

Abstract
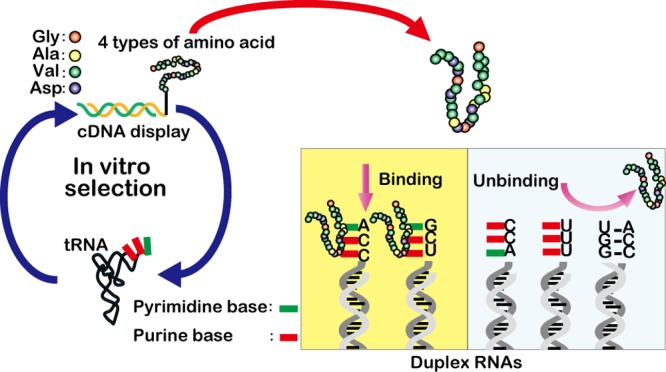
RNA–protein interactions have a central role in the living world. In this article, we examined whether primitive peptides (30 residues) consisting of four types of amino acid (Gly, Ala, Asp, and Val) could interact with tRNA as a model of primitive RNAs in the RNA world. By in vitro selection of binding peptides using the cDNA display method, a characteristic peptide was selected from a random peptide library and assayed by electrophoretic mobility shift and pull-down assays. Interestingly, the selected peptide bound to a single-stranded region including a loop structure of an RNA molecule with some sequence specificity.
Introduction
RNA–protein interactions are essential biological phenomena that maintain the viability of living cells.1,2 In particular, RNA binding proteins play an important role in controlling all major steps of the lifecycle of mRNA, including translation.3 Thus, one interesting question regarding the early stages of protein evolution on earth is how could a primitive protein (peptide) interact with RNA. In the origin of life, the “RNA world” hypothesis describes RNA molecules as performing self-replication and preserving genetic information,4 and a change toward the RNA–protein world (RNP world) occurred with the emergence of primitive ribosomes.5 So what function did the first protein have? Previous theoretical examination describing the emergence of the translation system suggested that if the first protein could interact with RNA and this RNA–protein complex could enhance the activity of a replicase-ribozyme then the early translation systems could have evolved by a virus-like strategy based on Eigen’s hypercycle theory.6 An RNA–protein enzyme has also been suggested to have functioned in the transition period from the RNA world to the RNP world.7 Thus, determining whether a primitive protein (peptide) can interact with RNA is of intrinsic importance. Primitive proteins probably consisted of a smaller subset of amino acids when compared with the 20 possible amino acids used presently because the translation system and amino acid synthesis pathways were likely primitive. A small set of amino acids, such as Gly, Ala, Asp, and Val, could have been abundantly synthesized by electric discharges under conditions of the primitive atmosphere on earth and by the impact of comets and meteorites on the surface of the primitive earth.8 According to Eigen’s theory, the first codons were GNC (N: A, C, G, and T) that encoded Gly, Ala, Asp, and Val.9 Interestingly, from the viewpoint of the present protein analysis, Ikehara suggested that primitive proteins also consisted of those four amino acids.10 Thus, we investigated whether peptides consisting of these four amino acids could interact with RNA by in vitro selection from a random peptide library consisting of Gly (G), Ala (A), Asp (D), and Val (V) using cDNA display.
The cDNA display method is a genotype–phenotype linking technology for in vitro selection of peptides and proteins using a cell-free translation system.11−16 The cDNA–protein linkage is very stable in comparison with that of similar methods (e.g., ribosome display, mRNA display) because a cDNA molecule is covalently bonded to its coded protein via a puromycin linker. Furthermore, any unwanted interactions originating from secondary structures of the mRNA can be prevented because of the formation of a cDNA/mRNA duplex with reverse transcription using a primer region in the puromycin linker. In this study, we performed in vitro selection using cDNA display with a 30-residue random peptide library against tRNA (a mixture of E. coli tRNAs), which should represent a model of structured RNA molecules in the RNA world.
Results and Discussion
In Vitro Selection of Primitive Peptides Consisting of Four Types of Amino Acid
The in vitro selection process using cDNA display with a peptide library composed of four amino acids (G, A, D, V) from the GNC random DNA library is shown in Scheme 1. The initial DNA library was constructed with eXact tag (Bio-Rad) and GADV regions. The eXact tag region was removed after translation and reverse transcription, i.e., following the synthesis of the cDNA display library. The resulting peptide region consisted of only G, A, D, and V. This truncated cDNA display library was then incubated with tRNA immobilized resin beads. After washing with selection buffer, the remaining cDNA display molecules were collected by elution after degradation of the peptide region with proteinase K. The cDNA moieties of the collected molecules were amplified by PCR for the next round of in vitro selection. The theoretical complexity of the pool is around 430 (= 1018). On the other hand, the initial size of the cDNA display library for in vitro selection was on the order of 1014 sequences. Our library covered only a part (<1/104) of the whole sequence space, but this number would be quite high compared to that of most other in vitro selections. After three rounds of selection, the library DNA molecules were cloned and sequenced (Table S1). Although 46 clones were sequenced, only six clones were obtained with a correct sequence that coded the four amino acids without frame shifts. Many deletions and frame shifts were also found in the sequences of the eXact tag region. This suggests that errors were made during the selection process resulting from PCR. PCR of GC-rich DNA with the normal Taq polymerase used herein is problematic owing to its poor amplification efficiency and replication fidelity because stable secondary structures resist melting and promote nonspecific product formation. Comparatively, the six selected peptides had many Val residues (Table 1).
Scheme 1. Schematic Representation of the in Vitro Selection of tRNA Binding Peptides.
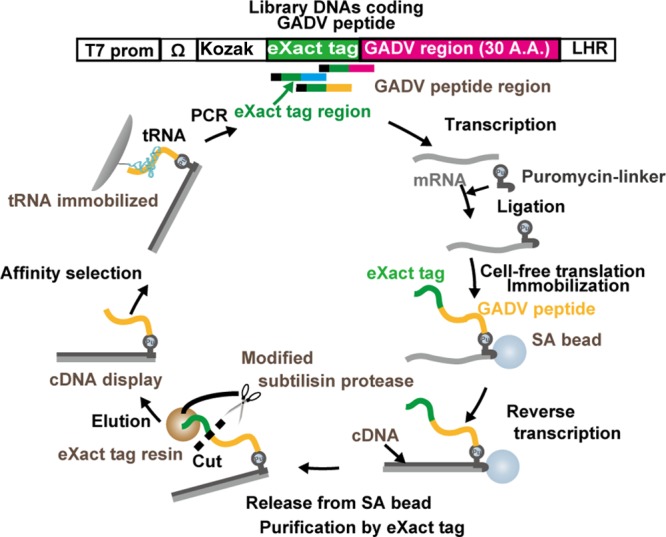
Table 1. Selected Peptide Sequences.
| amino acid sequence (N → C) | size (aa) | |
|---|---|---|
| GADV1 | GVVVVVVVAAVADVDGDAVVVDVGDVDVVV | 30 |
| GADV2 | VGVDAVVDVDVGGDVVDVDDVVAGVVGVVV | 30 |
| GADV3 | AVVAGVVDAVVVVVVVVDDDDAVDDVAGA | 29 |
| GADV4 | VVGVVAVDVVVVDDAVVVVDGVV | 23 |
| GADV5 | VDAVVGVGDDVVVVVAGVGA | 20 |
| GADV6 | AVVDVVVAVDVGDVGDVVDV | 20 |
Electrophoretic Mobility Shift Assay (EMSA)
Interactions between the selected peptide GADV1 and tRNA molecules were analyzed by EMSA (Figure 1) using a chemically synthesized fluorescently labeled peptide. The dissociation constant of the peptide and tRNA was estimated by plotting the ratio of shifted GADV1 peptide bands against the unbound GADV1 peptide band over a range of tRNA concentrations (12.5–200 μM). The results indicate that the GADV1 peptide has a moderate affinity toward tRNA with an apparent equilibrium dissociation constant (KDapp) of 66 ± 3 μM. The interaction was also confirmed qualitatively by fluorescence depolarization experiments (Figure S1). On the other hand, a control peptide lacking several valines, GADV1–4×Val peptide, interacts with tRNA with only a very weak affinity. The GADV1 peptide can interact with tRNA at two regions of one tRNA molecule, or higher tRNA concentrations may result in two tRNAs being bound for one peptide because two shifted bands were observed on the gel at 100 and 200 μM tRNA (Figure 1). Additional experiments would be necessary to investigate the interaction between GADV1 and tRNA in more detail. Next, four short control peptides, DAVV, VVDV, VGDA, and DAVVVDVG, were also tested for tRNA interaction. However, the interaction between the short peptides and tRNA could not be detected clearly (Figure S2). These results indicate that, although N-terminal Val residues seem to play an important role, the full-length GADV1 peptide is required for binding to tRNA with a KDapp in the micromolar range.
Figure 1.

(a) Fluorescence images of the gels loaded with GADV1 peptide (upper panel) and GADV1–4×Val peptide (lower panel) with different concentrations of tRNA. The loading sample buffer that included bromophenol blue (BPB) was loaded into the empty lanes to visually confirm alternate lanes in the gel. The position of BPB is indicated by an arrow in the figure. Each lane was loaded with the same amount of tRNA. However, the fluorescence of FAM was quenched at high concentrations of tRNA. (b) Binding curve plotted to estimate the affinity of GADV1 peptide for tRNA. The fraction of bound peptide was estimated as follows: [sum of the intensity of each upshifted GADV1 band at each concentration of tRNA]/([sum of the intensity of each upshifted GADV1 band at each concentration of tRNA] + [intensity of the free GADV1 band at each concentration of RNA]) × 100. The concentration of GADV1 was lower than that of tRNA; thus,the apparent dissociation constant (KDapp) can be approximated by the concentration of tRNA that results in one-half of the GADV1 being mobility-shifted. Curve fitting gave KDapp = 66 ± 3 μM (mean ± standard error; n = 3).
Pull-Down Assay Using Short RNA Immobilized Beads
The hydrophobic character of the GADV1 peptide and the absence of positively charged residues suggest that this peptide could interact with the accessible bases of RNA. To confirm this conjecture, pull-down assays were performed using a “Minihelix” RNA (a part of tRNA)17 with a loop region and a UCCA single-stranded region, a Minihelix-UCCA RNA that lacked the UCCA single-stranded region from the Minihelix, a Duplex RNA UCCA that lacked the loop region from the Minihelix, a Duplex RNA that lacked both the loop region and the UCCA single-stranded region from the Minihelix, and a single-stranded RNA that had no defined secondary structure (Table 2 and Figure S3). These RNAs were immobilized on magnetic beads, and a pull-down assay with the GADV1 peptide was performed. All immobilized RNAs, except the Duplex RNA, were observed to bind the GADV1 peptide (Figure 2a). Minihelix-UCCA and Duplex RNA UCCA could also bind the GADV 1 peptide (as could Minihelix). These results indicate that the GADV1 peptide can interact with the bases of loop and single-stranded regions of RNA. The GADV1 peptide obviously binds to the single-stranded RNA most strongly in comparison with the above Minihelix RNA, Minihelix-UCCA, and Duplex RNA UCCA. The sequence specificity of GADV1 binding must be weak because there are no identical exposed base sequence motifs among the Minihelix-UCCA, Duplex RNA UCCA, and single-stranded RNA. The target molecule in the in vitro selection experiment was a mixture of E. coli tRNA, and all of the tRNAs have only one exposed identical sequence, which is CCA at the 3′ terminus. GADV1 may be selected as a ligand for this universally conserved sequence. When considering that the loop of Minihelix has a base sequence of UCG, the sequence specificity might be decreased from CCA to YYR (Y = pyrimidine, R = purine). In order to confirm this conjecture, several pull-down assays using Duplex RNA UCCA, Duplex RNA UUCG (with a YYR single-stranded region), Duplex RNA UACC (with a RYY single-stranded region), and Duplex RNA UUUU (with a YYY single-stranded region) were performed. The results showed that the GADV1 peptide could bind to both the UCCA and UUCG RNAs, but it did not bind to the UACC or UUUU RNAs. (Figure 2b). The results suggest that the GADV1 peptide can interact with YYR single-stranded regions of RNA.The single-stranded RNA has three YYR regions, which may explain why the interaction between the GADV1 peptide and the single-stranded RNA was observed to be the strongest. As mentioned earlier, in the binding assay of Figure 1, the formation of a (tRNA)1(GADV1)2 or [(tRNA)1(GADV1)1]2 complex is not likely to take place because tRNA was in excess over the GADV1 peptide and the concentration of GADV1 was low. Thus, two up-shifted bands may correspond to (tRNA)2(GADV1)1 or two types of (tRNA)1(GADV1)1 complex, reflecting the presence of multiple putative binding sites in the tRNA (the loop and UCCA regions). In addition, the binding site at the loop regions in tRNA may be the anticodon loop because the bases in the anticodon loop in tRNA are more accessible than the bases in the T-loop and D-loop.
Table 2. RNA Sequences Used in the Binding Assay of GADV1a.

Figure 2.

Pull-down assay of GADV1 peptide against several types of model RNAs. Schematic RNA sequences, gel images, and the quantified band intensities are shown. Letters in blue indicate YYR (Y = pyrimidine, R = purine) base sequences in the single-stranded and loop regions. (a) Binding against Minihelix and its derivatives. The single-stranded RNA is derived from Minihelix and is unlikely to form secondary structure (see Figure S3). (b) Binding against several types of single-stranded regions. The band intensity of each lane was measured using analysis software, and the results are shown at the bottom. Values are shown as the mean ± standard error (n = 3). Full gel images are shown in Figure S4.
RNP World and Solubility of GADV1
Although a primitive peptide is unlikely to adopt a specific structure, a variety of conceivable structures containing two antiparallel β-strands and possibly a single α-helix can be predicted for GADV1 using computer simulations (Figure S5). In general, present day functional small proteins (except membrane proteins) are soluble and have stable conformations; however, the GAVD1 peptide has low water solubility (<4 μM) because it has many Val residues and probably adopts a dynamic ensemble of conformations. In the early stage of the RNP world, it is assumed that the main functional molecules were still RNAs and that proteins played a role in enhancing the function of these RNAs. Therefore, the solubility of RNA–peptide complexes may be more important than that of peptides alone when considering the functional activity of such primitive peptides. A previous study showed that proteins (>100 amino acids in length) consisting of 5 or 12 types of amino acid could acquire solubility by in vitro selection.18,19 However, peptides consisting of a smaller set of amino acids like GADV1 might have obtained solubility by forming RNA–peptide complexes in the last stage of the RNA world. Alternatively, there might be very little Val in the RNA world, assuming the conditions (simulated neutral atmospheres containing N2 and CO2) tested by Cleaves et al.20 A decrease in the number of Val residues in a peptide should markedly increase its water solubility. In the future, it would be very interesting to perform in vitro selection using the GAD peptide library against tRNA. Recently, intrinsically disordered proteins (IDPs) have been shown to possess many functions, including post-transcriptional gene regulation, cell signaling, and control of metabolic pathways. In particular, intrinsically disordered regions of RNA binding proteins often protect RNA molecules from nucleases and act as RNA chaperones.21−23 Primitive peptides (proteins) like GADV1 may have interacted with RNA molecules in the early stages of the RNP world in a similar manner as that observed for IDPs.
In this study, we have shown that a peptide (selected by cDNA display) consisting of four types of amino acid (G, A, V, and D) is able to interact with single-stranded regions of RNA with a KDapp of 66 ± 3 μM. The selected GADV1 peptide can interact with RNAs containing the single-stranded sequence YYR (Y = pyrimidine, R = purine). Early peptides (proteins) such as GADV1 could have interacted with RNA molecules in the early stages of the RNP world in a similar manner as that observed for IDPs.
Materials and Methods
Peptide Synthesis
The selected GADV1 peptide (GVVVVVVVAAVADVDGDAVVVDVGDVDVVV) was chemically synthesized by Toray Research Center (Tokyo, Japan). FAM (fluorescein amidite modification) was introduced at the N-terminus of GADV1 for the ease of fluorescence detection. The synthesized peptide with low solubility was examined by tricine sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) using a fluorescence image analyzer to confirm of peptide concentration (Figure S6). Reversed-phase HPLC analysis showed that the fraction of full-length FAM-GADV1 was 3% of the total synthesized peptides (Figure S7). Other control peptides (DAVV, VVDV, VGDA, and DAVVVDVG) were chemically synthesized and modified with FAM (SCRUM Inc., Tokyo, Japan). GADV1–4×Val (GVVVAAVADVDGDAVVVDVGDVDVVV) was also chemically synthesized and modified with ATTO 488 instead of FAM (Sigma-Aldrich Japan, Tokyo, Japan).
DNA Library Construction
For affinity selection against tRNA, random 30-mer peptides composed of Gly, Ala, Val, and Asp were designed while facilitating the formation and purification of cDNA-displayed proteins. The eXact tag was obtained from the Profinity eXact pPAL Supercoiled expression vector (Bio-Rad, Hercules, CA, USA) by PCR. The amplified eXact tag was joined to the 5′ UTR fragment DNA consisting of a T7 promoter, the tobacco mosaic virus “omega” UTR, a Kozak sequence, and an ATG start codon by overlap extension polymerase chain reaction (OE-PCR). DNA encoding the random 30-mer peptides was composed of the GNC codon triplet, where N indicates equimolar nucleotide mixtures of A, C, G, and T. The synthesized DNA coding this random peptide was joined with the above-mentioned UTR-eXact fragment by overlap PCR, yielding the DNA library for transcription.
Immobilization of tRNA on Agarose Beads
tRNA (total mixture) from E. coli. (Sigma-Aldrich Co., Saint Louis, MO, USA, catalog number R1753) was dissolved in water (30 μM). NaIO4 (0.1 M, 150 μL) was added to this tRNA (50 μ), and the mixture was incubated at 4 °C for 10 min to oxidize the 3′ terminus of the tRNA. 3′-Dialdehyde tRNA was isolated by precipitation with 1.4 mL of 2% LiClO4 in acetone followed by washing with 200 μL of acetone. The pellet was dissolved in 100 μL of 0.1 M sodium acetate, pH 5.2, and mixed with 100 μL of adipic acid dihydrazide-agarose resin (Sigma-Aldrich Co.) that was prewashed three times with ultrapure water (200 μL) using a spin column (GE Healthcare, Pittsburgh, PA, USA). The reaction mixture was shaken at room temperature for 3 h. d,l-Glyceraldehyde (67 mM, 100 μL) was added, and the mixture was shaken at room temperature for another 1 h. The resulting imine moiety of the tRNA-resin was reduced by adding 250 μL of 1 M NaCNBH3 and incubation at room temperature for 30 min. The agarose was washed with 500 μL of W1 buffer (0.1 M sodium acetate, pH 5.2, 300 mM NaCl, 8 M urea, and 0.1% SDS) and then suspended in 200 μL of W1 buffer. The immobilization efficiency was estimated by measuring the amount of recovered tRNA in the flow-through based on UV absorbance at 260 nm using a NanoDrop instrument (Thermo Fisher Scientific, Waltham, MA, USA). The immobilization amount of tRNA was 2–3 pmol per 10 μL of bead material.
In Vitro Selection against tRNA with a GADV Peptide Library against tRNA
The DNA library was transcribed into mRNA using the T7 RiboMAX Express large scale RNA production system (Promega, Madison, WI, USA), and the synthesized mRNA was purified with an RNA purification kit (FavorPrep After Tri-18 reagent RNA clean-up kit, Favorgen, Ping-Tung, Taiwan). Purified mRNA was hybridized to a short biotin segment puromycin linker (SBP linker)13 under annealing conditions (heating at 90 °C for 1 min followed by incubation at 70 °C for 1 min and subsequent cooling to 25 °C) in T4 ligase buffer (50 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 10 mM dithiothreitol, and 1 mM ATP) and ligated by T4 RNA ligase (0.4–2.0 U/pmol mRNA, Takara bio, Otsu, Japan) and polynucleotide kinase (0.5 U/pmol, Toyobo, Osaka, Japan) at 25 °C overnight. Translationof the linker-conjugated mRNAs was performed by an in vitro translation system with a Retic Lysate IVT kit (Retic Lysate IVT kit, Ambion, Austin, TX, USA) at 30 °C for 30 min. We did not modify this kit in order to synthesize our peptides composed of four species of amino acid. To synthesize an mRNA–linker–protein fusion, KCl and MgCl2 were added to the mixture (final concentrations of 800 and 80 mM, respectively), and the mixture was incubated at 37 °C for 40 min. The fusion library was immobilized on streptavidin-coated magnetic beads (SA-beads) (Dynabeads MyOne streptavidin C1 streptavidin magnetic beads, Invitrogen, Carlsbad, CA, USA) at 25 °C for 30 min. The beads were washed three times with 1× binding buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA, 1 M NaCl, 0.1% Tween 20). The immobilized fusions were reverse transcribed by Super Script III reverse transcriptase (Life Technologies, Carlsbad, CA, USA) at 40 °C for 30 min. To release the mRNA/cDNA–protein fusion molecules from the beads, Endonuclease V (Ambion) was added at 37 °C for 30 min. Supernatant containing cDNA display molecules was collected. To purify and cleave at the C-terminus of the eXact tag peptide residue, the eXact tag purification was performed using a Profinity eXact Mini spin column (Bio-Rad) to remove mRNA/cDNA fusions and cleave the eXact tag at its C-terminus. In the initial round, a cDNA display library was prepared from 40 pmol of library mRNA in 200 μL of selection buffer (50 mM Tris-HCl, pH 7.0, 467 mM NaCl, 57 mM MgCl2, 13 mM CaCl2, 9 mM KCl, 0.2% Tween 20). This mixture was incubated at room temperature for 30 min using a tube rotator (AS ONE, Osaka, Japan) with 100 μL of tRNA-agarose beads, which was prewashed using the selection buffer. The beads were washed 10 times using selection buffer. The bound cDNA display molecules were eluted from the beads with elution buffer (50 mM Tris-HCl, 6 M Urea, 2% SDS) at 37 °C for 20 min. The eluted cDNA display molecules were precipitated with ethanol and the coprecipitant (Quick-precip Plus, Edge BioSystems, Gaithersburg, MD, USA) and were dissolved in 20 μL of water. To prepare library DNAs for the next round of in vitro selection, T7 promoter reconstructed library DNAs were prepared from the above precipitated cDNA display molecules by PCR using Ex Taq HS (Takara Bio Inc., Shiga, Japan) and primers 5′-GATCCCGCGAAATTAATACGACTCACTATAGGGGAAGTATTTTTACAACAATTACCAACAACAACAACAAACAACAACAACATTACATTTTACATTCTACAACTACAAGCCACCATG-3′ and 5′-TTTCCCCGCCGCCCCCCGTCCTATCACCTCCATCTCCCCC-3′ for 25 cycles consisting of denaturation at 95 °C for 25 s, annealing at 69 °C for 20 s, and elongation at 72 °C for 30 s. The amplified product was analyzed by denaturing gel electrophoresis and visualized afterward via staining with SYBR Gold (Invitrogen). A total of three rounds of in vitro selection were performed according to the above protocol with minor changes as follows: the cDNA display library was prepared from 20 pmol of mRNA in the third round, the beads were washed 20 times using selection buffer in the second and third rounds, and the bound cDNA display molecules were eluted from the beads by proteinase K (500 μg/μL) (Wako, Tokyo, Japan) in proteinase K reaction buffer (10 mM Tris-HCl, pH 7.6, 10 mM EDTA, 0.5% SDS) at 37 °C for 30 min in the third round. After the third round, the library DNAs were the cloned using the pGEM-T easy vector system (Promega) and NEB 5-alpha competent E. coli (high efficiency) (New England Biolabs, Ipswich, MA, USA), and 46 DNA clones were sequenced by Operon Biotechnologies (Tokyo, Japan).
EMSA for GADV1–tRNA Interaction Analysis
GADV1 and GADV1–4×Val was dissolved in 2× selection buffer to 1 μM. The tRNA was dissolved in water (25, 50, 100, 200, and 400 μM). Then, 1 μL of each peptide solution was mixed with 1 μL of each tRNA solution, and the mixtures were incubated at 25 °C for 1 h. Native PAGE sample buffer (0.5× TBE, 30% glycerol) was added to the mixtures, and the samples were loaded immediately onto a 4% native PAGE that had undergone pre-electrophoresis at 100 V for 1 h. BPB dye was loaded to confirm alternate lanes. Electrophoresis was performed at 100 V for 90 min at 4 °C, and the gel was visualized with a fluorescence image analyzer (Pharos FX, Bio-Rad).
Pull-Down Assay for GADV1–tRNA Interaction Analysis Using Short RNA Immobilized Beads
Each RNA based on the sequence of the minihelix of E. coli tRNAGly (Table 2)17 was chemically synthesized by Hokkaido System Science Co., Ltd. (Hokkaido, Japan). Minihelix RNA, Minihelix-UCCA, Minihelix_duplex-B, and mixtures of Minihelix_duplex-B and Minihelix_duplex_UCCA(−) or Minihelix_duplex(−) were annealed in binding buffer (heating at 90 °C for 1 min and then cooling to 25 °C). A total of 50 pmol of each RNA sample was immobilized onto 10 μL of SA beads at 25 °C for 30 min. The mixtures of Minihelix_duplex_UUCG-B, Minihelix_duplex_UUUU-B, or Minihelix_duplex_UACC-B with Minihelix_duplex_UCCA(−) were annealed and immobilized in the same way. A total of 1000 pmol of biotin was then added to each sample to block free streptavidin. The beads were subsequently washed two times with 1× binding buffer and 1× selection buffer. The GADV1 peptide in selection buffer (10 μL, 3 μM concentration based on the intensity of FAM fluorescence) was incubated with each bead type at 25 °C for 1 h. The beads were washed three times with 200 μL of selection buffer, and the remaining peptides were eluted after incubation at 90 °C for 3 min in 20 μL of SDS sample buffer (62.5 mM Tris-HCl, pH 6.8, 4 M urea, 2% (w/v) SDS, 3% (w/v) sucrose, optimal amount of xylene cyanol). The eluates were subjected to 20% SDS-PAGE and visualized with a fluorescence image analyzer.
Acknowledgments
We are grateful to Profs. K. Nishigaki and M. Suzuki (Saitama Univ.) and Prof. M. Kinjo (Hokkaido Univ.) for helpful discussions and comments. We are also thankful to Dr. T. Terai (Saitama Univ.) for carefully reading and commenting on the revision of this manuscript.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.6b00015.
Results of clonal DNA sequencing, analysis of fluorescence depolarization, RNA secondary structure prediction, full gel images of pull-down assay, peptide structure prediction, tricine SDS-PAGE analysis, and reversed phase HPLC analysis (PDF)
This work was supported by JSPS KAKENHI (grant numbers 25008919 and 26350970) and the Core Research for Evolutional Science and Technology (CREST) program of the Japan Science and Technology Agency (JST).
The authors declare no competing financial interest.
Supplementary Material
References
- Hieronymus H.; Silver P. A. A systems view of mRNP biology. Genes Dev. 2004, 18, 2845–2860. 10.1101/gad.1256904. [DOI] [PubMed] [Google Scholar]
- Glisovic T.; Bachorik J. L.; Yong J.; Dreyfuss G. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008, 582, 1977–1986. 10.1016/j.febslet.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal N.; Scherrer T.; Gerber A. P.; Janga S. C. Interplay between posttranscriptional and posttranslational interactions of RNA-binding proteins. J. Mol. Biol. 2011, 409, 466–479. 10.1016/j.jmb.2011.03.064. [DOI] [PubMed] [Google Scholar]
- Gilbert W. Origin of life: The RNA world. Nature 1986, 319, 618. 10.1038/319618a0. [DOI] [Google Scholar]
- Weiner A. M. Eukaryotic nuclear telomeres: molecular fossils of the RNP world?. Cell 1988, 52, 155–157. 10.1016/0092-8674(88)90501-6. [DOI] [PubMed] [Google Scholar]
- Nemoto N.; Husimi Y. A model of the virus-type strategy in the early stage of encoded molecular evolution. J. Theor. Biol. 1995, 176, 67–77. 10.1006/jtbi.1995.0177. [DOI] [PubMed] [Google Scholar]
- Ahmad S.; Muthukumar S.; Kuncha S. K.; Routh S. B.; Yerabham A. S.; Hussain T.; Kamarthapu V.; Kruparani S. P.; Sankaranarayanan R. Specificity and catalysis hardwired at the RNA-protein interface in a translational proofreading enzyme. Nat. Commun. 2015, 6, 7552. 10.1038/ncomms8552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaia D. A.; Zaia C. T.; de Santana H. Which amino acids should be used in prebiotic chemistry studies?. Origins Life Evol. Biospheres 2008, 38, 469–488. 10.1007/s11084-008-9150-5. [DOI] [PubMed] [Google Scholar]
- Eigen M.; Schuster P.. The Hypercycle:A Principle of Natural Self-Organization; Springer-Verlag: Berlin, 1979; pp 60–88. [Google Scholar]
- Ikehara K. Possible steps to the emergence of life: the [GADV]-protein world hypothesis. Chem. Rec. 2005, 5, 107–118. 10.1002/tcr.20037. [DOI] [PubMed] [Google Scholar]
- Yamaguchi J.; Naimuddin M.; Biyani M.; Sasaki T.; Machida M.; Kubo T.; Funatsu T.; Husimi Y.; Nemoto N. cDNA display: a novel screening method for functional disulfide-rich peptides by solid-phase synthesis and stabilization of mRNA-protein fusions. Nucleic Acids Res. 2009, 37, e108. 10.1093/nar/gkp514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki Y.; Biyani M.; Tsuji-Ueno S.; Suzuki M.; Nishigaki K.; Husimi Y.; Nemoto N. One-pot preparation of mRNA/cDNA display by a novel and versatile puromycin-linker DNA. ACS Comb. Sci. 2011, 13, 478–485. 10.1021/co2000295. [DOI] [PubMed] [Google Scholar]
- Ueno S.; Kimura S.; Ichiki T.; Nemoto N. Improvement of a puromycin-linker to extend the selection target varieties in cDNA display method. J. Biotechnol. 2012, 162, 299–302. 10.1016/j.jbiotec.2012.09.003. [DOI] [PubMed] [Google Scholar]
- Ueno S.; Yoshida S.; Mondal A.; Nishina K.; Koyama M.; Sakata I.; Miura K.; Hayashi Y.; Nemoto N.; Nishigaki K.; Sakai T. In vitro selection of a peptide antagonist of growth hormone secretagogue receptor using cDNA display. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 11121–11126. 10.1073/pnas.1203561109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki Y.; Nishigaki K.; Nemoto N. Amino group binding peptide aptamers with double disulphide-bridged loops selected by in vitro selection using cDNA display. Chem. Commun. 2014, 50, 5608–5610. 10.1039/c4cc00799a. [DOI] [PubMed] [Google Scholar]
- Mochizuki Y.; Suzuki T.; Fujimoto K.; Nemoto N. A versatile puromycin-linker using cnvK for high-throughput in vitro selection by cDNA display. J. Biotechnol. 2015, 212, 174–180. 10.1016/j.jbiotec.2015.08.020. [DOI] [PubMed] [Google Scholar]
- Schimmel P.; Giegé R.; Moras D.; Yokoyama S. An operational RNA code for amino acids and possible relationship to genetic code. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 8763–8768. 10.1073/pnas.90.19.8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi N.; Kakukawa K.; Oishi Y.; Yanagawa H. High solubility of random-sequence proteins consisting of five kinds of primitive amino acids. Protein Eng., Des. Sel. 2005, 18, 279–284. 10.1093/protein/gzi034. [DOI] [PubMed] [Google Scholar]
- Tanaka J.; Doi N.; Takashima H.; Yanagawa H. Comparative characterization of randomsequence proteins consisting of 5, 12, and 20 kinds of amino acids. Protein Sci. 2010, 19, 786–795. 10.1002/pro.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleaves H. J.; Chalmers J. H.; Lazcano A.; Miller S. L.; Bada J. L. A reassessment of prebiotic organic synthesis in neutral planetary atmospheres. Origins Life Evol. Biospheres 2008, 38, 105–115. 10.1007/s11084-007-9120-3. [DOI] [PubMed] [Google Scholar]
- Kucera N. J.; Hodsdon M. E.; Wolin S. L. An intrinsically disordered C terminus allows the La protein to assist the biogenesis of diverse noncoding RNA precursors. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 1308–1313. 10.1073/pnas.1017085108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa P.; Csermely P. The role of structural disorder in the function of RNA and protein chaperones. FASEB J. 2004, 18, 1169–1175. 10.1096/fj.04-1584rev. [DOI] [PubMed] [Google Scholar]
- van der Lee R.; Buljan M.; Lang B.; Weatheritt R. J.; Daughdrill G. W.; Dunker A. K.; Fuxreiter M.; Gough J.; Gsponer J.; Jones D. T.; Kim P. M.; Kriwacki R. W.; Oldfield C. J.; Pappu R. V.; Tompa P.; Uversky V. N.; Wright P. E.; Babu M. M. Classification of Intrinsically Disordered Regions and Proteins. Chem. Rev. 2014, 114, 6589–6631. 10.1021/cr400525m. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.