

Abstract
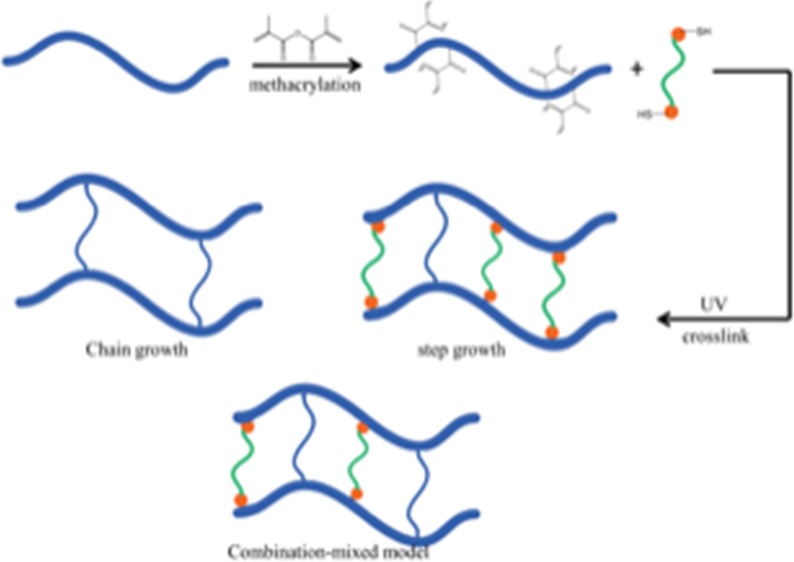
Gellan gum is a naturally occurring polymer that can cross-link in the presence of divalent cations to form biocompatible hydrogels. However, physically cross-linked gellan gum hydrogels lose their stability under physiological conditions, thus restricting the applications of these hydrogels in vivo. To improve the mechanical strength of the gels, we incorporated methacrylate into the gellan gum and chemically cross-linked the hydrogel through three polymerization methods: step growth through thiol–ene photoclick chemistry, chain-growth via photopolymerization, and mixed model in which both mechanisms were employed. Methacrylation was confirmed and quantified by proton nuclear magnetic resonance (1H NMR) and Fourier transform infrared spectroscopy. The mechanical properties and chemistry of the cross-linked gels were systematically altered by varying the reaction conditions. The compression moduli of the resulting hydrogels ranged between 6.4 and 17.2 kPa. The swelling ratios of the hydrogels were correlated with the compression moduli and affected by the addition of calcium. In vitro enzymatic degradation rate was found to depend on the degree of methacrylation. NIH/3T3 fibroblast cell proliferation and morphology were related to substrate stiffness, with a high stiffness leading generally to higher proliferation. The proliferation is further affected by the thiol–ene ratio. These results suggest that a hydrogel platform based on the gellan gum can offer versatile chemical modifications and tunable mechanical properties. The influence of these substrates on cell behavior suggests that the gellan gum hydrogels have the flexibility to be engineered for a variety of biomaterials applications.
1. Introduction
The overriding design principles for tissue engineering scaffolds are that the scaffold should best simulate the properties of the native tissue.1 Namely, the scaffolds should have similar mechanical properties to the tissue it will replace, the scaffold should be biocompatible, and the degradation rate of the scaffold should match that of the surrounding tissue.1 The native extracellular matrix (ECM) is a scaffold for cells to attach and proliferate. This structure has a high water content and a three-dimensional network that can be mimicked by hydrogels. Therefore, research efforts on designing hydrogels have focused on improving the biocompatibility and controllable mechanical properties to match those of native tissues.
Gellan gum is a natural polysaccharide derived from the bacteria and has potential applications in this area owing to its biocompatibility and flexibility for modification. The repeating unit of the gellan gum is a tetrasaccharide comprising l-rhamnose, d-glucuronic acid, and two d-glucose residues. As with other polysaccharides, lysozyme secreted by monocytes and neutrophils2 can degrade the backbone.3 The kinetics of degradation can be controlled by altering the degree of cross-linking, as well as the type of cross-linking bond, which can be achieved by altering the physical or chemical linkages.4 As with changing degradation profiles, the mechanical properties can also be altered by modifying the type and degree of cross-links.4 The mechanical property design requirements for scaffolds depend on the type of tissue, with softer (<2 kPa) substrates yielding the desired outcomes for liver and harder (>40 kPa) implants required for bone tissue engineering.1
Cations can be used to cross-link gellan gum gels by charge-shielding the polymer chains, promoting physical cross-links and aggregation. More specifically, multivalent cations provide bridges between carboxyl groups and reduce electrical repulsion.5 Although monovalent cations can also screen the charges on gellan molecules, the interaction is much weaker than the ionic bonds formed between the carboxylic acids and the divalent cations.5 Therefore, physically cross-linked gellan gum hydrogels will lose their stability in physiological conditions as the divalent cations are replaced by monovalent cations that are present at much higher concentrations in vivo.4
One way to improve the stability of gellan gum hydrogels is to covalently cross-link the gel.6−8 Carbodiimide amidation and esterification are commonly employed to conjugate the abundant carboxylic acid or hydroxyl groups present on the polysaccharide to a moiety capable of chemical cross-linking. Glycidyl methacrylate9 and methacrylic anhydride6,8,10 have been successfully used to methacrylate gellan gum.11,12 Enzymatic degradation of the hydrogel is retained as lysozyme attacks the backbone and can be tuned based on the degree of modification with acrylates and thiol concentrations. Likewise, the mechanical properties of these gels can be similarly tuned.13−15
The thiol–ene photoclick chemistry provides another free-radical-mediated method to synthesize hydrogels using cross-linkers containing multiple thiol groups.16−18 Specifically, a free radical abstracts a proton from a thiol, generating a thiyl radical. Subsequent propagation steps occur in a step-growth mechanism between the thiyl radical and the vinyl group.19 In this work, we examine the relationship between the mechanical properties and the cross-linking mechanism with cell proliferation using covalently cross-linked gellan gum. Through the introduction of carbon–carbon double bonds to the gellan gum, the polymer can be photo-cross-linked through chain growth, step growth, or a combination of the two, termed mixed model, to form gels with varying compressive moduli. Thiol–ene step-growth cross-linking is not inhibited by oxygen, therefore chain-growth cross-linking can be controlled by purging oxygen from the precursor solution. By adjusting the cross-linking conditions, the mechanical properties of the gel were systematically varied. The degradation profiles of these polymers in the presence of lysozyme was also investigated. Correlations between the mechanical properties and the cell proliferation were explored.
2. Methods and Materials
2.1. Materials
Gellan gum was purchased from Spectrum Chemical Mfg. Corp. (New Brunswick, NJ); methacrylate anhydride (MA) was obtained from BeanTown Chemical (Hudson, NH); 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone (Irgacure 2959) was obtained from Sigma-Aldrich (St. Louis, MO); dithiothreitol (DTT) was supplied by VWR Chemical (Batavia, IL). Fresh deionized (DI) water (Milli-Q Nanopure, Thermo Scientific, Waltham, MA) was used throughout this study.
2.2. Gellan Gum Modification
Methacrylated gellan gum was synthesized via substitution of the hydroxyl groups in the gellan gum repeating units with MA. Briefly, 1 g gellan gum was dissolved in 100 mL DI water in a round-bottom flask and heated to 90 °C for 30 min under constant stirring. The mixture was slowly cooled to 60 °C and either 3.5 mL or 8.5 mL MA was added for a low modified gellan gum (LMGG) or a high modified gellan gum (HMGG), respectively. The pH was maintained between 8 and 9.5. After 4 h, the product was dialyzed (12 000–14 000 molecular weight cutoff) against DI water for at least 3 days. The DI water was refreshed daily. The final product was lyophilized (Labconco, Kansas City, MO, 4.5 L).
2.3. Characterization of Modification of Gellan Gum
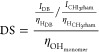
To quantify the percent modification of synthesized LMGG and HMGG, NMR was used to characterize the polymers. The modified polymers (10 mg) were dissolved in 1 mL D2O. The 1H NMR spectra were recorded on a Bruker Avance III spectrometer with a sweep width of 6602.1 Hz, a 90° pulse, and an acquisition time of 2.48 s. All of the spectra were obtained at room temperature. A total of 16 repetitive scans with 64 points were acquired and the data were processed in MNova with 128 k-points, zero filling, and exponential line broadening of 1.0 Hz. The methyl group on the rhamnose structural unit on gellan gum was used as a reference (δ = 1.45 ppm). The degree of substitution was calculated using eq 1.
 |
1 |
where IDB is the integration of the double bond proton peak, ICHl3rham is the integration of the reference peak, ηHDB and ηHCH3rham are the number of protons in the double bond and in the methyl group on the rhamnose, respectively, and ηOHmonomer is the number of reactive hydroxyl sites on the gellan gum.
2.4. Synthesis of Hydrogels
In 1 mL DI water, 10 mg modified gellan gum was dissolved at 55 °C. To this mixture, 1 mg Irgacure 2959 was added. For mixed-model LMGG hydrogels, either 3.4 or 6.8 μL 10 wt % DTT was added for a thiol–ene ratio of 0.5 or 1, then 2 μL of 0.5 M CaCl2 were added to solution before degassing for 20 min. For step-growth and mixed-model HMGG hydrogels, 10 or 20 μL of 10 wt % DTT was added such that the thiol-to-alkene mole ratio was 0.5 or 1. To these mixtures, either 0 or 6 μL of 0.5 M CaCl2 was added to yield a final concentration of 0 or 1 mM Ca2+. For chain-growth hydrogels, DTT was not added. Chemically cross-linked hydrogels were obtained by exposing the precursor solution to UV light (15 W, 365 nm, UVP, Upland, CA) for 10 min. Both chain-growth and mixed-model solutions were degassed before cross-linking.
2.5. Compressive Modulus
The compressive modulus for each experimental condition was obtained through manual compression measurements. Obtained hydrogel pegs (n = 3) approximately 15 × 15 × 6 mm3 were placed between two microscope slides and measured as weight was added. A stress–strain curve was created using distances between the microscope slides measured through Image J (NIH, Bethesda, MD). The linear regions of the curve under 5–15% strain were used to report the compressive moduli.
2.6. Swelling Ratio
The swelling ratio was measured for the hydrogels in DI water. Each experimental condition was tested at a concentration of 1% w/v modified gellan gum. The obtained hydrogels weighed (w0). The hydrogels were subsequently immersed in DI water until equilibrium was reached and weighed (ws). The wet swelling ratio (S) was calculated using eq 2.
| 2 |
2.7. In Vitro Degradation Profile
The effect of the different cross-linking mechanisms on the degradation profiles of LMGG and HMGG hydrogels was examined. Hydrogels (n = 3) were immersed in 0.5 mg/mL lysozyme and phosphate buffered saline (PBS, diluted from 10× solution to 0.1 M, pH 7.4, Fisher Scientific, Pittsburgh, PA) and weighed daily.
2.8. Cell Culture
NIH/3T3 fibroblasts (ATCC, Manassas, VA) were cultured in complete medium (CM, Dulbecco’s modified Eagle’s medium supplemented with 10% bovine calf serum, 100 U/L penicillin, and 100 μg/L streptomycin) at 37 °C in 5% CO2. The cells were passaged every 3–5 days using 0.025% trypsin–ethylenediaminetetraacetic acid (Mediatech, Tewksbury, MA) detachment and subcultured at 6.7 × 103 cells/cm2.
2.9. Cell Proliferation
Cell proliferation was tested on all gelation formulations for both LMGG and HMGG. A solution of 10 mg/mL LMGG or HMGG in CM was prepared at 37 °C. Solutions for the mixed and chain-growth gelation procedures were degassed for 15 min. To each well, 100 μL of each modified gellan gum solution was added to 48-well plates (KSE Scientific, Durham NC). The hydrogels were exposed to UV light for 10 min. Cells were seeded at a concentration of 1.25 × 105 cells/cm2 on each modified gellan gum hydrogel and 200 μL CM without phenol red was added the plate was incubated for 48 h. Live and dead controls were generated by culturing cells without the gel. After 48 h, the dead control supernatant was aspirated and replaced with 95% ethanol for 10 min. After this incubation period, the supernatant in each well was aspirated. To each well, 150 μL of working solution (2 μM calcein AM (AnaSpec, Fremont, CA) and 7.5 μM 7-aminoactinomycin D (Tonbo Biosciences, San Diego, CA) in PBS) was added and the plates were incubated to 30–40 min at 37 °C in 5% CO2. The live cells were quantified using an excitation/emission of 485/590 nm. The dead cells were measured using an excitation/emission of 528/645 nm using a plate reader (BioTek Synergy HT Multidetection Microplate Rreader BioTek, Winooski, VT).
Cells stained using the live/dead assay were imaged using an EVOS FLoid Imaging Station (Life Technologies, Grand Island, NY) using the red channel (excitation/emission 586/646 nm) and the green channel (482/532 nm).
2.10. Statistics and Data Analysis
Statistical analysis was performed using JMP statistical software (SAS Institute Inc., Cary, NC). Statistical significance of the mean comparisons was determined by a two-way analysis of variance (ANOVA). Pair-wise comparisons were analyzed with Tukey’s honest significance difference test. Differences were considered statistically significant for p < 0.05.
3. Results and Discussion
3.1. Gellan Gum Modification and Characterization
Methacrylation of gellan gum is schematically illustrated in Figure 1A. The NMR spectrum of methacrylated gellan gum is shown in Figure 1B. The 1H NMR spectra of unmodified gellan gum (Figure 1B top) and modified gellan gum (Figure 1B middle and bottom) were obtained at 25 °C. All three spectra showed the characteristic methyl peak from rhamnose (δ = 1.3 ppm). Methacrylation was verified and quantified by the characteristic carbon–carbon double bond peaks (δ = 5.7 and 6.2 ppm) and a peak corresponding to the methyl group on the newly methacrylated moiety (δ = 1.95 ppm). The degree of modification was calculated by comparing the integrated proton peaks from the methyl group on the methacrylate and the methyl group on rhamnose. The average number of methacrylate groups per LMGG repeating unit was 0.5 ± 0.06 (Figure 1B middle), which increased to 2.0 ± 0.11 for HMGG (Figure 1B bottom).
Figure 1.

Synthesis and characterization of methacrylated gellan gum. (A) Schematic illustration of the methacrylation reaction. (B) 1H NMR of gellan gum (top), low-modified (middle), and high-modified (bottom) gellan gum. (C) Fourier transform infrared spectroscopy (FTIR) spectra of gellan gum and low- and high-modified gellan gum.
Methacrylation of gellan gum was further confirmed by FTIR (Figure 1C). The carbon–carbon double bond peak is expected to appear at 1640 cm–1, which overlaps with other vibrational modes in the gellan gum. However, the characteristic peak at 1720 cm–1 corresponding to the carbonyl present on the methacrylate group appeared after chemical modification and grew in intensity with increasing degree of methacrylation.
The viscosity average molecular weight of GG as well as modified GG were also measured (Figure S1). Unmodified GG has a viscosity average molecular weight of 212.2 ± 21.0 kDa. LMGG and HMGG were significantly different from unmodified GG (266.1 ± 6.78 and 383.7 ± 38.4 kDa, respectively). The increase in the viscosity average molecular weight after modification was higher (25.5 ± 8.9% for LMGG and 80.1 ± 15.8% for HMGG) than that calculated via the degree of methacrylation (4.8% for LMGG and 19% for HMGG). This likely resulted from the change in the solubility of the polymer that contributed to the increased viscosity.
The resultant gels are labeled by numbers according to their unique formula shown under each graph. Gels 1–4 are LMGG gels, whereas 5–14 are HMGG gels.
3.2. Compression Modulus
The compressive moduli of the 14 different formulations of hydrogels were obtained by analyzing the stress–strain curves in the region of 5–15% strain. The modulus of the hydrogels ranged from 6.4 to 17.2 kPa. A higher degree of methacrylation resulted in more rigid substrates, with LMGG gels ranging from 6.4 to 11.3 kPa and HMGG gels ranging from 7.4 to 17.2 kPa (Figure 2). The increased mechanical strength in HMGG was expected due to a higher degree of modification and cross-linking.
Figure 2.

Mechanical properties of modified gels. Compressive moduli of low- and high-modified gellan gum hydrogels cross-linked through different mechanisms. Data are presented as mean ± standard deviation (SD). n = 5. Statistical analysis through two-way ANOVA and Tukey’s HSD post hoc test. Bars with the same letter (A–G) are not statistically different (p < 0.05).
The mechanical properties of the gellan gum hydrogels synthesized here were altered by three factors: the presence of calcium, the chemical cross-linking mechanism, and the thiol–ene ratio. We first explore the effect of physical cross-linking on the mechanical properties of hydrogels by adding divalent cations. Calcium was found to either increase or have no significant influence on the compressive moduli of the gels. LMGG gels were not able to form in the absence of calcium. For HMGG gels, the addition of calcium had little effect on the hydrogels cross-linked through mixed model. The hydrogels cross-linked through the step-growth mechanism of equimolar thiol–ene together with calcium exhibited an increased compression modulus (9.9 ± 0.6 kPa) compared to their counterparts in the absence of calcium (7.3 ± 0.8 kPa). A similar trend was observed when half-molar equivalents of thiol were added, with the modulus increasing from 9.8 ± 0.7 to 12.8 ± 0.9 kPa. The compressive modulus of the gels formed using chain-growth cross-linking was also improved through the addition of calcium (14.6 ± 0.8 to 16.5 ± 1.0 kPa).
Divalent cations such as calcium form bridges between carboxyl groups and reduce electrical repulsion, resulting in physical cross-links.20 These junctions allow gellan gum chains to bundle to form helical structures16 and may increase the mechanical properties. However, the interaction between calcium cations with the gellan gum hydroxyl and carboxyl groups reduces the hydrogen bond donors and acceptors available to interact with water,17 thus the strength between the network and the water matrix decreased. As a result, the compressive modulus was not significantly influenced in some hydrogels.
We then investigated how the cross-linking mechanism influences the hydrogel stiffness. The results showed that the cross-linking mechanisms significantly affected the mechanical properties for all of the gels studied here. For LMGG gels, the compression modulus was increased from 6.4 ± 0.7 to 8.1 ± 0.7 kPa when the cross-linking mechanism was changed from step growth to mixed model using equimolar thiol–ene gels. The same trend was observed for half molar thiol–ene LMGG gels, where the modulus increased from 9.6 ± 0.7 to 11.3 ± 0.8 kPa. Similarly, for HMGG gels, the compression modulus increased from 7.4 ± 0.8 to 12.4 ± 0.8 kPa in the absence of calcium and from 9.9 ± 0.6 to 13.0 ± 0.7 kPa in its presence for equimolar thiol–ene gels. This trend was also observed for the half molar thiol–ene hydrogels in which the compression modulus changes from 9.8 ± 0.7 to 16.4 ± 0.9 kPa in the absence of calcium and from 12.8 ± 0.9 to 17.2 ± 0.1 kPa in the presence of calcium. Surprisingly, the chain-growth cross-linked gels had the highest compression modulus of 14.6 ± 0.8 and 16.5 ± 1.0 kPa in the absence and presence of calcium, respectively.
The chain-growth hydrogels were cross-linked without dithiol cross-linkers and expected to have the lowest cross-linking density and thus the lowest compression modulus. However, the results showed that chain-growth hydrogels have the highest compressive modulus among the hydrogels studied here. Two reasons contribute to the high compression moduli of chain-growth hydrogels compared with the mixed-model and step-growth hydrogels in this study. First, homopolymerization of acrylate yields heterogeneous chains and causes a high degree of chain entanglements, which contribute to a higher modulus.18 Second, there is a parabolic relationship between the thiol–ene ratio and the resultant hydrogel shear modulus, with the shear modulus initially increasing and then decreasing with increasing thiol content.21 The two ratios used here may not be optimal to yield the highest compressive modulus. The higher modulus for the chain-growth hydrogels over the step-growth hydrogel has been previously observed by Lin and co-workers.18 In their study, the chain-growth methacrylated gelatin had a shear modulus more than double that of the step-growth norbornene functionalized gelatin (0.9 vs 0.4 kPa).18
The higher moduli observed in the mixed-model hydrogels compared with the step-growth counterparts may result from the oxygen level in the precursor solutions. The presence of oxygen in the precursor solutions inhibits radical polymerization and reduces cross-linking density.22,23 The dissolved oxygen in the mixed-model hydrogel precursor solutions was reduced via degassing to promote cross-linking.
The thiol–ene ratio also plays an important role in modulating the compressive modulus of the hydrogels. Equimolar thiol–ene hydrogels were initially expected to yield the highest compression modulus because each thiol group was expected to react with one vinyl group and consume all of the available vinyls.24 However, carbon–carbon bonds can homopolymerize, resulting in excessive thiols, which react to form dangling dithiol bonds.18,24,25 In addition, increased dithiol results in increased chain transfer, which terminates chain propagation and reduces the degree of polymerization.21,26,27 For these reasons, half-molar thiol–ene hydrogels had higher compressive moduli compared with equimolar gels. For LMGG hydrogels, the half-molar thiol–ene gels yielded higher compressive moduli than the equimolar gels for both step-growth and mixed-model cross-linkings (8.1 ± 0.7 and 11.3 ± 0.8 vs 6.4 ± 0.7 and 9.6 ± 0.7 kPa, respectively). This trend continued for HMGG gels, with the half-molar thiol–ene gels having higher moduli than their equimolar counterparts. This relationship between the thiol content and the cross-linking density for step-growth and mixed-model gels has been observed previously.21,24 Homopolymerization of norbornene is difficult due to steric hindrance and was still found to have the highest cross-linking density at a lower than equimolar thiol–ene ratio.24 Defects in cross-linking such as formation of loops and dangling structures are believed to be responsible for this phenomenon.25
3.3. Swelling Ratio
The swelling ratio was measured in DI water as an indirect method for assessing the cross-linking density, as a high swelling ratio generally corresponds to a low cross-linking density and vice versa. In this work, the wet swelling ratios of the 14 types of gels (Figure 3) were negatively correlated with the compressive moduli (R = −0.55). The swelling ratios of the gels ranged from 2.46 ± 0.11 to 4.28 ± 0.14 for LMGG gels and from 0.98 ± 0.01 to 1.95 ± 0.34 for HMGG gels. A higher compressive modulus generally resulted in less water uptake. In addition to the cross-linking density, the formation of ionic bonds and the hydrophilicity of the network influence swelling.
Figure 3.

Swelling behavior of gellan gum hydrogels. Hydrogels cross-linked through different mechanisms were swelled in PBS. Data presented as mean ± SD. n = 3. Statistical analysis through two-way ANOVA and Tukey’s HSD post hoc test. Bars with the same letter (A–J) are not statistically different (p < 0.05).
Calcium ions not only form ionic bonds that increase the cross-linking density and lower the swelling ratio but also reduce water uptake. Because all LMGG hydrogels were strengthened with calcium, this discussion will focus on HMGG hydrogels. The influence of calcium on the swelling ratio was significant for thiol cross-linked gels. For equimolar thiol–ene ratio hydrogels, the swelling ratio of the step-growth gels decreased from 1.94 ± 0.34 to 1.29 ± 0.058 when calcium was added. Similar decreases in the swelling ratio were observed for equimolar mixed-model hydrogels when calcium was added (1.73 ± 0.13 to 1.26 ± 0.035). This trend continued for half-molar thiol–ene gels formed through step-growth cross-linking (1.57 ± 0.033 in the absence and 1.07 ± 0.030 in the presence of calcium) and mixed-model cross-linking (1.46 ± 0.031 in the absence and 1.09 ± 0.044 in the presence of calcium). Chain-growth gels, on the other hand, did not have significant differences with or without calcium (0.98 ± 0.009 and 1.1 ± 0.030, respectively). This could be caused by the rigid network of chain-growth cross-links, which may have reduced the effect of the calcium physical cross-links.28
An interesting finding regarding the swelling measurements was on the mixed-mode hydrogels. Those hydrogels had similar compression moduli with or without the addition of calcium, thus they were expected to have similar cross-linking densities and swelling ratios. However, mixed-mode hydrogels with calcium had a lower water uptake when compared to their counterparts without calcium. This phenomenon was caused by multiple mechanisms regarding how ionic bonds can influence the swelling ratio. First, the formation of ionic bonds increases the cross-linking density and reduces the swelling capacity.4 Second, interactions between the carboxylic acids on gellan gum and calcium cations decrease the available hydrogen bond donors and acceptors available for interaction with water,17 thus decreasing the amount of water associated with the hydrogel network. Third, compared with covalently cross-linked polymeric networks, ionic bonds are less flexible, which reduces their ability to swell.29
All LMGG gels showed significantly higher swelling ratios compared to HMGG gels and all LMGG gels were strengthened with ionic cross-links. For LMGG gels, the compressive modulus was negatively correlated with the swelling ratio (R = −0.97). Hydrophilicity affects the swelling response of hydrogels, with hydrophilic networks taking up more water.30 LMGG has one methacrylate group on every two tetrasaccharide repeating unit, whereas HMGG has two methacrylates on each repeating unit. This suggests that LMGG will have a stronger affinity for water than HMGG.
The swelling ratio of gellan gum hydrogels cross-linked via chain-growth polymerization has been studied previously. The dual cross-linked gellan gum hydrogel (chain-growth and ionic cross-linking) exhibited significantly lower swelling ratios (∼100% of original weight), which is similar to our results.4 The same group also investigated the gellan gum swelling behavior in PBS. The dual cross-linked gellan gum hydrogels shrank when immersed in PBS solution due to the formation of helical structures and cross-links.4 Reis and co-workers found 2500% water uptake in photo-cross-linked gellan gum hydrogel in PBS after 30 days.9 The same group further investigated and compared water uptake in photo-cross-linked and ionically cross-linked gellan gum hydrogels, where both showed ∼2500% water uptake after 90 days.10 However, the swelling ratios of gellan gum hydrogels in both DI water and PBS were highly dependent on the degree of methacrylation and the final concentration of the cations. Due to this reason, our results here cannot be directly compared with the literature.
3.4. In Vitro Degradation
Lysozyme, an antibacterial enzyme released by macrophages during the wound-healing process,31 breaks down the cell walls of bacteria and releases N-acetyl-d-glucosamine.32 Previous studies have shown that polysaccharides can undergo enzymatic hydrolysis in the presence of lysozyme.11,33−36 It is believed that lysozyme breaks ether bonds connecting the structural backbone of the polysaccharide.37 The in vitro degradation profile of hydrogels in this study was obtained by exposing the hydrogels to PBS or 0.5 mg/mL lysozyme (Figure 4). The hydrogels immersed in pH 7.4 PBS were stable, with 80% mass remaining after 42 days (Figure 4A). This weight loss likely results from the dehydration of the gel, as the ionic concentration of the PBS solution is higher than that in the hydrogels.11 In lysozyme, all of the gels degraded to 20% of their initial weight after 16 days (Figure 4B), with HMGG gels degrading slightly faster than LMGG gels. The differences in the degradation rates for LMGG and HMGG hydrogels may result from differences in network hydrophilicity. The HMGG backbone contains more hydrophobic methacrylate groups than LMGG. These methacrylate groups can form hydrophobic kinetic chains during cross-linking,23,38 which may retard the diffusion of lysozyme solution into the hydrogel.
Figure 4.

Gellan gum hydrogel degradation. Hydrogels formed through different cross-linking methods were immersed in (A) PBS and (B) 0.5 mg/mL lysozyme at 37°C. Data presented as mean ± SD. n = 3.
The stability of gellan gum hydrogels in PBS has been confirmed through several studies. Reis and co-workers immersed the gellan gum hydrogel for 30 days in PBS, and the hydrogels showed less than 20% weight loss.9 In a later study, the same group conducted a PBS stability test for both ionic cross-linked and photo-cross-linked gellan gum gels up to 90 days, which resulted in nearly 20% weight loss.10 The stability of another polysaccharide, chitosan, in PBS was also found to retain 80% of its original weight after 18 days,11 which is in line with our findings.
The degradation rate of polysaccharide hydrogels in lysozyme is highly dependent on both concentration of lysozyme and the type of polysaccharide. The photo-cross-linked chitosan hydrogels were found to completely disintegrated in 1 mg/mL lysozyme after 8 days.11 Methacrylated chitin hydrogels retained 50% of their original weight after 60 h in 1 mg/mL lysozyme, but completely degraded in 50 mg/mL lysozyme within 10 h.39 The density of cross-links in the methacrylated chitin gels also alters the degradation rate, with a higher cross-linking density leading to a slower degradation rate.39 These results combined with the observations in our study provide cues for controlling the degradation rates for future applications using hydrogels as tissue engineering scaffolds.
3.5. Cell Proliferation
Fibroblasts are key players in both physiological repair of tissues after injury and in maintaining skin homeostasis.40,41 They are recruited to the wound site 2–10 days after injury and differentiate into myofibroblast.42 The differentiation of fibroblasts to myofibroblasts was found to be sensitive to substrate stiffness.43 Myofibroblasts are contractile cells that bring wound edges together and deposit and remodel the ECM.42 Therefore, the interaction between hydrogels, which are used to mimic native ECM, and fibroblasts is important in designing tissue engineering scaffolds.
The cell proliferation of NIH/3T3 fibroblasts seeded on 14 various substrates was visualized by live/dead fluorescence staining (Figure 5A). No dead cells were observed for any of the hydrogels. All LMGG hydrogels and the chain-growth HMGG hydrogels showed the highest cell proliferation. LMGG mixed-model gels showed lower cell proliferation than LMGG step-growth gels. Cell proliferation on HMGG hydrogels was well correlated with the hydrogel compressive modulus (R = 0.80). The softest HMGG gels—those formed using equimolar thiol–ene ratios—yielded the lowest cell proliferation (4.6 ± 0.5% in the absence and 5.7 ± 0.4% in the presence of calcium). The stiffest HMGG substrates—those formed through chain-growth cross-linking—had the highest cell proliferation (46.5 ± 5.7% in the absence and 52.9 ± 5.8 in the presence of calcium).
Figure 5.

Cytocompatibility of gellan gum hydrogels. NIH/3T3 fibroblasts were seeded on hydrogels and tissue culture plastic (control). (A) Representative micrographs of live (green) and dead (red) cells cultured for 48 h. (B) Quantification of live and dead cells. Data presented as mean ± SD. n = 4. Statistical analysis through two-way ANOVA and Tukey’s HSD post hoc test. Bars with the same letter (A–G) are not statistically different (p < 0.05).
Mechanical signals play an important role in myofibroblast differentiation, activation, and adhesion.44 Fibroblasts differentiate into a contractile myofibroblast phenotype when exposed to substrates with elastic moduli corresponding to pathologically stiff fibrotic tissue with microfilament bundles or mature focal adhesions,45 whereas those cultured on softer substrates show little development of stress fibers.43,46 Fluorescent images of cells stained with a live-dead assay were used to visualize cell morphology. Here, fibroblasts seeded on the stiffest hydrogel (gel 5) had some stress fibers and microfilament bundles (Figure 5A). The cells seeded on softer substrates (gels 9 and 11) formed clusters of cells that did not spread on the surfaces. Conversely, the cells seeded on stiffer hydrogels (gels 12 and 13) spread more evenly across the surface of the gel.
In addition to mechanical stress, the surface chemistry can influence fibroblast morphology and differentiation. As shown in Figure 5, the fibroblasts cultured on half-molar thiol–ene step-growth HMGG hydrogels in the absence of calcium showed microfilament bundles even though the gel was softer than the similarly formed chain-growth gel 6. Microfilament bundles were only observed on the substrates fabricated with half-molar thiol–ene ratios. These microfilaments disappeared when the modulus of gel 11 was increased via either more chemical cross-linking (gel 12) or ionic cross-linking (gel 13).
Several studies have demonstrated that cell adhesion, spreading, and proliferation depend on the surface chemistry, topology, and wettability.47−54 Combinatorial approaches for developing nonfouling biomaterials have revealed that chemistry plays a role in cell activation55 and mitigating the foreign body response.56 A multiarm poly(ethylene glycol)–norbornene macromer was developed via step-growth method, and the resultant thiol–ene hydrogel contained more homogeneous networks. Encapsulated MIN6 β-cells had significantly higher proliferation in the hydrogel formed with thiol–ene chemistry. Additionally, the encapsulated cells in the thiol–ene hydrogels grew in clusters.57 Here, the effect of the thiol content was found to be the major parameter responsible for fibroblast morphology differences. Cell behavior in relation to the thiol–ene group has not been studied extensively. More experiments are necessary to decouple the effect of surface chemistry and mechanical properties on fibroblast differentiation and morphology.
Cell responses and proliferation on biomaterial surfaces are crucial to biomedical applications and tissue engineering. No dead cells were observed for any of the hydrogels fabricated here. Cell proliferation data indicate that both LMGG and HMGG hydrogels are biocompatible and bioactive. In addition, cell proliferation of HMGG was controlled, in part, by the tunable mechanical properties of the hydrogel. The design principles of tissue engineering constructs greatly depend on the native mechanical properties of the organ in which the biomaterial will be implanted. Owing to their range of compressive moduli (6.4–17.2 kPa), the gellan gum hydrogels fabricated here have the potential to be used as dermal implants. During the wound-healing process, fibroblasts differentiate into myofibroblasts and deposit newly synthesized ECM 2 weeks after injury.58 Cell proliferation, activation of growth factors, and mechanics of ECM are the known contributors to fibrosis.59 A higher elastic moduli (16–20 kPa) of granulation tissue during the repairing stage leads to a significantly higher level of fibroblast differentiation.60 Skin stiffness changes during the proliferation and regenerative stages and becomes stiffer in fibrotic tissue (∼50 kPa). Thus, a soft tissue scaffold for dermal should be in a relatively lower range of 1–20 kPa.60 Our hydrogels have the potential to be applied as biologically active wound dressings and mimic the mechanical properties and microenvironment of the ECM.
4. Conclusions
We successfully modified gellan gum by reacting it with methacrylic anhydride. Both low-modified and high-modified gellan gum hydrogels were synthesized by varying the amount of methacrylic anhydride. Taking advantage of the thiol–ene click chemistry, the hydrogels were fabricated through chain-growth, step-growth, and mixed-model mechanisms. We demonstrated that the addition of calcium and thiol–ene ratio can significantly alter the compressive modulus of the resultant hydrogels. The swelling ratios of hydrogels were negatively correlated with hydrogel compression modulus. The formation of physical cross-links through the addition of calcium reduced the network flexibility and suppressed the hydrogel swelling. NIH/3T3 fibroblasts proliferated on the hydrogels and had a positive correlation with substrate stiffness. Cell morphology and spreading were found to be related to substrate chemistry. In general, the addition of calcium, an optimized thiol–ene ratio, and mixed-mode cross-linking mechanisms can yield stiffer hydrogels, which are related to increased fibroblast proliferation. Over all, this study demonstrated the ability to tune the mechanical properties of gellan gum hydrogels through the chemical modification and the addition of divalent ions. The data presented here will help build a gellan gum platform for future applications in tissue engineering.
Acknowledgments
This work was supported by the Roy J. Carver Charitable Trust Grant No. 13-4265. S.J. would like to thank start-up fund support from the Iowa State University.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b00683.
Viscosity average molecular weight of GG, LMGG, and HMGG (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Boddupalli A.; Zhu L.; Bratlie K. M. Methods for Implant Acceptance and Wound Healing: Material Selection and Implant Location Modulate Macrophage and Fibroblast Phenotypes. Adv. Healthc. Mater. 2016, 5, 2575–2594. 10.1002/adhm.201600532. [DOI] [PubMed] [Google Scholar]
- Gordon S.; Todd J.; Cohn Z. A. In vitro synthesis and secretion of lysozyme by mononuclear phagocytes. J. Exp. Med. 1974, 139, 1228–1248. 10.1084/jem.139.5.1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suri S.; Banerjee R. In vitro evaluation of in situ gels as short term vitreous substitutes. J. Biomed. Mater. Res., Part A 2006, 79, 650–664. 10.1002/jbm.a.30917. [DOI] [PubMed] [Google Scholar]
- Coutinho D. F.; Sant S. V.; Shin H.; Oliveira J. T.; Gomes M. E.; Neves N. M.; Khademhosseini A.; Reis R. L. Modified Gellan Gum hydrogels with tunable physical and mechanical properties. Biomaterials 2010, 31, 7494–7502. 10.1016/j.biomaterials.2010.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsukawa S.; Tang Z.; Watanabe T. Hydrogen-bonding behavior of gellan in solution during structural change observed by 1H NMR and circular dichroism methods. Prog. Colloid Polym. Sci. 1999, 114, 15–24. 10.1007/3-540-48349-7_3. [DOI] [Google Scholar]
- Shin H.; Olsen B. D.; Khademhosseini A. The mechanical properties and cytotoxicity of cell-laden double-network hydrogels based on photocrosslinkable gelatin and gellan gum biomacromolecules. Biomaterials 2012, 33, 3143–3152. 10.1016/j.biomaterials.2011.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjizadeh A.; Doillon C. J. Directional migration of endothelial cells towards angiogenesis using polymer fibres in a 3D co-culture system. J. Tissue Eng. Regener. Med. 2010, 4, 524–531. 10.1002/term.269. [DOI] [PubMed] [Google Scholar]
- Nichol J. W.; Koshy S. T.; Bae H.; Hwang C. M.; Yamanlar S.; Khademhosseini A. Cell-laden microengineered gelatin methacrylate hydrogels. Biomaterials 2010, 31, 5536–5544. 10.1016/j.biomaterials.2010.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva-Correia J.; Oliveira J. M.; Caridade S. G.; Oliveira J. T.; Sousa R. A.; Mano J. F.; Reis R. L. Gellan gum-based hydrogels for intervertebral disc tissue-engineering applications. J. Tissue Eng. Regener. Med. 2011, 5, e97–e107. 10.1002/term.363. [DOI] [PubMed] [Google Scholar]
- Silva-Correia J.; Zavan B.; Vindigni V.; Silva T. H.; Oliveira J. M.; Abatangelo G.; Reis R. L. Biocompatibility evaluation of ionic- and photo-crosslinked methacrylated gellan gum hydrogels: In vitro and in vivo study. Adv. Healthc. Mater. 2013, 2, 568–575. 10.1002/adhm.201200256. [DOI] [PubMed] [Google Scholar]
- Hong Y.; Song H.; Gong Y.; Mao Z.; Gao C.; Shen J. Covalently crosslinked chitosan hydrogel: Properties of in vitro degradation and chondrocyte encapsulation. Acta Biomater. 2007, 3, 23–31. 10.1016/j.actbio.2006.06.007. [DOI] [PubMed] [Google Scholar]
- Shih H.; Lin C. C. Cross-linking and degradation of step-growth hydrogels formed by thiol-ene photoclick chemistry. Biomacromolecules 2012, 13, 2003–2012. 10.1021/bm300752j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rydholm A. E.; Bowman C. N.; Anseth K. S. Degradable thiol-acrylate photopolymers: Polymerization and degradation behavior of an in situ forming biomaterial. Biomaterials 2005, 26, 4495–4506. 10.1016/j.biomaterials.2004.11.046. [DOI] [PubMed] [Google Scholar]
- Xu K.; Fu Y.; Chung W.; Zheng X.; Cui Y.; Hsu I. C.; Kao W. J. Thiol-ene-based biological/synthetic hybrid biomatrix for 3-D living cell culture. Acta Biomater. 2012, 8, 2504–2516. 10.1016/j.actbio.2012.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toepke M. W.; Impellitteri N. A.; Theisen J. M.; Murphy W. L. Characterization of thiol-ene crosslinked PEG hydrogels. Macromol. Mater. Eng. 2013, 298, 699–703. 10.1002/mame.201200119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris C. J.; Gilmore K. J.; Wallace G. G.; Panhuis M. Modified gellan gum hydrogels for tissue engineering applications. Soft Matter 2013, 9, 3705–3711. 10.1039/c3sm27389j. [DOI] [Google Scholar]
- Huang Y.; Yu H.; Xiao C. pH-sensitive cationic guar gum/poly (acrylic acid) polyelectrolyte hydrogels: Swelling and in vitro drug release. Carbohydr. Polym. 2007, 69, 774–783. 10.1016/j.carbpol.2007.02.016. [DOI] [Google Scholar]
- Mũnoz Z.; Shih H.; Lin C.-C. Gelatin hydrogels formed by orthogonal thiol-norbornene photochemistry for cell encapsulation. Biomater. Sci. 2014, 2, 1063–1072. 10.1039/C4BM00070F. [DOI] [PubMed] [Google Scholar]
- Cramer N. B.; Bowman C. N. Kinetics of thiol-ene and thiol-acrylate photopolymerizations with real-time Fourier transform infrared. J. Polym. Sci., Part A: Polym. Chem. 2001, 39, 3311–3319. 10.1002/pola.1314. [DOI] [Google Scholar]
- Tako M.; Teruya T.; Tamaki Y.; Konishi T. Molecular origin for rheological characteristics of native gellan gum. Colloid Polym. Sci. 2009, 287, 1445–1454. 10.1007/s00396-009-2112-2. [DOI] [Google Scholar]
- Hao Y.; Lin C. C. Degradable thiol-acrylate hydrogels as tunable matrices for three-dimensional hepatic culture. J. Biomed. Mater. Res., Part A 2014, 102, 3813–3827. 10.1002/jbm.a.35044. [DOI] [PubMed] [Google Scholar]
- Ouyang L.; Highley C. B.; Sun W.; Burdick J. A. A Generalizable Strategy for the 3D Bioprinting of Hydrogels from Nonviscous Photo-crosslinkable Inks. Adv. Mater. 2017, 29, 1604983 10.1002/adma.201604983. [DOI] [PubMed] [Google Scholar]
- Roberts J. J.; Bryant S. J. Comparison of photopolymerizable thiol-ene PEG and acrylate-based PEG hydrogels for cartilage development. Biomaterials 2013, 34, 9969–9979. 10.1016/j.biomaterials.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gramlich W. M.; Kim I. L.; Burdick J. A. Synthesis and orthogonal photopatterning of hyaluronic acid hydrogels with thiol-norbornene chemistry. Biomaterials 2013, 34, 9803–9811. 10.1016/j.biomaterials.2013.08.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miquelard-Garnier G.; Demeures S.; Creton C.; Hourdet D. Synthesis and rheological behavior of new hydrophobically modified hydrogels with tunable properties. Macromolecules 2006, 39, 8128–8139. 10.1021/ma061361n. [DOI] [Google Scholar]
- Jemal A.; Siegel R.; Ward E.; Hao Y.; Xu J.; Murray T.; Thun M. J. Cancer Statistics, 2008. Ca-Cancer J. Clin. 2008, 58, 71–96. 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- Grover G. N.; Rao N.; Christman K. L. Myocardial matrix-polyethylene glycol hybrid hydrogels for tissue engineering. Nanotechnology 2014, 25, 014011 10.1088/0957-4484/25/1/014011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairbanks B. D.; Schwartz M. P.; Halevi A. E.; Nuttelman C. R.; Bowman C. N.; Anseth K. S. A versatile synthetic extracellular matrix mimic via thiol-norbornene photopolymerization. Adv. Mater. 2009, 21, 5005–5010. 10.1002/adma.200901808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihaila S. M.; Gaharwar A. K.; Reis R. L.; Marques A. P.; Gomes M. E.; Khademhosseini A. Photocrosslinkable kappa-carrageenan hydrogels for tissue engineering applications. Adv. Healthc. Mater. 2013, 2, 895–907. 10.1002/adhm.201200317. [DOI] [PubMed] [Google Scholar]
- Qiu Y.; Park K. Environment-sensitive Hydrogels for Drug Delivery, Adv. Drug Deliv. Adv. Drug Delivery Rev. 2001, 53, 321–339. 10.1016/S0169-409X(01)00203-4. [DOI] [PubMed] [Google Scholar]
- Frohm M.; Gunne H.; Bergman A. C.; Agerberth B.; Bergman T.; Boman a; Lidén S.; Jörnvall H.; Boman H. G. Biochemical and antibacterial analysis of human wound and blister fluid. Eur. J. Biochem. 1996, 237, 86–92. 10.1111/j.1432-1033.1996.0086n.x. [DOI] [PubMed] [Google Scholar]
- Charernsriwilaiwat N.; Opanasopit P.; Rojanarata T.; Ngawhirunpat T. Lysozyme-loaded, electrospun chitosan-based nanofiber mats for wound healing. Int. J. Pharm. 2012, 427, 379–384. 10.1016/j.ijpharm.2012.02.010. [DOI] [PubMed] [Google Scholar]
- Pangburn S. H.; Trescony P. V.; Heller J. Lysozyme degradation of partially deacetylated chitin, its films and hydrogels. Biomaterials 1982, 3, 105–108. 10.1016/0142-9612(82)90043-6. [DOI] [PubMed] [Google Scholar]
- Tomihata K.; Ikada Y. In vitro and in vivo degradation of films of chitin and its deacetylated derivatives. Biomaterials 1997, 18, 567–575. 10.1016/S0142-9612(96)00167-6. [DOI] [PubMed] [Google Scholar]
- De Jong S. J.; Van Eerdenbrugh B.; Van Nostrum C. F.; Kettenes-van den Bosch J. J.; Hennink W. E. Physically crosslinked dextran hydrogels by stereocomplex formation of lactic acid oligomers: Degradation and protein release behavior. J. Controlled Release 2001, 71, 261–275. 10.1016/S0168-3659(01)00228-0. [DOI] [PubMed] [Google Scholar]
- Jin R.; Moreira Teixeira L. S.; Dijkstra P. J.; Karperien M.; van Blitterswijk C. A.; Zhong Z. Y.; Feijen J. Injectable chitosan-based hydrogels for cartilage tissue engineering. Biomaterials 2009, 30, 2544–2551. 10.1016/j.biomaterials.2009.01.020. [DOI] [PubMed] [Google Scholar]
- Blake C. C. F.; Johnson L. N.; Mair G. A.; North A. C. T.; Phillips D. C.; Sarma V. R. Crystallographic Studies of the Activity of Hen Egg-White Lysozyme. Proc. R. Soc. B 1967, 167, 378–388. 10.1098/rspb.1967.0035. [DOI] [PubMed] [Google Scholar]
- Custódio C. A.; Reis R. L.; Mano J. F. Photo-Cross-Linked Laminarin-Based Hydrogels for Biomedical Applications. Biomacromolecules 2016, 17, 1602–1609. 10.1021/acs.biomac.5b01736. [DOI] [PubMed] [Google Scholar]
- Kang W.; Bi B.; Zhuo R.; Jiang X. Photocrosslinked methacrylated carboxymethyl chitin hydrogels with tunable degradation and mechanical behavior. Carbohydr. Polym. 2017, 160, 18–25. 10.1016/j.carbpol.2016.12.032. [DOI] [PubMed] [Google Scholar]
- Hinz B. Formation and function of the myofibroblast during tissue repair. J. Invest. Dermatol. 2007, 127, 526–537. 10.1038/sj.jid.5700613. [DOI] [PubMed] [Google Scholar]
- Darby I. A.; Laverdet B.; Bonté F.; Desmoulière A. Fibroblasts and myofibroblasts in wound healing. Clin., Cosmet. Invest. Dermatol. 2014, 7, 301–311. 10.2147/CCID.S50046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurtner G. C.; Werner S.; Barrandon Y.; Longaker M. T. Wound repair and regeneration. Nature 2008, 453, 314–321. 10.1038/nature07039. [DOI] [PubMed] [Google Scholar]
- Balestrini J. L.; Chaudhry S.; Sarrazy V.; Koehler A.; Hinz B. The mechanical memory of lung myofibroblasts. Integr. Biol. 2012, 4, 410. 10.1039/c2ib00149g. [DOI] [PubMed] [Google Scholar]
- Hinz B. The myofibroblast: Paradigm for a mechanically active cell. J. Biomech. 2010, 43, 146–155. 10.1016/j.jbiomech.2009.09.020. [DOI] [PubMed] [Google Scholar]
- Hinz B.; Mastrangelo D.; Iselin C. E.; Chaponnier C.; Gabbiani G. Mechanical tension controls granulation tissue contractile activity and myofibroblast differentiation. Am. J. Pathol. 2001, 159, 1009–1020. 10.1016/S0002-9440(10)61776-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Discher D. E.; Janmey P.; Wang Y.-L. Tissue cells feel and respond to the stiffness of their substrate. Science 2005, 310, 1139–1143. 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]
- Ahsan F.; Rivas I. P.; Khan M. A.; Torres Suarez A. I. Targeting to macrophages: role of physicochemical properties of particulate carriers--liposomes and microspheres--on the phagocytosis by macrophages. J. Controlled Release 2002, 79, 29–40. 10.1016/S0168-3659(01)00549-1. [DOI] [PubMed] [Google Scholar]
- Bratlie K. M.; Dang T. T.; Lyle S.; Nahrendorf M.; Weissleder R.; Langer R.; Anderson D. G. Rapid biocompatibility analysis of materials via in vivo fluorescence imaging of mouse models. PLoS One 2010, 5, e10032 10.1371/journal.pone.0010032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champion J. A.; Mitragotri S. Role of target geometry in phagocytosis. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 4930–4934. 10.1073/pnas.0600997103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabata Y.; Ikada Y. Effect of the size and surface charge of polymer microspheres on their phagocytosis by macrophage. Biomaterials 1988, 9, 356–362. 10.1016/0142-9612(88)90033-6. [DOI] [PubMed] [Google Scholar]
- Gristina A. Biomaterial-centered infection: microbial adhesion versus tissue integration. Science 1987, 237, 1588–1595. 10.1126/science.3629258. [DOI] [PubMed] [Google Scholar]
- Stevens M. M.; George J. H. Exploring and engineering the cell surface interface. Science 2005, 310, 1135–1138. 10.1126/science.1106587. [DOI] [PubMed] [Google Scholar]
- Akilbekova D.; Philiph R.; Graham A.; Bratlie K. M. Macrophage reprogramming: Influence of latex beads with various functional groups on macrophage phenotype and phagocytic uptake in vitro. J. Biomed. Mater. Res., Part A 2015, 103, 262–268. 10.1002/jbm.a.35169. [DOI] [PubMed] [Google Scholar]
- Wang D.; Phan N.; Isely C.; Bruene L.; Bratlie K. M. Effect of surface modification and macrophage phenotype on particle internalization. Biomacromolecules 2014, 15, 4102–4110. 10.1021/bm5011382. [DOI] [PubMed] [Google Scholar]
- Brocchini S.; James K.; Tangpasuthadol V.; Kohn J. Structure-property correlations in a combinatorial library of degradable biomaterials. J. Biomed. Mater. Res. 1998, 42, 66–75. . [DOI] [PubMed] [Google Scholar]
- Vegas A. J.; Veiseh O.; Doloff J. C.; Ma M.; Tam H. H.; Bratlie K.; Li J.; Bader A. R.; Langan E.; Olejnik K.; et al. Combinatorial hydrogel library enables identification of materials that mitigate the foreign body response in primates. Nat. Biotechnol. 2016, 34, 345–352. 10.1038/nbt.3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C. C.; Raza A.; Shih H. PEG hydrogels formed by thiol-ene photo-click chemistry and their effect on the formation and recovery of insulin-secreting cell spheroids. Biomaterials 2011, 32, 9685–9695. 10.1016/j.biomaterials.2011.08.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko R. V.; James S. L.; James S. E. A review of tissue-engineered skin bioconstructs available for skin reconstruction. J. R. Soc., Interface 2010, 7, 229–258. 10.1098/rsif.2009.0403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells R. G. Tissue mechanics and fibrosis. Biochim. Biophys. Acta, Mol. Basis Dis. 2013, 1832, 884–890. 10.1016/j.bbadis.2013.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffin J. M.; Pittet P.; Csucs G.; Lussi J. W.; Meister J. J.; Hinz B. Focal adhesion size controls tension-dependent recruitment of alpha-smooth muscle actin to stress fibers. J. Cell Biol. 2006, 172, 259–268. 10.1083/jcb.200506179. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.