

Abstract
A combined biochemical, structural, and cell biology characterization of dictyostatin is described, which enables an improved understanding of the structural determinants responsible for the high-affinity binding of this anticancer agent to the taxane site in microtubules (MTs). The study reveals that this macrolide is highly optimized for MT binding and that only a few of the structural modifications featured in a library of synthetic analogues resulted in small gains in binding affinity. The high efficiency of the dictyostatin chemotype in overcoming various kinds of clinically relevant resistance mechanisms highlights its potential for therapeutic development for the treatment of drug-resistant tumors. A structural explanation is advanced to account for the synergy observed between dictyostatin and taxanes on the basis of their differential effects on the MT lattice. The X-ray crystal structure of a tubulin–dictyostatin complex and additional molecular modeling have allowed the rationalization of the structure–activity relationships for a set of synthetic dictyostatin analogues, including the highly active hybrid 12 with discodermolide. Altogether, the work reported here is anticipated to facilitate the improved design and synthesis of more efficacious dictyostatin analogues and hybrids with other MT-stabilizing agents.
Introduction
Microtubule-stabilizing agents (MSAs) are an important class of clinically used anticancer drugs that target tubulin, the main component of the microtubule (MT) cytoskeleton that is responsible for the segregation of chromosomes during cell division, among other functions. MSAs inhibit cell division by virtue of their preferential binding to assembled MTs relative to unassembled αβ-tubulin heterodimers.1 Although the prototypical paclitaxel and docetaxel have demonstrated great efficacy in inhibiting the growth of solid tumors, solubility problems and development of resistance have stimulated the search for other chemotypes with similar modes of action but improved pharmacological profiles. The main mechanisms of resistance to MSAs are (i) overexpression of βIII-tubulin isotypes and/or membrane efflux transporters from the ATP-binding cassette (ABC) superfamily, for example, P-glycoprotein (P-gp), which is primarily responsible for the multidrug resistance (MDR) phenotype and (ii) mutations in the β-tubulin gene.
This ongoing quest to identify new chemotypes acting as MSAs has resulted in the discovery of several other important antimitotic natural products (e.g., epothilones, sarcodictyins, cyclostreptin, peloruside, laulimalide, zampanolide, dictyostatin, and discodermolide) targeting at least three different binding sites2 on β-tubulin but displaying similar biological activities. Of these, the dictyostatin and discodermolide chemotypes, which are complex marine sponge-derived polyketides, show the highest affinity for MTs3 and have enhanced water solubility. These two properties apparently make these MSAs more prone to circumventing P-gp-mediated MDR because their high affinities for an abundant protein should result in low intracellular concentrations of the free drug and decreased interaction with the efflux transporter.
We have previously shown that it is possible to structurally optimize an MSA by studying the influence of selected chemical modifications in critical pharmacophore regions and by combining into a single molecular entity those changes that result in favorable contributions to the binding free energy. This rational approach has been successfully used to optimize the binding to the MTs of several epothilone4 and taxane analogues.5
Interest in the dictyostatin chemotype6 has driven the chemical synthesis of a variety of analogues to probe the structural determinants of tubulin binding affinity, ability to act as an MSA, and cytotoxic activity.7,8 We have used a library of 16 structurally diverse compounds (Figure 1) arising from this synthetic effort to investigate the following: (1) the influence of selected chemical modifications on the tubulin binding constants, stoichiometry, and MT structural properties using several biochemical and biophysical methods; (2) the binding epitopes and bioactive conformations of these compounds by means of X-ray crystallography and molecular modeling techniques; and (3) their biological effects on a representative panel of tumor cell lines that are either sensitive or resistant to other MSAs. As a long-term objective, this unified approach may lead to the rational design of anticancer drug candidates with significantly increased cytotoxicity and reduced resistance relative to the parent natural product.
Figure 1.

Chemical structures of the compounds used in this study.
In the present study, the structural modifications encompass a large part of the dictyostatin (1, Figure 1) scaffold, along with designed hybrids with discodermolide (3).9−14 Although some limited information about the cytotoxicity of these compounds in selected cancer cell lines has already been reported, little is known about the details of their interactions with MTs and tubulin, the contribution of the structural modifications to complex formation, and the adoption of the bioactive conformation.
Extensive nuclear magnetic resonance (NMR) studies have been performed to propose a bioactive conformation for dictyostatin and guide the synthesis of new and simplified analogues (reviewed by Larsen et al.8). However, a crystal structure of dictyostatin, either alone or in complex with tubulin, was not previously available to enable the rational structure-based design of improved analogues. To address this deficiency and help elucidate the pharmacophore region of this important MSA, we solved the structure of the resultant T2R-TTL–dictyostatin complex at 2.3 Å resolution using X-ray crystallography. We then simulated a macromolecular ensemble, incorporating this experimentally determined protein–ligand complex in the context of a reduced representation of a MT, using molecular dynamics (MD). In silico evaluation of the modeled complexes with the available synthetic dictyostatin analogues was then used to rationalize the structure–activity relationships (SARs). Taken together, these new results clarify the long-standing issue of defining the pharmacophore for the dictyostatin chemotype and shed additional light on longitudinal and lateral interactions in MSA-bound tubulin. They also provide a powerful tool for the rational design of simplified dictyostatin analogues and hybrids, including linker-modified targeted versions for incorporation as payloads in antibody–drug conjugates.
Results
Cytotoxicity of Dictyostatin and Analogues in Resistant Cells and Synergy with Paclitaxel and Peloruside A
To initially explore the therapeutic potential of the dictyostatin chemotype as an anticancer drug candidate, the cytotoxicities of the compounds in Figure 1 were measured in a panel of cancer cell lines that are resistant to other MSAs through three different mechanisms. The synergistic effect of their combination with either paclitaxel or peloruside A was also evaluated.
In the A2780, A2780AD, and A549 cell lines, all compounds that were examined arrested the cell cycle in the G2/M phase, as expected (Figure S1). In this regard, all of them, with the exception of analogues 2 and 7, were also shown to enhance tubulin polymerization in vitro in the absence of MT-associated proteins (MAPS) and to reduce the critical concentration (Cr) of tubulin required for assembly from 3.3 ± 0.1 μM in the absence of drug to 0.4 ± 0.1 μM in the presence of parent dictyostatin (Table 1).
Table 1. Cytotoxicity of the Compounds (Chemical Structures in Figure 1) in Ovarian Tumor Cell Lines Sensitive to MSA (A2780) and MSA-Resistant due to P-gp Overexpression (A2780AD) and in Cervical Tumor Cell Lines Sensitive to MSA (HeLa S3) and MSA-Resistant due to βIII-Tubulin Overexpression (HeLa-βIII).
| compound | A2780 (nM) | A2780AD (nM) | R/S | HeLa-S3 (nM) | HeLa βIII (nM) | R/S | Crb (μM) |
|---|---|---|---|---|---|---|---|
| dimethyl sulfoxide (DMSO, vehicle) | 3.3 ± 0.1a | ||||||
| paclitaxel | 1.8 ± 0.4 | 1491 ± 284 | 828.2 | 0.8 ± 0.2 | 8.6 ± 1.4 | 10.8 | 0.5 ± 0.1a |
| dictyostatin | 2.55 ± 1 | 3.9 ± 0.2 | 1.5 | 1.5 ± 0.3 | 4.4 ± 0.8 | 2.9 | 0.4 ± 0.1 |
| 2 | 1203 ± 52 | 1423 ± 39 | 1.2 | 1050 ± 145 | 1160 ± 115.2 | 1.1 | 3.3 ± 0.3 |
| discodermolide | 28 ± 3 | 37.9 ± 5.2 | 1.35 | 6 ± 0.8 | 13.2 ± 1.25 | 2.2 | 0.6 ± 0.1 |
| 4 | 3.6 ± 1.5 | 5.1 ± 0.85 | 1.4 | 3.5 ± 0.7 | 10.2 ± 1.5 | 3.0 | 1.2 ± 0.1 |
| 5 | 5.5 ± 1.2 | 4.7 ± 1.6 | 0.85 | 9.7 ± 1.9 | 35 ± 2 | 3.6 | 1.4 ± 0.1 |
| 6 | 33.3 ± 11.6 | 16 ± 0.7 | 0.5 | 13 ± 3 | 18.5 ± 1.1 | 1.4 | 2.2 ± 0.2 |
| 7 | 7000 ± 300 | 8575 ± 125 | 1.2 | 2627.5 ± 182 | 7275 ± 625 | 2.8 | 3.4 ± 0.2 |
| 8 | 30.6 ± 8.2 | 17.6 ± 0.45 | 0.6 | 16.7 ± 3.2 | 15.3 ± 1.4 | 0.9 | 2.4 ± 0.1 |
| 9 | 4.9 ± 0.8 | 6.9 ± 0.4 | 1.4 | 1.9 ± 0.1 | 6.15 ± 0.8 | 3.2 | 0.5 ± 0.1 |
| 10 | 42.95 ± 4.7 | 94.4 ± 2.7 | 2.2 | 18.3 ± 1.9 | 21 ± 1.5 | 1.2 | 1.4 ± 0.2 |
| 11 | 66.6 ± 8.9 | 260 ± 43 | 3.9 | 25.85 ± 1.3 | 31.35 ± 4 | 1.2 | 0.8 ± 0.1 |
| 12 | 1.9 ± 0.9 | 0.9 ± 0.1 | 0.5 | 0.5 ± 0.02 | 1.8 ± 0.2 | 3.6 | 1.1 ± 0.1 |
| 13 | 69 ± 1.7 | 413.5 ± 21.6 | 6 | 32.6 ± 4.65 | 31.2 ± 6.5 | 1.0 | 1.2 ± 0.1 |
| 14 | 101.5 ± 3.95 | 89.3 ± 3.9 | 0.9 | 34 ± 3 | 67 ± 5.4 | 1.9 | 1.5 ± 0.2 |
| 15 | 488 ± 35 | 551.5 ± 53.3 | 1.1 | 719 ± 39 | 1047.5 ± 120 | 1.5 | 0.9 ± 0.1 |
| 16 | 2933 ± 219 | 4733 ± 437 | 1.6 | 2360 ± 133 | 1450 ± 104.1 | 0.6 | 1.8 ± 0.1 |
Data taken from ref (3).
Cr for tubulin assembly in the GAB buffer.
The compounds were evaluated in A2780 ovarian tumor cells and their resistant A2780AD counterparts (Table 1). As previously shown,15 dictyostatin is active in this P-gp-overexpressing resistant cell line. All of the compounds tested, including those incorporating hydrophobic side chains, were found to be able to overcome P-gp-mediated resistance. The finding that all of the compounds show resistance indices between 0.5 and 3.9 (for 11) indicates that they are not substrates for this membrane drug efflux pump. Evaluation against HeLa-S3 and HeLa-βIII cervical tumor cells, the latter resistant to paclitaxel through overexpression of the βIII isotype of tubulin, also showed that all of the compounds in the library were equally active against these two cell lines (Table 1).
Finally, the compounds were assayed in paclitaxel-resistant PTX-10 and PTX-22 ovarian cancer cells harboring β-tubulin variants F272V and A366T, respectively, and epothilone-resistant A8 cells containing the β-tubulin variant T276I (Table S1). Most of the compounds were not affected by the F272V mutation, as shown by much lower R/S indices than that of paclitaxel (R/S = 37), although high values were obtained for 5, 8, 10, 11, 13, and 14. By contrast, only 11 and 14 showed R/S indices 3 times lower than that of paclitaxel in cells expressing A366T β-tubulin. Furthermore, A8 cells harboring T276I β-tubulin did not show resistance to any of the compounds tested. As anticipated from earlier work,12 the synthetic hybrid 12 of dictyostatin with discodermolide was generally more potent (low or sub-nanomolar) in the cell lines tested than either natural product itself.
Previous studies had indicated that although discodermolide and paclitaxel share the same binding site on β-tubulin, their combination is synergistic in tumor cell lines16−18 and in xenograft tumor-implanted animals.19 All of the compounds tested, with the exception of 10, were shown to act synergistically with paclitaxel (Table S2), as previously observed for discodermolide16,17,20 and two other analogues.18 Significantly, when peloruside A, an MSA targeting a different binding site on β-tubulin,21 was used in the same cell line in place of paclitaxel, the synergy was only observed at low ligand concentrations, and none was observed with 12 (Table S2).
Morphological Effects of the Compounds on the Cell Cytoskeleton
The effect of the compounds on the cell cytoskeleton was evaluated in human lung carcinoma A549 cells at different ligand concentrations (Figure 2). The immunofluorescence experiments show that the analogues potently induce the emergence of abnormal MT bundles, as for the parent compounds dictyostatin and discodermolide, and also for paclitaxel, but with some differences in the morphology. In the case of taxanes, the bundles are long and thin, whereas in the case of the compounds tested here, they are shorter and thicker. Nonetheless, all compounds induced a somewhat parallel alignment of MTs (straighter MTs) in the absence of MT bundles, in contrast to the organized MT network that was observed in the untreated (DMSO) cells. Dictyostatin and 12 were especially potent in inducing MT bundles, already apparent at concentrations of ∼50 nM, as well as discodermolide, 6, and 9, for which this effect was visible at concentrations similar to those of paclitaxel (100 nM). Compounds 4, 5, and 8 required concentrations of ∼250 nM, whereas the other compounds assayed (e.g., 11, 13, 14, and 15) were significantly less active, requiring μM concentrations to induce the MT bundles.
Figure 2.

Effect of the compounds on the MT network and the nuclear morphology of A549 cells. Effect of dictyostatin analogues 4, 5, 6, 8, 9, 10, and 11 and hybrid molecules 12, 13, 14, and 15 on the MT network, the mitotic spindle, and the nuclear morphology of A549 cells in comparison with untreated cells (DMSO), cells treated with paclitaxel, and the reference parent compounds dictyostatin and discodermolide. Cells were incubated for 24 h in the presence of DMSO (A, A′), paclitaxel 100 nM (B, B′), dictyostatin 50 nM (C, C′), discodermolide 100 nM (D, D′), 4 250 nM (E, E′), 5 200 nM (F, F′), 6 100 nM (G, G′), 8 200 nM (H, H′), 9 100 nM (I, I′), 10 800 nM (J, J′), 11 2 μM (K, K′), 12 60 nM (L, L′), 13 1.8 μM (M, M′), 14 6 μM (N, N′), and 15 8 μM (O, O′). MTs (A, B, C, D, E, F, G, H, I, J, K, L, M, N, and O) are labeled with the DM1A antibody against α-tubulin, whereas DNA (A′, B′, C′, D′, E′, F′, G′, H′, I′, J′, K′, L′, M′, N′, and O′) is marked with Hoechst 33342. The insets (A, A′, B, B′, C, C′, the latest representative for all ligands examined) show the mitotic spindles in the same preparations. The bar at O′ represents 10 μm, with all panels showing the same magnification.
Another morphological difference observed was the presence of a large number of micronucleated cells, multipolar mitotic spindles, and multiple asters (Figure 2).
Affinity of the Compounds for the Taxane-Binding Site
The binding of the compounds to the paclitaxel site in the MTs was tested using the displacement of the bona fide fluorescent paclitaxel analogue Flutax-24,5 (Table 2). Because 4, 6, 9, and 12 completely displaced Flutax-2 at equimolar concentrations, we also carried out a direct competition experiment with epothilone B (binding affinity = 7.5 × 108 M–1 at 35 °C) (Table 2) to measure their high binding affinities more accurately.22
Table 2. Affinity Constants (Ka) of the Compounds for Binding to MTs and Effect of Single-Point Ligand Changes on the Binding Affinity of the Ligands for MTs.
|
Ka (107 M–1) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| ligand | 26 °C | 27 °C | 30 °C | 32 °C | 35 °C | 37 °C | 40 °C | 42 °C | ΔG 35 °C (kJ mol–1) |
| paclitaxel3 | 2.64 ± 0.2 | 2.19 ± 0.05 | 1.83 ± 0.1 | 1.81 ± 0.2 | 1.43 ± 0.2 | 1.07 ± 0.1 | 0.96 ± 0.1 | 0.94 ± 0.2 | –42.2 ± 0.4 |
| docetaxel3 | 6.95 ± 0.4 | 6.57 ± 0.5 | 5.42 ± 0.4 | 4.89 ± 0.4 | 3.93 ± 0.3 | 3.09 ± 0.2 | 2.89 ± 0.2 | 2.38 ± 0.1 | –44.8 ± 0.2 |
| dictyostatin3 | 35.5 ± 7 | 32.0 ± 5.5 | 24.8 ± 3 | 23.1 ± 3 | 19.2 ± 2.5 | 16.8 ± 2 | 14.1 ± 2 | 14.2 ± 2 | –48.8 ± 0.4 |
| 2 | 0.0051 ± 0.003 | 0.0050 ± 0.003 | 0.0047 ± 0.003 | 0.0048 ± 0.0035 | 0.0041 ± 0.0026 | 0.0035 ± 0.002 | 0.0037 ± 0.001 | 0.0035 ± 0.002 | –27.2 ± 1.2 |
| discodermolide3 | 872 ± 82 | 837 ± 77 | 958 ± 134 | 704 ± 95 | 555 ± 111 | 526 ± 72 | 547 ± 60 | 321 ± 73 | –57.4 ± 0.5 |
| 5 | 2.4 ± 1.2 | 2.5 ± 1.1 | 2.6 ± 1.2 | 2.4 ± 1.2 | 2.2 ± 0.4 | 2.2 ± 0.45 | 1.9 ± 0.25 | 1.87 ± 0.4 | –43.3 ± 0.4 |
| 7 | 0.06 ± 0.02 | 0.05 ± 0.02 | 0.06 ± 0.01 | 0.07 ± 0.01 | 0.06 ± 0.01 | 0.06 ± 0.02 | 0.04 ± 0.01 | 0.04 ± 0.2 | –34.4 ± 0.4 |
| 8 | 2.1 ± 0.4 | 1.9 ± 0.5 | 1.7 ± 0.3 | 1.7 ± 0.4 | 1.6 ± 0.2 | 1.4 ± 0.2 | 1.4 ± 0.2 | 1.2 ± 0.3 | –42.5 ± 0.3 |
| 10 | 0.62 ± 0.3 | 0.66 ± 0.3 | 0.57 ± 0.2 | 0.62 ± 0.25 | 0.54 ± 0.2 | 0.51 ± 0.2 | 0.49 ± 0.2 | 0.45 ± 0.2 | –39.7 ± 0.8 |
| 11 | 0.33 ± 0.05 | 0.36 ± 0.1 | 0.29 ± 0.09 | 0.27 ± 0.07 | 0.28 ± 0.05 | 0.25 ± 0.01 | 0.3 ± 0.02 | 0.23 ± 0.02 | –38.0 ± 0.4 |
| 13 | 1.13 ± 0.3 | 1.12 ± 0.3 | 0.98 ± 0.2 | 0.95 ± 0.15 | 0.73 ± 0.1 | 0.71 ± 0.1 | 0.56 ± 0.1 | 0.58 ± 0.08 | –40.4 ± 0.3 |
| 14 | 0.07 ± 0.03 | 0.055 ± 0.02 | 0.043 ± 0.01 | 0.04 ± 0.006 | 0.033 ± 0.005 | 0.025 ± 0.004 | 0.029 ± 0.004 | 0.028 ± 0.01 | –32.5 ± 0.4 |
| 15 | 0.29 ± 0.1 | 0.29 ± 0.1 | 0.3 ± 0.1 | 0.33 ± 0.07 | 0.32 ± 0.1 | 0.33 ± 0.2 | 0.4 ± 0.2 | 0.28 ± 0.05 | –38.3 ± 0.7 |
| 16 | 0.32 ± 0.2 | 0.4 ± 0.2 | 0.4 ± 0.1 | 0.4 ± 0.1 | 0.35 ± 0.08 | 0.35 ± 0.1 | 0.3 ± 0.1 | 0.25 ± 0.07 | –38.6 ± 0.5 |
|
Ka (107 M–1) |
|||||
|---|---|---|---|---|---|
| ligand | 25 °C | 30 °C | 35 °C | 40 °C | ΔG 35 °C (kJ mol–1) |
| 4 | 119.3 ± 3 | 68.5 ± 8 | 53.8 ± 4.8 | 34 ± 1.3 | 51.5 ± 0.2 |
| 6 | 88.1 ± 14 | 55 ± 3 | 32.6 ± 3.4 | 26.4 ± 2.5 | 50.2 ± 0.3 |
| 9 | 229.7 ± 22 | 175 ± 16.5 | 113.8 ± 3.25 | 63.4 ± 2 | 53.4 ± 0.1 |
| 12 | 106.4 ± 32 | 75.1 ± 4 | 65.2 ± 2.6 | 32.2 ± 1.5 | 51.9 ± 0.1 |
| change | description | ΔΔG 35 °C (kJ mol–1) |
|---|---|---|
| 1 → 4 | removal of C6 methyl | –2.7 |
| 1 → 5 | reduction of C10=C11 double bond | +5.5 |
| 12 → 13 | +11.5 | |
| 1 → 6 | reduction of C2=C3 double bond | –1.4 |
| 1 → 10 | removal of C16 methyl | +9.1 |
| 11 → 7 | +3.6 | |
| 1 → 11 | epimerization of C9 hydroxyl | +10.8 |
| 10 → 7 | +5.3 | |
| 15 → 16 | –0.3 | |
| 6 → 8 | reduction of C4=C5 double bond | +6.1 |
| 1 → 9 | methylation of C9 hydroxyl | –4.6 |
| 12 → 15 | reduction of C4=C5 double bond and hydroxylation at C5 | +13.6 |
| 12 → 14 | introduction of docetaxel side chain at C7 | +19.4 |
| 1 → 14 | esterification of C7 hydroxyl | +9.7 |
Effects of the Compounds on the MT Structure
The effect of the compounds on the MT structure was studied using small-angle X-ray scattering (SAXS), which allows the measurement of the average number of protofilaments of the induced MTs, as determined by the lateral contacts between adjacent tubulin dimers.23 The largest effect was exerted by dictyostatin and discodermolide, which induced 14.5 ± 0.3 protofilament MTs, whereas the number of protofilaments in the presence of analogues 5, 6, and 11 (13.7 ± 0.1, 13.5 ± 0.3, and 13.2 ± 0.5, respectively) was similar to that observed in the absence of drug (13.6 protofilaments).24 This number was even smaller in the presence of dictyostatin–discodermolide hybrids 12, 14, and 15 (13.2 ± 1, 13.1 ± 0.2, and 12.9 ± 0.3, respectively).
Crystal Structure of the T2R-TTL–Dictyostatin Complex
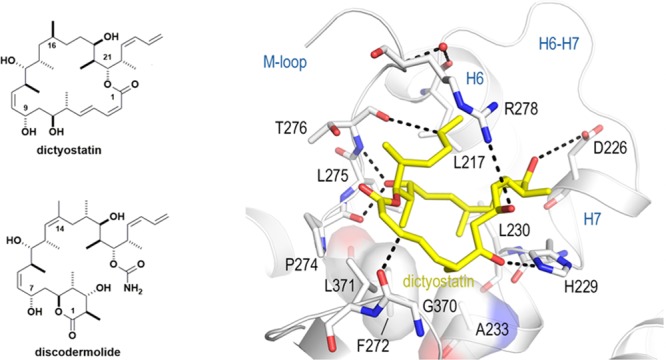
The X-ray crystal structure of the dictyostatin-bound tubulin was determined to 2.3 Å resolution and is shown in Figure 3. As expected, dictyostatin occupies the taxane-binding site on β-tubulin, and in agreement with previous proposals,25,26 its C19-hydroxyl forms two hydrogen bonds with both the main chain carbonyl of Pro274 and the main chain amide of Thr276. This central anchoring point is equivalent to that of the oxetane oxygen in paclitaxel27 and that of the carbonyl C1–O in epothilone A.28 Two additional hydrogen bonds are formed by the hydroxyls at C7 and C13 in dictyostatin to the side chains of His229 and Asp226, respectively. The interaction at C13 of dictyostatin is equivalent to the C7–OH–Asp226 interaction observed in the tubulin–epothilone A complex.28 The methyl substituent of the Thr276 side chain is in hydrophobic contact with the C22 methyl group of dictyostatin, whereas the hydroxyl substituent on the amino acid residue is not involved in the recognition of the ligand. This observation satisfactorily accounts for the resilience of dictyostatin to the T276I mutation, which severely compromises binding of the epothilone chemotype. Furthermore, the olefins at C3 and C25 form two weak C=O···HC= hydrogen bonds30 to the main chain carbonyls of Gly370 and Thr276, respectively. The dictyostatin–tubulin binding arrangement is completed by a cation−π interaction between the olefinic C23–C26 moiety and the guanidinium of Arg278 in the M-loop, and by van der Waals and hydrophobic interactions with Leu217, Leu230, Ala233, Phe272, Leu275, and Leu371 (Figure 3A,B), distinct from most of the epothilone contacts (Figure 3C). Notably, the F272V replacement does not affect the binding affinity of dictyostatin, although it has a strongly deleterious effect for some of its analogues, for example, 5, 8, 10, 11, 13, 14, and 16 (Table 2).
Figure 3.
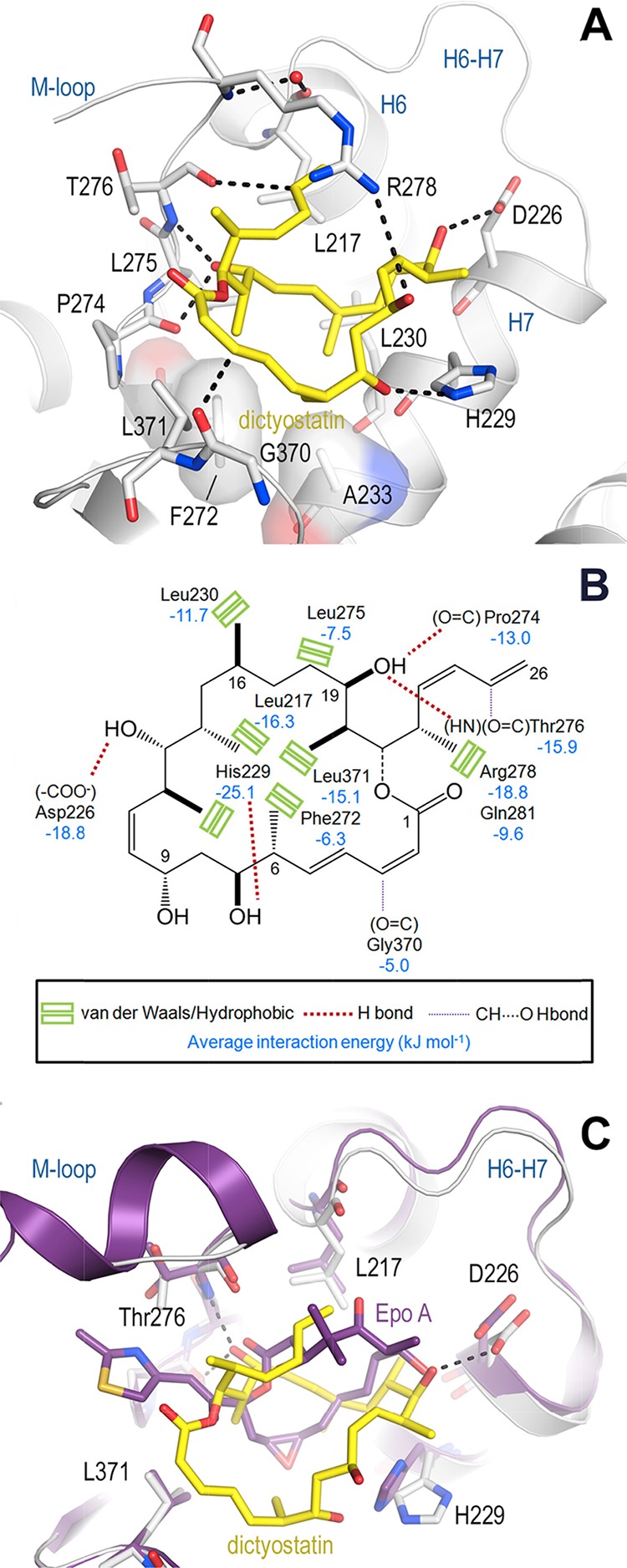
Structure of the tubulin–dictyostatin complex. (A) Close-up view of the tubulin–dictyostatin complex. Dictyostatin is shown as a yellow stick representation. β-Tubulin is displayed as a light gray ribbon. Key residues forming the interaction with the ligand are shown as a stick representation and are labeled. Hydrogen bonds are highlighted as dashed black lines. (B) Schematic 2D representation of the interactions responsible for the affinity of dictyostatin for the taxane-binding site in β-tubulin. Green rectangles represent hydrophobic contacts. Standard and C=O···HC= hydrogen bonds are shown as thick and thin dotted lines, respectively. Average interaction energies (kJ mol–1), as calculated from the MD simulations,29 are given for the most important interacting β-tubulin residues. (C) Superimposition of the tubulin–dictyostatin (yellow sticks, light gray ribbon) and tubulin–epothilone A (violet-purple sticks and ribbon) structures. Hydrogen bonds of the tubulin–dictyostatin complex are highlighted as dashed black lines.
Arguably, the most novel and striking feature of the T2R-TTL–dictyostatin complex reported here is the presence of two (=CH···O=) hydrogen bonds involving the olefinic carbons at C3 and C25 and the carbonyl oxygens of Gly370 and Thr276, respectively. This type of interaction is considered weaker in energy30 than the other hydrogen bonds described above but nonetheless appears to be significant for proper ligand orientation and stabilization within the binding site.
The tubulin-bound conformation of dictyostatin in this complex is remarkably similar to both the solid state (deposited in the Cambridge Structural Database with code Vintan0131) and solution32 structures of its closest MSA relative, discodermolide. Furthermore, this same conformation is also observed in an independently solved structure of discodermolide bound to the T2R-TTL complex (Figure S2). This is particularly noteworthy in view of the ∼30 times greater binding affinity of discodermolide for this β-tubulin site compared with that of its 22-membered macrolide relative, dictyostatin3 and the fact that the entropic contribution to binding is more favorable for discodermolide, which argues against a loss of conformational freedom for this compound upon binding.
Simulation of Dictyostatin Binding to a Reduced Representation of an MT
The importance of studying the binding of an MSA in the context of a realistic representation of an MT34 led us to expand the size of our previous tubulin model system beyond that of a simple dimer.35 A short MT stretch was built by taking two α1-β1-α2 segments from two vicinal protofilaments34 and incorporating one bound drug molecule (either dictyostatin or epothilone A were used here for comparative purposes) into each central β-tubulin subunit28 (see Experimental Procedures). The conformational stability of each of these assemblies, in which both M-loops are structurally ordered as α-helices at the beginning, was then examined by means of unrestrained MD simulations in an explicit aqueous solvent. By using this computational protocol, two distinct MSA-binding sites could be monitored in parallel: site 1, at the interface between two neighboring protofilaments and site 2, devoid of any protein lateral contacts but exposed to the bulk solvent (Figure S3). Because no major differences are found in the binding site between the straight and curved conformation,28 the large differences in the binding affinity that are consistently observed experimentally between the unassembled and assembled tubulin must reflect the relevance of the lateral contacts for the binding.
When the T2R-TTL–dictyostatin complex was simulated in the context of our reduced representation of a MT, the hydrogen bonds initially present between OG1(Thr276) and both NE2(Gln281) and NH1(Arg284) could not be stabilized by the bound ligand (Figure 4), unlike the situation observed in the epothilone A complex28,35 and (Figure S4). This is a direct consequence of the lack of pharmacophore interactions in dictyostatin equivalent to those involving the side-chain thiazole ring nitrogen and the C3–OH group attached to the macrocycle in epothilone A (Figure 3C). Instead, transient hydrogen bonds were observed between NE2(Gln281) and the lactone carbonyl oxygen of dictyostatin and also between NH1(Arg284) and the Asp90 carboxylate of the neighboring β1 subunit (Figure 4). The weak O(Thr276)–C25 hydrogen bond was the most affected in the absence of neighboring protofilaments (site 2). By contrast, the hydrogen bond between OD1/OD2(Asp226) and OH at C13 was reinforced in both sites for the full duration of the simulation and that established between NE2(His229) and the hydroxyl at C7 was maintained during most of the simulated trajectory. The structural rearrangements of the M-loops were more noticeable in the tubulin complex with dictyostatin relative to that with epothilone A, and also in site 2 relative to site 1, as expected from the larger solvent exposure of the former.
Figure 4.

Model of dictyostatin bound to a MT. (A) Superimposition of a representative structure of each major cluster of dictyostatin conformers (C atoms colored in orange) bound to β-tubulin in subunit B (C atoms colored in blue, site 1) and F (C atoms colored in yellow, site 2) along the MD simulation (100 ns) onto subunit B of the crystal structure of the dimeric tubulin in the complex with dictyostatin (C atoms colored in gray) reported in this work. (B) Time evolution of distances (Å) relevant to the pharmacophore along the MD simulation in the sites of B and F tubulin monomers.
Discussion
In 2001, discodermolide entered phase I clinical trials as an anticancer drug candidate, with Novartis.36 However, it did not advance into phase II because of unexpected lung toxicity.37 Until now, no other MSA belonging to either the discodermolide or dictyostatin chemotypes has entered clinical trials despite the high affinity of these compounds for tubulin,3 as well as their potent cytotoxicity and ability to overcome P-gp-mediated MDR.5 Nevertheless, these promising biological attributes have inspired us to further study the dictyostatin chemotype to identify the structural determinants responsible for their high binding affinities and their potent effects in inhibiting the growth of tumor cells.
SARs for the Dictyostatin Analogues
Out of all analogues of dictyostatin that we examined (the open-chain discodermolide notwithstanding), only 4, 6, 9, and 12 showed a greater affinity for tubulin than the parent compound (Table 2) indicating that, in contrast to paclitaxel and epothilones,4,5 dictyostatin is highly optimized for binding to MTs, and there is scope to prepare even more efficacious analogues based on the further evolution and optimization of this novel macrocyclic scaffold.
Our modeling studies provide a rationale for these findings and for some earlier SAR results.38 Thus, the dramatic loss of affinity found for the ring-contracted analogue 2 and similar 20-membered macrolides resulting from C21 to C19 translactonization14 is a direct consequence of the pronounced conformational changes relative to that in dictyostatin (Figure 5A) to the extent that the pharmacophore hotspots essential for a strong tubulin binding are missing altogether. Removal of the methyl group at C6, as in analogue 4,14 is likely to introduce some additional conformational flexibility in the macrocycle, leading to an improved interaction between the C1–C5 conjugated dienoate system and the Gly370 backbone, which is reflected in a gain in the free energy of binding of −2.7 kJ mol–1. By contrast, removal of the methyl group at C16, as in analogue 10, results in a weaker binding (ΔΔG +9.1 kJ mol–1), most likely due to the loss of favorable interactions with the hydrophobic pocket formed by the side chains of Leu230, Ala233, Phe272, and Leu275, as anticipated from the previous modeling work.39 Replacement of the hydroxyl group at C9 with a methoxy group (as found in the highest-affinity compound 9) probably shields the hydrogen bond between OH at C7 of dictyostatin and NE2(His229) from disrupting the interactions with water molecules (ΔΔG −4.6 kJ mol–1). However, epimerization at C9,15 as featured in both 7 and 11, disturbs the interaction involving OH at C7 and promotes conformational changes in the macrocycle that preclude an optimum fit into the binding site and translate into considerable losses of affinity (ΔΔG +5.3 and +10.8 kJ mol–1). Selective hydrogenation of the C2–C3 double bond in 6 slightly increases the binding affinity (ΔΔG −1.4 kJ mol–1), whereas hydrogenation of both C2–C3 and C4–C5 double bonds in 8(14) increases the flexibility of the macrocycle and leads to a lower affinity for tubulin (ΔΔG +6.1 kJ mol–1). By contrast, introduction of a trisubstituted double bond at C15–C16, together with additional methylation at C18 so as to incorporate some of the structural features present in discodermolide,40 yields the most cytotoxic hybrid compound 12, which possibly owes its higher affinity to increased van der Waals interactions with the side chains of Leu217 and Leu230. Nevertheless, other hybrids such as 13, 14, and 16, or more sophisticated constructs merging features from dictyostatin, discodermolide, and taxanes, such as 14 and 17, have considerably less affinity than the parent compound, essentially because the extra substituents are not properly oriented to improve the fit into the taxane-binding site of β-tubulin (Figure 5B).
Figure 5.
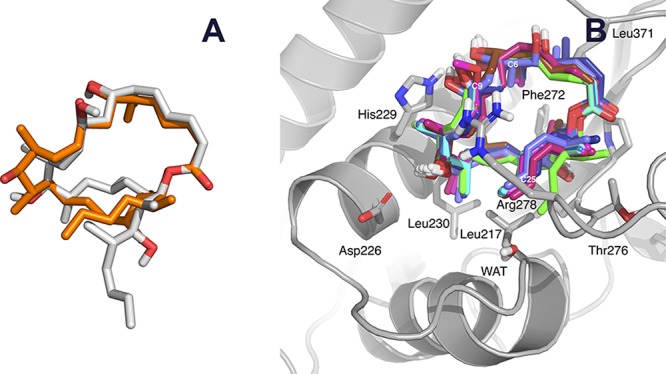
Comparison between the predicted structures of the analogues. (A) Superimposition of a representative structure of the major conformer of isodictyostatin (2, C atoms colored in gray), as simulated using MD for 100 ns in a box of TIP3P water molecules, onto the crystal structure conformation of dictyostatin (dictyostatin, C atoms colored in orange). (B) Molecular model showing the best docking poses of compounds 4–16 bound to the taxane-binding site of β-tubulin. The very weak binder isodictyostatin (2) and hybrid 14, which incorporates a fragment derived from docetaxel, are not shown.
Furthermore, this bound conformation also accounts for SAR results recently reported for some other active dictyostatin analogues41 designed as putative payloads for antibody–drug conjugates. By inspection of the tubulin-bound conformation, it is apparent that the C6 methyl group of dictyostatin is located deep in the binding pocket, such that a functionalized alkyl linker placed at this position will extend toward H1 or point inside of the binding pocket and occupy the space that is otherwise filled by the C13 side chain of paclitaxel (Figure S5A,B). By contrast, inclusion of a functionalized alkyl linker in place of the C12 methyl group will cause full exposure of this group to the solvent, and the flexible linker may perturb the interaction of the ligand by interfering with the elements surrounding the binding site (Figure S5C).
Comparison of Epothilone A and Dictyostatin Chemotypes
When the β1 subunits of [epothilone A:(α1-β1-α2)]2 and [dictyostatin:(α1-β1-α2)]2 complexes were best-fit superimposed (Figure S6), these two MSAs were revealed to share only two anchoring points that involved hydrogen bonds (Table S3), namely, the carboxylate of Asp226 (OH at C3 of epothilone A vs OH at C13 of dictyostatin) and the backbone amide nitrogen of Thr276 (O1 of epothilone A vs OH at C19 of dictyostatin). Notably, only the latter interaction is shared with paclitaxel (PDB entry 1JFF),27 the prototypical MSA that engages its oxetane oxygen in a hydrogen bond. In the case of dictyostatin, the strength of this anchoring point is augmented because the OH group at C19 simultaneously donates a hydrogen bond to O(Pro274). This common polar pharmacophore is complemented by distinct van der Waals interactions involving the side chains of His229 and a number of hydrophobic residues (Figure 3B).
A unique structural feature of dictyostatin relative to other MSAs is the presence of two conjugated diene regions that give rise to two weak C=O···HC= hydrogen bonds30 involving the olefinic carbons at C3 and C25 on the one side and the backbone carbonyl groups of Gly370 and Thr276 on the other side (Figure 3B). These distinctive interactions are maintained throughout the MD simulations. In addition, the alkene at C2–C3 in dictyostatin interacts with the hydrophobic side chain of Leu371, whereas the C22–C26 conjugated diene system can establish a stabilizing π–cation interaction with the guanidinium group of Arg278.
In summary, dictyostatin makes use of some common binding hotspots on the tubulin protein employed by epothilone A (Asp226, His229, Phe272, Thr276, and Arg278) in a way that is compatible with maintaining an α-helical conformation of the M-loop, even though this is not present in the crystal structure. Nonetheless, the Thr276 and Gln281 side chains of this structural element are found to be more disordered because of the lack of highly directional hydrogen-bonding interactions with the bound ligand.
Compounds of the Dictyostatin Chemotype are Effective in All Types of Taxane-Resistant Cells
From a potential therapeutic standpoint, another remarkable property of the synthetic analogues based on the dictyostatin chemotype is their pronounced ability to overcome all kinds of known resistance mechanisms related to tubulin-targeting anticancer drugs (i.e., P-gp overexpression, mutations in their binding site, and expression of the βIII tubulin isotype).
Significantly, the dictyostatin chemotype shows potent (low nanomolar) activity (Table 1) against P-gp-overexpressing resistant cells (A2780AD), independent of the binding affinity. This effect is probably not related to the competition between the target and the drug efflux pump, as is the case for other high-affinity tubulin modulators,5,42 but is likely due to a low affinity for the pump, which prefers compounds with high lipophilicity (e.g., uncharged and possessing aromatic rings).43
With respect to the resistant cell lines harboring mutations, the relative efficacy depends on the compound tested (Table S1). Although most of the analogues tested are not affected by the F272V mutation, as shown by R/S indices much lower than that for paclitaxel (R/S = 37), the high values obtained for a few of them, namely, 5, 8, 10, 11, 13, and 14, suggest that intraprotein or ligand–protein interactions involving this aromatic residue are highly sensitive to relatively subtle conformational changes in the macrocycle and/or particular variations in the side chain replacement. By contrast, the finding that only 11 and 14 show R/S indices around 4.0 (i.e., three times lower than that of paclitaxel) in cells expressing the A366T β-tubulin variant indicates a much smaller influence of this residue on the binding of this chemotype, which is in agreement with the present modeling studies. Likewise, the observation that A8 cells harboring T276I β-tubulin do not show resistance to any of the compounds tested concurs with the lack of direct interactions of dictyostatin and its analogues with the side chain hydroxyl of this amino acid residue, as was also found for paclitaxel. Finally, the finding that the side chain of Ser277 in the tubulin/dictyostatin complexes is directed away from the bound ligand (as is the case for the tubulin/epothilone complexes28) satisfactorily accounts for the fact that the resistant cells overexpressing the βIII isotype are equally sensitive to these MSA chemotypes.44
Synergy between Paclitaxel and Dictyostatin Chemotypes
The results obtained in this study (Table S2) are in accordance with the previous observation that although discodermolide and paclitaxel share the same tubulin-binding site, their combination is synergistic in both tumor cell cultures16−18 and tumor-implanted animals.19
Such a synergy has also been described between paclitaxel and laulimalide or peloruside A,45−47 which are MSAs targeting a different binding site. A crosstalk between the laulimalide/peloruside and taxane sites via the M-loop of β-tubulin has been advanced as an explanation for this synergy, which is based on comparing the conformation of the Gln294–Phe296 segment in the presence and absence of bound drugs because it adopts a helical, polymerization-competent, MT-stabilizing “on-state” or a turn-type III, “off-state”.21 However, this kind of mechanism would not be possible for compounds that share a common binding site.
However, this mechanism implies that a single tubulin molecule should be ligated by both ligands to experience this synergy, and this is not the case. It is important to realize that both the synergies between paclitaxel and dictyostatin and between paclitaxel-site and laulimalide-site ligands in the inhibition of cell proliferation occur at low nanomolar drug concentrations, far below those needed to bind tubulin stoichiometrically within the cell. At such low nanomolar IC50 values, the percentage of tubulin bound by either a paclitaxel-site or a laulimalide-site ligand is in the range of 2–5% of all of the available proteins.5 Considering that binding to the paclitaxel and the laulimalide sites is independent (binding of laulimalide does not significantly modify the binding affinity of the taxane-site ligand48), these figures imply that the probability that a single tubulin protein is bound by both taxane-site and laulimalide-site ligands is between 0.04 and 0.25%. This makes it unlikely that an individual tubulin protein has both taxane and laulimalide molecules simultaneously bound to it. Thus, both in the taxane-site/laulimalide site and in the dictyostatin and taxane chemotypes, the observed synergy should be attributed to the interdimer effects caused by cooperativity of their differential effects when bound at different tubulins of the same MT lattice as opposed to an intradimer effect.
Effects on the MT Structure
In contrast to what would be expected from consideration of an earlier model for a binding pose for discodermolide25 far from the M-loop, a significant effect of both dictyostatin and discodermolide was observed here on the MT protofilament number. Because these two MSAs induce 14.5 ± 0.1 protofilament MTs, they must exert a strong influence on the interprotofilament lateral interactions that stabilize the MTs. Interestingly, this influence has the opposite effect to that resulting from paclitaxel binding, which reduces this number.24 Reduction in the protofilament number arises from a smaller interprotofilament angle and vice versa. The fact that these two opposite effects can take place in different β-tubulin subunits within the same MT provides a possible structural explanation for the synergistic effect observed.
Intriguingly, the number of protofilaments does not change in the MTs stabilized by epothilone A, an MSA for which the M-loop appears structurally defined as an α-helix in the X-ray tubulin–ligand complexes.21,28 This M-loop conformation, which is also observed in the complex with zampanolide,28 correlates with the conformation observed in the MTs,34 despite the fact that the (α1-β1:α2-β2) tetramer in the T2R-TTL–ligand complexes is bent and most of the M-loop is solvent exposed. The interprotofilament lateral interactions are present, however, in our modeled [dictyostatin/(α1-β1-α2)]2 and [epothilone A/(α1-β1-α2)]2 complexes, and some of the features observed can be important to account for the stabilization of the lateral contacts brought about by drug binding beyond the well-established role of the side chain of Tyr283 in the M-loop:34 first, the stabilizing role of the water molecule that bridges a hydrogen-bonding interaction between the main chain NH(Arg278) at the plus end of the M-loop’s helical dipole and the backbone O(Leu217); second, a hydrogen bond between O(Tyr283) at the minus end of the M-loop helix and NH(Arg88) in the neighboring β-tubulin subunit; third, another hydrogen bond between O(Gln282) in the M-loop and NH(Ala57) in the 56–59 twisted β-hairpin of the neighboring β-tubulin subunit; and finally, NH of Arg284 at the minus end of the M-loop helix is engaged in a hydrogen bond with O(Ser280) within the helix, whereas its carbonyl oxygen is exposed to the solvent and its guanidinium side chain is extended to allow the establishment of multiple electrostatic interactions with the carboxamide oxygen of Gln294, the hydroxyl of Thr276, and the carboxylate of Glu290.
Altogether, we believe that the network of interactions just described is sensitive to the presence of a bound ligand in the taxane-binding site in such a way that the spatial relation between the M-loop (R278GSQQY283) in one β-tubulin subunit and the twisted β-hairpin (Y53NEAA̲G̲NKYV62) of the β-subunit in the neighboring protofilaments changes slightly but sufficiently to act as a wedge that can either increase or decrease the interprotofilament angle and thereby lead to MTs with a higher or lower number of protofilaments. An important exception is epothilone A that binds to MTs without affecting their overall structure most likely because its rather unique set of interactions gives rise to stabilization of the side chains of Thr276 and Gln281. Additional work is in progress to provide further support for this hypothesis.
Experimental Procedures
Protein and Ligands
Purified calf-brain tubulin and chemicals were obtained as previously described.49 Stabilized, moderately cross-linked MTs were prepared as reported earlier.50 The recombinant expression and purification of the stathmin-like domain of RB3 and chicken TTL in bacteria, and the reconstitution of the T2R-TTL complex was performed as described previously.28
Peloruside A was kindly provided by Dr. Peter T. Northcote (Victoria University of Wellington), Flutax-2 was kindly provided by Dr. Wei-Shuo Fang (Institute Materia Medica Beijing), and epothilone B was kindly provided by Prof. Karl-Heinz Altmann (ETH, Zürich). Paclitaxel was kindly provided by the National Cancer Institute (Bethesda, USA).
Dictyostatin (1), isodictyostatin (2), discodermolide (3), 6-desmethyldictyostatin (4), 10,11-dihydrodictyostatin (5), 2,3-dihydrodictyostatin (6), 16-desmethyl-9-epi-dictyostatin (7), 2,3,4,5-tetrahydrodictyostatin (8), 9-methoxydictyostatin (9), 16-desmethyldictyostatin (10), 9-epi-dictyostatin (11), and the discodermolide/dictyostatin hybrids (12–15) were synthesized as previously described: dictyostatin,51 (2) (A. Meyer, Ph.D. thesis, University of Cambridge, 2005), discodermolide,52 (4, 6, 8),14 (5),13 (7, 9, 10, 11),10 (12),12 (13),53 (14),54 and (15, 16).11
The ring-contracted analogue 2 was separated from residual dictyostatin before each experiment using a depletion method. A sample of 2 in the desired buffer was incubated for 30 min at 25 °C with a 5% molar ratio of stabilized MTs. The stabilized MTs were then removed by pelleting in a TLA 100.2 rotor at 50 000 rpm for 20 min in an Optima TLX ultracentrifuge.
Biochemical Characterization
The binding constants of the compounds to stabilized MTs were measured as previously described.5 The Cr of the tubulin assembly in the presence of compounds in the GAB buffer (3.4 M glycerol, 10 mM sodium phosphate (NaPi), 1 mM EGTA, 6 mM MgCl2, and 1 mM GTP, pH 6.7) was measured as reported earlier.3
Crystallization, Data Collection, and Structure Solution
Crystals of T2R-TTL were grown as previously described,28,55 except that tubulin was not subjected to a cycle of polymerization/depolymerization before complex assembly and crystallization. Crystals were soaked overnight in a reservoir solution (10% PEG 4K, 16% glycerol, 30 mM CaCl2, 30 mM MgCl2, 0.1 M MES/imidazole pH 6.7, 1 mM AmPPcP, 10 mM DTT, and 0.1 mM GDP) supplemented with 1 mM dictyostatin and were collected directly from the drop and flash-cooled in liquid nitrogen. Data collection at beamline X06DA at the Swiss Light Source (Paul Scherrer Institut, Villigen, Switzerland) and data processing and structure solution were performed as previously described.55 Details of the data collection and refinement statistics are given in Table S4.
The chains in the T2R-TTL–dictyostatin complex are defined as follows: chain A, α1-tubulin; chain B, β1-tubulin; chain C, α2-tubulin; chain D, β2-tubulin; chain E, RB3; and chain F, TTL. Chain D was used throughout for the structural analysis and figure preparation. Structure visualization, molecular editing, and figure preparation were performed with PyMOL (the PyMOL molecular graphics system, version 1.5.0.5. Schrödinger, LLC). The atomic coordinates and structure factors have been deposited in the Protein Data Bank (www.rcsb.org) under accession code 5MF4 (T2R-TTL–dictyostatin).
In Silico Model Building and Molecular Simulations
Our reduced representation of an MT for simulation purposes consisted of the subunits α1:β1:α2 from one protofilament together with the closely interacting α1′:β1′:α2′ subunits from a neighboring protofilament (Figure S3). Details of the construction of the macromolecular ensemble are described in Supporting Information.
X-Ray Scattering Measurements
Ligand-induced MTs in the presence of the desired ligands were prepared and their SAXS profiles were collected on a Bruker Nanostar system as described.23
Cell Biology
Cytotoxicity evaluation was performed on A2780,56 P-gp-overexpressing A2780AD,57 1A9, PTX10, PTX22, and A8 (A2780-derived)58 human ovarian carcinoma cell lines, as well as HeLa and βIII-transfected HeLa cervical cell lines59,60 using a modified MTT assay.61 Synergy between drugs was evaluated using the combination index (CI), as previously defined,62,63 which indicates additivity, antagonism, and synergy for values of 1, >1, and <1, respectively. Indirect immunofluorescence was performed in A549 cells, as previously described.64 The cell cycle analysis was performed as previously reported.3
Acknowledgments
We thank Peter T. Northcote for peloruside A, W.-S. Fang for Flutax-2, K. H. Altmann for epothilone D, Dr. Paraskevi Giannakakou (Weill Cornell Medical Center, New York) for the 1A9, PTX10, PTX22, and A8 cell lines, and Prof. Richard Ludueña (University of Texas) for the HeLa βIII-transfected cells. We thank Matadero INCOVA (Segovia) for the calf brains for tubulin purification. This work was supported in part by grants BIO2013-42984-R (J.F.D.) and SAF2012-39760-C02-02 (F.G.) from Ministerio de Economía y Competitividad, grant S2010/BMD-2457 BIPEDD2 from Comunidad Autónoma de Madrid (F.G. and J.F.D.), and the Swiss National Science Foundation grants 310030B_138659 and 31003A_166608 (M.O.S.). The authors acknowledge networking contribution by the COST Action CM1407 “Challenging organic syntheses inspired by nature—from natural products chemistry to drug discovery” and the COST action CM1470. I.P. thanks the EPSRC and AstraZeneca for funding, Dr. John Leonard (AstraZeneca) for useful discussions, Dr. Stuart Mickel (Novartis) for the provision of chemicals, and the EPSRC UK National Mass Spectrometry Facility at Swansea University for mass spectra.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.6b00317.
Results: Simulation of epothilone A binding to a reduced representation of a MT. Methods: In silico model building and molecular simulations. Tables: (S1) Cytotoxicity of the compounds in ovarian cell lines both sensitive (1A9) and resistant to MSA (PTX10, PTX22 and A8) because of amino acid replacements in β-tubulin, (S2) Values calculated for the Cl between paclitaxel or peloruside A and the ligands studied, (S3) Average binding energy per residue (kJ mol–-1) of dictyostatin and epothilone A with β1-tubulin along the MD simulation of the [MSA:(α1-β1-α2)]2 complexes. (S4) Data collection and refinement statistics of the dictyostatin crystal. Figures (S1) Effect of the ligands on the cell cycle of A2780 ovarian carcinoma cells. (S2) Comparison of the bound structures of dictyostatin and discodermolide. (S3) Reduced representation of a MT used for the simulation. (S4) Model of epothilone bound to a MT. (S5) Modeled effect of substitutions at C6 and C12 in the ligand, and Figure with the superposition of the modeled binding modes of dictyostatin and epothilone A (PDF)
Author Present Address
# Department of Biochemistry, University of Zurich, Zürich, Switzerland (K.B.).
The authors declare no competing financial interest.
Supplementary Material
References
- Diaz J. F.; Menendez M.; Andreu J. M. Thermodynamics of ligand-induced assembly of tubulin. Biochemistry 1993, 32, 10067–10077. 10.1021/bi00089a023. [DOI] [PubMed] [Google Scholar]
- Field J. J.; Díaz J. F.; Miller J. H. The Binding Sites of Microtubule-Stabilizing Agents. Chem. Biol. 2013, 20, 301–315. 10.1016/j.chembiol.2013.01.014. [DOI] [PubMed] [Google Scholar]
- Buey R. M.; Barasoain I.; Jackson E.; Meyer A.; Giannakakou P.; Paterson I.; Mooberry S.; Andreu J. M.; Diaz J. F. Microtubule interactions with chemically diverse stabilizing agents: Thermodynamics of binding to the paclitaxel site predicts cytotoxicity. Chem. Biol. 2005, 12, 1269–1279. 10.1016/j.chembiol.2005.09.010. [DOI] [PubMed] [Google Scholar]
- Buey R. M.; Díaz J. F.; Andreu J. M.; O’Brate A.; Giannakakou P.; Nicolaou K. C.; Sasmal P. K.; Ritzén A.; Namoto K. Interaction of Epothilone Analogs with the Paclitaxel Binding Site; Relationship between Binding Affinity, Microtubule Stabilization, and Cytotoxicity. Chem. Biol. 2004, 11, 225–236. 10.1016/s1074-5521(04)00025-0. [DOI] [PubMed] [Google Scholar]
- Matesanz R.; Barasoain I.; Yang C.-G.; Wang L.; Li X.; de Inés C.; Coderch C.; Gago F.; Jiménez-Barbero J.; Andreu J. M.; Fang W.-S.; Díaz J. F. Optimization of taxane binding to microtubules: Binding affinity dissection and incremental construction of a high-affinity analog of paclitaxel. Chem. Biol. 2008, 15, 573–585. 10.1016/j.chembiol.2008.05.008. [DOI] [PubMed] [Google Scholar]
- Isbrucker R. A.; Cummins J.; Pomponi S. A.; Longley R. E.; Wright A. E. Tubulin polymerizing activity of dictyostatin-1, a polyketide of marine sponge origin. Biochem. Pharmacol. 2003, 66, 75–82. 10.1016/s0006-2952(03)00192-8. [DOI] [PubMed] [Google Scholar]
- Florence G. J.; Gardner N. M.; Paterson I. Development of practical syntheses of the marine anticancer agents discodermolide and dictyostatin. Nat. Prod. Rep. 2008, 25, 342–375. 10.1039/b705661n. [DOI] [PubMed] [Google Scholar]
- Larsen E. M.; Wilson M. R.; Taylor R. E. Conformation–activity relationships of polyketide natural products. Nat. Prod. Rep. 2015, 32, 1183–1206. 10.1039/C5NP00014A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson I.; Gardner N. M.; Naylor G. J. Total Synthesis of Novel Dictyostatin Analogs and Hybrids as Microtubule-Stabilizing Anticancer Agents. Pure Appl. Chem. 2009, 81, 169–180. 10.1351/pac-con-08-09-17. [DOI] [Google Scholar]
- Paterson I.; Gardner N. M.; Poullennec K. G.; Wright A. E. Synthesis and biological evaluation of novel analogues of dictyostatin. Bioorg. Med. Chem. Lett. 2007, 17, 2443–2447. 10.1016/j.bmcl.2007.02.031. [DOI] [PubMed] [Google Scholar]
- Paterson I.; Gardner N. M. Design, synthesis and biological evaluation of a macrocyclic discodermolide/dictyostatin hybrid. Chem. Commun. 2007, 49–51. 10.1039/b615122a. [DOI] [PubMed] [Google Scholar]
- Paterson I.; Naylor G. J.; Wright A. E. Total synthesis of a potent hybrid of the anticancer natural products dictyostatin and discodermolide. Chem. Commun. 2008, 4628–4630. 10.1039/b811575c. [DOI] [PubMed] [Google Scholar]
- Paterson I.; Gardner N. M.; Poullennec K. G.; Wright A. E. Synthesis and biological evaluation of 10,11-dihydrodictyostatin, a potent analogue of the marine anticancer agent dictyostatin. J. Nat. Prod. 2008, 71, 364–369. 10.1021/np070547s. [DOI] [PubMed] [Google Scholar]
- Paterson I.; Gardner N. M.; Guzmán E.; Wright A. E. Total synthesis and biological evaluation of novel C2–C6 region analogues of dictyostatin. Bioorg. Med. Chem. 2009, 17, 2282–2289. 10.1016/j.bmc.2008.10.084. [DOI] [PubMed] [Google Scholar]
- Zanato C.; Pignataro L.; Ambrosi A.; Hao Z.; Trigili C.; Díaz J. F.; Barasoain I.; Gennari C. Highly Stereoselective Total Synthesis of (+)-9-epi-Dictyostatin and (−)-12,13-Bis-epi-dictyostatin. Eur. J. Org. Chem. 2011, 2643–2661. 10.1002/ejoc.201100244. [DOI] [Google Scholar]
- Martello L. A.; McDaid H. M.; Regl D. L.; Yang C. P.; Meng D.; Pettus T. R.; Kaufman M. D.; Arimoto H.; Danishefsky S. J.; Smith A. B. III; Horwitz S. B. Taxol and discodermolide represent a synergistic drug combination in human carcinoma cell lines. Clin. Cancer Res. 2000, 6, 1978–1987. [PubMed] [Google Scholar]
- Honore S.; Kamath K.; Braguer D.; Horwitz S. B.; Wilson L.; Briand C.; Jordan M. A. Synergistic suppression of microtubule dynamics by discodermolide and paclitaxel in non-small cell lung carcinoma cells. Cancer Res. 2004, 64, 4957–4964. 10.1158/0008-5472.can-04-0693. [DOI] [PubMed] [Google Scholar]
- Vollmer L. L.; Jiménez M.; Camarco D. P.; Zhu W.; Daghestani H. N.; Balachandran R.; Reese C. E.; Lazo J. S.; Hukriede N. A.; Curran D. P.; Day B. W.; Vogt A. A Simplified Synthesis of Novel Dictyostatin Analogues with in Vitro Activity against Epothilone B–Resistant Cells and Antiangiogenic Activity in Zebrafish Embryos. Mol. Cancer Ther. 2011, 10, 994–1006. 10.1158/1535-7163.MCT-10-1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G. S.; Lopez-Barcons L.; Freeze B. S.; Smith A. B. III; Goldberg G. L.; Horwitz S. B.; McDaid H. M. Potentiation of taxol efficacy and by discodermolide in ovarian carcinoma xenograft-bearing mice. Clin. Cancer Res. 2006, 12, 298–304. 10.1158/1078-0432.CCR-05-0229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakakou P.; Fojo T. Discodermolide: Just another microtubule-stabilizing agent? No! A lesson in synergy. Clin. Cancer Res. 2000, 6, 1613–1615. [PubMed] [Google Scholar]
- Prota A. E.; Bargsten K.; Northcote P. T.; Marsh M.; Altmann K.-H.; Miller J. H.; Diaz J. F.; Steinmetz M. O. Structural basis of microtubule stabilization by laulimalide and peloruside A. Angew. Chem., Int. Ed. 2014, 53, 1621–1625. 10.1002/anie.201307749. [DOI] [PubMed] [Google Scholar]
- Díaz J. F.; Buey R. M.. Characterizing Ligand-Microtubule Binding by Competition Methods. In Methods in Molecular Medicine; Zhou J., Ed.; Humana Press Inc.: Totowa, NJ, 2007; Vol. 137, pp 245–260. [DOI] [PubMed] [Google Scholar]
- Matesanz R.; Rodríguez-Salarichs J.; Pera B.; Canales Á.; Andreu J. M.; Jiménez-Barbero J.; Bras W.; Nogales A.; Fang W.-S.; Díaz J. F. Modulation of microtubule interprotofilament interactions by modified taxanes. Biophys. J. 2011, 101, 2970–2980. 10.1016/j.bpj.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreu J. M.; Bordas J.; Diaz J. F.; García de Ancos J.; Gil R.; Medrano F. J.; Nogales E.; Pantos E.; Towns-Andrews E. Low resolution structure of microtubules in solution: Synchrotron X-ray scattering and electron microscopy of taxol-induced microtubules assembled from purified tubulin in comparison with glycerol and MAP-induced microtubules. J. Mol. Biol. 1992, 226, 169–184. 10.1016/0022-2836(92)90132-4. [DOI] [PubMed] [Google Scholar]
- Canales A.; Matesanz R.; Gardner N. M.; Andreu J. M.; Paterson I.; Diaz J. F.; Jiménez-Barbero J. The Bound Conformation of Microtubule-Stabilizing Agents: NMR Insights into the Bioactive 3D Structure of Discodermolide and Dictyostatin. Chem.—Eur. J. 2008, 14, 7557–7569. 10.1002/chem.200800039. [DOI] [PubMed] [Google Scholar]
- Jogalekar A. S.; Damodaran K.; Kriel F. H.; Jung W.-H.; Alcaraz A. A.; Zhong S.; Curran D. P.; Snyder J. P. Dictyostatin Flexibility Bridges Conformations in Solution and in the β-Tubulin Taxane Binding Site. J. Am. Chem. Soc. 2011, 133, 2427–2436. 10.1021/ja1023817. [DOI] [PubMed] [Google Scholar]
- Löwe J.; Li H.; Downing K. H.; Nogales E. Refined structure of αβ-tubulin at 3.5 Å resolution. J. Mol. Biol. 2001, 313, 1045–1057. 10.1006/jmbi.2001.5077. [DOI] [PubMed] [Google Scholar]
- Prota A. E.; Bargsten K.; Zurwerra D.; Field J. J.; Diaz J. F.; Altmann K.-H.; Steinmetz M. O. Molecular mechanism of action of microtubule-stabilizing anticancer agents. Science 2013, 339, 587–590. 10.1126/science.1230582. [DOI] [PubMed] [Google Scholar]
- Klett J.; Núñez-Salgado A.; Santos H. G. D.; Cortés-Cabrera Á.; Perona A.; Gil-Redondo R.; Abia D.; Gago F.; Morreale A. MM-ISMSA: An Ultrafast and Accurate Scoring Function for Protein–Protein Docking. J. Chem. Theory Comput. 2012, 8, 3395–3408. 10.1021/ct300497z. [DOI] [PubMed] [Google Scholar]
- Desiraju G. R. Hydrogen Bridges in Crystal Engineering: Interactions without Borders. Acc. Chem. Res. 2002, 35, 565–573. 10.1021/ar010054t. [DOI] [PubMed] [Google Scholar]
- Smith A. B. III; Kaufman M. D.; Beauchamp T. J.; LaMarche M. J.; Arimoto H. Gram-scale synthesis of (+)-discodermolide. Org. Lett. 1999, 1, 1823–1826. 10.1021/ol9910870. [DOI] [PubMed] [Google Scholar]
- Smith A. B. III; LaMarche M. J.; Falcone-Hindley M. Solution structure of (+)-discodermolide. Org. Lett. 2001, 3, 695–698. 10.1021/ol006967p. [DOI] [PubMed] [Google Scholar]
- Alushin G. M.; Lander G. C.; Kellogg E. H.; Zhang R.; Baker D.; Nogales E. High-Resolution Microtubule Structures Reveal the Structural Transitions in αβ-Tubulin upon GTP Hydrolysis. Cell 2014, 157, 1117–1129. 10.1016/j.cell.2014.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canales A.; Nieto L.; Rodríguez-Salarichs J.; Sanchez-Murcia P. A.; Coderch C.; Cortés-Cabrera A.; Paterson I.; Carlomagno T.; Gago F.; Andreu J. M.; Altmann K.-H.; Jiménez-Barbero J.; Diaz J. F. Molecular recognition of epothilones by microtubules and tubulin dimers revealed by biochemical and NMR approaches. ACS Chem. Biol. 2014, 9, 1033–1043. 10.1021/cb400673h. [DOI] [PubMed] [Google Scholar]
- Mita A.; Lockhart A. C.; Chen T.-L.; Bochinski K.; Curtright J.; Cooper W.; Hammond L.; Rothenberg M.; Rowinsky E.; Sharma S. A phase I pharmacokinetic (PK) trial of XAA296A (discodermolide) administered every 3 wks to adult patients with advanced solid malignancies. J. Clin. Oncol. 2004, 22, 2025. 10.1200/jco.2004.22.90140.2025. [DOI] [Google Scholar]
- Dumontet C.; Jordan M. A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discovery 2010, 9, 790–803. 10.1038/nrd3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung W.-H.; Harrison C.; Shin Y.; Fournier J.-H.; Balachandran R.; Raccor B. S.; Sikorski R. P.; Vogt A.; Curran D. P.; Day B. W. Total Synthesis and Biological Evaluation of C16 Analogs of (−)-Dictyostatin. J. Med. Chem. 2007, 50, 2951–2966. 10.1021/jm061385k. [DOI] [PubMed] [Google Scholar]
- Shin Y.; Fournier J.-H.; Balachandran R.; Madiraju C.; Raccor B. S.; Zhu G.; Edler M. C.; Hamel E.; Day B. W.; Curran D. P. Synthesis and Biological Evaluation of (−)-16-Normethyldictyostatin: A Potent Analogue of (−)-Dictyostatin. Org. Lett. 2005, 7, 2873–2876. 10.1021/ol050808u. [DOI] [PubMed] [Google Scholar]
- Gunasekera S. P.; Gunasekera M.; Longley R. E.; Schulte G. K. Discodermolide: A new bioactive polyhydroxylated lactone from the marine sponge Discodermia dissoluta. J. Org. Chem. 1990, 55, 4912–4915. 10.1021/jo00303a029. [DOI] [Google Scholar]
- Ho S.; Sackett D. L.; Leighton J. L. A “Methyl Extension” Strategy for Polyketide Natural Product Linker Site Validation and Its Application to Dictyostatin. J. Am. Chem. Soc. 2015, 137, 14047–14050. 10.1021/jacs.5b09869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Díez M.; Guillén-Navarro M. J.; Pera B.; Bouchet B. P.; Martinez-Leal J. F.; Barasoain I.; Cuevas C.; Andreu J. M.; García-Fernández L. F.; Diaz J. F.; Avilés P.; Galmarini C. M. PM060184, a new tubulin binding agent with potent antitumor activity including P-glycoprotein over-expressing tumors. Biochem. Pharmacol. 2014, 88, 291–302. 10.1016/j.bcp.2014.01.026. [DOI] [PubMed] [Google Scholar]
- Nobili S.; Landini I.; Mazzei T.; Mini E. Overcoming tumor multidrug resistance using drugs able to evade P-glycoprotein or to exploit its expression. Med. Res. Rev. 2012, 32, 1220–1262. 10.1002/med.20239. [DOI] [PubMed] [Google Scholar]
- Dumontet C.; Jordan M. A.; Lee F. F. Y. Ixabepilone: Targeting βIII-tubulin expression in taxane-resistant malignancies. Mol. Cancer Ther. 2009, 8, 17–25. 10.1158/1535-7163.MCT-08-0986. [DOI] [PubMed] [Google Scholar]
- Hamel E.; Day B. W.; Miller J. H.; Jung M. K.; Northcote P. T.; Ghosh A. K.; Curran D. P.; Cushman M.; Nicolaou K. C.; Paterson I.; Sorensen E. J. Synergistic effects of peloruside A and laulimalide with taxoid site drugs, but not with each other, on tubulin assembly. Mol. Pharmacol. 2006, 70, 1555–1564. 10.1124/mol.106.027847. [DOI] [PubMed] [Google Scholar]
- Wilmes A.; Bargh K.; Kelly C.; Northcote P. T.; Miller J. H. Peloruside A synergizes with other microtubule stabilizing agents in cultured cancer cell lines. Mol. Pharm. 2007, 4, 269–280. 10.1021/mp060101p. [DOI] [PubMed] [Google Scholar]
- Wilmes A.; O’Sullivan D.; Chan A.; Chandrahasen C.; Paterson I.; Northcote P. T.; La Flamme A. C.; Miller J. H.; Osullivan D. Synergistic interactions between peloruside A and other microtubule-stabilizing and destabilizing agents in cultured human ovarian carcinoma cells and murine T cells. Cancer Chemother. Pharmacol. 2011, 68, 117–126. 10.1007/s00280-010-1461-3. [DOI] [PubMed] [Google Scholar]
- Pryor D. E.; O’Brate A.; Bilcer G.; Diaz J. F.; Wang Y.; Wang Y.; Kabaki M.; Jung M. K.; Andreu J. M.; Ghosh A. K.; Giannakakou P.; Hamel E. The microtubule stabilizing agent laulimalide does not bind in the taxoid site, kills cells resistant to paclitaxel and epothilones, and may not require its epoxide moiety for activity. Biochemistry 2002, 41, 9109–9115. 10.1021/bi020211b. [DOI] [PubMed] [Google Scholar]
- Diaz J. F.; Andreu J. M. Assembly of purified GDP-tubulin into microtubules induced by taxol and taxotere: Reversibility, ligand stoichiometry, and competition. Biochemistry 1993, 32, 2747–2755. 10.1021/bi00062a003. [DOI] [PubMed] [Google Scholar]
- Díaz J. F.; Strobe R.; Engelborghs Y.; Souto A. A.; Andreu J. M. Molecular recognition of taxol by microtubules. Kinetics and thermodynamics of binding of fluorescent taxol derivatives to an exposed site. J. Biol. Chem. 2000, 275, 26265–26276. 10.1074/jbc.m003120200. [DOI] [PubMed] [Google Scholar]
- Paterson I.; Britton R.; Delgado O.; Meyer A.; Poullennec K. G. Total synthesis and configurational assignment of (−)-dictyostatin, a microtubule-stabilizing macrolide of marine sponge origin. Angew. Chem., Int. Ed. 2004, 43, 4629–4633. 10.1002/anie.200460589. [DOI] [PubMed] [Google Scholar]
- Paterson I.; Lyothier I. Total synthesis of (+)-discodermolide: An improved endgame exploiting a Still–Gennari-type olefination with a C1–C8 β-ketophosphonate fragment. Org. Lett. 2004, 6, 4933–4936. 10.1021/ol0478842. [DOI] [PubMed] [Google Scholar]
- Paterson I.; Naylor G. J.; Gardner N. M.; Guzmán E.; Wright A. E. Total Synthesis and Biological Evaluation of a Series of Macrocyclic Hybrids and Analogues of the Antimitotic Natural Products Dictyostatin, Discodermolide, and Taxol. Chem.—Asian J. 2011, 6, 459–473. 10.1002/asia.201000541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson I.; Naylor G. J.; Fujita T.; Guzman E.; Wright A. E. Total synthesis of a library of designed hybrids of the microtubule-stabilising anticancer agents taxol, discodermolide and dictyostatin. Chem. Commun. 2010, 46, 261–263. 10.1039/b921237j. [DOI] [PubMed] [Google Scholar]
- Prota A. E.; Magiera M. M.; Kuijpers M.; Bargsten K.; Frey D.; Wieser M.; Jaussi R.; Hoogenraad C. C.; Kammerer R. A.; Janke C.; Steinmetz M. O. Structural basis of tubulin tyrosination by tubulin tyrosine ligase. J. Cell Biol. 2013, 200, 259–270. 10.1083/jcb.201211017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louie K. G.; Behrens B. C.; Kinsella T. J.; Hamilton T. C.; Grotzinger K. R.; McKoy W. M.; Winker M. A.; Ozols R. F. Radiation survival parameters of antineoplastic drug-sensitive and -resistant human ovarian cancer cell lines and their modification by buthionine sulfoximine. Cancer Res. 1985, 45, 2110–2115. [PubMed] [Google Scholar]
- Kowalski R. J.; Giannakakou P.; Gunasekera S. P.; Longley R. E.; Day B. W.; Hamel E. The microtubule-stabilizing agent discodermolide competitively inhibits the binding of paclitaxel (taxol) to tubulin polymers, enhances tubulin nucleation reactions more potently than paclitaxel, and inhibits the growth of paclitaxel-resistant cells. Mol. Pharmacol. 1997, 52, 613–622. 10.1124/mol.52.4.613. [DOI] [PubMed] [Google Scholar]
- Giannakakou P.; Sackett D. L.; Kang Y.-K.; Zhan Z.; Buters J. T. M.; Fojo T.; Poruchynsky M. S. Paclitaxel-resistant human ovarian cancer cells have mutant β-tubulins that exhibit impaired paclitaxel-driven polymerization. J. Biol. Chem. 1997, 272, 17118–17125. 10.1074/jbc.272.27.17118. [DOI] [PubMed] [Google Scholar]
- Joe P. A.; Banerjee A.; Ludueña R. F. The roles of cys124 and ser239 in the functional properties of human βIII tubulin. Cell Motil. Cytoskeleton 2008, 65, 476–486. 10.1002/cm.20274. [DOI] [PubMed] [Google Scholar]
- Stengel C.; Newman S. P.; Leese M. P.; Potter B. V. L.; Reed M. J.; Purohit A. Class III β-tubulin expression and in vitro resistance to microtubule targeting agents. Br. J. Cancer 2010, 102, 316–324. 10.1038/sj.bjc.6605489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C.-G.; Barasoain I.; Li X.; Matesanz R.; Liu R.; Sharom F. J.; Yin D.-L.; Díaz J. F.; Fang W.-S. Overcoming Tumor Drug Resistence Mediated by P-Glycoprotein Overexpression with High Affinity Taxanes: A SAR Study of C-2 Modified 7-Acyl-10-Deacetil Cephalomannines. ChemMedChem 2007, 2, 691–701. 10.1002/cmdc.200700002. [DOI] [PubMed] [Google Scholar]
- Chou T.-C.; Talalay P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Berenbaum M. C. The expected effect of a combination of agents: The general solution. J. Theor. Biol. 1985, 114, 413–431. 10.1016/S0022-5193(85)80176-4. [DOI] [PubMed] [Google Scholar]
- de Ines C.; Leynadier D.; Barasoain I.; Peyrot V.; Garcia P.; Briand C.; Rener G. A.; Temple C. Jr. Inhibition of microtubules and cell cycle arrest by a new 1-deaza-7,8-dihydropteridine antitumor drug, CI 980, and by its chiral isomer, NSC 613863. Cancer Res. 1994, 54, 75–84. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.