

Abstract

Two new bromotyrosine derivatives, anomoian B (1) and aplyzanzine B (2), were isolated, respectively, from the organic extracts of a Verongida sponge belonging to the Hexadella genus and from a two-sponge association (Jaspis sp. and Bubaris sp.), both collected off the coast of Indonesia. The planar structure of 1 and 2 was determined by 1D and 2D NMR experiments and by high-resolution mass spectrometry, while their absolute stereochemistry was assigned by comparison with optical rotation values of known bromotyrosines and by chemical degradation. Both compounds showed moderate antiproliferative activity against a panel of different cancer cell lines. Their cytotoxic activity is facilitated through the induction of apoptosis, which is mediated neither by the generation of reactive oxygen species nor by the inhibition of histone deacetylases in these cell lines.
1. Introduction
Marine sponges belonging to the order Verongida are devoid of spicules, and their characterization is a real challenge in terms of taxonomy.1 However, the bromotyrosine derivatives that are distinctive of this type of sponges2 have been used as chemotaxonomic markers and have been proved to be a useful tool to facilitate taxonomic identification.3 Since the first report of (+)-aeroplysinin-1,3 marine sponges of the order Verongida have been found to produce many bromotyrosine derivatives such as aerothionin,4 purealidins,5,6 purpurealidins,7 purpuramines,6,8 aplysamines,3 pseudoceralidinone A,9 and purealines.10 Despite this, Verongida is not the only order of sponges where bromotyrosine derivatives have been found. Two-sponge associations, where at least one of the members belongs to the genus Jaspis, are also known to contain this kind of metabolites.11
From an ecological point of view, this type of compounds has evolved as a defense system against natural predators, with the exception of some nudibranchs and mollusks that can feed on these sponges.12
Some of these alkaloids have been described as antiproliferative, apolipoprotein E modulators or cytotoxic and antibacterial agents.2 Previous studies of their molecular mechanism of action revealed that psammaplin13,14 is an inhibitor of histone deacetylases (HDACs), whereas the small bromotyrosine derivatives15 are potent apoptotic effectors.
2. Results and Discussion
In the course of our ongoing drug discovery program to find new antitumor agents from marine sponges,16,17 extracts of two Indonesian samples were fractionated, guided by their cytotoxic activities, leading to the isolation of two new bromotyrosine derivatives. The first sample, an unknown species of the genus Hexadella (order Verongida) collected at Para Island in Indonesia, was extracted, and the soluble materials were subjected to reversed-phase chromatography to yield anomoian B (1, 0.037%, wet wt) as a colorless, amorphous solid together with the known bromotyrosine alkaloid aplysamine 4 (3) (Chart 1).14 Previous chemical studies of marine sponges of the genus Hexadella reported the isolation of bromotyrosine derivatives related to aerothionin and bisindole alkaloids.19−21
Chart 1.

The second sample, a two-sponge association of Jaspis sp. and Bubaris sp. collected in Pulau Saujung in Indonesia, was extracted to give an organic extract, which after bioguided fractionation yielded aplyzanzine B (2, 0.057%, wet wt) as a brown amorphous solid. Although there are earlier reports of chemical studies of Jaspis genus sponges associated with another sponge genus,11 this work constitutes the first study of a two-sponge association of Jaspis sp. and Bubaris sp. As far as we know, there is a just one chemical study of a sponge belonging to Bubaris genus that describes the isolation of a sesquiterpenoid isocyanide.22,23
The (+)-low-resolution electrospray ionization mass spectra (LRESIMS) data of 1 [[α]D + 10.5° (c 1.0, CH3OH)] showed the [M + H]+ ion as a cluster at m/z 740, 742, 744, 746, and 748 in a 1:4:6:4:1 ratio, indicating the presence of four bromine atoms in the molecule. This was verified by subsequent (+)-high-resolution electrospray ionization time-of-flight mass spectrometry (HRESITOFMS) of the first isotopic peak at m/z 739.9355 [M + H]+ (calcd for C25H3479Br4N3O3m/z 739.9328), which in combination with its NMR data (Table 1) allowed us to propose the molecular formula of 1 as C25H33Br4N3O3.
Table 1. NMR Data of Anomoian B (1) in CD3OD and Aplyzanzine B (2) in CDCl3.
|
1 |
2 |
||||
|---|---|---|---|---|---|
| pos. | δC type | δHm (J in Hz) | pos. | δC type | δHm (J in Hz) |
| 1 | 134.5 C | 1 | 133.4 C | ||
| 2 | 135.2 CH | 7.51s | 2 | 133.6 CH | 7.40s |
| 3 | 119.3 C | 3 | 118.5 C | ||
| 4 | 155.1 C | 4 | 153.9 C | ||
| 5 | 119.3 C | 5 | 118.5 C | ||
| 6 | 135.2 CH | 7.51s | 6 | 133.6 CH | 7.40s |
| 7 | 34.3 CH2 | 3.36m | 7 | 34.1 CH2 | 3.22dd (12.4, 3.6) |
| 3.00m | 3.09dd (12.4, 11.3) | ||||
| 8 | 70.5 CH | 3.91dd (11.2, 4.6) | 8 | 60.4 CH | 4.97dd (11.3, 3.6) |
| 9 | 167.2 C | 9 | 166.8 C | ||
| 10 | 38.0 CH2 | 3.51m | 10 | 54.9 CH2 | 5.06d (17.5) |
| 3.28m | 4.19d (17.5) | ||||
| 11 | 30.6 CH2 | 1.84m | 11 | 189.3 C | |
| 1.70m | 12 | 132.5 C | |||
| 12 | 71.6 CH2 | 3.87m | 13 | 132.7 CH | 8.03s |
| 3.75m | 14 | 118.9 C | |||
| 13 | 153.4 C | 15 | 157.0 C | ||
| 14 | 119.4 C | 16 | 118.9 C | ||
| 15 | 134.4 CH | 7.57s | 17 | 132.7 CH | 8.03s |
| 16 | 135.1 C | 18 | 69.9 CH2 | 4.13t (5.5) | |
| 17 | 134.4 CH | 7.57s | 19 | 25.3 CH2 | 2.32dq (11.2, 5.5) |
| 18 | 119.4 C | 20 | 55.5 CH2 | 3.42m | |
| 19 | 30.3 CH2 | 2.99m | 21 | 40.0 CH3 | 2.97s |
| 20 | 59.1 CH2 | 3.33m | 22 | 40.0 CH3 | 2.97s |
| 21 | 42.5 CH3 | 2.99s | 23 | 42.9 CH3 | 2.88s |
| 22 | 42.5 CH3 | 2.99s | 24 | 42.9 CH3 | 2.88s |
| 23 | 43.6 CH3 | 2.93s | 25 | 60.7 CH3 | 3.85s |
| 24 | 43.6 CH3 | 2.93s | 26 | 37.6 CH3 | 2.75s |
| 25 | 61.1 CH3 | 3.77s | |||
Maximum UV absorptions at 276 and 284 nm indicated a possible substituted benzoic system, whereas the diagnostic IR vibration at 1673 cm–1 suggested the presence of an amide carbonyl group. The 13C NMR spectrum (see Table 1) disclosed signals owing to nine sp2 quaternary carbons including an amide (δC 167.2), four sp2 methines, one sp3 methine, six sp3 methylenes, and five methyls.
Analysis of the 1H–1H correlation spectroscopy (COSY) along with the Distortionless Enhancement by Polarization Transfer (DEPT)-135-edited heteronuclear single-quantum correlation (HSQC) spectra of 1 revealed the presence of three spin systems: (a) 2-aminoethyl (δC7 34.3/δH7 3.36/3.00 and δC8 70.5/δH8 3.91), (b) 3-aminopropanol (δC10 38.0/δH10 3.51/3.28, δC11 30.6/δH11 1.84/1.70, and δC12 71.6/δH12 3.87/3.75), and (c) 2-aminoethyl (δC19 30.3/δH22 2.99 and δC20 59.1/δH23 3.33) (Figure 1A). Additionally, singlets observed in the 1H NMR spectrum of 1 were assigned to a methoxy group (δOMe 61.1/δOMe 3.77), two dimethylamino groups (δC21,22 42.5/δH21,22 2.99 and δC23,24 43.6/δH23,24 2.93), and four aromatic protons (δC2,6 135.2/δH2,6 7.51 and δC15,17 134.4/δH15,17 7.57, 2H each), characteristic of two symmetrically dibromine-substituted aromatic rings. The three spin systems and the other groups were connected by the aid of a heteronuclear multiple-bond correlation (HMBC) experiment (see Figure 1A): (a) The methoxy group was located at C4 of one of the dibromine aromatic rings by the HMBC correlation between the Me25 protons at δH 3.77 and the quaternary aromatic carbon C4 at δC 155.1. Furthermore, the HMBC correlations between the aromatic protons H-2/6 and the C4 and C7 carbons connected the aromatic ring to the spin system a; correlations between the dimethylamino protons H21/H22 at δH 2.99 and C8 at δC8 70.5 and between the proton H8 at δH8 3.91 and the carbonyl amide C9 allowed us to identify an N,N′-dimethylamino 4-methoxy-3,5-dibrominetyrosine unit, or “tyrosine” fragment, in 1. (b) 3-Aminopropanol (spin b) was linked on one side to the former tyrosine fragment by an HMBC correlation between the methylene H210 protons at δH 3.51/3.28 and the carbonyl amide C9 and on the other side to the second dibromine-substituted aromatic ring by the key long-range correlation between the methylene H212 at δH 3.87/3.75 and the aromatic oxygenated carbon C13 at δC 153.4. (c) Finally, the second 2-aminoethyl group (spin c) was connected on one side to the second dibromine-substituted aromatic ring by HMBC correlations between the aromatic protons H15/H17 at δH 7.57 and the methylene carbon C19 (δC 30.3) and on the other side to the dimethylamino moiety through the HMBC correlation between methyl protons H23/24 at δH 2.93 and the methylene carbon C20. These correlations allowed us to identify an N,N′-dimethylamino-3,5-dibrominetyramine unit (the “tyramine” fragment).
Figure 1.
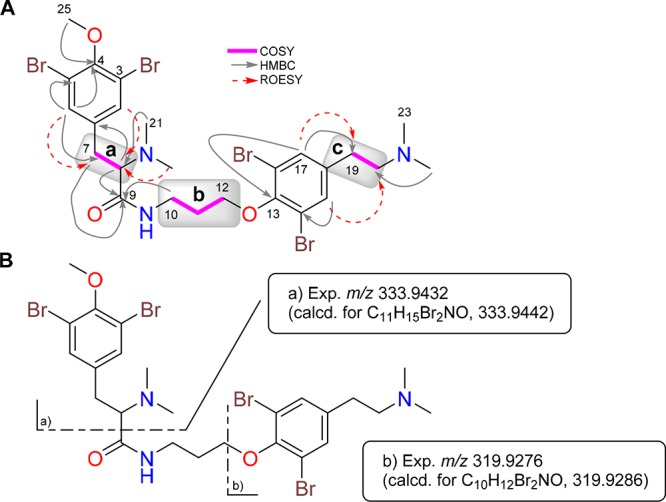
A) Selected two-dimensional (2D) NMR correlations of compound 1. (B) (+)-HRESIMS of the fragments detected in a MS/MS experiment of 1.
Nuclear Overhauser effect (NOE) correlations between protons H2/6 and H7/8, H21/22 and H8, and H15/17 and H19/20 observed in a rotating-frame Overhauser effect spectroscopy (ROESY) experiment of 1 were consistent with the proposed structure (Figure 1A). Moreover, the proposed planar structure of 1 was further confirmed by the presence of diagnostically important peaks at m/z 333.9432 and 319.9276 detected in the (+)-HRESIMS/MS experiment of 1 (Figure 1B), corresponding to selective fragmentation arising from the loss of both the tyrosine and tyramine fragments. All of this information allowed us to establish the structure of 1, named anomoian B, as having a framework (tyrosine)–(“3-aminopropanol”)–(tyramine), similar to that found in the purpuramines.6,8
A similar approximation was chosen for the structure elucidation of 2, namely, as aplyzanzine B. The molecular formula of aplyzanzine B (2) was determined as C26H34Br4N3O4 based on the [M + H]+ ion peak observed at m/z 767.9285 in its (+)-HRESITOF mass spectrum and on the NMR data (Table 1).
The 1H and 13C NMR spectral data (Table 1) of 2 showed the presence of the same N,N′-dimethylamino-4-methoxy-3,5-dibrominetyrosine unit (spin a), a 3-aminopropanol entity (δC18 69.9/δH18 4.13, δC19 25.3/δH19 2.32, and δC20 55.5/δH20 3.42) (spin c), a second dimethylamino group, and another symmetrically dibromine-substituted aromatic ring as in 1. Furthermore, the NMR spectra of 2 showed the presence of additional diastereotopic methylene protons (δC10 54.9/δH10 5.06 and 4.19) (spin b), an N-methylamide group (δC26 37.6/δH26 2.75), and a ketone carbonyl group (δC 189.3) instead of the second 2-aminoethyl group present in 1.
Key HMBC correlations allowed us to identify the presence of a −N(Me)–CH2–CO– unit linked to the N,N′-dimethylamino-4-methoxy-3,5-dibrominetyrosine unit (tyrosine fragment) as follows:
-
(1)
HMBC cross peaks from the methylamide protons at δH26 2.75 to the carbonyl amide carbon C9 (δC 166.8) and the methylene carbon C10 (δC 54.9) and
-
(2)
HMBC correlations from the diastereotopic methylene protons H210 (δH 5.06 and 4.19) (spin b) to the carbon C9 and the ketone carbonyl carbon C11 (δC 189.3).
Moreover, long-range correlations from the aromatic protons H13/17 to the ketone carbonyl carbon C11 allowed us to link the −N(Me)–CH2–CO– unit to the second dibromine aromatic ring on the other side. Thus, an N-methylamino-3,5-dibrominetyramine unit (tyramine fragment) was identified as a substructure of 2.
The second N,N′-dimethylamino group was linked to the 3-aminopropanol unit (spin c) by the HMBC correlations from methyl protons H23/24 at δH 2.88 to the methylene carbon C20 (δC 55.5). Finally, spin c was in turn connected to the second dibromine aromatic ring by the long-range correlations from the methylene protons H218 (δH 4.13) and from the aromatic protons H17/18 (δH 8.03) to the same aromatic carbon C15 (δC 157.0), completing the planar structure of compound 2 (Figure 2A).
Figure 2.

A) Selected 2D NMR correlations of compound 2. (B) (+)-HRESIMS of the fragments detected in a MS/MS experiment of 2.
Again, NOE correlations between aromatic protons H2/6 and H7/8, the methylamide protons H26 and H8, the diastereotopic methylene protons H210 and aromatic protons H13/17, and the methylene protons H220 and methyl protons H23/24 observed in a ROESY experiment of 2 agreed with the proposed structure (Figure 2A). Furthermore, the fragmentation pathway detected in the (+)-HRESIMS/MS experiment of 2, displayed in Figure 2B, confirmed the proposed planar structure for 2. In this way, the structure of compound 2 was shown to have a framework (tyrosine)–(tyramine)–(3-aminopropanol) similar to that present in aplysamine 4 (3). This compound was also isolated in this study and was identified by comparison of its spectral data with those reported by Scheuer and co-workers for aplysamine 4 (3)18 isolated from the Verongida sponge Psammaplysilla purpurea.
The S absolute stereochemistry at C8 in both compounds was determined by comparing their [α]D values (+10.5° for 1 and +47.5° for 2) to those of similar compounds reported previously in the bibliography such as iso-anomoian ([α]D + 4.5°)12 and suberedamine A ([α]D + 19.5°),10 all containing an l-tyrosine residue. Bromotyrosines bearing a d-tyrosine moiety, such as ianthesine A ([α]D – 118°) and its degradation tyrosine derivative N,N-dimethyl-d-tyrosine ([α]D – 73°),25 have been reported to have a negative sign [α]D value.15 It is important to notice that both compounds were subjected to high-performance liquid chromatography (HPLC) chiral analysis and just one peak was observed for each compound, ruling out a possible racemization at C8 position during the isolation and purification processes.
Aplyzanzine B (2) was hydrolyzed at 150 °C under acid conditions to afford bromotyrosines (S)-3,5-dibromo-N,N-dimethyltyrosine (4) and (S)-3-(3,5-dibromo-4-methoxyphenyl)-2-(dimethylamino)propanoic acid (5) (Figure 3), whose structures were deduced by NMR spectral data and by (+)-HRESI mass spectra displaying molecular formulae of C12H15Br2NO3 and C11H13Br2NO3, respectively. The positive [α]D optical rotation values of +31° and +33° for 4 and 5, respectively, confirmed the same absolute stereochemistry of C8 as established for 2. (R)-3,5-Dibromo-N,N-dimethyltyrosine was obtained previously by acid hydrolysis of ianthesine A,25 whereas compound 5 was reported as an intermediate in the synthesis of several bromotyrosine derivatives.26,27
Figure 3.
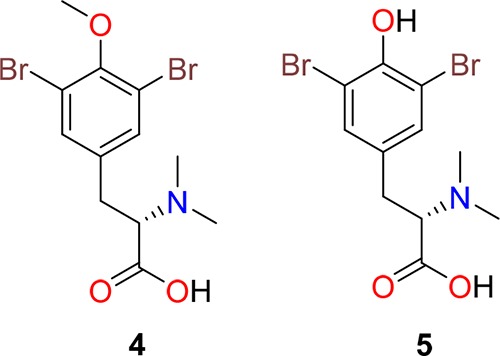
Compounds 4 and 5 obtained by hydrolysis of 2.
Aplyzanzine B (1) and anomoian B (2) exhibit significant cytotoxic activity against three human tumor cell lines from different tissues: A549 (lung carcinoma), HT-29 (colorectal adenocarcinoma), and MDA-MB-231 (breast adenocarcinoma). IC50 values for anomoian B (1) in these cell lines are 5.1, 3.2, and 5.3 μM. IC50 values for aplyzanzine B (2) are 6.1, 1.6, and 7.8 μM. The bromotyrosines 4 and 5 were also evaluated under the same conditions as 2 with the aim of determining a structure–activity relationship for this type of compounds. The lack of cytotoxic activity showed by compounds 4 and 5 seems to indicate that the rest of the molecule plays an important role in the cytotoxic activity of compounds 1 and 2.
In order to get some insights into the mechanism of action of the cytotoxic activity of these compounds, a further study of cell death induction was performed.
The data published by Su et al.15 indicated that the cytotoxic activity of small bromotyrosine derivatives is mediated through the induction of apoptosis, also known as programmed cell death. During apoptosis, some proteins are cleaved by caspase-3 and they can be used as markers of cell death. Poly (ADP-ribose) polymerase (PARP-1) is a target of caspase-3, and its cleavage is considered to be a hallmark of apoptosis. PARP-1 cleavage can be easily detected by Western blot. In addition, the apoptotic program leads to DNA internucleosomal fragmentation; histone H2AX becomes rapidly phosphorylated when DNA is damaged (known as γH2AX) and plays an important role in the execution of apoptosis. To investigate whether the antiproliferative effect of aplyzanzine B (1) and anomoian B (2) was due to the induction of apoptosis, three human tumor cell lines (the same ones in which cytotoxicity had been previously demonstrated, namely, A549, HT-29, and MDA-MB-231) were incubated with both compounds at 10 and 20 μg/mL for 24 h (also 48 h for A549).
HT-29 was the cell line with higher sensitivity to both compounds at 24 h. Morphological alteration characteristic of apoptosis, such as dense chromatin surrounded by a halo, was observed in the white-field microscopy images (Figure 4). Protein analysis by Western blot confirmed that, in HT-29 cells, both aplyzanzine B (1) and anomoian B (2) exerted their antiproliferative effect through the induction of apoptosis. Increasing concentrations of both compounds led to PARP cleavage and γH2AX induction (Figure 5). It is important to notice that γH2AX induction is not an effect of direct binding to DNA by these compounds. Band-shift assays have shown that, after incubation with these compounds, the electrophoretic migration of a 200 bp DNA fragment is not affected (data not shown).
Figure 4.

Images of human tumor cell lines HT-29, MDA-MB-231, and A-549 under different conditions assayed as indicated in the figure. Images were obtained at 20× magnification with a white-field microscope.
Figure 5.

Detection of PARP and γH2AX by Western blot in HT-29 at 24 h.
We further investigated whether reactive oxygen species (ROS) were involved in the apoptosis induced by aplyzanzine B and anomoian B, as previously established for the dibromotyrosine derivative (1′R,5′S,6′S)-2-(3′,5′-dibromo-1′,6′-dihydroxy-4′-oxacyclohex-2′-enyl) acetonitrile (DT).15 ROS play a critical role in cellular homeostasis, and their overproduction may induce apoptosis or other types of cell death. ROS scavenger NAC (N-acetylcysteine) was used to pretreat the cells in order to diminish the production of ROS. Pretreatment with 1.5 mM NAC did not avoid PARP cleavage and γH2AX induction, indicating that ROS are not the main mediators of the apoptotic process triggered by these compounds in HT-29 cells.
MDA-MB-231 and A549 cells are more resistant to both compounds under the assay conditions used. In MDA-MB-231 cells, PARP processing is faintly detected at 24 h, as is also the case of γH2AX induction (Figure 6). In A549 cells, incubation for 48 h with both compounds was necessary to detect PARP cleavage and γH2AX induction (Figure 7). ROS scavenger NAC did not affect γH2AX induction in these cell lines.
Figure 6.

Detection of PARP and γH2AX by Western blot in MDA-MB-231 at 24 h.
Figure 7.

Detection of PARP and γH2AX by Western blot in A549 at 48 h.
Previous studies have shown that the psammaplins, also bromotyrosine derivatives, inhibit HDACs.16 HDAC inhibitors are a promising new class of anticancer agents that act by inhibiting cell proliferation and inducing cell cycle arrest in several cancer cell types. To analyze whether the antiproliferative effect of aplyzanzine B (1) and anomoian B (2) was related to the HDAC inhibition, A549 cells were treated with both compounds at different concentrations. As a consequence of HDAC inhibition, acetylated histone 4 is accumulated in cells. The results, shown in Figure 8, indicated that neither of the compounds inhibited HDACs. Compounds 1 and 2 did not induce acetyl-H4 accumulation as seen for the positive control trichostatin A, a known HDAC inhibitor.
Figure 8.
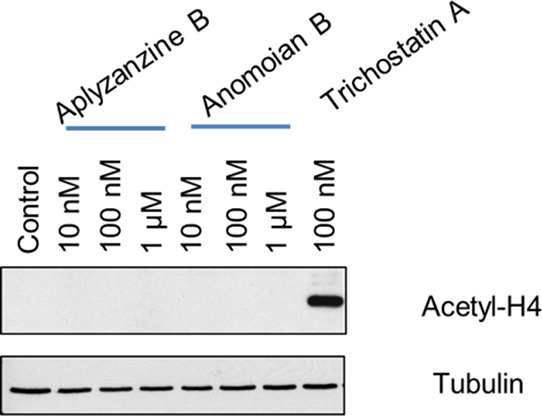
Acetyl-H4 Western blot in A549 cells treated with different concentrations of aplyzanzine B (1) and anomoian B (2). Trichostatin A was used as a positive control, and tubulin is shown as a loading control.
3. Conclusions
In summary, the new bromotyrosine derivatives anomoian B (1) and aplyzanzine B (2) were isolated from Indonesian sponges and form part of the few examples of bromotyrosine derivatives with a chiral center at C8. Although the oxime group of the α-carbon in tyrosine residues is present in most bromotyrosine derivatives, this group has been replaced in other related analogues by a primary amine as in hexadellin A,8,20 an N-methylamine group as in anomoian A,23,27 suberedamines A and B,24 or an N,N′-dimethylamine group as in aplyzanzine A.28
Aplyzanzine B (2) is the first compound of the bromotyrosine family bearing a ketone carbonyl group adjacent to the dibromobenzene ring. Biogenetically, the ketone functionality could arise through the oxidation of the benzylic position of the tyramine unit (see Figure 9). Compounds bearing a OH group at that position have already been reported such as hemifistularin 3,29 aplysamine A,9 and fistularin-3.30 A further oxidation of that benzylic OH group could be the origin of the ketone carbonyl group present in 2.
Figure 9.

Biogenesis proposal for the ketone functionality present in aplyzanzine B (2).
The two new compounds described herein have shown moderate antitumor activity against a panel of three human tumor cell lines (A549, HT-29, and MDA-MB-231), being most active in the colon adenocarcinoma HT-29 cell line. Both compounds induce the apoptotic cell death of the tumor cell lines assayed, although none of the mechanisms described for this family of compounds, namely, the induction of ROS and the inhibition of HDACs, seem to be involved in the induction of apoptosis caused by the new compounds.
4. Materials and Methods
4.1. General Experimental Procedures
Optical rotations were determined using a Jasco P-1020 polarimeter. UV spectra were recorded using an Agilent 8453 ultraviolet–visible spectrometer. IR spectra were obtained with a PerkinElmer Spectrum 100 Fourier transform infrared spectrometer with attenuated total reflection sampling. NMR spectra were recorded on a Varian “Unity 500” spectrometer and a Bruker ADVANCE 500 spectrometer at 500/125 MHz (1H/13C). Chemical shifts were reported in parts per million using residual CD3OH (δ 3.31 ppm for 1H and 49.0 ppm for 13C) as an internal reference. HRESITOFMS was performed on an Agilent 6230 TOF LC/MS chromatograph spectrometer. (+)-ESIMS spectra were recorded using an Agilent 1100 Series LC/MSD spectrometer. All ESIMS/MS measurements were recorded on a QqQ-TOF Applied Biosystems QSTAR Elite. Chiral HPLC analyses were completed in a Chiralpak AD-H column (5 × 150 mm, H2O + 0.04% CF3 CO2 H/CH3 CN + 0.04% CF3CO2H gradient from 20 to 84% in 16 min, flow rate: 3 mL/min).
4.2. Animal Material
The sponge Hexadella cf indica Dendy (47 g) was collected in May 2010 by hand using rebreather diving near Para Island, Indonesia (03°03.603′ N/125°30.496′ E) at a depth of 81 m and was frozen immediately after collection. The two-sponge association of Jaspis sp. and Bubaris sp. (229 g) samples was collected in September 2012 by hand using rebreather diving in Pulau Saujung, Indonesia (07°20.375′ S/117°32.288′ E) at a depth of 100 m and was frozen immediately after collection. Voucher specimens were deposited at PharmaMar.
4.3. Extraction and Isolation
Hexadella cf indica Dendy (47 g) was triturated and exhaustively extracted with CH3OH/CH2Cl2 (1:1, 3 × 400 mL). The combined extracts were concentrated to yield a crude mass of 3.2 g. The crude product was subjected to vacuum liquid chromatography (VLC) on LiChroprep RP-18 with a stepped gradient from H2O to CH3OH to CH2Cl2. The fraction that eluted with CH3OH (565 mg) was subjected to HPLC Prep (XBridge Prep C18 5 μm, 19 × 150 mm, gradient H2O/CH3CN from 25 to 76% CH3CN in 11 min and then 100% in 2 min, flow rate: 15 mL/min, UV detection) to yield a crude fraction (263 mg). This fraction was further purified by semipreparative HPLC (SunFire C18 5 μm, 10 × 150 mm, H2O + 0.1% CF3CO2H/CH3CN + 0.1% CF3CO2H gradient from 30 to 70% in 13 min, flow rate: 3 mL/min, UV detection) to obtain anomoian B (1, 17.6 mg, 0.037% wet wt).
A two-sponge association of Jaspis sp. and Bubaris sp. (229 g) was triturated and exhaustively extracted with CH3OH/CH2Cl2 (1:1, 3 × 400 mL). The combined extracts were concentrated to yield a crude mass of 9.7 g. The crude product was subjected to VLC on LiChroprep RP-18 with a stepped gradient from H2O to CH3OH to CH2Cl2. The fraction that eluted with CH3OH gave a fraction of 1.88 g, which was subjected to reversed-phase flash chromatography gradient (H2O/CH3CN from 5 to 100% in 25 min) to yield a crude fraction (257 mg). This fraction was further purified by semipreparative HPLC (XBridge Prep C18 5 μm, 10 × 250 mm, H2O + 0.04% CF3CO2H/CH3CN + 0.04% CF3CO2H gradient from 20 to 84% in 16 min, flow rate: 3 mL/min, UV detection) to obtain aplyzanzine B (2, 130 mg, 0.057% wet wt). Chiral HPLC analysis of 1 and 2 was performed in a Chiralpak AD-H column (5 × 150 mm, H2O + 0.04% CF3CO2H/CH3CN + 0.04% CF3CO2H gradient from 20 to 84% in 16 min, flow rate: 3 mL/min, MS detection).
4.4. Anomoian B (1)
Colorless amorphous solid; [α]D + 10.5 (c 0.1, CH3OH); CD spectrum, see the Supporting Information; IR νmax: 2929, 1673, 1473, 1261, 1201, 1133, 834, 800, and 722 cm–1; UV (CH3OH) λmax 276 and 284 nm. 1H NMR (500 MHz) and 13C NMR (125 MHz) (see Table 1); HREITOFMS m/z 739.9355 [M + H]+ (calcd for C25H3479Br4N3O3m/z 739.9328). CD spectrum (MeOH) (see the Supporting Information).
4.5. Aplyzanzine B (2)
Brown amorphous solid; [α]D + 47.5 (c 0.1, CH3OH); CD spectrum, see the Supporting Information; IR νmax: 2972, 1684, 1544, 1474, 1424, 1261, 1200, and 1130 cm–1; UV (CH3OH) λmax 259 nm. 1H NMR (500 MHz) and 13C NMR (125 MHz) (see Table 1); HREITOFMS m/z 767.9285 [M + H]+ (calcd for C26H3479Br4N3O4m/z 767.9277). CD spectrum (MeOH) (see the Supporting Information).
4.6. Acidic Hydrolysis of Aplyzanzine B
Compound 2 was hydrolyzed in 6 N HCl at 150 °C for 72 h. The hydrolysate was dried under nitrogen and reconstituted in methanol. The residue was purified by HPLC {symmetry C18, 5 μm, 4.6 × 150 mm; linear gradient from 5 to 100% CH3CN [0.04% trifluoroacetyl (TFA)] in H2O [0.04% TFA] over 40 min, flow rate: 0.8 mL/min} to give compounds 4 and 5.
4.7. (S)-3,5-Dibromo-N,N-Dimethyltyrosine (4)
Colorless amorphous solid; [α]D + 30 (c 0.1, CH3OH) HREITOFMS m/z: 378.9422 [M + H]+ (calcd for C12H1579Br2NO3m/z: 378.9419). 1H NMR (500 MHz, D2O): δ 7.53 (s, 2H), 3.88 (s, 3H), 3.73 (dd, J = 9.5, 5.6 Hz, 1H), 3.26 (dd, J = 13.7, 5.6 Hz, 1H), 3.00 (dd, J = 13.7, 9.5 Hz, 1H), 2.88 (s, 6H).
4.8. (S)-3-(3,5-Dibromo-4-methoxyphenyl)-2-(dimethylamino)-propanoic Acid (5)
Colorless amorphous solid; [α]D + 33 (c 0.1, CH3OH). HREITOFMS m/z: 365.9355 [M + H]+ (calcd for C11H1479Br2NO3m/z: 365.9335). 1H NMR (500 MHz, D2O): δ 7.37 (s, 2H), 3.74 (dd, J = 8.0, 6.3 Hz, 1H), 3.12 (dd, J = 14.4, 6.3 Hz, 1H), 2.99 (dd, J = 14.4, 8.0 Hz, 1H), 2.88 (s, 6H).
4.9. Evaluation of Cytotoxic Activity
The cytotoxic activity of the isolated compounds was evaluated using a panel of three human tumor cell lines: A549 (lung), HT-29 (colon), and MDA-MB-231 (breast). Cells were maintained at 37 °C and 5% CO2 in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 2 mM l-glutamine, and 100 units/mL penicillin–streptomycin. To determine the cell viability, cells were washed twice with phosphate-buffered saline (PBS), fixed for 15 min in 1% glutaraldehyde solution, rinsed twice in PBS, and stained in 0.4% sulforhodamine B solution for 30 min at room temperature. Cells were then rinsed several times with 1% acetic acid solution and were air-dried. SRB was then extracted with a 10 mM Trizma base solution, and the absorbance was measured at 490 nm. GI50 values (concentration of drug that produces a 50% inhibition of cell growth) were calculated using Prism v5.02 software (GraphPad, La Jolla, USA). Experiments were performed in triplicate.
4.10. Western Blotting Analysis
Cell protein extracts were prepared following standard procedures in a radioimmunoprecipitation assay buffer in the presence of protease inhibitors (Complete, Roche Diagnostics) and phosphatase inhibitors (PhosSTOP, Roche Diagnostics). After quantification with the BCA protein assay kit (Thermo Scientific, Waltham, MA, USA), 20 μg of protein was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto poly(vinylidene difluoride) membranes (Immobilon-P, Millipore, Billerica, MA, USA). After hybridizing with the appropriated primary [PARP (Santa Cruz), γH2AX and acetyl-histone H4 (Upstate, Millipore), and tubulin (Sigma)] and secondary antibodies (Santa Cruz), blots were developed by a peroxidase reaction using the electrochemical luminescence detection system (Amersham-G.E. Healthcare, Little Chalfont, UK).
Acknowledgments
The authors thank Paloma Fernández López for her excellent technical assistance. Sponges were collected with the permission of the RISKET authorities under permit no. 0118/SIP/FRP/IV/212. The authors would also like to thank Dr. Ir. Gembong Baskoro, M.Sc., and Widya Kartika University (Kota SBY, Jawa Timur, Indonesia) for the Research Collaboration Agreement to do sampling in Indonesia. The authors gratefully acknowledge the help of their PharmaMar colleagues C. de Eguilior for collecting the marine samples, S. Bueno for determining the sponge taxonomies, S. Cascajares for conducting the biological assays, S. Munt for revising the manuscript, and S. González for performing the MS experiments. The present research was financed in part by Grants from Ministerio de Economía y Competitividad of Spain (AGL2015-63740-C2-2-R and RTC-2016-4611-1).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b00417.
1H, 13C, gHSQC, gCOSY, gHMBC, TOCSY, ROESY NMR, and CD spectra of anomoian B (1) and aplyzanzine B (2) and LRESIMS and 1H NMR spectra of 4 and 5 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Ehrlich H.; Ilan M.; Maldonado M.; Muricy G.; Bavestrello G.; Kljajic Z.; Carballo J. L. Int. J. Biol. Macromol. 2010, 47, 132–140. 10.1016/j.ijbiomac.2010.05.007. [DOI] [PubMed] [Google Scholar]
- Proksch P.; Putz A.; Ortlepp S.; Kjer J.; Bayer M. Phytochem. Rev. 2010, 9, 475–489. 10.1007/s11101-010-9178-9. [DOI] [Google Scholar]
- Gotsbacher M.; Karuso P. Mar. Drugs 2015, 13, 1389–1409. 10.3390/md13031389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulmor W.; Van Lear G. E.; Morton G. O.; Mills R. D. Tetrahedron Lett. 1970, 52, 4551–4552. 10.1016/s0040-4039(00)89414-9. [DOI] [PubMed] [Google Scholar]
- McMillan J. A.; Paul I. C.; Goo Y. M.; Rinehart K. L.; Krueger W. C.; Pschigoda L. M. Tetrahedron Lett. 1981, 22, 39–42. 10.1016/0040-4039(81)80035-4. [DOI] [Google Scholar]
- Tabudravu J. N.; Jaspars M. J. Nat. Prod. 2002, 65, 1798–1801. 10.1021/np020275n. [DOI] [PubMed] [Google Scholar]
- Tilvi S.; Rodrigues C.; Naik C. G.; Parameswaran P. S.; Wahidhulla S. Tetrahedron 2004, 60, 10207–10215. 10.1016/j.tet.2004.09.009. [DOI] [Google Scholar]
- Dai J.; Parrish S. M.; Yoshida W. Y.; Yip M. L. R.; Turkson J.; Kelly M.; Williams P. Bioorg. Med. Chem. Lett. 2016, 26, 499–504. 10.1016/j.bmcl.2015.11.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran T. D.; Pham N. B.; Fechner G.; Hooper J. N. A.; Quinn R. J. J. Nat. Prod. 2013, 76, 516–523. 10.1021/np300648d. [DOI] [PubMed] [Google Scholar]
- Salim A. A.; Khalil Z. G.; Capon R. J. Tetrahedron 2012, 68, 9802–9807. 10.1016/j.tet.2012.09.008. [DOI] [Google Scholar]
- Shinde P. B.; Lee Y. M.; Dang H. T.; Hong J.; Lee C. O.; Jung J. H. Bioorg. Med. Chem. Lett. 2008, 18, 6414–6418. 10.1016/j.bmcl.2008.10.082. [DOI] [PubMed] [Google Scholar]
- Reverter M.; Perez T.; Ereskovsky A. V.; Banaigs B. J. Chem. Ecol. 2016, 42, 60–70. 10.1007/s10886-015-0664-9. [DOI] [PubMed] [Google Scholar]
- Kottakota S. K.; Benton M.; Evangelopoulos D.; Guzman J. D.; Bhakta S.; McHugh T. D.; Gray M.; Groundwater P. W.; Marrs E. C. L.; Perry J. D.; Harburn J. J. Org. Lett. 2012, 14, 6310–6313. 10.1021/ol303057a. [DOI] [PubMed] [Google Scholar]
- Jiménez C.; Crews P. Tetrahedron 1991, 47, 2097–2102. 10.1016/s0040-4020(01)96120-4. [DOI] [Google Scholar]
- Su J.-H.; Chen Y.-C.; El-Shazly M.; Du Y.-C.; Su C.-W.; Tsao C.-W.; Liu L.-L.; Chou Y.; Chang W.-B.; Su Y.-D.; Chiang M.; Yeh Y.-T.; Lu M.-C. Mar. Drugs 2013, 11, 3168–3185. 10.3390/md11093168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes F.; Ardá A.; Martín R.; Fernández R.; Rueda A.; Montalvo D.; Gómez C.; Jiménez C.; Rodríguez J.; Sánchez-Puelles J. M. J. Nat. Prod. 2004, 67, 1190–1192. 10.1021/np049903m. [DOI] [PubMed] [Google Scholar]
- Poza J. J.; Fernández R.; Reyes F.; Rodríguez J.; Jiménez C. J. Org. Chem. 2008, 73, 7978–7984. 10.1021/jo801198u. [DOI] [PubMed] [Google Scholar]
- Jurek J.; Yoshida W. Y.; Scheuer P. J.; Kelly-Borges M. J. Nat. Prod. 1993, 56, 1609–1612. 10.1021/np50099a025. [DOI] [Google Scholar]
- Matsunaga S.; Kobayashi H.; Van Soest R. W. M.; Fusetani N. J. Org. Chem. 2005, 70, 1893–1896. 10.1021/jo048203j. [DOI] [PubMed] [Google Scholar]
- Morris S. A.; Andersen R. J. Can. J. Chem. 1989, 67, 677–681. 10.1139/v89-102. [DOI] [Google Scholar]
- Morris S. A.; Andersen R. J. Tetrahedron 1990, 46, 715–720. 10.1016/s0040-4020(01)81355-7. [DOI] [Google Scholar]
- Wright A. E.; McCarthy P.; Cross S. S.; Rake J. B.; McConnell O. EP 285302 A1 19881005, 1988.
- Kernan M. R.; Cambie R. C.; Bergquist P. R. J. Nat. Prod. 1990, 53, 720–723. 10.1021/np50069a033. [DOI] [Google Scholar]
- Tsuda M.; Sakuma Y.; Kobayashi J. J. Nat. Prod. 2001, 64, 980–982. 10.1021/np010077g. [DOI] [PubMed] [Google Scholar]
- Okamoto Y.; Ojika M.; Kato S.; Sakagami Y. Tetrahedron 2000, 56, 5813–5818. 10.1016/s0040-4020(00)00544-5. [DOI] [Google Scholar]
- Marner F. J.; Moore R. E.; Hirotsu K.; Clardy J. J. Org. Chem. 1977, 42, 2815–2819. 10.1021/jo00437a005. [DOI] [Google Scholar]
- Kottakota S. K.; Evangelopoulos D.; Alnimr A.; Bhakta S.; McHugh T. D.; Gray M.; Groundwater P. W.; Marrs E. C. L.; Perry J. D.; Spilling C. D.; Harburn J. J. J. Nat. Prod. 2012, 75, 1090–1101. 10.1021/np300102z. [DOI] [PubMed] [Google Scholar]
- Evan T.; Rudi A.; Ilan M.; Kashman Y. J. Nat. Prod. 2001, 64, 226–227. 10.1021/np000383e. [DOI] [PubMed] [Google Scholar]
- Kubo H.; Matsui K.; Saitoh T.; Nishiyama S. Tetrahedron 2014, 70, 6392–6397. 10.1016/j.tet.2014.07.049. [DOI] [Google Scholar]
- Zhao J.; Liu H. Zhongnan Yaoxue 2013, 11, 185–186. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.