

Abstract
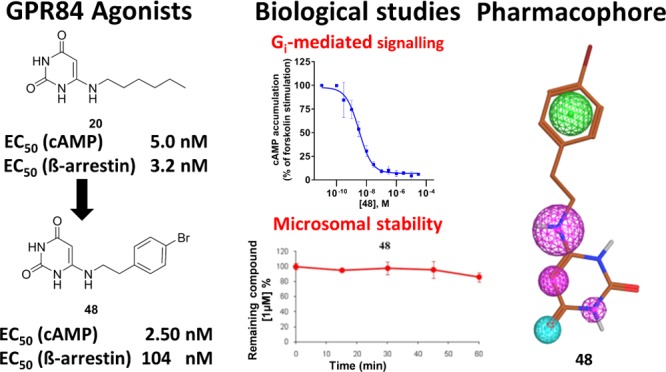
GPR84, a Gi protein-coupled receptor that is activated by medium-chain (hydroxy)fatty acids, appears to play an important role in inflammation, immunity, and cancer. Recently, 6-octylaminouracil (4) has been reported to act as an agonist at GPR84. Here, we describe the synthesis of 69 derivatives and analogs of 4, 66 of which represent new compounds. They were evaluated in (a) cyclic adenosine monophosphate accumulation and (b) β-arrestin assays in human GPR84-expressing cells. Potent nonbiased as well as G protein-biased agonists were developed, e.g., 6-hexylamino-2,4(1H,3H)-pyrimidinedione (20, PSB-1584, EC50 5.0 nM (a), 3.2 nM (b), bias factor: 0) and 6-((p-chloro- and p-bromo-phenylethyl)amino)-2,4(1H,3H)-pyrimidinedione (47, PSB-16434, EC50 7.1 nM (a), 520 nM (b), bias factor: 1.9 = 79-fold Gi pathway-selective; 48, PSB-17365, EC50 2.5 nM (a), 100 nM (b), bias factor 1.3 = 20-fold selective), which were selective versus other free fatty acid-activated receptors. Compounds 20 and 48 were found to be metabolically stable upon incubation with human liver microsomes. A pharmacophore model was created on the basis of structurally diverse lipidlike GPR84 agonists.
Introduction
G protein-coupled receptors (GPCRs) are seven transmembrane receptors that constitute one of the largest gene families in the human genome.1,2 They are considered attractive targets for the development of drugs for many human diseases, and it has been estimated that about 30% of modern drugs target GPCRs, including many of the world’s best-selling pharmaceuticals.3,4 More than 100 GPCRs represent orphan receptors, whose natural ligands are still unknown or unconfirmed.5 GPR84 is a so far poorly investigated GPCR that is activated by micromolar concentrations of medium-chain fatty acids with carbon chain lengths of 9–14, which couples to the Gi/o pathway.6−8 The receptor is predominantly expressed in immune system-related tissues and cells, such as bone marrow, spleen, lung, lymph nodes,9 and brain microglia. Moreover, it is expressed in adipose tissue.10,11 GPR84 expression is upregulated in macrophages upon lipopolysaccharide (LPS) stimulation, where it regulates the production of interleukin-12 (IL-12), a proinflammatory cytokine that controls the balance of T helper (Th1/Th2) responses and plays an important role in inflammatory diseases such as rheumatoid arthritis and inflammatory bowel disease.12 The potential role of GPR84 in this context has been supported by the finding that knockout of GPR84 in T cells leads to augmented production of the anti-inflammatory cytokine IL-4.12 It has been demonstrated that the zebrafish GPR84 (zGPR84) is involved in the accumulation of lipid droplets, and undecanoic acid was shown to amplify lipopolysaccharide-induced production of the proinflammatory cytokine IL-12p40 through GPR84, indicating that GPR84 may play a role in directly linking fatty acid metabolism to immune responses.13 In animal models of endotoxemia and multiple sclerosis (experimental autoimmune encephalomyelitis), it was found that GPR84 is upregulated in microglia, suggesting that GPR84 may also regulate neuroinflammatory processes.10 A recent study reported that GPR84 may act as a sensor in amyloid pathology, and its deficiency led to reduced microgliosis and dendritic homeostasis in a mouse model of Alzheimer’s disease.14 Thus, GPR84 is a proinflammatory receptor.8 Potent and selective GPR84 agonists and antagonists are required to further study its (patho)physiological roles and its potential as a future drug target. GPR84 agonists might be useful for the treatment of cancer by activating the immune response (immuno-oncology), whereas desensitization of GPR84 by agonists might lead to a functional blockade of the receptor, resulting in anti-inflammatory effects and possibly antiproliferative effects on acute myeloid leukemia.15
In 2006, GPR84 was shown to be activated by medium-chain free fatty acids with chain lengths of C9–14. In particular, decanoic acid (C10, 1, Figure 1), undecanoic acid (C11) and lauric acid (C12) were found to be among the most potent agonists of GPR84.7 Other studies by Suzuki et al. reported 2- or 3-hydroxy fatty acids to be comparatively more potent than the nonhydroxylated fatty acids,8 and recently, Hoffmann et al. have found that 3-hydroxytetradecanoic acid effectively activates GPR84 (compounds 2 and 3, see Figure 1).16 Moreover, 6-octylaminouracil (4, Figure 1)8 and the structurally related (hexylthio)pyrimidine-4,6-diol (5, Figure 1) and its derivatives were identified as synthetic GPR84 agonists.17,18 Compound 4, which displays a lipid-mimetic structure, was reported to activate human GPR84 with an EC50 value of 512 nM determined in [35S]GTPγS binding assays.8 Compound 5 exhibited GPR84 agonistic activity with an EC50 of 139 nM determined in a calcium mobilization assay in HEK293 cells recombinantly expressing the human GPR84.17,18
Figure 1.

Structures of reported GPR84 agonists.
Embelin (6, Figure 1), a natural product isolated from the plant Embelia ribes (Myrsinaceae), also activates GPR84 (EC50 795 nM, cyclic adenosine monophosphate (cAMP) assay, see Table 1).19 Embelin was reported to have anticancer, antioxidant, anthelmintic, antifertility, antitumor, antiapoptotic, antimicrobial, analgesic, and anti-inflammatory activities.20−22 The safety and toxicity profile of embelin in rodents and nonrodents was investigated and the compound was found to be safe up to 3 g/kg (orally) when tested in rodents after acute exposure. Embelin was reported to act as an X-linked inhibitor of apoptosis (IC50 4.1 μM) besides interaction with other targets,23 but it is even more potent as an agonist of GPR84. Diindolylmethane (DIM, 7),8,24 another natural product-derived compound that activates GPR84, also possesses anticancer activity.25 In a previous study, we investigated the structure–activity relationships (SARs) of DIM derivatives and analogues and developed a potent and selective GPR84 agonist derived from DIM, compound 8 (PSB-16671), with an EC50 value of 41.3 nM (Figure 1).26 DIM derivatives were characterized as agonists with an ago-allosteric mechanism of action as compared to fatty acids.26,27 In the present study, we used the potent and selective GPR84 agonist 4 as a lead structure. Our goal was to study the SARs of 6-aminouracil derivatives and analogues as GPR84 agonists, to improve their potency, optimize their properties, including selectivity and metabolic stability, and develop Gi protein-biased agonists.
Table 1. Agonistic Activity of 6-Alkylaminouracils and Standard Agonists at GPR84a.

| human
GPR84 |
||||
|---|---|---|---|---|
| cAMP assayb | β-arrestin assay | |||
| compound | R1 | EC50 ± SEM (μM) (or percent receptor activation at 10 μM)b [efficacy]c | EC50 ± SEM (μM) (or percent receptor activation at 10 μM) [efficacy]d | bias factor ΔΔlog(Emax/EC50)e |
| Standard Agonists (For Structures, See Figure 1) | ||||
| decanoic acid (1) | 7.4 ± 0.4 [100%] | 6.1 ± 0.4 [92%] | 0.0 | |
| (R,S)-3-hydroxydodecanoic acid (2) | 1.3 ± 0.15 [92%] | 4.4 ± 1.30 [90%] | 0.0 | |
| embelin (6) | 0.80 ± 0.17 [86%] | 0.42 ± 0.19 [100%] | –0.3 | |
| urapidil (for structure see above) | >10 (7%) | n.d.f | n.d. | |
| 2-hexylthiopyrimidine-4,6-diol (5)g | 0.0066 ± 0.0041 [153%] | 0.078 ± 0.049 [242%] | 0.9 | |
| Structure A: 6-Alkylaminouracils | ||||
| 4(8,30) | –CH2(CH2)6–CH3 | 0.017 ± 0.003 [104%] | 0.11 ± 0.04 [189%] | 0.6 |
| 13 | –CH2CH3 | >10 (15%) | n.d. | n.d. |
| 14 | –CH2CH2CH3 | ≫10 (−9%) | n.d. | n.d. |
| 15 | –CH2CH(CH3)2 | >10 (11%) | n.d. | n.d. |
| 16 | –CH2C(CH3)3 | >10 (4%) | n.d. | n.d. |
| 17 | –CH2CH2CH(CH3)2 | >10 (14%) | n.d. | n.d. |
| 18 | –CH2CH2C(CH3)3 | >10 (18%) | n.d. | n.d. |
| 19 | –CH2(CH2)3–CH3 | 0.46 ± 0.13 [106%] | 4.8 ± 1.3 [147%] | 0.9 |
| 20 (PSB-1584) | –CH2(CH2)4–CH3 | 0.0050 ± 0.0013 [127%] | 0.0032 ± 0.0011 [98%] | 0.0 |
| 21 | –CH2(CH2)5–CH3 | 0.012 ± 0.006 [113%] | 0.39 ± 0.07 [250%] | 1.2 |
| 22 | –CH2(CH2)7–CH3 | 0.030 ± 0.010 [118%] | 0.034 ± 0.007 [142%] | 0.0 |
| 23 | –CH2(CH2)8–CH3 | 0.021 ± 0.002 [109%] | 0.36 ± 0.01 [109%] | 1.3 |
| 24 | –CH2CH(CH2CH3)–(CH2)3CH3 | >10 (14%) | n.d. | n.d. |
| 25 | –CH2(CH2)4–CH(CH3)2 | 5.9 ± 2.6 [99%] | n.d. | n.d. |
| 26 | –CH2–(CH2)2–S–CH3 | 23 ± 3 [100%] | n.d. | n.d. |
| 27 | –CH2(CH2)5–OH | ≥10 (51 ± 8%) | n.d. | n.d. |
| 28 | –CH2(CH2)6–OH | 2.0 ± 0.5 [105%] | 2.1 ± 0.6 [211%] | –0.2 |
| 29 | –CH2(CH2)4–COOH | >10 (25%) | n.d. | n.d. |
| 30 | –CH2(CH2)6–COOH | >10 (13%) | n.d. | n.d. |
| Structure B: 3-Methyl-Substituted Uracil Derivatives | ||||
| 31 | –CH2(CH2)6–CH3 | 0.72 ± 0.27 [95%] | >10 (32%) | >1.2 |
| 32 | –CH2(CH2)7–CH3 | 2.0 ± 0.5 [74%] | n.d. | n.d. |
| 33 | –CH2(CH2)8–CH3 | 1.9 ± 0.68 [72%] | >10 (8%) | >0.6 |
| Structure C: 1-Methyl-Substituted Uracil Derivatives | ||||
| 34 | –CH2(CH2)4–CH3 | >10 (32%) | n.d. | n.d. |
| 35 | –CH2(CH2)6–CH3 | >10 (34%) | n.d. | n.d. |
| Structure D: 1,3-Dimethyl-Substituted Uracil Derivative | ||||
| 36 | –CH2(CH2)6–CH3 | >10 (25%) | n.d. | n.d. |
| Structure E: N6-Methyl-Substituted Uracil Derivatives | ||||
| 37 | –CH2(CH2)4–CH3 | 0.11 ± 0.04 [106%] | 4.2 ± 0.7 [278%] | 1.2 |
| 38 | –CH2(CH2)6–CH3 | 0.038 ± 0.020 [107%] | 0.64 ± 0.02 [309%] | 0.8 |
| Structure F: 5-Alkylamino-Substituted Uracil Derivative | ||||
| 39 | –CH2(CH2)6–CH3 | >10 (6%) | n.d. | n.d. |
EC50 values are shown in bold. Efficacy is in italic.
Inhibition of forskolin (10 μM)-induced decrease in cAMP accumulation.
Efficacy (Emax) relative to the max. effect of decanoic acid (100 μM) (=100%).
Efficacy (Emax) relative to the max. effect of embelin (10 μM) (=100%).
Bias factor was calculated as described in Experimental Section.
n.d., not determined.
A literature value of 0.144 μM was reported for this compound, determined in a homogeneous time-resolved fluorescence assay (Cisbio) on HEK293 cells stably expressing human GPR84.18
Results and Discussion
Compound Design
To get insights into the SARs of uracil derivatives as GPR84 agonists, the following structural modifications of the lead agonist 4 were made (Figure 2): (A) optimization of the alkyl chain length; (B) introduction of aromatic residues into the side chain, probing the importance of the hydrogen bond donors: (C) N1–H, (D) N3–H, and (E) N6–H; and (F) introduction of a substituent at position 5 of the uracil moiety.
Figure 2.

Structural modifications of lead compound 4.
Chemistry
All target compounds were synthesized by coupling of the 6-chlorouracil derivative 12 or 13 with the appropriate amines. The commercially available barbituric acid 9 was converted to 2,4,6-trichloropyrimidine (10) by treatment with phosphorus oxychloride in the presence of dimethylaniline. The key intermediate 6-chlorouracil (11) was obtained by heating 10 to reflux with aqueous sodium hydroxide solution. 6-Chloro-1-methyluracil (12a) and 6-chloro-3-methyluracil (12b) were synthesized by treatment of 11 with methyl iodide in the presence of potassium carbonate, followed by silica gel column chromatography using methanol (1%) in dichloromethane. 1,3-Dimethyl-6-chlorouracil (12c) was synthesized by treating 11 with an excess of methyl iodide. Finally, 6-chlorouracils 12a–c and 5-bromouracil 13, respectively, were treated with the appropriate amine in 1-butanol under reflux overnight to produce the desired products 4 and 14–71. Bromination was achieved by treatment of the appropriate uracil derivative with N-bromosuccinimide (NBS) to yield 72–75, whereas reacting 6, 20, 21, 42, or 43 with sodium nitrite under acidic conditions led to the 5-nitrosouracil derivatives 76–80. The structures of all synthesized final products were confirmed by 1H and 13C NMR, or attached proton test (13Capt) NMR spectroscopy. The purity of the compounds was determined by high-performance liquid chromatography (HPLC) coupled to UV and electrospray ionization mass spectrometry (ESI-MS), confirming a purity of at least 95% (Scheme 1).
Scheme 1. Synthesis of 5-Alkylamino or 6-(Ar)Alkylamino-Substituted Uracil Derivatives (4 and 14–80).

Reagents and condition: (i) POCl3, dimethylaniline, 50 °C, 7 h and 125 °C, 20 min; (ii) aqueous NaOH, reflux, 3 h; (iii) K2CO3, CH3I, dimethyl sulfoxide (DMSO), room temperature (rt), 2 h or K2CO3, N,N′-dimethylformamide (DMF), 6 h for 12a and 12b or K2CO3, CH3I, DMF, 50 °C, 12 h for 12c; (iv) amines, 1-butanol, reflux, 12 h; (v) pyridine, N-bromosuccinimide (NBS), 80 °C, 2 h for 72–75 from 20, 6, 47, and 43; or NaNO2, CH3COOH/H2O (1:1), 60–70 °C, 1 h and cooled to 4 °C for 76–80 from 6, 20, 21, 42, and 43, 67–79%; (vi) hexylamine, 1-butanol, reflux, 12 h.
Pharmacological Evaluation
All synthesized compounds were initially investigated in cAMP accumulation assays at a concentration of 10 μM for their potency to inhibit forskolin (10 μM)-induced cAMP accumulation. Chinese hamster ovary (CHO) cells stably expressing the Gi protein-coupled human GPR84 were employed. Full concentration–response curves were determined, and EC50 values were calculated for compounds that showed more than 50% inhibition of cAMP accumulation in preliminary tests (see Tables 1–3). Efficacy of the compounds was determined by comparing their maximal effects to the maximal signal induced by decanoic acid (100 μM; EC50 7.42 μM). Standard GPR84 agonists were tested under the same conditions for comparison (see Table 1). Selected compounds that did not activate the receptor were screened in antagonist assays versus decanoic acid (20 μM) at a concentration of 10 μM. GPR84 specificity of the observed effects was confirmed for the most potent compounds by testing them in the same assay, but in nontransfected CHO cells lacking GPR84 expression. Selected compounds were also investigated in β-arrestin 2 recruitment assays using the β-galactosidase fragment complementation technology (PathHunter, DiscoverX)28,29 (see Tables 1–3). Potent GPR84 agonists were additionally studied at human free fatty acid receptors FFAR1 and FFAR4 to explore their selectivity (see Tables S1 and S2).26
Table 3. Various 6-Substituted and 5,6-Disubstituted Uracil Derivatives.

Inhibition of forskolin (10 μM)-induced decrease in cAMP accumulation.
Efficacy (Emax) relative to the max. effect of decanoic acid (100 μM) (=100%).
Efficacy (Emax) relative to the max. effect of embelin (10 μM) (=100%).
Bias factor was calculated as described in Experimental Section.
n.d., not determined.
Structure–Activity Relationships
cAMP Accumulation Assays
Previously, 6-octylaminouracil (4) was identified as a GPR84 agonist displaying an EC50 value of 512 nM determined in [35S]GTPγS binding assays using Sf9 cell membranes expressing a human GPR84-Gαi fusion protein.8 This relatively high EC50 value may be due to the highly artificial test system that was employed. In our cAMP accumulation assay using CHO cells transfected with the human GPR84, 4 induced an inhibition of forskolin-induced cAMP accumulation with an EC50 value of 17 nM (see Figure 6). It was about 440-fold more potent than the standard agonist decanoic acid (EC50 7400 nM, p = 0.0003). We confirmed that the effect seen with 4 in the cAMP assay was clearly due to GPR84 activation as it had no effect in nontransfected CHO cells (see Figure 4).
Figure 6.

Concentration–response curves of selected compounds at human GPR84 in cAMP assays (A, C) and in enzyme fragment complementation β-arrestin recruitment assays (B, D). Mean values ± SEM from three to four independent experiments performed in duplicates are shown. For EC50 values, see Tables 1 and 2.
Figure 4.
Evaluation of selected compounds at wild-type (wt) CHO cells in cAMP accumulation assays. CHO wt cells were preincubated with the respective test compounds at the indicated concentrations for 5 min. Then, 10 μM forskolin was added and the cells were incubated for additional 15 min. The maximal forskolin-induced cAMP accumulation in the absence of test compound stimulation was defined as 100%. Mean values ± standard error of the mean (SEM) from three independent experiments performed in duplicates are shown.
To gain deeper insights into the SARs of uracil derivatives as agonists of GPR84, we initially focused on modifying the hydrophobic alkyl tail: a change in alkyl chain length ranging from C1 to C7 and C9 to C10 (see Table 1) demonstrated that the right alkyl chain length was essential for high potency of the compounds at GPR84. A short chain length of C2–3 as in 13 and 14, as well as a branched alkyl chain as in 15–18, yielded inactive uracil derivatives, whereas 19 with an alkyl chain length of five carbon atoms displayed moderate agonistic activity with an EC50 of 460 nM. Increasing the chain length by one more methylene unit to hexyl (20) led to a highly potent agonist displaying an EC50 value of 5.0 nM, 92-fold more potent than 19 (p = 0.0391). Further extension of the alkyl chain length to C7 (21, EC50 12 nM), C9 (22, EC50 30 nM), or C10 (23, EC50 21 nM) led to slightly reduced activities. Branching of the alkyl chain as in N6-(R,S)-(2-ethyl)hexyluracil (24) abolished activity, indicating limited space. N6-Methylthiopropyluracil (26, EC50 23 000 nM), an analogue of N6-pentyluracil (19, EC50 460 nM), in which a methylene group was exchanged for a (lipophilic) sulfur atom, was surprisingly 50-fold less potent than 19. The rank order of potency for the length of the alkyl chain attached to the uracil core was as follows: C6 (20, EC50 5.0 nM, vs 4, p = 0.0185) ≥ C7 (21, EC50 12 nM) ≥ C8 (4, EC50 17 nM) ≥ C10 (23, EC50 21 nM) ≥ C9 (22, EC50 30 nM) > C5 (19, EC50 460 nM, p = 0.0268). Next, polar groups such as hydroxy or carboxy were introduced at the end of the alkyl chain yielding compounds 27, 28 or 29, 30. Among them, only the hydroxyheptyl derivative 28 (EC50 2000 nM) showed moderate activity; its potency was significantly decreased in comparison to the lead compound 4 (p = 0.0130); the other polar derivatives were all inactive, again indicating that a highly lipophilic pocket harbored the alkyl chain.
Our next effort was to investigate the importance of the NH functions, N1–H, N3–H, and N6–H of the 6-aminouracil derivatives. Methylation of N3 reduced the agonistic potency by more than 40-fold (compare 31 (EC50 720 nM) with 4 (EC50 17 nM, 42-fold difference), 32 (EC50 2000 nM) with 22 (EC50 30 nM, 67-fold difference), and 33 (EC50 1900 nM) with 23 (EC50 21 nM, 90-fold difference)). Methylation of N1 (34–35) or N1,N3-dimethylation (36) virtually abolished potency of the compounds. Thus, both NH atoms are important and may serve as hydrogen bond donors, with N1–H being more important than the N3–H atom. Substitution of the hydrogen atom at the 6-amino group (N6–H) of the uracil core with a methyl group also led to a reduction in potency of the hexyl-substituted derivative (compare 37 (EC50 110 nM) with 20 (EC50 5.0 nM), 22-fold reduction). However, surprisingly, N6-methylation of the octyl-substituted lead structure 4 only led to an insignificant (2-fold) decrease in potency (compare 38 (EC50 38 nM) with 4 (EC50 17 nM)). Taken together, these studies suggest that all NH functions in the 6-aminouracil derivatives, N1–H, N3–H, and N6–H, should be ideally unsubstituted, but a free NH function appears to be particularly important at the N1-position.
Next, we moved the octylamino substituent of lead compound 4 from the 6- to the 5-position of the uracil core, resulting in 39, which turned out to be completely inactive (EC50 > 10 μM). This confirms that the position of the hydrophobic tail is very important for its interaction with the receptor.
Subsequently, we introduced a large variety of aromatic residues attached to the N6-alkyl chain (see Table 2). Benzyl (40) and (1-naphthyl)methyl substitution (41) led to inactive compounds, whereas longer alkyl or alkoxy linkers of two to four atoms between N6 of the aminouracil core and the aromatic ring resulted in moderately to highly potent GPR84 agonists. A phenylethyl substituent led to moderate activity (42, EC50 200 nM). Further elongation of the carbon chain in the phenylpropyl derivative 43 reduced activity by 9-fold (EC50 1700 nM); however, an additional methylene group (in the phenylbutyl derivative 44, EC50 24 nM) dramatically increased potency to a value similar to that determined for the octyl-substituted lead structure 4. The lipophilic chains of both compounds, 4 and 44, have about the same length. Encouraged by these results, our next effort was to introduce substituents on the phenyl ring of the relatively potent phenylethyl-substituted compound 42 with the goal to improve its potency. The following substituents were introduced at the p-position of the phenyl ring: p-methyl (45, EC50 13 nM), p-fluoro (46, EC50 65 nM), p-chloro (47, EC50 7.1 nM), p-bromo (48, EC50 2.5 nM), p-ethyl (49, EC50 4.3 nM), p-methoxy (50, EC50 57 nM), and p-tert-butyl (51, EC50 44 nM). The obtained results showed that the substituents led to an increase in potency, and hydrophobic residues were particularly favorable. For example, the bulky p-bromo substituent in 48 displayed a 23-fold increased potency compared to the corresponding p-methoxy derivative 50, and in fact, 48 (EC50 2.5 nM) was the most potent GPR84 agonist of the present series. The rank order of potency for substituents at the p-position of the phenyl ring in N6-phenethyluracil derivatives was as follows: p-bromo (48, EC50 2.5 nM) ≥ p-ethyl (49, EC50 4.3 nM) ≥ p-chloro (47, EC50 7.1 nM) > p-methyl (45, EC50 13 nM, p = 0.0166) > p-tert-butyl (51, EC50 44 nM; p = 0.0008) ≥ p-methoxy (50, EC50 57 nM) ≥ p-fluoro (46, EC50 65 nM) ≥ p-unsubstituted (42, EC50 200 nM).
Table 2. Agonistic Activity of 6-Arylalkylamino-Substituted Uracil Derivatives.

Inhibition of forskolin (10 μM)-induced decrease in cAMP accumulation.
Efficacy (Emax) relative to the max. effect of decanoic acid (100 μM) (=100%).
Efficacy (Emax) relative to the max. effect of embelin (10 μM) (=100%).
Bias factor was calculated as described in Experimental Section.
n.d., not determined.
Subsequently, the effects of substituents at the o- or m-position of the phenyl moiety were investigated. The results showed that substituents in these positions generally reduced potency compared to compounds with substituents at the p-position. The rank order of potency for substituents at the m-position of the phenyl ring in N6-phenethyluracil derivatives was as follows: m-bromo (55, EC50 64 nM) ≥ m-methyl (52, EC50 79 nM) ≥ o-fluoro (57, EC50 100 nM, p = 0.0299 compared to 55) = o-chloro (58, EC50 100 nM) ≥ m-chloro (54, EC50 130 nM) ≥ o,m-unsubstituted (42, EC50 200 nM) > m-fluoro (53, EC50 390 nM, p = 0.0007 vs 54) > o-methyl (56, EC50 3200 nM, p < 0.0001 vs 53) ≫ o-bromo (59, EC50 > 10 000 nM). m-Methoxy substitution was not well tolerated as demonstrated by the moderate potency of the m,p-dimethoxyphenyl derivative 60 (EC50 810 nM, compared to that of the p-methoxy derivative 50, EC50 57 nM, p = 0.0327). Dichloro substitution in the m- and p-positions (61, EC50 44 nM) combined the positive effect of the p-chloro substituent and the negative effect of the m-chloro substituent. Bioisosteric replacement of the phenyl ring in 42 (EC50 200 nM) with a 2-thienyl ring (63, EC50 190 nM) was well tolerated, whereas exchange for a 3-indolyl residue significantly decreased potency (62, EC50 1400 nM, p = 0.0052). Introducing an ether linkage into the side chain to increase polarity was well tolerated (compare the phenoxypropyl derivative 65, EC50 73 nM, with its phenylbutyl analog 44, EC50 24 nM) and even led to 17-fold enhanced potency in case of the phenoxyethyl derivative 64 (EC50 100 nM) compared to the analogous phenylpropyl derivative 43 (1700 nM).
In a next small series of compounds, we rigidified the N6-substitutent by integrating the 6-amino group into cyclic structures, piperidine or piperazine (see Table 3). The 4-phenylpiperidinyl derivative 66 was inactive, whereas the corresponding, somewhat more flexible, 4-benzylpiperidinyl-substituted compound 67 (EC50 130 nM) was equipotent to the N6-phenylethyl-substituted analogue 42 and 13-fold more potent than the N6-phenylpropyl-substituted compound 43. Four 6-piperazinyluracil derivatives were obtained, which were substituted on the free piperazine N-atom (68–71). All four compounds showed submicromolar potency with IC50 values between 38 and 150 nM, indicating that various residues are tolerated ranging from substituted phenyl (68, 69) to indanyl (70) and even to (1-naphthyl)methyl (71).
Finally, we investigated the effect of substituting the uracil 5-position of 5-(ar)alkylaminouracil derivatives (72–80, Table 3). We selected potent agonists discussed above (4, 20, 21, 42, 43, and 48) and introduced a bromo or nitroso substituent at position 5. The resulting derivatives showed only moderate to weak potency compared to their parent compounds (compare for 5-bromo substitution 72 vs 20 (1500-fold difference, p = 0.0094), 73 vs 4 (119-fold difference, p = 0.0287), 74 vs 48 (148-fold difference, p < 0.00001), and 75 vs 43 (>6-fold difference) and for 5-nitroso substitution 76 vs 20 (1570-fold difference, p = 0.0001), 77 vs 21 (40-fold difference, p = 0.0006), 78 vs 4 (36-fold difference, p = 0.0022), and 79 vs 42 (>51-fold difference, p < 0.0001)).
Selected compounds that were inactive as agonists were subsequently tested for potential blockade of the receptor in cAMP assays. However, none of them was found to be an antagonist of GPR84 (data not shown).
The N6-substituted 1,3-dimethyl-6-aminouracil derivative uradipil (see Table 1), an α1-adrenoreceptor antagonist and serotonin 5-HT1A receptor agonist, which is therapeutically used as an antihypertensive drug in Europe, neither activated nor inhibited GPR84 as determined in cAMP accumulation assays.
Figure 3 summarizes the structure–activity relationships of the investigated compounds as agonists of the human GPR84: (i) the length of the alkyl chain plays an important role in determining the potency; an optimal chain length of six carbon atoms was determined. Introducing an aromatic residue into the alkyl chain was well tolerated and in some cases led to increased potency. However, branching of the alkyl chain or attachment of polar groups (−OH or −COOH) at its terminus reduced or abolished the activity; (ii) free N3–H and particularly N1–H functions in the uracil core structure were found to be very important for activity; (iii) the substitution at the 6-position of uracil was crucial, whereas 5-substitution abolished the activity.
Figure 3.

Structure–activity relationships of uracil derivatives at human GPR84 as determined in cAMP assays.
Efficacy in cAMP Assays
The maximal effect of the physiological agonist decanoic acid (1) was set at 100% and compared to the maximal effects observed for the investigated uracil derivatives (see Tables 1–3). Lead structure 4 displayed a similar efficacy (104%), and most of the potent uracil derivatives showed the same or even higher efficacy. For example, the most potent agonists 20, 47, and 48 had efficacies of 127–137%. Our results showed that potent uracil-derived GPR84 agonists fully activate the Gi protein-coupled pathway.
Specificity of Effects Determined in cAMP Accumulation Assays
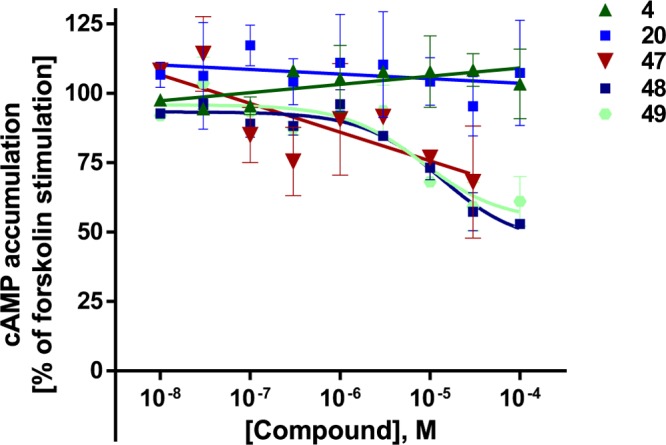
Selected potent GPR84 agonists, namely, 20, 47, 48, 49, as well as the lead compound 4, were investigated for their ability to inhibit forskolin-induced cAMP accumulation in CHO wild-type (wt) cells, which do not express GPR84. None of the tested compounds showed any significant effect at concentrations of up to 1 μM. At high concentrations of up to 100 μM, 4 and 20 showed no effect, whereas the other investigated compounds induced a minor inhibition, which never exceeded 50% at a very high concentration of 100 μM (Figure 4). This indicates that the effects measured in cAMP assays on GPR84-transfected cells (Tables 1–3) were clearly due to GPR84 activation.
β-Arrestin Recruitment Assays
Selected agonists that had shown potency in the cAMP assays were further evaluated in β-arrestin recruitment assays using the β-galactosidase complementation assay technology (DiscoverX) (see Tables 1–3 and Figure 6B,D). The standard agonist embelin (6, Figure 1) was used in these assays as a reference compound. Embelin is more suitable than decanoic acid (1) because it is more potent and has a somewhat higher efficacy in the β-arrestin assays than the physiological standard agonist 1. Both lipidic agonists, embelin and decanoic acid, were unbiased, showing nearly identical EC50 values in the two different assays (decanoic acid: 7420 and 6080 nM, respectively; embelin: 795 and 424 nM, respectively; see Table 1). In contrast, lead structure 4 and the structurally related (hexylthio)pyrimidine-4,6-diol (5) were nearly 7- to 10-fold more potent in the Gi-dependent cAMP assay compared to the β-arrestin recruitment assay (4, 17 vs 110 nM; 5, 6.6 vs 78 nM). As a direct comparison of EC50 values obtained in different assays may be misleading due to different degrees of signal amplification, we calculated the bias factors. The Hill slope of the curves was in most cases close to 1, between 0.9 and 1.3. Because both assays were conducted in the same cellular background, the reference compound will control for any systematic bias.33 For each of the two intracellular pathways, Δlog(Emax/EC50) was computed, followed by calculating the pathway bias factor as ΔΔlog(Emax/EC50) according to a recently described and validated method.33 A bias factor of 0 means no bias, whereas a factor of 1 corresponds to a 10-fold preference, and a factor of 2 to a 100-fold selectivity for the Gi-coupled pathway. For many compounds, we observed an ca. 8- to 20-fold preference for the cAMP pathway (bias factor of 0.9–1.3) compared to β-arrestin recruitment. Nevertheless, the SARs of the uracil derivatives determined in β-arrestin assays were quite similar to those observed in cAMP assays (see Figure 5). Among the tested compounds, three very potent unbiased agonists could be identified, revealing almost identical EC50 values in both assays (see Figures 5 and 6A,B): 20, EC50 cAMP 5.0 nM, β-arrestin 3.2 nM, bias factor: 0.0; 22, EC50 cAMP 30.0 nM, β-arrestin 34.0 nM (bias factor: 0.0); 51, EC50 cAMP 44.0 nM, β-arrestin 39.0 nM (bias factor −0.1). The unbiased compound 20 with a C6 alkyl tail was the most potent agonist of the present series in the β-arrestin assay. The rank order of potency in the β-arrestin assay with regard to the length of the alkyl chain at the uracil core was as follows: C6 (20, 3.2 nM) > C9 (22, EC50 34 nM, p = 0.0113) ≥ C8 (4, EC50 110 nM) > C10 (23, EC50 360 nM, p = 0.0033) ≈ C7 (21, EC50 390 nM) > C5 (19, EC50 4800 nM, p = 0.0292). A compound with a polar hydroxy group at the end of the lipophilic tail exhibited identical potency in both assay systems (28, EC50 1980 vs 2120 nM, bias factor: −0.2). In contrast, compounds 37 (EC50 110 nM (cAMP) vs 4200 nM (β-arrestin)) and 38 (EC50 38 nM (cAMP) vs 640 nM (β-arrestin)) displayed bias factors of 1.2 and 0.6, respectively. The most biased agonists displaying a bias factor (ΔΔlog(Emax/EC50)) ≥ 1.3 and an EC50 < 10 nM were 47, 48, and 49 (Figure 6), all of which represent p-substituted 6-(phenylethylamino)uracil derivatives. The most pathway-selective agonist of the whole series was 47 with an IC50 value in the cAMP assay of 7.1 nM and a bias factor of 1.9 corresponding to a 79-fold preference for Gi coupling versus β-arrestin recruitment (see Figure 5B).
Figure 5.

(A) Correlation between the pEC50 values determined in cAMP assays and pEC50 values determined in β-arrestin assays. (Number of pairs: 34; p value (two-tailed) = 0.0008). The most potent unbiased GPR84 agonists 20, 22, and 51 are marked in red; the most potent Gi-biased agonists 47, 48, and 49 are marked in turquois. (B) Bias factors [ΔΔlog(Emax/EC50)] calculated for the most potent biased and unbiased GPR84 agonists; positive values indicate bias for the Gi protein-dependent over β-arrestin recruitment pathway.
Concentration–response curves for selected agonists including the lead structure 4 are depicted in Figure 6A–D.
Efficacy in β-Arrestin Assays
The maximal effect of embelin (6) in the β-arrestin assays was set at 100% efficacy. Decanoic acid (1) was slightly less efficacious (92%), whereas lead structure 4 showed a higher efficacy of 189%. Most of the potent uracil derivatives displayed high efficacies of around 150–250%, with a few exceptions. Particularly high efficacy was observed for the N6-methylated 6-aminouracil derivatives 37 (278%) and 38 (309%) and 6-(p-bromophenethylamino)uracil (48, 283%), whereas the 6-alkylaminouracil derivatives 20, 23, 77, and 78 showed efficacies of only around 100%. For comparison, the previously published GPR84 agonist 5 displayed a high efficacy of 242%. The availability of compounds with a range of efficacies in β-arrestin assays will be useful to study its pharmacological significance.
Receptor Selectivity
Because the investigated uracil derivatives can be envisaged as mimics of fatty acids such as dodecanoic acid, their selectivities versus the G protein-coupled free fatty acid receptors FFAR1 and FFAR4 were subsequently studied (see Tables S1 and S2 for the entire set of compounds). In both, agonist and antagonist assays, none of the compounds activated or inhibited FFAR1 or FFAR4. These results suggest that the developed uracil derivatives can be considered as selective GPR84 agonists versus FFAR1 and FFAR4.
Metabolic Stability
For subsequent in vivo studies, metabolically stable drugs will be required. Therefore, we studied selected potent compounds, namely, 20, 22, 42, and 48, for their metabolic stability in human liver microsomes. Although 6-nonylaminouracil (22) showed a very short half-life of 2 min (Clint 880 μL/min mg protein), 6-phenethylaminouracil (42) and the two most potent GPR84 agonists of the present series, 6-hexylaminouracil (20) and 6-(p-bromophenethylamino)uracil (48), proved to be highly stable (see Figure 7 for 20, 22, and 48 and Figure S1 for 42). Even after an incubation period of 60 min, degradation of 20, 42, and 48 was negligible. In addition, we investigated the metabolic stability of 2-hexylthiopyrimidine-4,6-diol (5), a GPR84 agonist recently reported by Liu et al.18 (Figure 7). Compound 5 was metabolized, and after 30 min of incubation in human microsomes, almost 40% of the drug had disappeared. In comparison, after the same incubation time, no degradation of 6-aminouracil derivatives 20, 42, and 48 could be observed. This indicates that the potent GPR84 agonists 20 and 48 are metabolically stable and should be useful tools for in vivo studies.
Figure 7.

Metabolic stability of 6-aminouracil derivatives 20, 22, and 48, and 2-hexylthiopyrimidine-4,6-diol (5) in human liver microsomes (0.5 mg/mL, mixed gender, pooled). Compounds were tested at a concentration of 1 μM. Data points represent mean values ± SD (for details, see Experimental Section).
Pharmacophore Modeling
To understand and rationalize important structural features of lipidlike GPR84 agonists, we compared the uracil derivatives 4 (lead structure) and 48 (potency in the low nanomolar range) to those of the known GPR84 agonists 3-hydroxydodecanoic acid (2) and embelin (6), which are >100-fold less potent than 48. Figure 8A shows an overall flexible alignment of the selected GPR84 agonists 2, 4, 6, and 48. All four structures overlay very well with several features designated F1–F5 that likely contribute to high GPR84 potency:
-
(i)
A hydrogen bond acceptor (F1) is found in all structures: the carbonyl of the carboxylic acid in 2, the C1-carbonyl group in 6, and the C2-carbonyl group in the uracil derivatives 4 and 48. This pharmacophore feature is shown the cyan-colored sphere in Figure 8A,B.
-
(ii)
Three hydrogen bond donors (F2, F3, and F5 shown as the magenta-colored spheres in Figure 8A,B) are found in the most potent agonists, 4 and 48, namely, N1–H (F2), N6–H (F3), and N3–H (F5), whereas agonist 2 has F3 (3-OH group), but not F2 and F5, and agonist 6 features only F2 (2-OH group).
-
(iii)
A long aliphatic chain or an arylalkyl residue (F4 shown as the green-colored sphere in Figure 8A,B). This feature is found in all agonists. The aromatic ring replacing the terminal part of the aliphatic chain improves hydrophobic interactions and therefore increases potency.
Figure 8.

Overall alignment of the selected GPR84 agonists 2 (green), 4 (cyan), 6 (blue), and 48 (orange). (A) Left: five pharmacophore features were identified (F1: H bond acceptor; F2, F3, and F5: H bond donor; and F4: hydrophobic). Right: the distances between the pharmacophore features are shown as red lines, and the distance (Å) between the features is reported in the table. (B) Each individual GPR84 agonist is shown with the five identified pharmacophore features (F1–F5); oxygen atoms are colored red, nitrogen atoms blue, bromine atoms dark red, and hydrogen atoms silver white; nonpolar hydrogen atoms are omitted. (C) Pharmacophore features are indicated in two-dimensional (2D) representation for 2, 4, 6, and 48 (cyan, hydrogen bond acceptor; magenta, hydrogen bond donor; green, hydrophobic/aromatic domain). (D) Alignment of the previously published agonist 5 with the created pharmacophore model in 2D representation.
The steric and electronic fit of the four structures is excellent and in agreement with the observed SARs. The additional hydrogen bond donors in the aminouracil derivatives 4 and 48 may be the reason for their improved GPR84 potency as compared to the standard agonists 2 and 6. The uracil ring, which lacks flexibility, keeps the hydrogen bond acceptor and donor features in a fixed position for interaction with the amino acid residues of GPR84.
We subsequently investigated whether the published GPR84 agonist 2-hexylthiopyrimidine-4,6-diol (5) matches the developed pharmacophore model (see Figure 8D). In fact, it can be superimposed displaying most of the identified interaction features.
Conclusions
In conclusion, a series of 69 6-aralkylamino- and alkylamino-substituted uracil derivatives was synthesized, of which 66 (13–39, 41–65, 66–68, and 70–80) are new compounds that are not previously reported in the literature. Starting from 6-octylaminouracil (4) as a lead structure, our goal was to study the SARs of this class of GPR84 agonists, improve their potency determined by Gi protein-dependent inhibition of intracellular cAMP formation, and obtain metabolically stable compounds suitable for in vivo studies. Many 6-(ar)alkylamino-substituted uracil derivatives showed high GPR84 agonistic activity, whereas substitution at the 5-position reduced or abolished activity. The length of the carbon chain attached at the 6-position of the uracil core determined the potency of compounds. Moreover, the introduction of an aromatic residue into the alkyl chain further improved potency. 6-Hexylamino-2,4(1H,3H)-pyrimidinedione (20, EC50 5.0 nM), 6-((p-chlorophenylethyl)amino)-2,4(1H,3H)-pyrimidinedione (47, EC50 7.1 nM), and 6-((p-bromophenylethyl)amino)-2,4(1H,3H)-pyrimidinedione (48, EC50 2.5 nM) were found to be the most potent GPR84 agonists of the present series showing high efficacy. Potencies of the selected compounds were further determined in β-arrestin assays, which indicated that the phenethyl-substituted 6-aminouracil derivatives 47–49 are biased toward Gi-mediated adenylate cyclase inhibition (20- to 79-fold), whereas the alkyl-substituted 20 displayed the same EC50 values in cAMP and β-arrestin assays and was unbiased. Biased and unbiased agonists may display different pharmacological profiles. For example, it had been suggested that the μ-opioid receptor agonist morphine and related opioids display their severe side effects, such as fatal respiratory depression, by signaling through the β-arrestin pathway. Therefore, agonists that are Gi-protein-biased and do not induce β-arrestin recruitment have been developed.34 Gi-protein-biased GPR84 agonists may be devoid of inducing receptor desensitization and thus exhibit longer-lasting effects, or they might trigger different signaling pathways compared to nonbiased agonists.
The new compounds showed high selectivity for GPR84 versus the related GPCRs FFAR1 and FFAR4 that display an overlapping ligand preference regarding fatty acids. The new GPR84 agonists, which exhibit excellent metabolic stability, will be useful tool compounds for elucidating the physiologic roles and therapeutic potential of GPR84
Experimental Section
General Methods
All commercially available reagents were used as purchased (Acros, Alfa Aesar, Sigma-Aldrich, ABCR or TCI). Solvents were used without additional purification or drying except for dichloromethane, which was distilled over calcium hydride. The reactions were monitored by thin-layer chromatography (TLC) using aluminum sheets with silica gel 60 F254 (Merck). Column chromatography was performed with 0.060–0.200 mm silica gel with pore diameter of ca. 6 nm. All synthesized compounds were finally dried in vacuum at 8–12 Pa (0.08–0.12 mbar) using a sliding vane rotary vacuum pump (Vacuubrand GmbH). 1H and 13C NMR data were collected on a Bruker Avance 500 MHz NMR spectrometer at 500 and 126 MHz, respectively. If indicated, NMR data were collected on a Bruker Ascend 600 MHz NMR spectrometer at 600 MHz (1H) and 151 MHz (13C). DMSO-d6 was employed as a solvent at 303 K, unless otherwise noted. Chemical shifts are reported in parts per million (ppm) relative to the deuterated solvent, that is, DMSO, δ 1H: 2.49 ppm; 13C: 39.7 ppm. Coupling constants J are given in hertz, and spin multiplicities are given as s (singlet), d (doublet), t (triplet), q (quartet), sext. (sextet), m (multiplet), and br (broad). Melting points were determined on a Büchi 530 melting point apparatus and are uncorrected. The purities of isolated products were determined by ESI-mass spectra obtained on an liquid chromatography–mass spectrometry (LC–MS) instrument (Applied Biosystems API 2000 LCMS/MS, HPLC Agilent 1100) using the following procedure: the compounds were dissolved at a concentration of 1.0 mg/mL in acetonitrile containing 2 mM ammonium acetate. Then, 10 μL of the sample was injected into an HPLC column (Macherey-Nagel Nucleodur 3 μ C18, 50 × 2.00 mm2). Elution was performed with a gradient of water/acetonitrile (containing 2 mM ammonium acetate) from 90:10 to 0:100 for 20 min at a flow rate of 300 μL/min, starting the gradient after 10 min. UV absorption was detected from 200 to 950 nm using a diode array detector. Purity of all compounds was determined at 254 nm. The purity of the compounds was generally ≥95%.
Compounds 13(8) and 69(31,32) have previously been described. Compound 5 was synthesized according to a reported synthetic procedure.18 The synthesis and structural characterization of the new compounds are described below. Data for compounds 14–18, 24–27, 29, 30, 34–36, 39–41, 47, 59, 66, 72, 75, 76, and 79 are reported in Supporting Information.
General Synthetic Procedure for the Preparation of 4 and 13–71
A suspension of 6-chlorouracil (10 mmol) and the appropriate amine (50 mmol, 5 equiv) in 1-butanol (20 mL) was refluxed at 125 °C for 12 h. The reaction was cooled to rt, and half of the solvent was removed by evaporation under reduced pressure. The precipitated solid was filtered off, washed with 1-butanol (∼20 mL), followed by diethyl ether (∼20 mL), and dried under vacuum.
General Synthetic Procedure for the Preparation of 72–75
To a solution of the appropriate 6-alkylaminouracil derivative (10 mmol) in dry pyridine (2 mL) was added N-bromosuccinimide (NBS, 10 mmol) under an Ar atmosphere. The solution was heated to 80 °C for 2 h and cooled to rt. The precipitated solid was filtered off, washed with diethyl ether (∼25 mL), and subjected into column chromatography to afford the desire product.
General Synthetic Procedure for the Preparation of 76–80
To a suspension of 5-alkylaminouracil (5 mmol) in water/acetic acid (6.0:6.0 mL) heated at 60–70 °C was added a solution of sodium nitrite (10 mmol, 2.0 equiv) in water (2 mL). The resulting solution was stirred at the same temperature for 1 h and then cooled to 4 °C. The resulting suspension was filtered off and washed with cold water (2 × 50 mL) to afford the product.
6-Pentylamino-2,4(1H,3H)-pyrimidinedione (19)
Compound 19 was synthesized using 6-chlorouracil (11, 10 mmol) and 1-aminopentane (50 mmol, 5 equiv) and was isolated as an off-white solid (99% yield). 1H NMR (600 MHz, DMSO-d6) δ (ppm) 6.63 (s, 1H, 1-NH), 4.34 (s, 1H, 5-H), 2.95 (td, J = 7.1, 4.9 Hz, 2H, NHCH2), 1.47 (p, J = 7.2 Hz, 2H, CH2), 1.37 (p, J = 7.3 Hz, 2H, CH2), 1.27–1.24 (m, 4H, 2 × −CH2), 0.86 (t, J = 6.9, 4.1 Hz, 3H, −CH3); 13C NMR (151 MHz, DMSO-d6) δ 164.54 (4-C), 155.67 (2-C), 152.44 (6-C), 72.38 (5-C), 31.53 (NH–C), 28.66 (CH2), 22.08 (CH2), 21.92 (CH2), 14.06 (CH3); LC–MS positive mode: 197 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: 98%. mp: 259–261 °C.
6-Hexylamino-2,4(1H,3H)-pyrimidinedione (20)
Compound 20 was synthesized using 6-chlorouracil (11, 10 mmol) and 1-aminohexane (50 mmol, 5 equiv) and was isolated as a yellow solid (89% yield). 1H NMR (500 MHz, DMSO-d6) δ (ppm) 10.09 (s, 1H, 1-NH), 9.77 (s, 1H, 3-NH), 6.03 (t, J = 5.7 Hz, 1H, 6-NH), 4.38 (d, J = 1.5 Hz, 1H, 5-H), 2.97 (dd, J = 7.1, 5.5 Hz, 2H, NH–CH2), 1.46 (m, 2H, CH2), 1.39–1.11 (m, 6H, 3 × CH2), 0.99–0.61 (m, 3H, CH3); 13C NMR (126 MHz, DMSO-d6) δ (ppm) 164.27 (4-C), 154.12 (2-C), 150.87 (6-C), 72.54 (5-C), 30.96 (NH–CH2), 28.20 (CH2), 26.00 (2 × CH2), 22.12 (CH2), 13.97 (CH3); LC–MS (m/z): positive mode 291 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: 98%. mp: 263–265 °C.
6-Heptylamino-2,4(1H,3H)-pyrimidinedione (21)
Compound 21 was synthesized using 6-chlorouracil (11, 10 mmol) and 1-aminoheptan (50 mmol, 5 equiv) and was isolated as a yellow solid (94% yield). 1H NMR (600 MHz, DMSO-d6) δ (ppm) 6.50 (s, 1H, 6-NH), 4.35 (s, 1H, 5-H), 2.95 (q, J = 6.6 Hz, 2H, NHCH2), 1.46 (m, 4H, CH2), 1.33–1.06 (m, 6H, 3 × CH2), 0.85 (t, J = 6.9 Hz, 3H, CH3); 13C NMR (151 MHz, DMSO-d6) δ 164.38 (4-C), 154.59 (2-C), 151.12 (6-C), 72.36 (5-C), 31.34 (NH–CH2), 31.26 (CH2), 28.71 (CH2), 28.45 (2 × CH2), 22.14 (CH2), 14.07 (CH3); LC–MS (m/z): positive mode 225 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: 98%. mp: 196–198 °C.
6-Nonylamino-2,4(1H,3H)-pyrimidinedione (22)
Compound 22 was synthesized using 6-chlorouracil (11, 10 mmol) and 1-aminononane (50 mmol, 5 equiv) and was isolated as an off-white solid (99% yield). 1H NMR (600 MHz, DMSO-d6) δ (ppm) 6.57 (s, 1H, 6-NH), 4.34 (s, 1H, 5-H), 2.95 (q, J = 6.5 Hz, 2H, NHCH2), 1.46 (p, J = 7.0 Hz, 2H, CH2), 1.36 (p, J = 7.0 Hz, 2H, CH2), 1.30–1.07 (m, 10H, 5 × CH2), 0.85 (t, J = 6.8 Hz, 3H, CH3); 13C NMR (151 MHz, DMSO-d6) δ 164.50 (4-C), 155.53 (2-C), 152.29 (6-C), 72.38 (5-C), 31.90 (NH–CH2), 31.39, 29.11, 29.06, 29.02, 28.81, 28.26, 26.40, 22.21 (7 × CH2), 14.06 (CH3); LC–MS (m/z): positive mode 253 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: 98%. mp: 161–263 °C.
6-Decylamino-2,4(1H,3H)-pyrimidinedione (23)
Compound 23 was synthesized using 6-chlorouracil (11, 10 mmol) and 1-aminodecane (50 mmol, 5 equiv) and was isolated as an off-white solid (92% yield). 1H NMR (500 MHz, DMSO-d6) δ (ppm) 10.09 (s, 1H, 1-NH), 9.77 (s, 1H, 3-H), 6.02 (t, J = 5.0 Hz, 1H, 6-NH), 4.37 (d, J = 1.5 Hz, 1H, 5-H), 3.10–2.71 (m, 2H, NHCH2), 1.46 (p, J = 6.9 Hz, 2H, CH2), 1.25 (d, J = 12.8 Hz, 14H, 7 × CH2), 0.95–0.50 (m, 3H, CH3); 13C NMR (126 MHz, DMSO-d6) δ 164.29 (4-C), 154.14 (2-C), 150.90 (2-C), 72.57 (5-C), 31.40 (NHCH2), 29.08 (CH2), 29.03 (CH2), 28.79, 28.76, 28.24, 26.35, 22.19 (5 × CH2), 14.05 (CH3); LC–MS (m/z): positive mode 267 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: 98%. mp: 270–272 °C.
6-(7-Hydroxyheptylamino)-2,4(1H,3H)-pyrimidinedione (28)
Compound 28 was synthesized using 6-chlorouracil (11, 10 mmol) and 1-amino-7-hydroxyheptane (50 mmol, 5 equiv) and was isolated as a white solid (89% yield). 1H NMR (600 MHz, DMSO-d6) δ (ppm) 10.10 (s, 1H, 1-NH), 9.78 (s, 1H, 1-NH), 6.06 (s, 1H, 6-NH), 4.38 (d, J = 1.4 Hz, 1H, 5-H), 4.29 (t, J = 5.2 Hz, 1H, OH), 3.36 (dd, J = 6.5, 4.9 Hz, 2H, OCH2), 2.97 (dd, J = 7.0, 5.4 Hz, 2H, NH–CH2), 1.39 (m, 2H, CH2), 1.26 (d, J = 6.0 Hz, 6H, 3 × CH2); 13C NMR (151 MHz, DMSO-d6) δ 164.34 (4-C), 154.19 (2-C), 150.95 (6-C), 72.59 (5-C), 60.86 (O–CH2), 32.67 (NHCH2), 29.03 (CH2), 28.86 (CH2), 28.28 (CH2), 26.38 (CH2), 25.59 (2 × CH2); LC–MS (m/z): positive mode 257 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: 99.7%. mp: 238–240 °C.
6-Heptylamino-3-methyl-2,4(1H,3H)-pyrimidinedione (31)
Compound 31 was synthesized using 6-chloro-3-methyluracil (12a, 10 mmol) and 1-aminoheptane (50 mmol, 5 equiv) and was isolated as a white solid (86% yield). 1H NMR (600 MHz, DMSO-d6) δ (ppm) 10.10 (s, 1H, 1-NH), 6.15 (s, 1H, 6-NH), 4.54 (s, 1H, 5-H), 3.01 (s, 3H, 3–CH3), 2.98 (td, J = 7.0, 5.5 Hz, 2H, NH–CH2), 1.46 (p, J = 6.8 Hz, 2H, CH2), 1.35–1.16 (m, 10H, 5 × CH2), 0.85 (t, J = 6.8 Hz, 3H, CH3); 13C NMR (151 MHz, DMSO-d6) δ 163.29 (4-C), 157.66 (2-C), 153.22 (6-C), 72.52 (5-C), 41.36 (N–CH3), 31.36 (NHCH2), 28.77 (CH2), 28.31 (CH2), 26.38 (CH2), 26.38 (CH2), 22.22 (CH2), 14.09 (CH3); LC–MS (m/z): positive mode 254 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: 98.5%. mp: 207–209 °C.
6-Nonylamino-3-methyl-2,4(1H,3H)-pyrimidinedione (32)
Compound 32 was synthesized using 6-chloro-3-methyluracil (12a, 10 mmol) and 1-aminononane (50 mmol, 5 equiv) and was isolated as a yellow solid (97% yield). 1H NMR (500 MHz, DMSO-d6) δ (ppm) 6.50 (m, 1H, 6-NH), 4.51 (s, 1H, 5-H), 3.01 (s, 3H, CH3), 2.97 (q, J = 6.6 Hz, 2H, NHCH2), 1.53 (m, 4H, 2 × CH2), 1.46 (m, 4H, 2 × CH2), 0.91–0.85 (m, 9H, 3 × CH2 and CH3); 13C NMR (126 MHz, DMSO-d6) δ 163.28 (4-C), 152.76 (2 × C), 150.99 (6-C), 72.29 (5-C), 31.38 (NHCH2), 29.04 (N–CH3), 28.90 (CH2), 28.78 (2 × CH2), 28.65 (CH2), 28.26 (CH2), 27.08 (CH2), 26.36 (CH2), 26.00 (CH2), 22.20 (CH2), 14.05 (CH3); LC–MS (m/z): positive mode 267 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: 96.9%. mp: 143–145 °C.
6-Decylamino-3-methyl-2,4(1H,3H)-pyrimidinedione (33)
Compound 33 was synthesized using 6-chloro-3-methyluracil (12a, 10 mmol) and 1-aminodecane (50 mmol, 5 equiv) and was isolated as a white solid (98% yield). 1H NMR (500 MHz, DMSO-d6) δ (ppm) 10.19 (s, 1H, 3-NH), 6.01 (s, 1H, 6-NH), 4.54 (s, 1H, 5-H), 3.01 (s, 3H, N–CH3), 3.00–2.88 (m, 2H, NH–CH2), 1.46 (p, J = 7.1 Hz, 2H, CH2), 1.33–1.11 (m, 14H, 7 × CH2), 0.93–0.71 (m, 3H, CH3); 13C NMR (126 MHz, DMSO-d6) 163.23 (4-C), 152.56 (2-C), 151.08 (6-C), 72.49 (5-C), 31.38 (N–CH3), 29.06 (N–CH2), 28.77 (CH2), 28.25 (CH2), 26.31 (CH2), 26.00 (CH2), 22.18 (CH2), 14.04 (CH3); LC–MS (m/z): positive mode 281 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: 95.3%. mp: 210–212 °C.
6-(N-Methylhexylamino)-2,4(1H,3H)-pyrimidinedione (37)
Compound 37 was synthesized using 6-chlorouracil (11, 10 mmol) and 1-methylaminohexane (50 mmol, 5 equiv) and was isolated as a yellow solid (92% yield). 1H NMR (500 MHz, DMSO-d6) δ (ppm) 10.16 (s, 1H, 1-NH), 4.39 (s, 1H, 5-H), 3.29 (s, 3H, N–CH3), 2.85 (s, 3H, N–CH2), 1.55 (p, J = 8.0, 7.6 Hz, 2H, CH2), 1.44 (p, J = 7.3 Hz, 2H, CH2), 1.26–121 (m, 6H, 3 × CH2), 0.86 (td, J = 6.8, 3.9 Hz, 3H, CH3); 13C NMR (126 MHz, DMSO-d6) δ 163.90 (4-C), 154.43 (2-C), 151.60 (6-C), 74.65 (5-C), 37.10 (N–CH3), 32.79 (N–CH2), 26.65 (CH2), 25.76 (2 × CH2), 22.18 (2 × CH2), 13.96 (CH3); LC–MS (m/z): positive mode 226 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: >99%. mp: 162–164 °C.
6-(N-Methyloctylamino)-2,4(1H,3H)-pyrimidinedione (38)
Compound 38 was synthesized using 6-chlorouracil (11, 10 mmol) and N-methyloctylamine (50 mmol, 5 equiv) and was isolated as pale yellow solid (93% yield). 1H NMR (500 MHz, DMSO-d6) δ (ppm) 8.86 (s, 1H, 3-NH), 4.41 (s, 1H, 5-H), 2.86–2.65 (m, 2H, NHCH2), 1.79–1.43 (m, 2H, CH2), 1.35–1.11 (m, 10H, 5 × CH2), 0.95–0.68 (m, 3H, CH3); 13C NMR (126 MHz, DMSO-d6) δ 164.00 (4-C), 155.13 (2-C), 151.63 (6-C), 73.65 (5-C), 51.32 (N–CH3), 31.87 (N–CH2), 26.61 (CH2), 25.66 (CH2), 24.32 (CH2), 22.28 (CH2), 21.99 (CH2), 13.91 (CH3); LC–MS (m/z): positive mode 253 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: >99%. mp: 182–184 °C.
6-(2-Phenylethylamino)-2,4(1H,3H)-pyrimidinedione (42)
Compound 42 was synthesized using 6-chlorouracil (11, 10 mmol) and 2-phenethylamine (50 mmol, 5 equiv) and was isolated as a white solid (95% yield). 1H NMR (500 MHz, DMSO-d6) δ (ppm) 10.03 (s, 1H, 1-NH), 9.15 (s, 1H, 6-NH), 7.44–7.28 (m, 2H, 2 × Ar–H), 7.28–7.17 (m, 3H, 3 × Ar–H), 4.46 (s, 1H, 5-H), 3.26–3.10 (m, 2H, NH–CH2), 2.79 (t, J = 7.2 Hz, 2H, Ar–CH2); 13C NMR (126 MHz, DMSO-d6) δ 164.39 (4-C), 154.20 (2-C), 151.06 (6-C), 138.87 (Ar–C), 128.87 (Ar–C), 128.78 (Ar–C), 128.63 (Ar–C), 128.54 (Ar–C), 126.45 (Ar–C), 72.84 (5-C), 42.74 (NHCH2), 34.26 (Ar–CH2); LC–MS (m/z): positive mode 231 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: >98%. mp: 292–294 °C.
6-(3-Phenylpropylamino)-2,4(1H,3H)-pyrimidinedione (43)
Compound 43 was synthesized using 6-chlorouracil (11, 10 mmol) and 3-phenylpropylamine (50 mmol, 5 equiv) and was isolated as a white solid (91% yield). 1H NMR (500 MHz, DMSO-d6) δ (ppm) 7.30–7.24 (m, 2H, 2 × Ar–H), 7.21–7.13 (m, 3H, 3 × Ar–H), 6.69 (s, 1H, 6-NH), 4.33 (s, 1H, 5-H), 2.98 (td, J = 7.1, 4.9 Hz, 2H, NHCH2), 2.65–2.58 (m, 2H, Ar–CH2), 1.91–1.45 (m, 2H, CH2); 13C NMR (126 MHz, DMSO-d6) δ 164.57 (4-C), 155.62 (2-C), 152.40 (6-C), 141.57 (Ar–C), 128.48 (Ar–C), 128.45 (Ar–C), 128.40 (Ar–C), 125.97 (Ar–C), 125.87 (Ar–C), 72.51 (5-C), 33.03 (NHCH2), 32.52 (Ar–CH2), 30.14 (CH2). LC–MS (m/z): positive mode 245 [M + H]1+; purity by HPLC UV (254 nm)-ESI-MS: >99%. mp: 283–285 °C.
6-(4-Phenylbutylamino)-2,4(1H,3H)-pyrimidinedione (44)
Compound 44 was synthesized using 6-chlorouracil (11, 10 mmol) and phenylbutylamine (50 mmol, 5 equiv) and was isolated as a pale yellow solid (97% yield). 1H NMR (500 MHz, DMSO-d6) δ (ppm) 7.26 (t, J = 7.5 Hz, 2H, 2 × Ar–H), 7.21–7.11 (m, 3H, 3 × Ar–H), 6.60 (s, 1H, 6-NH), 4.35 (s, 1H, 5-H), 2.99 (q, J = 6.6 Hz, 2H, NHCH2), 2.60–2.55 (m, 2H, Ar–CH2), 1.58 (m, 2H, CH2), 1.52–1.44 (m, 2H, CH2); 13C NMR (126 MHz, DMSO-d6) δ 164.52 (4-C), 155.57 (2-C), 152.36 (6-C), 142.14 (Ar–C), 128.41 (Ar–C), 128.37 (Ar–C), 128.34 (Ar–C), 125.81 (Ar–C), 125.75 (Ar–C), 72.46 (5-C), 34.84 (NH–CH2), 31.54 (Ar–CH2), 28.38 (CH2CH2); LC–MS (m/z): positive mode 259 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: >98%. mp: 165–167 °C.
6-(p-Methylphenylethylamino)-2,4(1H,3H)-pyrimidinedione (45)
Compound 45 was synthesized using 6-chlorouracil (11, 10 mmol) and 1-(p-methylphenyl)ethylamine (50 mmol, 5 equiv) and was isolated as a white solid (96% yield). 1H NMR (500 MHz, DMSO-d6) δ (ppm) 10.12 (s, 1H, 1-NH), 9.35 (s, 1H, 3-NH), 7.29–6.76 (m, 4H, 4 × Ar–H), 6.09 (s, 1H, 6-NH), 4.46 (s, 1H, 5-H), 3.25–3.05 (m, 2H, NHCH2), 2.74 (t, J = 7.2 Hz, 2H, Ar–CH2), 2.26 (s, 3H, Ar–CH3); 13C NMR (126 MHz, DMSO-d6) δ 164.35 (4-C), 154.02 (2-C), 150.90 (6-C), 135.69 (Ar–C), 135.42 (Ar–C), 129.12 (Ar–C), 128.74 (Ar–C), 72.83 (5-C), 42.80 (NHCH2), 33.80 (Ar–CH2), 20.75 (Ar–CH3); LC–MS (m/z): positive mode 245 [M + H]1+; purity by HPLC UV (254 nm)-ESI-MS: 95.7%. mp: >300 °C.
6-(p-Fluorophenylethylamino)-2,4(1H,3H)-pyrimidinedione (46)
Compound 46 was synthesized using 6-chlorouracil (11, 10 mmol) and 1-(p-fluorophenyl)ethylamine (50 mmol, 5 equiv) and was isolated as a pale light yellow solid (79% yield). 1H NMR (500 MHz, DMSO-d6) δ (ppm) 7.25–7.23 (m, 2H, 2 × Ar–H), 7.18–6.98 (m, 2H, 2 × Ar–H), 4.45 (s, 1H, 5-H), 3.23 (dd, J = 7.2, 5.4 Hz, 2H, NHCH2), 2.79 (td, J = 7.1, 4.6 Hz, 2H, Ar–CH2); 13C NMR (126 MHz, DMSO-d6) δ 164.42 (4-C), 154.55 (2-C), 151.43 (6-C), 135.05 (Ar–C), 130.72 (Ar–C), 130.66 (Ar–C), 130.48 (Ar–C), 115.26 (Ar–C), 114.96 (Ar–C), 72.83 (5-C), 42.76 (NHCH2), 33.41 (Ar–CH2). LC–MS (m/z): positive mode 249 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: 95.5%. mp: 280–282 °C.
6-(p-Bromophenylethylamino)-2,4(1H,3H)-pyrimidinedione (48)
Compound 48 was synthesized using 6-chlorouracil (11, 10 mmol) and 1-(p-bromophenyl)ethylamine (50 mmol, 5 equiv) and was isolated as a light brown solid (87% yield). 1H NMR (500 MHz, DMSO-d6) δ (ppm) 9.41 (s, 1H, 1-NH), 7.51–7.47 (m, 2H, 2 × Ar–H), 7.24–7.19 (m, 2H, 2 × Ar–H), 6.03 (s, 1H, 3-NH), 4.47 (s, 1H, 5-H), 3.25 (dd, J = 7.2, 5.6 Hz, 2H, NHCH2), 2.77 (t, J = 7.1 Hz, 2H, Ar–CH2); 13C NMR (126 MHz, DMSO-d6) δ 164.34 (4-C), 154.09 (2-C), 151.00 (6-C), 138.35 (Ar–C), 131.35 (Ar–C), 131.30 (Ar–C), 131.21 (Ar–C), 119.57 (Ar–C), 72.91 (5-C), 42.39 (NHCH2), 33.57 (Ar–CH2); LC–MS (m/z): positive mode 291 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: >97%. mp: >300 °C.
6-(p-Ethylphenylethylamino)-2,4(1H,3H)-pyrimidinedione (49)
Compound 49 was synthesized using 6-chlorouracil (11, 10 mmol) and 1-(p-ethylphenyl)ethylamine (50 mmol, 5 equiv) and was isolated as a pale brown solid (74% yield). 1H NMR (600 MHz, DMSO-d6) δ (ppm) 7.21–6.83 (m, 4H, 4 × Ar–H), 4.44 (s, 1H, 5-H), 3.22 (td, J = 7.1, 5.0 Hz, 2H, NHCH2), 2.77 (dt, J = 23.0, 7.1 Hz, 2H, Ar–CH2), 2.55 (m, 2H, Ar–CH2), 1.15 (t, J = 7.6, Hz, 3H, CH3); 13C NMR (151 MHz, DMSO-d6) δ 164.48 (4-C), 154.87 (2-C), 151.76 (6-C), 141.80 (Ar–C), 136.09 (Ar–C), 128.78 (Ar–C), 128.70 (Ar–C), 127.92 (Ar–C), 127.82 (Ar–C), 72.73 (5-C), 43.02 (NH–CH2), 42.91 (Ar–CH2), 33.87 (Ar–CH2), 15.8 (CH3); LC–MS (m/z): positive mode 259 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: 95.5%. mp: 270–272 °C.
6-(p-Methoxyphenylethylamino)-2,4(1H,3H)-pyrimidinedione (50)
Compound 50 was synthesized using 6-chlorouracil (11, 10 mmol) and 1-(p-methoxyphenyl)ethylamine (50 mmol, 5 equiv) and was isolated as a brown solid (82% yield). 1H NMR (500 MHz, DMSO-d6) δ (ppm) 12.37 (s, 1H, 1-NH), 6.60 (dd, J = 9.4, 2.0 Hz, 2H, 2 × Ar–H), 6.44 (dd, J = 12.4, 2.1 Hz, 2H, 2 × Ar–H), 4.96 (s, 1H, 5-H), 3.88 (s, 3H, O–CH3), 2.98 (t, J = 6.5 Hz, 2H, NHCH2), 2.53 (t, J = 6.8 Hz, 2H, Ar–CH2); 13C NMR (126 MHz, DMSO-d6) δ 164.04 (4-C), 162.98 (2-C), 159.21 (6-C), 137.20 (Ar–C), 126.66 (Ar–C), 122.53 (Ar–C), 114.73 (Ar–C), 71.60 (5-C), 45.33 (OCH3), 35.90 (NHCH2), 21.20 (Ar–CH2); LC–MS (m/z): positive mode 261 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: 95.0%. mp: 282–284 °C.
6-(p-tert-Butylphenylethylamino)-2,4(1H,3H)-pyrimidinedione (51)
Compound 51 was synthesized using 6-chlorouracil (11, 10 mmol) and 1-(p-tert-butylphenyl)ethylamine (50 mmol, 5 equiv) and was isolated as a white solid (76% yield). 1H NMR (600 MHz, DMSO-d6) δ (ppm) 10.13 (s, 1H, 1-NH), 9.87 (s, 1H, 3-NH), 7.31 (d, J = 1.9 Hz, 2H, 2 × Ar–H), 7.19–7.13 (m, 2H, 2 × Ar–H), 6.01 (s, 1H, 6-NH), 4.47 (s, 1H, 5-H), 3.24 (td, J = 7.1, 5.4 Hz, 2H, NHCH2), 2.75 (t, J = 7.1 Hz, 2H, Ar–CH2), 1.25 (s, 9H, C(CH3)3); 13C NMR (151 MHz, DMSO-d6) δ 164.37 (4-C), 154.03 (2-C), 150.92 (6-C), 135.74 (Ar–C), 128.54 (Ar–C), 125.30 (Ar–C), 72.86 (5-C), 42.72 (NHCH2), 34.25 (Ar–CH2), 31.34 (C(CH3)3); LC–MS (m/z): positive mode 287 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: >98.0%. mp: >300 °C.
6-(m-Methylphenylethylamino)-2,4(1H,3H)-pyrimidinedione (52)
Compound 52 was synthesized using 6-chlorouracil (11, 10 mmol) and 1-(m-methylphenyl)ethylamine (50 mmol, 5 equiv) and was isolated as a white solid (83% yield). 1H NMR (600 MHz, DMSO-d6) δ (ppm) 7.61–7.57 (m, 2H, 2 × Ar–H), 7.40–7.30 (m, 1H, Ar–H), 7.18 (s, 1H, Ar–H), 6.39 (s, 1H, 6-NH), 4.46 (s, 1H, 5-H), 3.22 (q, J = 6.8 Hz, 2H, NHCH2), 2.75 (t, J = 7.2 Hz, 2H, Ar–CH2), 2.27 (s, 3H, Ar–CH3); 13C NMR (151 MHz, DMSO-d6) δ 166.12 (4-C), 154.97 (2-C), 153.32 (6-C), 141.94 (Ar–C), 127.81 (Ar–C), 122.37 (Ar–C), 121.34 (Ar–C), 125.81 (Ar–C), 125.75 (Ar–C), 72.46 (5-C), 34.84 (NHCH2), 31.54 (Ar–CH2), 28.38 (Ar–CH3). LC–MS (m/z): positive mode 291 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: 95.7%. mp: 229–231 °C.
6-(m-Fluorophenylethylamino)-2,4(1H,3H)-pyrimidinedione (53)
Compound 53 was synthesized using 6-chlorouracil (11, 10 mmol) and 1-(m-fluorophenyl)ethylamine (50 mmol, 5 equiv) and was isolated as a yellow solid (91% yield). 1H NMR (500 MHz, DMSO-d6) δ (ppm) 10.14 (s, 1H, 3-NH), 7.37–7.23 (m, 2H, 2 × Ar–H), 7.21–7.10 (m, 2H, 2 × Ar–H), 4.48 (s, 1H, 5-H), 3.26 (dd, J = 7.4, 6.0 Hz, 2H, NHCH2), 2.83 (t, J = 7.2 Hz, 2H, Ar–CH2); 13C NMR (126 MHz, DMSO-d6) δ 164.37 (4-C), 159.91 (2-C), 154.06 (6-C), 131.42 (Ar–C), 128.71 (Ar–C), 128.64 (Ar–C), 125.40 (Ar–C), 115.40 (Ar–C), 115.22 (Ar–C), 72.84 (5-C), 41.52 (NHCH2), 27.67 (Ar–CH2); LC–MS (m/z): positive mode 249 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: >98.0%. mp: >300 °C.
6-(m-Chlorophenylethylamino)-2,4(1H,3H)-pyrimidinedione (54)
Compound 54 was synthesized using 6-chlorouracil (11, 10 mmol) and 1-(m-chlorophenyl)ethylamine (50 mmol, 5 equiv) and was isolated as a white solid (94% yield). 1H NMR (600 MHz, DMSO-d6) δ (ppm) 10.13 (s, 1H, 3-NH), 9.38–7.30 (m, 2H, 2 × Ar–H), 7.27 (dd, J = 8.0, 2.2 Hz, 1H, Ar–H), 7.22 (s, 1H, Ar–H), 4.49 (s, 1H), 3.27 (dd, J = 7.2, 5.6 Hz, 2H, NHCH2), 2.80 (t, J = 7.1 Hz, 2H, Ar–CH2); 13C NMR (151 MHz, DMSO-d6) δ 164.36 (4-C), 154.02 (2-C), 150.94 (6-C), 141.53 (Ar–C), 133.15 (Ar–C), 130.33 (Ar–C), 128.82 (Ar–C), 127.71 (Ar–C), 126.47, 72.98 (Ar–C), 42.34 (NHCH2), 33.84 (Ar–CH2); LC–MS (m/z): positive mode 265 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: >98.0%. mp: 283–285 °C.
6-(m-Bromophenylethylamino)-2,4(1H,3H)-pyrimidinedione (55)
Compound 55 was synthesized using 6-chlorouracil (11, 10 mmol) and 1-(m-bromophenyl)ethylamine (50 mmol, 5 equiv) and was isolated as a white solid (93% yield). 1H NMR (600 MHz, DMSO-d6) δ (ppm) 7.56–7.47 (m, 1H, Ar–H), 7.47–7.36 (m, 3H, 3 × Ar–H), 6.21 (s, 1H, 3-NH), 4.49 (s, 1H, 5-H), 3.26 (d, J = 6.8 Hz, 2H, NH–CH2), 2.79 (t, J = 7.1 Hz, 2H, Ar–CH2); 13C NMR (151 MHz, DMSO-d6) δ 164.37 (4-C), 154.06 (2-C), 150.94 (6-C), 141.82 (Ar–C), 131.67 (Ar–C), 130.63 (Ar–C), 129.74 (Ar–C), 129.36 (Ar–C), 128.08 (Ar–C), 72.94 (5-C), 42.36 (NHCH2), 33.82 (Ar–CH2); LC–MS (m/z): positive mode 309 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: >98.0%. mp: 264–266 °C.
6-(o-Methylphenylethylamino)-2,4(1H,3H)-pyrimidinedione (56)
Compound 56 was synthesized using 6-chlorouracil (11, 10 mmol) and 1-(o-methylphenyl)ethylamine (50 mmol, 5 equiv) and was isolated as a white solid (89% yield). 1H NMR (600 MHz, DMSO-d6) δ (ppm) 10.12 (s, 1H, 1-NH), 7.27–7.07 (m, 4H, 4 × Ar–H), 4.47 (s, 1H, 5-H), 3.26–3.12 (m, 2H, NHCH2), 2.79 (t, J = 7.4 Hz, 2H, Ar–CH2), 2.28 (s, 3H, Ar–CH3); 13C NMR (151 MHz, DMSO-d6) δ 165.60 (4-C), 164.39 (2-C), 154.14 (6-C), 136.13 (Ar–C), 130.39 (Ar–C), 129.29 (Ar–C), 126.95 (Ar–C), 126.27 (Ar–C), 72.75 (5-C), 41.72 (NHCH2), 31.68 (Ar–CH2), 19.10 (Ar–CH3); LC–MS (m/z): positive mode 245 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: >96.0%. mp: 298–300 °C.
6-(o-Fluorophenylethylamino)-2,4(1H,3H)-pyrimidinedione (57)
Compound 57 was synthesized using 6-chlorouracil (11, 10 mmol) and 1-(o-methylphenyl)ethylamine (50 mmol, 5 equiv) and was isolated as a white solid (78% yield). 1H NMR (600 MHz, DMSO-d6) δ (ppm) 10.14 (s, 1H, 1-NH), 7.49–7.21 (m, 1H, Ar–H), 7.20–6.82 (m, 3H, Ar–H), 6.02 (s, 1H, 3-NH), 4.59 (s, 1H, 5-H), 3.27–3.26 (m, 2H, NHCH2), 2.81 (t, J = 7.1 Hz, 2H, Ar–CH2); 13C NMR (151 MHz, DMSO-d6) δ 164.36 (4-C), 154.02 (2-C), 150.93 (6-C), 141.83 (Ar–C), 130.33 (Ar–C), 125.08 (Ar–C), 115.73 (Ar–C), 115.60 (Ar–C), 113.17 (Ar–C), 72.95 (5-C), 42.33 (NHCH2), 33.92 (Ar–CH2); LC–MS (m/z): positive mode 249 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: 95.4%. mp: 285–287 °C.
6-(o-Chlorophenylethylamino)-2,4(1H,3H)-pyrimidinedione (58)
Compound 58 was synthesized using 6-chlorouracil (11, 10 mmol) and 1-(o-chlorophenyl)ethylamine (50 mmol, 5 equiv) and was isolated as a light yellow solid (88% yield). 1H NMR (500 MHz, DMSO-d6) δ (ppm) 10.13 (s, 1H, 1-NH), 7.48–7.22 (m, 4H, 4 × Ar–H), 6.12 (s, 1H, 6-NH), 4.50 (s, 1H, 5-H), 3.28–3.22 (m, 2H, NHCH2), 2.92 (dd, J = 7.9, 6.6 Hz, 2H, Ar–CH2); 13C NMR (126 MHz, DMSO-d6) δ 164.34 (4-C), 154.05 (2-C), 150.93 (6-C), 136.21 (Ar–C), 133.27 (Ar–C), 131.35 (Ar–C), 129.40 (Ar–C), 128.54 (Ar–C), 127.49 (Ar–C), 72.85 (5-C), 41.12 (NHCH2), 32.10 (Ar–CH2); LC–MS (m/z): positive mode 265 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: 96.7%. mp: 289–291 °C.
6-(m,p-Dimethoxyphenethylamino)-2,4(1H,3H)-pyrimidinedione (60)
Compound 60 was synthesized using 6-chlorouracil (11, 10 mmol) and 1-(m,p-dimethoxyphenyl)ethylamine (50 mmol, 5 equiv) and was isolated as a white solid (92% yield). 1H NMR (500 MHz, DMSO-d6) δ (ppm) 6.95–6.80 (m, 2H, 2 × Ar–H), 6.74 (dd, J = 8.2, 2.0 Hz, 1H, Ar–H), 6.09 (s, 1H, 6-NH), 4.48 (s, 1H, 5-H), 3.74 (s, 3H, OCH3), 3.71 (s, 3H, OCH3), 3.24 (td, J = 7.2, 5.6 Hz, 2H, NHCH2), 2.72 (t, J = 7.1 Hz, 2H, Ar–CH2); 13C NMR (126 MHz, DMSO-d6) δ 164.42 (4-C), 154.55 (2-C), 151.43 (6-C), 135.05 (Ar–C), 130.72 (Ar–C), 130.66 (Ar–C), 130.55 (Ar–C), 115.26 (Ar–C), 114.96 (Ar–C), 72.83 (5-C), 43.04 (OCH3), 42.76 (OCH3), 37.71 (NHCH2), 33.41 (Ar–CH2); LC–MS (m/z): positive mode 291 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: >99%. mp: 265–267 °C.
6-(o,p-Dichlorophenethylamino)-2,4(1H,3H)-pyrimidinedione (61)
Compound 61 was synthesized using 6-chlorouracil (11, 10 mmol) and 1-(o,p-dichlorophenyl)ethylamine (50 mmol, 5 equiv) and was isolated as a white solid (96% yield). 1H NMR (500 MHz, DMSO-d6) δ (ppm) 10.13 (s, 1H, 6-NH), 9.87 (s, 1H, 3-NH), 7.59 (t, J = 1.3 Hz, 1H, Ar–H), 7.39 (d, J = 1.3 Hz, 2H, 2 × Ar–H), 6.25 (s, 1H, 1-NH), 4.49 (d, J = 1.2 Hz, 1H, 5-H), 3.28–3.10 (m, 2H, NHCH2), 2.90 (t, J = 7.1 Hz, 2H, Ar–CH2); 13C NMR (126 MHz, DMSO-d6) δ 164.35 (4-C), 154.03 (2-C), 150.94 (6-C), 135.48 (Ar–C),134.25 (Ar–C), 132.68 (Ar–C), 132.11 (Ar–C), 128.81 (Ar–C), 127.60 (Ar–C), 72.92 (NHCH2), 31.53 (Ar–CH2); LC–MS (m/z): positive mode 299 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: >99.0%. mp: >300 °C.
6-(1H-Indol-3-ylethylamino)-2,4(1H,3H)-pyrimidinedione (62)
Compound 62 was synthesized using 6-chlorouracil (11, 10 mmol) and 1-(indol-3-yl)ethylamine (50 mmol, 5 equiv) and was isolated as a white solid (93% yield). 1H NMR (500 MHz, DMSO-d6) δ (ppm) 10.11 (s, 1H, 1-NH), 7.53 (dd, J = 8.1, 1.0 Hz, 1H, Ar–H), 7.34 (dt, J = 8.1, 0.9 Hz, 1H, Ar–H), 7.19 (d, J = 2.4 Hz, 1H, Ar–H), 7.07 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H, Ar–H), 6.98 (dd, J = 8.0, 6.9, Hz, 1H, Ar–H), 4.48 (d, J = 1.4 Hz, 1H, 5-H), 3.34–3.30 (m, 2H, NHCH2), 2.99–2.86 (m, 2H, Ar–CH2); 13C NMR (126 MHz, DMSO-d6) δ 164.38 (4-C), 154.08 (2-C), 150.91 (6-C), 136.45 (Ar−C), 127.20 (Ar−C), 123.32 (Ar−C), 121.16 (Ar−C), 118.48 (Ar−C), 118.34 (Ar−C), 111.56 (Ar−C), 110.96 (Ar−C), 72.72 (5-C), 42.01 (NHCH2), 24.20 (Ar–CH2); LC–MS (m/z): positive mode 270 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: >96.0%. mp: 270–272 °C.
6-(Thiophen-2-yl)ethylamino-2,4(1H,3H)-pyrimidinedione (63)
Compound 63 was synthesized using 6-chlorouracil (11, 10 mmol) and 2-thiopheneethylamine (50 mmol, 5 equiv) and was isolated as a yellow solid (97% yield). 1H NMR (500 MHz, DMSO-d6) δ (ppm) 7.32 (dd, J = 25.5, 5.1, Hz, 1H, Ar–H), 6.95 (dd, J = 14.4, 5.0, Hz, 2H, 2 × Ar–H), 6.29 (s, 1H, 3-NH), 4.45 (s, 1H, 5-H), 3.01 (t, J = 6.9 Hz, 2H, NHCH2), 2.90–2.65 (m, 2H, Ar–CH2); 13C NMR (126 MHz, DMSO-d6) δ 164.40 (4-C), 154.44 (2-C), 142.39 (6-C), 140.97 (Ar–C), 127.17 (Ar–C), 127.02 (Ar–C), 125.72 (Ar–C), 123.84 (Ar–C), 72.86 (5-C), 43.52 (NHCH2), 28.54 (Ar–CH2); LC–MS (m/z): positive mode 237 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: 95.5%. mp: 271–273 °C.
6-(2-Phenoxyethylamino)-2,4(1H,3H)-pyrimidinedione (64)
Compound 64 was synthesized using 6-chlorouracil (11, 10 mmol) and 2-phenoxyethylamine (50 mmol, 5 equiv) and was isolated as a pale yellow solid (87% yield). 1H NMR (600 MHz, DMSO-d6) δ (ppm) 10.17 (s, 1H, 3-NH), 7.33–7.25 (m, 2H, 2 × Ar–H), 6.97–6.90 (m, 3H, 3 × Ar–H), 4.52 (d, J = 1.6 Hz, 1H, 5-H), 4.08 (t, J = 5.2 Hz, 2H, OCH2), 3.40 (q, J = 5.3 Hz, 2H, NHCH2); 13C NMR (151 MHz, DMSO-d6) δ 164.36 (4-C), 158.32 (2-C), 154.19 (6-C), 150.87 (Ar–Ar–C), 129.71 (Ar–C), 129.60 (Ar–C), 121.07 (Ar–C), 114.69 (Ar–C), 73.09 (Ar–C), 65.77 (5-C), 41.11 (NHCH2), 40.99 (OCH2); LC–MS (m/z): positive mode 247 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: 97.0%. mp: >300 °C.
6-(3-Phenoxypropylamino)-2,4(1H,3H)-pyrimidinedione (65)
Compound 65 was synthesized using 6-chlorouracil (11, 10 mmol) and 3-phenoxypropylamine (50 mmol, 5 equiv) and was isolated as a light yellow solid (90% yield). 1H NMR (500 MHz, DMSO-d6) δ (ppm) 7.38–7.13 (m, 2H, 2 × Ar–H), 6.99–6.75 (m, 3H, 3 × Ar–H), 4.42 (s, 1H, 5-H), 4.01 (td, J = 6.2, 1.9 Hz, 2H, OCH2), 3.16 (q, J = 6.3 Hz, 2H, NHCH2), 1.88–1.87 (m, 2H, CH2); 13C NMR (126 MHz, DMSO-d6) δ 164.50 (4-C), 158.58 (2-C), 155.43 (6-C), 129.57 (Ar–C), 129.54 (Ar–C), 120.67 (Ar–C), 120.57 (Ar–C), 114.53 (Ar–C), 72.60 (Ar–C), 65.08 (5-C), 38.57 (NHCH2), 37.99 (OCH2), 28.06 (CH2); LC–MS (m/z): positive mode 261 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: 95.4%. mp: 180–182 °C.
6-(4-Benzylpiperidin-1yl)-2,4(1H,3H)-pyrimidinedione (67)
Compound 67 was synthesized using 6-chlorouracil (11, 10 mmol) and 4-benzylpiperidine (50 mmol, 5 equiv) and was isolated as a white solid (81% yield). 1H NMR (600 MHz, DMSO-d6) δ (ppm) 10.29 (d, J = 14.0 Hz, 2H, 2 × NH), 7.27 (t, J = 7.5 Hz, 2H, 2 × Ar–H), 7.21–7.01 (m, 3H, 3 × Ar–H), 4.60 (d, J = 1.5 Hz, 1H, 5-H), 3.70 (dt, J = 12.6, 2.3 Hz, 2H, N–CH2), 2.71 (dd, J = 12.8, 2.6 Hz, 2H, N–CH2), 2.50 (d, J = 7.0 Hz, 2H, CH2), 1.83–1.39 (m, 3H, CH and CH2), 1.37–0.87 (m, 2H, CH2); 13C NMR (151 MHz, DMSO-d6) δ 164.38 (4-C), 155.46 (2-C), 151.67 (6-C), 140.13 (Ar–C), 129.14 (Ar–C), 128.32 (Ar–C), 125.99 (Ar–C), 77.51 (5-C), 46.70 (N–CH2), 42.03 (N–CH2), 37.06 (Ar–CH2), 30.87 (2 × CH2); LC–MS (m/z): positive mode 285 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: >99.0%. mp: 277–279 °C.
6-[4-(p-Chlorophenyl)-1-piperazinyl]-2,4(1H,3H)-pyrimidinedione (68)
Compound 68 was synthesized using 6-chlorouracil (11, 10 mmol) and 1-(p-chlorophenyl)piperazine (50 mmol, 5 equiv) and was isolated as a white solid (76% yield). 1H NMR (500 MHz, DMSO-d6) δ (ppm) 10.41 (s, 1H, 3-NH), 7.30–7.18 (m, 2H, 2 × Ar–H), 7.03–6.93 (m, 2H, 2 × Ar–H), 4.70 (d, J = 1.3 Hz, 1H, 5-H), 3.40–3.32 (m, 4H, 2 × N–CH2), 3.18 (dd, J = 6.5, 3.8 Hz, 4H, 2 × N–CH2); 13C NMR (126 MHz, DMSO-d6) δ 164.33 (4-C), 155.68 (2-C), 151.56 (6-C), 149.48 (Ar–C), 149.01 (Ar–C), 128.94 (Ar–C), 128.81 (Ar–C), 122.94 (Ar–C), 117.68 (Ar–C), 78.24 (5-C), 47.47 (N–CH2), 46.05 (N–CH2), 45.49 (N–CH2), 42.65 (N–CH2). LC–MS (m/z): positive mode 307 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: 95.0%. mp: 279–281 °C.
6-[4-(2,3-Dihydro-1H-inden-2-yl)-1-piperazinyl]-2,4(1H,3H)-pyrimidinedione (70)
Compound 70 was synthesized using 6-chlorouracil (11, 10 mmol) and 4-(2,3-dihydro-1H-inden-2-yl)-1-piperazine (50 mmol, 5 equiv) and was isolated as a white solid (84% yield). 1H NMR (500 MHz, DMSO-d6) δ (ppm) 7.26 (dd, J = 7.1, 3.4 Hz, 1H, Ar–H), 7.22 (dd, J = 6.3, 2.3 Hz, 1H, Ar–H), 7.18 (dd, J = 7.6, 6.8, 3.9 Hz, 2H, 2 × Ar–H), 4.62 (s, 1H, 5-H), 4.46–3.97 (m, 1H, CH), 3.21 (dt, J = 6.2, 3.9 Hz, 4H, 2 × N–CH2), 2.81–2.79 (m, 2H, 2 × N–CH2), 2.38–2.36 (m, 2H, Ar–CH2), 1.99 (q, J = 7.2 Hz, 2H, Ar–CH2); 13C NMR (126 MHz, DMSO-d6) δ 164.32 (4-C), 155.76 (2-C), 151.57 (6-C), 143.84 (Ar–C), 142.32 (Ar–C), 127.59 (Ar–C), 126.23 (Ar–C), 125.12 (Ar–C), 124.76 (Ar–C), 77.91 (5-C), 69.05 (N–CH), 47.54 (N–CH2), 46.74 (N–CH2), 30.47 (2 × N–CH2), 24.33 (2 × Ar–CH2); LC–MS (m/z): positive mode 312 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: 98.0%. mp: 280–282 °C.
6-[4-(1-Naphthylmethyl)-1-piperazinyl]-2,4(1H,3H)-pyrimidinedione (71)
Compound 71 was synthesized using 6-chlorouracil (11, 10 mmol) and 1-(1-naphthylmethyl)piperazine (50 mmol, 5 equiv) and was isolated as a pale brown solid (82% yield). 1H NMR (500 MHz, DMSO-d6) δ (ppm) 8.27 (dd, J = 7.8, 1.5 Hz, 1H, Ar–H), 7.98–7.77 (m, 2H,2 × Ar–H), 7.63–7.48 (m, 2H, 2 × Ar–H), 7.48–7.37 (m, 2H, 2 × Ar–H), 4.63 (d, J = 1.3 Hz, 1H), 3.90 (s, 2H, Ar–CH2–N), 3.20 (t, J = 5.0 Hz, 4H, 2 × N–CH2), 2.47 (d, J = 4.9 Hz, 4H, 2 × N–CH2); 13C NMR (126 MHz, DMSO-d6) δ 164.32 (4-C), 155.79 (2-C), 151.56 (6-C), 133.62 (Ar–C), 132.15 (Ar–C), 128.38 (Ar–C), 128.01 (Ar–C), 127.64 (Ar–C), 125.97 (Ar–C), 125.83 (Ar–C), 125.31 (Ar–C), 124.87 (Ar–C), 78.04 (5-C), 60.02 (Ar–CH2–N), 52.07 (2 × N–CH2), 46.40 (2 × N–CH2); LC–MS (m/z): positive mode 336 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: 97.0%. mp: 240–242 °C.
5-Bromo-6-octylamino-2,4(1H,3H)-pyrimidinedione (73)
Compound 73 was synthesized using 6 (10 mmol) in pyridine (2 mL) and N-bromosuccinimide (12 mmol) and was isolated as a yellow solid (89% yield). 1H NMR (500 MHz, DMSO-d6) δ (ppm) 3.22 (q, J = 7.1 Hz, 2H, NHCH2), 1.41 (p, J = 6.8, 6.4 Hz, 2H, CH2), 1.31–1.15 (m, 10H, 5 × CH2), 0.85 (t, J = 6.8 Hz, 3H, CH3); 13C NMR (126 MHz, DMSO-d6) δ 164.15 (4-C), 159.30 (2-C), 152.20 (6-C), 97.76 (5-C), 31.33 (NH–CH2), 29.64 (CH2), 29.23 (CH2), 28.73 (CH2), 28.71 (CH2), 28.57 (CH2), 26.03 (CH2), 22.18 (CH2), 14.07 (CH3); LC–MS (m/z): positive mode 316 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: >97.0%. mp: > 300 °C.
5-Bromo-6-(p-bromophenylethylamino)-2,4(1H,3H)-pyrimidinedione (74)
Compound 74 was synthesized using 48 (10 mmol) in pyridine (2 mL) and N-bromosuccinimide (12 mmol) and was isolated as a yellow solid (97% yield). 1H NMR (600 MHz, DMSO-d6) δ (ppm) 8.30–8.18 (m, 2H, 2 × Ar–H), 7.26–7.15 (m, 2H, 2 × Ar–H), 3.47 (dt, J = 7.9, 6.3 Hz, 2H, NH–CH2), 2.70 (t, J = 7.3 Hz, 2H, Ar–CH2); 13C NMR (151 MHz, DMSO-d6) δ 160.17 (4-C), 150.14 (2-C), 147.19 (6-C), 137.96 (Ar–C), 131.36 (Ar–C), 129.12 (Ar–C), 119.67 (Ar–C), 97.95 (5-C), 42.40 (NH–CH2), 34.60 (Ar–CH2); LC–MS (m/z): positive mode 387 [M – 2H]2– and 389 [M – H]1–; purity by HPLC-UV (254 nm)-ESI-MS: 96.0%. mp: >300 °C.
6-Heptylamino-5-nitroso-2,4(1H,3H)-pyrimidinedione (77)
Compound 77 was synthesized using 21 (10 mmol) and was isolated as a pink solid (68% yield). 1H NMR (500 MHz, DMSO-d6) δ (ppm) 3.34–3.36 (m, 2H, NHCH2) 1.50–1.48 (m, 2H, CH2), 1.38–1.07 (m, 8H, 4 × CH2), 0.86 (t, J = 6.7 Hz, 3H, CH3); 13C NMR (126 MHz, DMSO-d6) δ 162.99 (4-C), 154.53 (2-C), 152.10 (6-C), 79.16 (5-C), 31.31 (NHCH2), 28.14 (CH2), 27.12 (CH2), 26.50 (CH2), 25.85 (CH2), 22.15 (CH2), 14.05 (CH3); LC–MS (m/z): positive mode 254 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: >96.0%. mp: 210–212 °C.
5-Nitroso-6-octylamino-2,4(1H,3H)-pyrimidinedione (78)
Compound 78 was synthesized using 6 (10 mmol) and was isolated as a pink solid (73% yield). 1H NMR (500 MHz, DMSO-d6) δ 10.74 (s, 1H, 1-NH), 7.59 (s, 1H, 3-NH), 3.35 (q, J = 6.7 Hz, 2H), 1.51 (p, J = 7.1 Hz, 2H), 1.35–1.08 (m, 10H), 0.85 (t, J = 6.7 Hz, 3H); 13C NMR (126 MHz, DMSO-d6) δ 161.74 (4-C), 151.88 (2-C), 149.19 (6-C), 138.22 (5-C), 31.32 (NHCH2), 28.31, 27.11, 26.33, 25.89, 22.18 (6 × CH2), 14.06 (CH3); LC–MS (m/z): positive mode 267 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: >99.0%. mp: 171–173 °C.
5-Nitroso 6-(3-phenylpropylamino)-2,4(1H,3H)-pyrimidinedione (80)
Compound 80 was synthesized using 43 (10 mmol) and was isolated as a light purple solid (72% yield). 1H NMR (500 MHz, DMSO-d6) δ 10.67 (s, 1H, 1-NH), 7.33–7.24 (m, 2H), 7.23–7.13 (m, 3H), 3.36 (q, J = 6.7 Hz, 2H), 2.62 (q, J = 7.7 Hz, 2H), 1.89–1.79 (m, 2H); 13C NMR (126 MHz, DMSO-d6) δ 163.58 (4-C), 161.90 (2-C), 141.20 (6-C), 138.32 (5-C), 128.59 (Ar–C), 128.49 (Ar–C), 128.40 (Ar–C), 128.36 (Ar–C), 126.21 (Ar–C), 126.03 (Ar–C), 41.21 (Ar–CH2), 30.03 (NHCH2), 29.41 (CH2); LC–MS (m/z): positive mode 274 [M + H]1+; purity by HPLC-UV (254 nm)-ESI-MS: 95.0%. mp: 212–214 °C.
Biological Assays
The recombinant CHO cell line expressing the human GPR84 (CHO-hGPR84 cells) with a β-galactosidase fragment and β-arrestin 2 containing the complementary fragment of the enzyme for performing β-arrestin recruitment (Pathhunter) was purchased from DiscoverX (Fremont, CA). The CHO-hGPR84 cells were cultured in F12 medium supplemented with 10% FCS, 100 units/mL penicillin G, 100 μg/mL streptomycin, 800 μg/mL G 418, 300 μg/mL hygromycin B, and 1% ultraglutamin (Invitrogen, Carlsbad, CA, or Sigma-Aldrich, St. Louis, MO). Stock solutions of compounds were prepared in DMSO. The final DMSO concentration in the assays did not exceed 1%. Data analysis was performed using GraphPad Prism (version 6.02). Concentration–response data were fitted by nonlinear regression to estimate EC50 values (Prism 6.02). The unpaired t test was used for statistical comparisons. Pathway bias was calculated as described by Winpenny et al.33 Δlog(Emax/EC50) was calculated by the following equation: Δlog(Emax/EC50) = log(EmaxB/EC50B) – log(EmaxA/EC50A), where A is the reference agonist decanoic acid and B is the test compound. ΔΔlog(Emax/EC50) was determined using the following equation: ΔΔlog(Emax/EC50) = Δlog(Emax/EC50)Gi pathway – Δlog(Emax/EC50)ß-arrestin pathway.
GPR84 cAMP Accumulation Assays
cAMP assays were performed as previously described.26,35 In short, CHO-hGPR84 cells were stimulated by the addition of forskolin (10 μM) in the absence (control) or presence of test compounds for 15 min. The reaction was stopped with hot (90 °C) lysis solution (4 mM ethylenediaminetetraacetic acid, 0.01% Triton X-100 in water). cAMP levels were quantified by a radioactive assay using [3H]cAMP (PerkinElmer, Rodgau, Germany) and a cAMP-binding protein prepared from bovine adrenal medulla. The forskolin-induced increase in cAMP concentration in the presence of agonists was expressed as a percentage of the response to forskolin in the absence of agonists (% of control). Three independent experiments, each in duplicate, were performed.
GPR84 β-Arrestin Recruitment Assays
β-Arrestin assays were performed as previously described.26 Briefly, CHO-hGPR84 cells (20 000 per well) were incubated with compound dilutions (in DMSO, final concentration: 1%) for 90 min before adding the detection reagent (DiscoverX). After 60 min of incubation at rt, the luminescence was measured using an NXT plate reader (PerkinElmer, Meriden, CT). Three to five independent experiments were performed, each in duplicate.
FFAR4 β-Arrestin Recruitment Assays
β-Arrestin assays were performed as previously described.26 In brief, CHO-hFFAR4 cells were incubated with test compound or the reference agonist 4-[(4-fluoro-4′-methyl[1,1′-biphenyl]-2-yl)methoxy]benzenepropanoic acid (TUG-891) for 90 min. In antagonist assays, the cells were preincubated with the antagonist for 30 min before adding the reference agonist. Next, the detection reagent was added and the mixture was incubated for 60 min at rt. The luminescence was measured using an NXT plate reader (PerkinElmer, Meriden, CT). All compounds were tested at a final concentration of 10 μM. Test results were normalized to values obtained by determining the background and the signal induced by 30 and 4 μM TUG-891 in agonist and antagonist assays, respectively. Three to four independent experiments were performed in duplicate.
FFAR1 Calcium Mobilization Assay
β-Arrestin assays were performed as previously described.26 In brief, 1321N1 astrocytoma cells recombinantly expressing human FFAR1 were incubated with Hank’s balanced salt solution buffer solution supplemented with 3 μM of the calcium dye Fluo-4-AM (Life Technologies, Darmstadt, Germany) and 0.06% Pluronic F-127 for 60 min. After exchanging the buffer, test compound solutions were added to each well using a FlexStation 3 plate reader (Molecular Devices, Sunnyvale, CA). In antagonist assays, cells were preincubated for 30 min with test compound solutions before adding the reference agonist 3-(4-(o-tolylethynyl)phenyl)propanoic acid (TUG-424, final concentration: 1 μM ≈ EC80). All compounds were tested at a final concentration of 10 μM. Signals induced by the test compounds were normalized to the signals induced by 1 and 10 μM TUG-424 in antagonist and agonist assays, respectively. Three to four independent experiments were performed in duplicate.
Metabolic Stability
Pooled human liver microsomes (0.5 mg/mL, mixed gender, pooled) were employed. Compounds were tested at a concentration of 1 μM. These experiments were performed by Pharmacelsus (Contract Research Organisation), Saarbrücken, Germany. Data points represent means of two separate experiments performed in duplicate. Standard deviations were around ±20%.
Pharmacophore Modeling
The selected GPR84 agonists 2, 4, 6, and 48 were flexibly aligned using the flexible alignment module implemented in Molecular Operating Environment (MOE 2014.09).36 The flexible alignment method utilizes the stochastic search procedure and simultaneously searches for the conformational space of the defined molecules and the space of alignment of those molecules.37 The method aligns the GPR84 agonists by maximizing steric and feature overlap using the MMFF94x force field.38 Each resulting alignment is given a score that quantifies the quality of the alignment in terms of both internal strain and overlap of molecular features. The similarity terms, including hydrogen bond donor, acceptor, hydrophobicity, and volume, were used in the flexible alignment with the default settings for the other parameters. On the basis of the overall score, strain energy, and visual inspection of the alignment, the presented flexible alignment of the GPR84 agonists was selected. This alignment was then used as a template for the pharmacophore model generation. The pharmacophore model was generated using the pharmacophore elucidator feature in MOE 2014.09. The pharmacophore features were calculated automatically with the consensus pharmacophore function. This function clusters the features into potential pharmacophore features, which are more conserved than a tolerance and threshold value. For the presented pharmacophore model, a tolerance distance of 1.5 Å and a threshold of 50% conservation were used.
Acknowledgments
T.P. is grateful to the Alexander von Humboldt (AvH) Foundation and to Bayer Pharma for a postdoctoral fellowship. G.B. acknowledges the CAPES Foundation and the Ministry of Education of Brazil for supporting an internship. Expert technical contributions by Marion Schneider (LC–MS analyses), as well as Sabine Terhart-Krabbe and Annette Reiner (NMR studies) are gratefully acknowledged.
Glossary
Abbreviations
- GPCR
G protein-coupled receptor
- cAMP
cyclic adenosine monophosphate
- GPR
G protein-coupled receptor
- FFA/R
free fatty acid/receptor
- CHO
Chinese hamster ovary
- SAR
structure–activity relationship
- DIM
diindolylmethane
- DMSO
dimethyl sulfoxide
- NMR
nuclear magnetic resonance
- Capt
C compensated attached proton test
- HPLC
high-performance liquid chromatography
- LC–MS
liquid chromatography–mass spectrometry
- ESI
electrospray ionization
- TLC
thin-layer chromatography
- DMF
N,N′-dimethylformamide
- mp
melting point
- NBS
N-bromosuccinimide
- PSB
Pharmaceutical Sciences Bonn
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b02092.
Metabolic stability of compound 42 in liver microsomes (Figure S1); potency of GPR84 agonists at FFAR1 (Table S1) and FFAR4 receptors (Table S2); pEC50 values of GPR84 agonists determined in cAMP and β-arrestin assays (Table S3); synthesis, analysis, and 1H and 13C NMR spectra of final products (Figures S1–S60) (PDF)
Details of human GPR84 (cAMP assay), human GPR84 (β-arrestin assay), human FFAR1 (agonistic potency), human FFAR1 (antagonistic potency), human FFAR4 (agonistic potency), and human FFAR4 (antagonistic potency) (CSV)
The authors declare no competing financial interest.
Supplementary Material
References
- Fredriksson R.; Schiöth H. B. The repertoire of G-protein-coupled receptors in fully sequenced genomes. Mol. Pharmacol. 2005, 67, 1414–1425. 10.1124/mol.104.009001. [DOI] [PubMed] [Google Scholar]
- Fredriksson R.; Lagerström M. C.; Lundin L.-G.; Schiöth H. B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- Lagerström M. C.; Schiöth H. B. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat. Rev. Drug Discovery 2008, 7, 339–357. 10.1038/nrd2518. [DOI] [PubMed] [Google Scholar]
- Overington J. P.; Al-Lazikani B.; Hopkins A. L. How many drug targets are there?. Nat. Rev. Drug Discovery 2006, 5, 993–996. 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- Alexander S. P. H.; Davenport A. P.; Kelly E.; Marrion N.; Peters J. A.; Benson H. E.; Faccenda E.; Pawson A. J.; Sharman J. L.; Southan C.; Davies J. A. The concise guide to pharmacology 2015/16: G protein-coupled receptors. Br. J. Pharmacol. 2015, 172, 5744–5869. 10.1111/bph.13348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittenberger T.; Schaller H. C.; Hellebrand S. An expressed sequence tag (EST) data mining strategy succeeding in the discovery of new G-protein coupled receptors. J. Mol. Biol. 2001, 307, 799–813. 10.1006/jmbi.2001.4520. [DOI] [PubMed] [Google Scholar]
- Wang J.; Wu X.; Simonavicius N.; Tian H.; Ling L. Medium chain fatty acids as ligands for orphan G protein-coupled receptor GPR84. J. Biol. Chem. 2006, 281, 34457–34464. 10.1074/jbc.M608019200. [DOI] [PubMed] [Google Scholar]
- Suzuki M.; Takaishi S.; Nagasaki M.; Onozawa Y.; Iino I.; Maeda H.; Komai T.; Oda T. Medium-chain fatty acid-sensing receptor, GPR84, is a proinflammatory receptor. J. Biol. Chem. 2013, 288, 10684–10691. 10.1074/jbc.M112.420042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimura A.; Hirasawa A.; Hara T.; Tsujimoto G. Free fatty acid receptors act as nutrient sensors to regulate energy homeostasis. Prostaglandins Other Lipid Mediators 2009, 89, 82–88. 10.1016/j.prostaglandins.2009.05.003. [DOI] [PubMed] [Google Scholar]
- Bouchard C.; Pagé J.; Bédard A.; Tremblay P.; Vallières L. G protein-coupled receptor 84, a microglia-associated protein expressed in neuroinflammatory conditions. Glia 2007, 55, 790–800. 10.1002/glia.20506. [DOI] [PubMed] [Google Scholar]
- Oh D. Y.; Talukdar S.; Bae E. J.; Imamura T.; Morinaga H.; Fan W.; Li P.; Lu W. J.; Watkins S. M.; Olefsky J. M. GPR120 is an Omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 2010, 142, 687–698. 10.1016/j.cell.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkataraman C.; Kuo F. The G-protein coupled receptor, GPR84 regulates IL-4 production by T lymphocytes in response to CD3 crosslinking. Immunol. Lett. 2005, 101, 144–153. 10.1016/j.imlet.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Huang Q.; Feng D.; Liu K.; Wang P.; Xiao H.; Wang Y.; Zhang S.; Liu Z. A medium-chain fatty acid receptor Gpr84 in zebrafish: expression pattern and roles in immune regulation. Dev. Comp. Immunol. 2014, 45, 252–258. 10.1016/j.dci.2014.03.017. [DOI] [PubMed] [Google Scholar]
- Audoy-Rémus J.; Bozoyan L.; Dumas A.; Filali M.; Cynthia L.; Lacroix S.; Rivest S.; Tremblay M.-E.; Vallières L. GPR84 deficiency reduces microgliosis, but accelerates dendritic degeneration and cognitive decline in a mouse model of Alzheimer’s disease. Brain, Behav., Immun. 2015, 46, 112–120. 10.1016/j.bbi.2015.01.010. [DOI] [PubMed] [Google Scholar]
- Dietrich P. A.; Yang C.; Leung H. H. L.; Lynch J. R.; Gonzales E.; Liu B.; Haber M.; Norris M. D.; Wang J.; Wang J. Y. GPR84 sustains aberrant β-catenin signaling in leukemic stem cells for maintenance of MLL leukemogenesis. Blood 2014, 124, 3284–3294. 10.1182/blood-2013-10-532523. [DOI] [PubMed] [Google Scholar]
- Hoffmann K.; Köse M.; Schiedel A.; Hartweg J. L.; Schneider M.; Foerch P.; Gillard M.; Müller C. E.; von Kügelgen I.. Activation of human GPR84 by hydroxy-fatty acids and preparations of bacterial lipopolysaccharides. Eur. J. Pharmacol. 2018. [Google Scholar]
- Zhang Q.; Yang H.; Li J.; Xie X. Discovery and characterization of a novel small molecule agonist for medium chain free fatty acid receptor GPR84. J. Pharmacol. Exp. Ther. 2016, 357, 337–344. 10.1124/jpet.116.232033. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Zhang Q.; Chen L.-H.; Yang H.; Lu W.; Xie X.; Nan F.-J. Design and synthesis of 2-alkylpyrimidine-4,6-diol and 6-alkylpyridine-2,4-diol as potent GPR84 agonists. ACS Med. Chem. Lett. 2016, 7, 579–583. 10.1021/acsmedchemlett.6b00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakak Y.; Unett D. J.; Gatlin J.; Liaw C. W.. Human G Protein-Coupled Receptor and Modulators Thereof for the Treatment of Atherosclerosis and Atherosclerotic Disease and for the Treatment of Conditions Related to MCP-1 Expression. WO2007027661 A3, April 26, 2007.
- Krishnaswamy M.; Purushothaman K. K. Antifertility properties of Embelia ribes: (embelin). Indian J. Exp. Biol. 1980, 18, 1359–1360. [PubMed] [Google Scholar]
- Chitra M.; Sukumar E.; Suja V.; Devi C. S. S. Antitumor, antiinflammatory and analgesic property of embelin, a plant product. Chemotherapy 1994, 40, 109–113. 10.1159/000239181. [DOI] [PubMed] [Google Scholar]
- Chitra M.; Devi C. S. S.; Sukumar E. Antibacterial activity of embelin. Fitoterapia 2003, 74, 401–403. 10.1016/S0367-326X(03)00066-2. [DOI] [PubMed] [Google Scholar]
- Kundap U. P.; Bhuvanendran S.; Kumari Y.; Othman I.; Shaikh M. F. Plant derived phytocompound, embelin in CNS Disorders: A systematic review. Front. Pharmacol. 2017, 8, 76. 10.3389/fphar.2017.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda S.; Yamamoto A.; Okada T.; Matsumura E.; Nose E.; Kogure K.; Kojima S.; Haga T. Identification of surrogate ligands for orphan G protein-coupled receptors. Life Sci. 2003, 74, 367–377. 10.1016/j.lfs.2003.09.030. [DOI] [PubMed] [Google Scholar]
- a Fares F. The anti-carcinogenic effect of indole-3-carbinol and 3,3′-diindolylmethane and their mechanism of action. Med. Chem. 2014, S1, 1–8. 10.4172/2161-0444.S1-002. [DOI] [Google Scholar]; b Kiselev V. I.; Drukh V. M.; Muyzhnek E. L.; Kuznetsov I. N.; Pchelintseva O. I.; Paltsev M. A. Preclinical antitumor activity of the diindolylmethane formulation in xenograft mouse model of prostate cancer. Exp. Oncol. 2014, 36, 90–93. [PubMed] [Google Scholar]; c Firestone G. L.; Bjeldanes L. F. Indole-3-carbinol and 3-3′-diindolylmethane antiproliferative signaling pathways control cell-cycle gene transcription in human breast cancer cells by regulating promoter–Sp1 transcription factor interactions. J. Nutr. 2003, 133, 2448S–2455S. 10.1093/jn/133.7.2448S. [DOI] [PubMed] [Google Scholar]
- Pillaiyar T.; Köse M.; Sylvester K.; Weighardt H.; Thimm D.; Borges G.; Förster I.; von Kügelgen I.; Müller C. E. Diindolylmethane derivatives: Potent agonists of the immunostimulatory orphan G protein-coupled receptor GPR84. J. Med. Chem. 2017, 60, 3636–3655. 10.1021/acs.jmedchem.6b01593. [DOI] [PubMed] [Google Scholar]
- Nikaido Y.; Koyama Y.; Yoshikawa Y.; Furuya T.; Takeda S. Mutation analysis and molecular modeling for the investigation of ligand-binding modes of GPR84. J. Biochem. 2015, 157, 311–320. 10.1093/jb/mvu075. [DOI] [PubMed] [Google Scholar]
- Southern C.; Cook J. M.; Neetoo-Isseljee Z.; Taylor D. L.; Kettleborough C. A.; Merritt A.; Bassoni D. L.; Raab W. J.; Quinn E.; Wehrman T. S.; Davenport A. P.; Brown A. J.; Green A.; Wigglesworth M. J.; Rees S. Screening β-arrestin recruitment for the identification of natural ligands for orphan G-protein-coupled receptors. J. Biomol. Screening 2013, 18, 599–609. 10.1177/1087057113475480. [DOI] [PubMed] [Google Scholar]
- https://www.discoverx.com/DiscoveRx/media/ContentFiles/DataSheets/95-0158C2.pdf.
- Seward E.; Diederich F. Redox-dependent complexation ability of flavin-hosts in aqueous solution. Tetrahedron Lett. 1987, 28, 5111–5114. 10.1016/S0040-4039(00)95604-1. [DOI] [Google Scholar]
- Dickens M. P.; Roxburgh P.; Hock A.; Mezna M.; Kellam B.; Vousden K. H.; Fischer P. M. 5-Deazaflavin derivatives as inhibitors of p53 ubiquitination by HDM2. Bioorg. Med. Chem. 2013, 21, 6868–6877. 10.1016/j.bmc.2013.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Emam A. A.; Massoud M. A. M.; El-Bendary E. R.; El-Sayed M. A. Synthesis of certain 6-(arylthio)uracils and related derivatives as potential antiviral agents. Bull. Korean Chem. Soc. 2004, 25, 991–996. 10.5012/bkcs.2004.25.7.991. [DOI] [Google Scholar]
- Winpenny D.; Clark M.; Cawkill D. Biased ligand quantification in drug discovery: from theory to high throughput screening to identify new biased μ opioid receptor agonists. Br. J. Pharmacol. 2016, 173, 1393–1403. 10.1111/bph.13441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A.; Lin H.; Aryal D. K.; McCorvy J. D.; Dengler D.; Corder G.; Levit A.; Kling R. C.; Bernat V.; Hübner H.; Huang X.-P.; Sassano M. F.; Giguère P. M.; Löber S.; Duan D.; Scherrer G.; Kobilka B. K.; Gmeiner P.; Roth B. L.; Shoichet B. K. Structure-based discovery of opioid analgesics with reduced side effects. Nature 2016, 537, 185–190. 10.1038/nature19112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimm D.; Knospe M.; Abdelrahman A.; Moutinho M.; Alsdorf B. B. A.; von Kügelgen I.; Schiedel A. C.; Müller C. E. Characterization of new G protein-coupled adenine receptors in mouse and hamster. Purinergic Signalling 2013, 9, 415–426. 10.1007/s11302-013-9360-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molecular Operating Environment (MOE), 2014.09; Chemical Computing Group Inc.: Montreal, QC, Canada, 2016. [Google Scholar]
- Labute P.; Williams C.; Feher M.; Sourial E.; Schmidt J. M. Flexible alignment of small molecules. J. Med. Chem. 2001, 44, 1483–1490. 10.1021/jm0002634. [DOI] [PubMed] [Google Scholar]
- Halgren T. A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. . [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.