) when compared with the separated MoCo and substrate species. Notably, the trends are exactly the same whether the reactants or separated species are used as reference (R2 = 0.98). These energies are high but on average agree within 1 kJ/mol with previously determined energies for the same substrates when comparing the TS to the separated MoCo and substrate species. With B3LYP-D3, that is, including dispersion, the activation energies are similar (within ∼8 kJ/mol) to those determined with B3LYP and the trends are the same as also observed by the correlation to the B3LYP energies (R2 = 0.99). The reactions are characterized by short OMoCo–CSub distances (∼1.5 Å) and rather long HSub–Csub distances (∼1.4 Å) in the TS (cf. Table S1A). In agreement with the observations by Alfaro and Jones, the lower activation barriers for the electron-withdrawing groups result in slightly shorter OMoCo–CSub and longer HSub–Csub distances, which is more similar to the product structures.5
Figure 2.
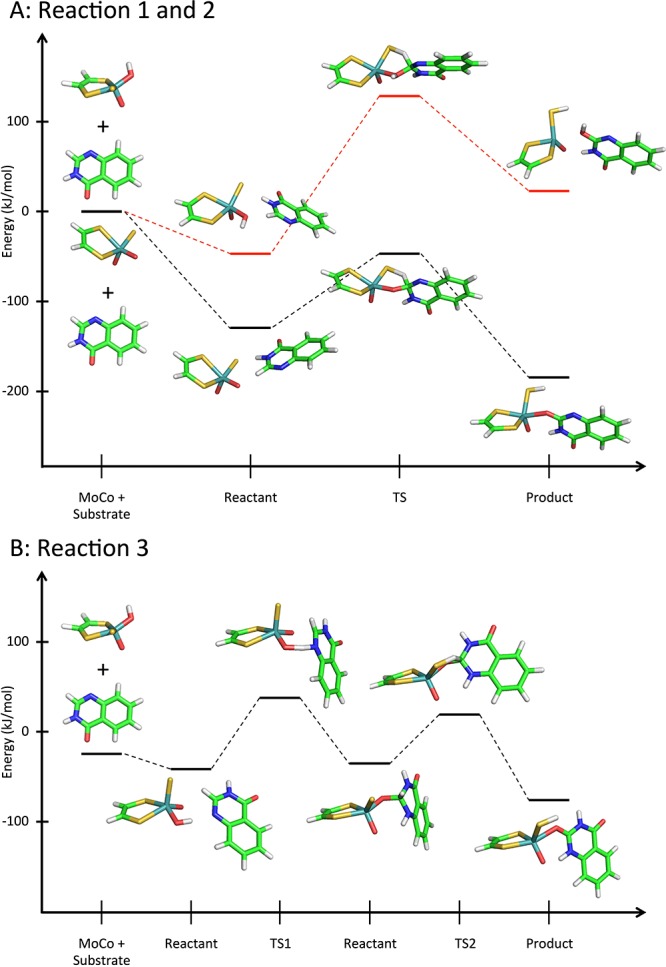
Reaction profiles for (A) reaction 1 (red lines) and 2 (black lines) and (B) reaction 3 for 4-quinazolinone.
Table 1. TS Energies (kJ/mol) for Reactions 1–3 Described in Figure 1a,b.
| B3LYP |
B3LYP-D3 |
|||||||
|---|---|---|---|---|---|---|---|---|
| reaction | 1 | 2 | 3 | 3 | 1 | 2 | 3 | 3 |
| substituent R | TS | TS | TS1 | TS2 | TS | TS | TS1 | TS2 |
| NO2 | 163.6 | 35.1 | 71.0 | 48.8 | 156.8 | 31.7 | 66.9 | 47.3 |
| CF3 | 170.0 | 56.9 | 77.0 | 56.6 | 163.2 | 53.4 | 71.0 | 54.5 |
| Cl | 175.2 | 73.7 | 79.2 | 61.1 | 167.5 | 69.7 | 72.3 | 58.8 |
| H | 175.8 | 83.6 | 79.7 | 61.2 | 166.8 | 79.0 | 72.6 | 58.7 |
| CH3 | 178.1 | 85.9 | 79.9 | 63.0 | 169.6 | 81.7 | 72.4 | 60.6 |
| OCH3 | 178.7 | 88.9 | 81.8 | 66.9 | 170.9 | 85.0 | 75.5 | 64.5 |
| NH2 | 181.1 | 93.0 | 84.3 | 69.8 | 172.2 | 89.1 | 76.3 | 67.2 |
| R2c | 0.98 | 0.96 | 0.97 | 0.99 | 0.98 | 0.90 | 0.96 | |
All TS energies are determined in vacuum with B3LYP or B3LYP-D3. The energies are given relative to the reactant complex.
Energies for the separated MoCo and substrate, the intermediates, and the products are given in Tables S1–S3.
Correlation to the activation energy for reaction 1 at the B3LYP level. The correlation with the activation energy for reaction 1 determined as the energy difference between the TS and the sum of the separated MoCo and substrate is R2 = 0.98. The correlation between TS1 and TS2 for reaction 3 is 0.98 at both B3LYP and B3LYP-D3 levels.
Near MoCo, there is a Glu residue, Glu1270 in human AO, which has been considered to be able to deprotonate the OH of MoCo in XO and AO.5,13 Initially, we included a model for this residue in the calculations to mimic the activation or the deprotonation of the OH group. However, we did not succeed in optimizing the TS. We then characterized the reaction with a deprotonated OH in MoCo (Figures 1B and 2A, reaction 2). By doing this, the possible energetic penalty for deprotonating OH is ignored. This significantly decreases the activation energies to 35–93 and 32–89 kJ/mol for B3LYP and B3LYP-D3, respectively (see Table 1), and a correlation of R2 = 0.98 with the concerted mechanism with an OH in MoCo is observed (Figure 1A, reaction 1). Including solvation increases these barriers because the MoCo with a formal charge of −2 is stabilized in the presence of solvent. For example, the activation barrier for 4-quinazolinone increases from 84 kJ/mol in vacuum to 110 and 123 kJ/mol for ε = 4 and 80, respectively (see Supporting Information, Table S2).
Both of these mechanisms involve a concerted oxidation of the substrate and a hydride transfer from the substrate to MoCo, a mechanism already suggested in 1987 by Skibo et al.14 based on kinetic studies on substituted quinazolinones by buttermilk XO and in agreement with more recent computational studies.5
Next, we studied the hydrogen transfer of the MoCo-bound OH to the N atom of the substrate, followed by the formation of a tetrahedral intermediate and a subsequent H transfer to the S atom of MoCo (Figure S1A, reaction 4).6−8,15 However, instead we observed that the other O atom bound to MoCo spontaneously reacted with the aromatic atom as shown in Figures 1C and 2B (reaction 3). The activation energies for the initial proton transfer are rate-limiting and range from 71 to 84 kJ/mol in vacuum and are systematically smaller with B3LYP-D3 by about 4–8 kJ/mol. The subsequent transfer of the H atom to the S atom of MoCo has a lower TS energy, and the trends in barrier height are similar to the first and rate-limiting step.
Because the TS along the path presented in Figure S1A could not be identified, it was then tested if the trends from a previous study about oxidation of xanthine could be reproduced.7 In this study, MoCo oxidizes a negatively charged xanthine (Figure S1B, reaction 4) because previous QM/MM studies had shown this to be a favorable reaction path. Despite differences in the methods (e.g., inclusion of relativistic effects and dispersion functional), we could reproduce trends from these TS energies (see Table S4), that is, the nucleophilic attack on the carbonyl C atom is lower in energy than the subsequent H transfer to the S atom of MoCo. Further examination of the binding pocket of AO (PDB entries 4UHW and 4UHX)16 reveals, however, that there is not a residue to abstract the proton from the substrates, like Glu802 in XO, and therefore, we did not pursue this reaction further.
Although the different reaction mechanisms studied with this small model system have very different activation energies in absolute numbers, the relative trends of the rate-limiting TS energies are the same with R2 of 0.98 and 0.96 for reactions 2 and 3 at the B3LYP level compared with reaction 1 (Table 1). Thus, all mechanisms are equally useful for the following studies because the ranking of the TS energies at different sites in a substrate would be the same. We decided to continue with the concerted mechanism because it agrees well with the observed large deuterium isotope effects, suggesting that the hydride transfer to the S atom of MoCo is rate-limiting.14 Moreover, reaction 3 would require a heteroatom accepting a proton as neighbor to the C atom that is oxidized, which is not always the case, for example, for a substrate such as N-[2-(dimethylamino)ethyl]acridine-4-carboxamide (DACA). Thus, in the following, the TSs were determined with the mechanism for reaction 1.
Activation Energies for AO Substrates
To explore the reactions for AO substrates, we investigated all possible AO sites in quinazoline, phthalazine, and quinoline using reaction 1 (Figure 3). For comparison, the most electron-withdrawing (NO2) or electron-donating (NH2) substituents at position 6 of quinazoline were included. Moreover, the observed AO SOMs of phenanthridine and DACA were included. The latter, DACA, was included because it has an unusual SOM opposite to the N atom of the aromatic ring. Finally, we also studied famciclovir as an example of a compound, where AO metabolism increases the bioavailability of the active species.
Figure 3.

AO substrates. Atom numbering according to Moss.17 DACA was represented by 4-methyl-acridine. Famciclovir was modeled as 2-aminopurine. */** indicate primary/secondary SOMs. Aromatic C atoms were colored according to TS energies (kJ/mol, at the B3LYP level) for the concerted reaction. TS energies are relative to the separated MoCo and substrate (cf. Tables S6–S11). The scale is from 131.9 kJ/mol (C4 of quinazoline) to 203.9 kJ/mol (C4 of phenanthridine). Nonaromatic C atoms and aromatic N atoms are colored white.
Quinazolines
The trends in activation energies for oxidations of C2 in 6-NO2 and 6-NH2 quinazolines were similar to the 6-substituted 4-quinazolidine compounds, that is, the electron-withdrawing group has a low barrier (Table S5). Considering all possible aromatic C atoms for quinazoline, C2 and C4, that is, the observed AO SOMs, have the lowest barriers of 154.2 and 131.9 kJ/mol, respectively (cf. Table S6, TS energies at the B3LYP level relative to separated MoCo and substrate). The primary SOM, C4, has the lowest barrier as clearly seen in Figure 4, where it is also observed that positions 5, 6, 7, and 8 have significantly higher TS energies of 164.9–191.1 kJ/mol.
Figure 4.

DFT-calculated transition energies (B3LYP-level ZPE-corrected BS2 energies) for the AO substrates. For each compound, the TS energies for the aromatic C atoms are listed. */** indicate primary/secondary SOMs.
Phthalazine
Phthalazine has three different C atoms, which in principle can be oxidized by AO. The reaction with the lowest TS energy of 152.1 kJ/mol oxidizes C4 and yields 1-phthalazinone (Figure 4 and Table S7), which also is the primary metabolite observed experimentally.18 Reactions on C5 and C6 have higher TS energies of 184.1 and 188.6 kJ/mol, respectively.
Quinoline
Although quinoline itself is not a drug compound, the quinoline moiety is present in numerous drug compounds and natural products.19 Examples on quinoline-containing compounds are the alkaloids such as quinine, quinidine, cinchonine, and cinchonidine, which all are oxidized at C2 in the quinoline moiety to give the corresponding lactams.20 The DFT calculations show that oxidation at the most reactive C atom C2 has a TS energy of 156.2 kJ/mol (Figure 4 and Table S8) and corresponds to the experimentally observed AO metabolite of quinoline.21 TS energies for reactions on the remaining C atoms range from 167.1 to 202.4 kJ/mol.
Famciclovir
Famciclovir is a prodrug of penciclovir with an improved oral bioavailability. The conversion of famciclovir to penciclovir requires hydrolyses of the ester functionalities and an AO-mediated oxidation of the 2-aminopurine moiety at position 6 forming the 6-oxo-metabolite. In addition to this major metabolite, the isomeric 8-oxo- and the 6,8-dioxo-metabolites are also formed in smaller amounts.22 The TS energy for forming the 8-oxo-metabolite of 151.9 kJ/mol is 20 kJ/mol (Figure 4 and Table S9), lower than that required for the formation of the 6-oxo-metabolite (with the primary SOM); however, both reactions have relatively low TS energies. Thus, the TS energies do not reflect the formation of the primary and secondary metabolites, which are most likely due to the steric hindrance at C8, an effect that is not taken into account when not considering the rest of the enzyme. It is also worth mentioning that the DFT calculations were not done on the actual famciclovir molecule, but on the parent moiety, 2-amino-9-methylpurine, that is, without considering the flexible substituent on N9.
N-[2-(Dimethylamino)ethyl]acridine-4-carboxamide
DACA has been included in this study because it is a substrate for AO, although it does not contain a cyclic −CH=N– moiety. Experimentally, it has been shown that metabolism takes place at position 9, yielding the 9(10H)-acridone metabolite.23 The DFT calculations were performed on 4-methylacridine and showed that oxidation at C9 has the lowest TS energy of 153.7 kJ/mol (Figure 4 and Table S10), whereas the other C oxidations are in the range of 170.0–190.0 kJ/mol and, thus, more unfavorable. Although not having a neighboring N atom, the geometry in the TS for oxidation of C9 is very similar to those on C atoms in substrates next to a N atom (comparing Tables S9 with S1–S8, S11). Other metabolic enzymes may also metabolize DACA, CYP450 may give N-demethylated metabolites and FMO may give N-oxide, and these metabolites may subsequently be oxidized at C9 by AO.24 The result for this atypical AO substrate suggests that the mechanism-based approach presented here is not limited to −CH=N– containing compounds but is generally applicable. This is in contrast to reaction 3 in which the H atom of the Mo-bound OH is transferred to a N atom next to the C atom of the substrate.
Phenanthridine
In phenanthridine, C6 is the most reactive site, and oxidation proceeds via the lowest TS energy (150.7 kJ/mol, cf. Table S11) in agreement with experimental data showing that phenanthridinone is the only metabolite formed by AO.25 This energy is significantly lower than that of TS energies of 171.3–203.9 kJ/mol for the other C atoms.
Considering the DFT-calculated TS energies for all of the relevant C atoms, the TS energies at the B3LYP level for the experimentally observed SOMs range from 131.9 (C4 of quinazoline) to 172.0 kJ/mol (C6 of famciclovir). For quinazoline, phthalazine, quinoline, DACA, and phenanthridine, the lowest TS energies are observed for the atoms that are the experimentally observed primary SOMs. For famciclovir, both of the experimentally observed primary and secondary SOMs correspond to atoms associated with low TS energies. Thus, based on these AO substrates, it seems that a threshold of ∼160 kJ/mol could be used to estimate whether AO oxidation takes place.
The present approach considers only the interactions between the MoCo and the substrate and, accordingly, ignores the effect of the surrounding protein. The substrate specificity between AO and XO metabolisms seems to be caused by two residues in the active site: Val811 and Met889 in AO and Glu802 and Arg880 in XO.26 Thus, the active site in AO is more hydrophobic than the XO active site, where the Glu802 and Arg880 residues in several X-ray structures are seen to fix the orientation of the substrate by several hydrogen bonds.27 One may therefore speculate that AO-mediated metabolism primarily is determined by reactivity and XO-mediated metabolism by reactivity and the possibility for establishing some specific hydrogen bonds. In both cases, the shape of the active sites may also play a role.
Fast Methods for the Prediction of SOM
Because the TS energies are time-consuming to determine, we investigated if other estimates of the reactivity could be used to indicate whether a site in the AO substrate is a likely SOM. The energies of the species formed along the reaction pathway were determined, that is, stability of the hydroxylated tetrahedral intermediate, the cation intermediate, and the product (Figure S2 and Table S12). We also considered the electronic properties of the aromatic C atoms via their electrostatic potential (ESP) and natural bond order (NBO) charges (Table S12). All calculated estimates of the intermediate or product stability were correlated with the TS energies. The best correlations are observed for the stability of the products and the ESP charges with R2 of 0.62 and 0.58, respectively. Slightly worse correlations are observed for the intermediate stabilities and NBO charges (cf. Table 2 and Figure S3). As shown in Figures 5 and S4, the stabilities of the intermediate, the cation, and the product are generally more favorable for the SOMs than the non-SOMs, whereas positively charged C atoms are characteristic for the atoms being SOMs.
Table 2. Correlation between the TS Energies and DFT-Calculated Propertiesa.
| stability of intermediate | stability of cation intermediate | stability of product | ESP charges | NBO charges | |
|---|---|---|---|---|---|
| TS energies | 0.52 | 0.15 | 0.62 | 0.58 | 0.47 |
| stability of intermediates | 0.06 | 0.31 | 0.43 | 0.25 | |
| stability of cation intermediates | 0.32 | 0.31 | 0.34 | ||
| stability of products | 0.71 | 0.79 | |||
| ESP charges | 0.72 |
The TS energies are given relative to the energies of separated substrate and MoCo at the B3LYP level.
Figure 5.

Box plot of calculated properties for the reaction. Red bars show the values for the SOMs and blue for the other aromatic C atoms.
More important than the correlation coefficient is the ability to distinguish between SOMs and non-SOMs. As already noted, the TS energies are lowest for the SOMs, and thus, they could be good predictors of where the AO reaction oxidizes the substrates. This is also clearly observed in the box plots in Figure 5 where the box and whiskers for the SOMs are in a narrow range of ∼150 kJ/mol with only two outliers (C4 of quinazoline and C6 of famciclovir, see also Figure 4). The product stabilities and ESP or NBO charges of the SOMs are separated from the other aromatic C atoms in Figure 5. Thus, for larger sets of compounds, it would be reasonable to consider these simpler properties as indicators of AO-mediated SOM than determining the more time-consuming TS energies.
Torres et al.10 investigated the relationship between the charge of a carbon atom near a nitrogen atom and the ability to predict the SOM in the compound. Their conclusion was that ESP charges were not as predictive as other reactivity measures because they were not always able to predict the primary SOM correctly. Although the ESP (and NBO) charges do not correlate well with the activation energy of the different sites, the most positive charges are the SOMs in all compounds, except for DACA. In DACA, C3 has a charge that is +0.13 compared with +0.11 for C9 (cf. Table S12C). Torres et al. instead used the stability of the tetrahedral intermediate, with OH added to the C atom and an H atom added to the N atom, resulting in a net addition of water, to predict >90% of the SOMs correctly. The stability of the intermediate was also used as a descriptor by Jones and Korzekwa and Xu et al.11,12 As seen in Figure 5, there is a separation between the SOM and the non-SOM stability for our estimates of the tetrahedral intermediates. Two sites are not correctly predicted (the intermediate generated from a reaction on C4 of quinoline and C7 of phenanthridine is more stable). The energy of the intermediate for C9 of DACA, on the other hand, is very low (214 kJ/mol compared with all other intermediates). Although the decision-tree model is an ultrafast method, the determination of the energy difference at the B3LYP level reduces the speed of the method and it could be interesting to develop a model based on the ESP charges, where only one calculation is necessary, instead of a calculation of both the substrate and the intermediate.
Conclusions
We have studied three different mechanisms for the AO-mediated oxidation of N-containing heterocyclic compounds and their trend in activation energies. We showed that the TS energies for all three reactions correlate. In addition, B3LYP and B3LYP-D3 energies correlate. For one of the mechanisms, the concerted mechanism, we have determined the activation energy for the formation of all possible metabolites showing that the reaction with the lowest activation barrier generally corresponds to the formation of the experimentally observed metabolite.
We have also shown that the SOM for AO-mediated metabolism can be derived from various molecular properties such as the ESP partial charges, which indicates that it will be possible to develop methods to predict SOM, which are considerably faster than the methods relying on identifying TSs.
Methods
The MoCo was modeled as a Mo–dithiolene complex, with an oxygen, a hydroxyl, and a sulfur substituent. All calculations were done at the DFT/B3LYP or DFT/B3LYP-D3 level of theory.28−31 Previous calculations on reactions with Mo-containing model systems with 1,2-dimethyldithiolene bound have shown that B3LYP can reproduce coupled cluster-derived energies.32,33 The TSs were identified by scanning the reaction coordinates before the TS optimizations were done. The TSs were verified by making a frequency calculation. All structures were optimized using the def2-SVP basis set, and the final energies were determined using the def2-larger TZVPP basis set.34 The energies were corrected for the zero-point vibrations. It is indicated that the energies are corrected for solvation with the COSMO model with dielectric constants of 4 and 80.35 The COSMO calculations were done with optimized radii of H, C, N, O, and S and 2.0 Å for Mo.7 All activation energies were given relative to the sum of the MoCo and substrate energies. ESP and NBO charges were calculated for each atom of the different substrates with single-point DFT calculations, using the def2-SVP basis set. For the stability of intermediates, cation and product geometry optimizations were conducted using the def2-SVP basis set.34 As shown in Figure S2, the difference in energy was calculated between the substrate and MoCo and (1) the sum of the product and the protonated and dehydroxylated MoCo; (2) the sum of the intermediate and the dehydroxylated MoCo; and (3) the sum of the cation and the MoCo + H–. TURBOMOLE was used for all DFT calculations.36
Glossary
Abbreviations
- AO
aldehyde oxidase
- DFT
density functional theory
- SOM
site of metabolism
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b00658.
Author Contributions
The manuscript was written through contributions from all authors. All authors have given approval to the final version of the manuscript.
This work was supported by the EU via the ARIADME project (607517).
The authors declare no competing financial interest.
Supplementary Material
References
- Terao M.; Romão M. J.; Leimkühler S.; Bolis M.; Fratelli M.; Coelho C.; Santos-Silva T.; Garattini E. Structure and function of mammalian aldehyde oxidases. Arch. Toxicol. 2016, 90, 753–780. 10.1007/s00204-016-1683-1. [DOI] [PubMed] [Google Scholar]
- Pryde D. C.; Dalvie D.; Hu Q.; Jones P.; Obach R. S.; Tran T.-D. Aldehyde Oxidase: An Enzyme of Emerging Importance in Drug Discovery. J. Med. Chem. 2010, 53, 8441–8460. 10.1021/jm100888d. [DOI] [PubMed] [Google Scholar]
- Garattini E.; Terao M. The role of aldehyde oxidase in drug metabolism. Expert Opin. Drug Metab. Toxicol. 2012, 8, 487–503. 10.1517/17425255.2012.663352. [DOI] [PubMed] [Google Scholar]
- Sodhi J. K.; Wong S.; Kirkpatrick D. S.; Liu L.; Khojasteh S. C.; Hop C. E. C. A.; Barr J. T.; Jones J. P.; Halladay J. S. A novel reaction mediated by human aldehyde oxidase: amide hydrolysis of GDC-0834. Drug Metab. Dispos. 2015, 43, 908–915. 10.1124/dmd.114.061804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfaro J. F.; Jones J. P. Studies on the mechanism of aldehyde oxidase and xanthine oxidase. J. Org. Chem. 2008, 73, 9469–9472. 10.1021/jo801053u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano T.; Ochi N.; Sato H.; Sakaki S. Oxidation reaction by xanthine oxidase. Theoretical study of reaction mechanism. J. Am. Chem. Soc. 2007, 129, 8131–8138. 10.1021/ja068584d. [DOI] [PubMed] [Google Scholar]
- Li J.; Ryde U. Comparison of the active-site design of molybdenum oxo-transfer enzymes by quantum mechanical calculations. Inorg. Chem. 2014, 53, 11913–11924. 10.1021/ic5010837. [DOI] [PubMed] [Google Scholar]
- Metz S.; Thiel W. QM/MM studies of xanthine oxidase: variations of cofactor, substrate, and active-site Glu802. J. Phys. Chem. B 2010, 114, 1506–1517. 10.1021/jp909999s. [DOI] [PubMed] [Google Scholar]
- Metz S.; Thiel W. A combined QM/MM study on the reductive half-reaction of xanthine oxidase: substrate orientation and mechanism. J. Am. Chem. Soc. 2009, 131, 14885–14902. 10.1021/ja9045394. [DOI] [PubMed] [Google Scholar]
- Torres R. A.; Korzekwa K. R.; McMasters D. R.; Fandozzi C. M.; Jones J. P. Use of density functional calculations to predict the regioselectivity of drugs and molecules metabolized by aldehyde oxidase. J. Med. Chem. 2007, 50, 4642–4647. 10.1021/jm0703690. [DOI] [PubMed] [Google Scholar]
- Jones J. P.; Korzekwa K. R. Predicting intrinsic clearance for drugs and drug candidates metabolized by aldehyde oxidase. Mol. Pharm. 2013, 10, 1262–1268. 10.1021/mp300568r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y.; Li L.; Wang Y.; Xing J.; Zhou L.; Zhong D.; Luo X.; Jiang H.; Chen K.; Zheng M.; Deng P.; Chen X. Aldehyde Oxidase Mediated Metabolism in Drug-like Molecules: A Combined Computational and Experimental Study. J. Med. Chem. 2017, 60, 2973–2982. 10.1021/acs.jmedchem.7b00019. [DOI] [PubMed] [Google Scholar]
- Coelho C.; Mahro M.; Trincão J.; Carvalho A. T. P.; Ramos M. J.; Terao M.; Garattini E.; Leimkühler S.; Romão M. J. The first mammalian aldehyde oxidase crystal structure: insights into substrate specificity. J. Biol. Chem. 2012, 287, 40690–40702. 10.1074/jbc.M112.390419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibo E. B.; Gilchrist J. H.; Lee C. H. Electronic probes of the mechanism of substrate oxidation by buttermilk xanthine oxidase: role of the active-site nucleophile in oxidation. Biochemistry 1987, 26, 3032–3037. 10.1021/bi00385a013. [DOI] [PubMed] [Google Scholar]
- Metz S.; Wang D.; Thiel W. Reductive half-reaction of aldehyde oxidoreductase toward acetaldehyde: a combined QM/MM study. J. Am. Chem. Soc. 2009, 131, 4628–4640. 10.1021/ja805938w. [DOI] [PubMed] [Google Scholar]
- Coelho C.; Foti A.; Hartmann T.; Santos-Silva T.; Leimkühler S.; Romão M. J. Structural insights into xenobiotic and inhibitor binding to human aldehyde oxidase. Nat. Chem. Biol. 2015, 11, 779–783. 10.1038/nchembio.1895. [DOI] [PubMed] [Google Scholar]
- Moss G. P. Nomenclature of Fused and Bridged Fused Ring Systems. Pure Appl. Chem. 1998, 70, 143–216. 10.1351/pac199870010143. [DOI] [Google Scholar]
- Barr J. T.; Jones J. P. Inhibition of human liver aldehyde oxidase: implications for potential drug-drug interactions. Drug Metab. Dispos. 2011, 39, 2381–2386. 10.1124/dmd.111.041806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afzal O.; Kumar S.; Haider M. R.; Ali M. R.; Kumar R.; Jaggi M.; Bawa S. A review on anticancer potential of bioactive heterocycle quinoline. Eur. J. Med. Chem. 2015, 97, 871–910. 10.1016/j.ejmech.2014.07.044. [DOI] [PubMed] [Google Scholar]
- Beedham C.; Al-Tayib Y.; Smith J. A. Role of guinea pig and rabbit hepatic aldehyde oxidase in oxidative in vitro metabolism of cinchona antimalarials. Drug Metab. Dispos. 1992, 20, 889. [PubMed] [Google Scholar]
- Beedham C.; Bruce S. E.; Critchley D. J.; Al-Tayib Y.; Rance D. J. Species variation in hepatic aldehyde oxidase activity. Eur. J. Drug Metab. Pharmacokinet. 1987, 12, 307–310. 10.1007/bf03189919. [DOI] [PubMed] [Google Scholar]
- Rashidi M. R.; Smith J. A.; Clarke S. E.; Beedham C. In Vitro Oxidation of Famciclovir and 6-Deoxypenciclovir by Aldehyde Oxidase From Human, Guinea Pig, Rabbit, and Rat Liver. Drug Metab. Dispos. 1997, 25, 805. [PubMed] [Google Scholar]
- Barr J. T.; Jones J. P. Evidence for Substrate-Dependent Inhibition Profiles for Human Liver Aldehyde Oxidase. Drug Metab. Dispos. 2012, 41, 24. 10.1124/dmd.112.048546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield P. C.; Robertson I. G. C.; Paxton J. W.; McCrystal M. R.; Evans B. D.; Kestell P.; Baguley B. C. Metabolism of N-[2-(dimethylamino)ethyl]acridine-4-carboxamide in cancer patients undergoing a phase I clinical trial. Cancer Chemother. Pharmacol. 1999, 44, 51–58. 10.1007/s002800050944. [DOI] [PubMed] [Google Scholar]
- Sorouraddin M.-H.; Fooladi E.; Naseri A.; Rashidi M.-R. A novel spectrophotometric method for determination of kinetic constants of aldehyde oxidase using multivariate calibration method. J. Biochem. Biophys. Methods 2008, 70, 999–1005. 10.1016/j.jbbm.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y.; Matsumura T.; Ichida K.; Okamoto K.; Nishino T. Human xanthine oxidase changes its substrate specificity to aldehyde oxidase type upon mutation of amino acid residues in the active site: roles of active site residues in binding and activation of purine substrate. J. Biochem. 2007, 141, 513–524. 10.1093/jb/mvm053. [DOI] [PubMed] [Google Scholar]
- Romão M. J.; Coelho C.; Santos-Silva T.; Foti A.; Terao M.; Garattini E.; Leimkühler S. Structural basis for the role of mammalian aldehyde oxidases in the metabolism of drugs and xenobiotics. Curr. Opin. Chem. Biol. 2017, 37, 39–47. 10.1016/j.cbpa.2017.01.005. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A: At., Mol., Opt. Phys. 1988, 38, 3098–3100. 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785–789. 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- Hu L.; Chen H. Assessment of DFT methods for computing activation energies of Mo/W-mediated reactions. J. Chem. Theory Comput. 2015, 11, 4601–4614. 10.1021/acs.jctc.5b00373. [DOI] [PubMed] [Google Scholar]
- van Severen M.-C.; Andrejić M.; Li J.; Starke K.; Mata R. A.; Nordlander E.; Ryde U. A quantum-mechanical study of the reaction mechanism of sulfite oxidase. J. Biol. Inorg. Chem. 2014, 19, 1165–1179. 10.1007/s00775-014-1172-z. [DOI] [PubMed] [Google Scholar]
- Weigend F.; Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Klamt A.; Schüürmann G. COSMO: a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc., Perkin Trans. 1 1993, 799–805. 10.1039/p29930000799. [DOI] [Google Scholar]
- TURBOMOLE, Version 6.3; TURBOMOLE GmbH, University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 2011.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.