

Formation of catalytically reactive metal oxide interfaces on the Pt3Ni(111) surface is revealed using operando surface analysis.
Abstract
The origin of the synergistic catalytic effect between metal catalysts and reducible oxides has been debated for decades. Clarification of this effect, namely, the strong metal-support interaction (SMSI), requires an understanding of the geometric and electronic structures of metal-metal oxide interfaces under operando conditions. We show that the inherent lattice mismatch of bimetallic materials selectively creates surface segregation of subsurface metal atoms. Interfacial metal-metal oxide nanostructures are then formed under chemical reaction environments at ambient pressure, which thus increases the catalytic activity for the CO oxidation reaction. Our in situ surface characterizations using ambient-pressure scanning tunneling microscopy and ambient-pressure x-ray photoelectron spectroscopy exhibit (i) a Pt-skin layer on the Pt-Ni alloyed surface under ultrahigh vacuum, (ii) selective Ni segregation followed by the formation of NiO1−x clusters under oxygen gas, and (iii) the coexistence of NiO1−x clusters on the Pt-skin during the CO oxidation reaction. The formation of interfacial Pt-NiO1−x nanostructures is responsible for a highly efficient step in the CO oxidation reaction. Density functional theory calculations of the Pt3Ni(111) surface demonstrate that a CO molecule adsorbed on an exposed Pt atom with an interfacial oxygen from a segregated NiO1−x cluster has a low surface energy barrier of 0.37 eV, compared with 0.86 eV for the Pt(111) surface.
INTRODUCTION
A long-standing question in heterogeneous catalysis is the exact role of the metal-metal oxide interface during catalytic reactions. In the decades since this synergistic catalytic effect [that is, the strong metal-support interaction (SMSI) effect] was discovered by Schwab (1), the cause of improved catalytic reaction efficiencies at catalyst interfaces has been continuously debated. Despite some controversial arguments, the concept of the SMSI effect has been used to explain many unusual catalytic behaviors at the metal-metal oxide interface. For example, low-temperature carbon monoxide (CO) oxidation over a few nanometer-sized gold particles deposited on a titanium dioxide (Au/TiO2) catalyst exhibits one important aspect of the metal-metal oxide interface along its perimeter region (2, 3). However, a comprehensive understanding of SMSI in heterogeneous catalysis is still lacking when explaining the role of the interfacial metal oxide nanostructure. A bimetallic platinum (Pt) alloy catalyst is an excellent platform to uncover the contentious role of the metal-metal oxide interface because the alloyed transition metal can coexist with the Pt surface layer in the form of an oxidized species on the bimetal surface during catalytic reactions.
Optimized tailoring of bimetallic catalysts composed of a transition metal alloyed with Pt is widely considered to be a promising way to overcome known problems preventing efficient energy conversion in modern industrial heterogeneous catalysis (4–7). Bimetallic materials have a unique surface structure because of their shifted “d-band center” and volcano plot, which are widely recognized as important properties for tuning catalytic activity, according to Sabatier’s principle (8). In addition, modification of the geometric and electronic structure of the bimetallic surface is a critical factor in controlling molecular behavior (5, 9). For example, a layer of Pt covering a bimetallic structure (Pt-skin) is known for facile charge transfer at its interface; in addition to this layer-structured surface playing a role in charge redistribution, it also protects against geometric deformation by leaching processes (8, 10). Thus, engineering the formation of a Pt-skin layer on a bimetallic surface is a smart strategy for designing high-performance heterogeneous catalysts (11, 12). Rational nanoscale control of platinum-nickel (Pt-Ni) bimetallic catalysts shows a highly enhanced catalytic activity for electrochemical and heterogeneous catalytic reactions, such as the oxygen reduction and CO oxidation reactions (13, 14). The unique surface properties of layer-by-layer alloyed materials have been investigated using model bimetallic Pt catalysts in ultrahigh vacuum (UHV) (10, 15), but current knowledge is still limited to fundamental material characterization instead of a full understanding of the molecular interactions on the modified surface at atmospheric pressure. The nature of the structural variation on the bimetallic layered surface leads to an inherent lattice mismatch, which reduces the surface free energy such that the modified structure is sensitive to changes in pressure or temperature. A representative phenomenon is the oxygen-induced segregation of subsurface transition metal atoms onto the topmost Pt-skin layer at catalytic reaction conditions that spontaneously form the metal-metal oxide interfacial nanostructure on the bimetallic Pt catalyst surface (16, 17).
Here, we report direct in situ observations as well as microscopic and spectroscopic evidence of the disintegration of the Pt-skin layer by segregated Ni oxide clusters on the Pt3Ni(111) surface at ambient pressure. Formation of the interfacial Pt-NiO1−x nanostructure is a decisive step in initiating catalytic reactions for CO oxidation (2CO + O2 → 2CO2) at room temperature. Density functional theory (DFT) calculations explain the thermodynamically favored reaction pathway on partially or fully formed NiO islands on a Pt3Ni(111) surface covered with Pt-skin, compared with a bare Pt(111) surface.
RESULTS
Figure 1A shows a scanning tunneling microscopy (STM) image of the wide terraces on the Pt3Ni(111) surface at UHV and 300 K. The inset in Fig. 1A shows an enlarged image of the clean surface identifying the nearest-neighbor distance (2.8 Å), which corresponds to that of the Pt(111) surface. The observed topmost layer is composed of Pt atoms because of selective segregation of the Pt. The latter Pt-skin layers are obtained at high annealing temperatures (1100 K), while annealing at lower temperatures yields chain-like features caused by incomplete atomic segregation (fig. S1). Although this flash-annealed Pt-skin layer has a geometry similar to the Pt(111) surface, the electronic structure is significantly different (15, 18). Thus, when exposing the Pt3Ni(111) surface to 120 mtorr of CO (Fig. 1B), locally formed bright spots are caused by CO molecule adsorption on the periodic lattice arrays of Pt atoms instead of the characteristic moiré pattern seen at high CO coverage (θCO > 0.5) (19). In contrast, the Pt-skin surface under 135 mtorr of O2 disintegrates by segregating the oxidized subsurface Ni atoms, as shown in Fig. 1C. This means that random subsurface Ni components affect molecular adsorption on the topmost Pt-skin layer, as is seen by the modification of the electronic structure of the Pt3Ni(111) surface. In addition, the poisoning effect of CO on the Pt-skin/Pt3Ni(111) surface is lower than that on the Pt(111) surface, as shown in the STM images under CO gas, which yields low CO coverage after CO exposure. Dissociative oxygen adsorption begins to cause surface restructuring at 3 mtorr of O2, and oxidized Ni clusters are pulled onto the Pt-skin layer at 120 mtorr of O2 (fig. S2). The corresponding oxygen-induced surface restructuring is strictly related to thermodynamic parameters (that is, pressure and temperature) such that the formation of oxidized Ni from Pt3Ni reliably follows the chemical potential of oxygen at a given temperature following this formula (17)
This distinctive surface segregation phenomenon arises mainly from the adsorbate-dependent modification of the electronic structure of the surface, but the effect of the “pressure gap” also plays a critical role in metastable subsurface Ni atom segregation on the bimetallic Pt-Ni catalyst surface. The huge pressure gap between UHV and atmospheric pressure causes a large discrepancy in the chemical potential corresponding to approximately 0.3 eV (20, 21). The exposed surface with a high coverage of molecular adsorbates results in restructuring behavior associated with a reduction of the surface free energy (22–24).
Fig. 1. STM images on the Pt3Ni(111) surface in UHV or ambient pressure condition at 300 K.

(A) Formation of a topmost Pt-skin layer after several cycles of Ar+ sputtering, followed by annealing at UHV and 1100 K (Vs = 0.32 V; It = 0.25 nA). (Inset) Atom-resolved STM image of the Pt-skin layer (Vs = 0.20 V; It = 0.20 nA). (B) CO adsorption on the terrace of the Pt-skin layer under 120 mtorr of CO (Vs = 0.45 V; It = 0.21 nA). (C) Disintegrated step and terrace structures of the Pt-skin layer under 135 mtorr of O2 (Vs = 1.40 V; It = 0.31 nA). (D) Evolution of the interfacial Pt-NiO1−x structure under 120 mtorr of mixed CO/O2 (1:5 ratio) (Vs = 1.25 V; It = 0.22 nA).
We obtained ambient-pressure STM (AP-STM) images during CO oxidation (Fig. 1D) at 120 mtorr of mixed CO/O2 (1:5 ratio) gas. Figure 1D exhibits a pronounced segregation of partially oxidized Ni clusters with the formation of interfacial Pt-NiO1−x nanostructures. The average size and height of the segregated clusters are 51.3 and 34.6% smaller, respectively, than those measured at 135 mtorr of O2 (fig. S3). Under the mixed CO/O2 gas, the observed clusters exhibit a hopping behavior on the wide terrace surface (see movie S1). After evacuating the reaction cell, the segregated clusters all remained stationary on the Pt3Ni(111) surface, as observed under both O2 alone and mixed CO/O2 gas at 300 K (figs. S2 and S4).
A series of AP-STM images taken under CO oxidation conditions confirm that the segregated nanoclusters are mobile during a time-lapse sequence of the measurements (see movie S1). The direction of the moving clusters is random, which indicates that the STM tip does not affect the motion of the clusters. Figure 2A shows spherical nanoscale particles lying on the wide terraces of the Pt3Ni(111) surface. With a 54-s time interval between the series of images in Fig. 2 (B and C), some particles (indicated with red arrows in the figures) are unstable, which is caused by chemical reactions taking place at the metal-metal oxide interface. The observed movement of the oxide clusters in the series of AP-STM images indicates an intrinsic relation between the surface redox process and the kinetic rates or reactivity of the surface.
Fig. 2. AP-STM images and AP-XPS spectra on the Pt3Ni(111) surface under mixed CO/O2 gas (1:5 ratio) at 300 K.
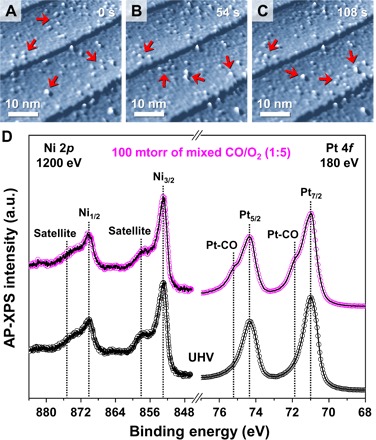
Time-lapse in situ AP-STM images of the segregated Ni oxide clusters on the Pt-skin layer (Vs = 1.40 V; It = 0.21 nA) at (A) 0 s, (B) 5 s, and (C) 108 s under 120 mtorr of mixed CO/O2 (1:5 ratio) gas at 300 K. (D) AP-XPS core-level spectra for Ni 2p (hν = 1200 eV) and for Pt 4f (hν = 180 eV) on the Pt-skin covered Pt3Ni(111) surface at 300 K under 100 mtorr of mixed CO/O2 (1:5 ratio) gas and at UHV. a.u., arbitrary units.
To investigate the chemically bonded species on the bimetal surfaces under reaction conditions, ambient-pressure x-ray photoelectron spectroscopy (AP-XPS) was carried out. AP-XPS plots of CO oxidation at 100 mtorr of mixed CO/O2 (1:5 ratio) gas show that the mixed CO and O2 adsorbate molecules create noticeable chemical bonds with the Pt or Ni species on the Pt3Ni(111) surface (Fig. 2D). The Pt 4f core-level spectrum only shows two distinguishable shoulder peaks (denoted as Pt-CO), compared with the well-defined clean Pt-skin surface in UHV (25). It is possible that dissociative oxygen adsorption also occurs immediately on the same surface (26); however, the amount of the chemisorbed p(2 × 2)-O measured is not nearly the amount of Pt-CO shown in the spectra (fig. S5). Likewise, the Ni 2p core-level AP-XPS spectrum exhibits a slightly broadened feature of the satellite peak, which is associated with Ni oxide formation (fig. S5) (17). A weak change in the residual gas profile [mass/charge ratio (m/z) = 44] is detected by CO2 evolution at these conditions (fig. S6), which implies that the segregated Ni oxide nanoclusters on the Pt3Ni(111) surface (Fig. 1D) play an important role in CO2 evolution by lowering the activation energy barrier. This is also consistent with results proposing a highly efficient catalyst using the synergistic effect of the surface and subsurface Ni species in the Pt-Ni bimetallic system (14). Furthermore, the results of these combined AP-STM and AP-XPS measurements reveal that the interfacial metal-metal oxide nanostructures act as active sites for facile intermediates that then follow a thermodynamically favorable reaction pathway (27). A possible explanation for this highly efficient energy conversion and catalytic activity enhancement is that hot electrons are initially exchanged between the reactant molecules and the metal-metal oxide interface (28). At this point, the spontaneously formed interfacial Pt-NiO1−x nanostructure, which is directly influenced by the effect of the pressure gap on the Pt3Ni(111) surface, significantly improves the catalytic reactivity for CO oxidation, in comparison with traditional Pt or Ni catalysts (29–31).
To explore the chemical interactions on the interfacial Pt-NiO1−x nanostructure during CO oxidation at the molecular level, we performed operando mass spectrometry combined with AP-XPS analysis for the O 1s core-level on the Pt3Ni(111) surface during CO oxidation. Figure 3A shows the evolution of the partial pressure measured using mass spectrometry during CO oxidation on the Pt3Ni (111) surface. The CO2 signal measured separately without a sample was one order of magnitude lower than that measured from Pt3Ni(111), indicating that the background reaction is negligible. Mass spectrometry results (Fig. 3A) and AP-XPS spectra (Fig. 3B) exhibit a correlation between the reaction species and intermediate formation at four temperatures in each region (i to iv) during the catalytic reaction. In region (i), the reactant molecules at 40 mtorr of CO and 100 mtorr of O2 (1:2.5 ratio) are introduced to the chamber at 300 K. As seen in previous AP-XPS studies (17, 25), five O 1s peaks are observed. Four are identified as molecular CO adsorption on the bridge site (light green; 531.2 eV) and on the atop site (red; 532.9 eV) of the Pt-skin layer, and as gas-phase CO(g) (pink; 537.0 eV) and O2(g) (olive; 537.8 and 539.1 eV). A new peak appears at 529.7 eV (violet) in region (ii) that is associated with the NiO species; the integrated peak area gradually increases with increasing temperature up to 543 K. Meanwhile, the integrated peak areas of the Pt-CO, CO(g), and O2(g) species decreased simultaneously in regions (ii) and (iii). Finally, in region (iv), catalytic conversion of the CO molecules is completed on the surface, as shown by the sudden surge in the CO2(g) concentration. Evolution of the CO2(g) species is confirmed by the rapid increase in the CO2 mass intensity (m/z = 44) and the appearance of the gas-phase CO2(g) peak at 535.7 eV in the XPS spectrum. The growth of NiO islands on the Pt3Ni(111) surface is further accelerated in this relatively O2-dominant environment, but dynamic defect states are unavoidable, as shown in the minor species at 531.4 eV during Ni oxidation (32). Pt 4f and Ni 2p core-level AP-XPS spectra at elevated temperature prove that chemical bonds between the Pt atoms and CO molecules formed on the surface, while the oxidation process is limited to the Ni atoms (fig. S7).
Fig. 3. Mass spectrometry profiles of the residual gas and AP-XPS spectra of the Pt3Ni(111) surface under 40 mtorr of CO and 100 mtorr of O2 mixed gas (1:2.5 ratio) at elevated temperatures.
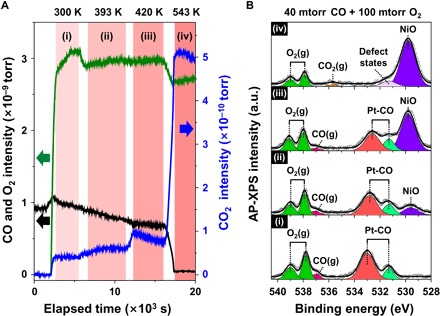
(A) Time-lapse mass fragment profiles for m/z = 28 (CO; black solid line), m/z = 32 (O2; green solid line), and m/z = 44 (CO2; blue solid line) in the second differential pumping stage of the photoelectron analyzer. (B) O 1s core-level AP-XPS spectra (hν = 650 eV) for (i) 300 K, (ii) 393 K, (iii) 420 K, and (iv) 543 K.
Significant consumption of gaseous CO and O2 molecules begins in region (ii) at a mild temperature (~400 K), while the subsurface Ni atoms spontaneously segregate onto the Pt-skin. This suggests that the adsorbed CO molecules on the topmost layer of Pt atoms could react with the dissociated oxygen on the segregated Ni oxide clusters. That is, the nanostructures on the Pt-skin layer could enhance the catalytic activity by forming metal-metal oxide interfaces. The measured CO2 evolution (m/z = 44) is affected by the degree of segregated NiO species at the elevated temperature; the calculated activation energy barriers of regions (iii) and (iv) are 0.25 and 0.46 eV, respectively. With Pt catalysts, CO poisoning is a significant problem and it is difficult to dissociate O2, which results in little activity for preferential CO oxidation with Pt catalysts (33). The incorporated Ni components successfully compensate for these drawbacks by changing the electronic and geometric structures on the surface (34). The formation of interfacial nanostructures between the Pt-skin layer and Ni oxides provides a great opportunity for improving reactant molecule conversion rates along their perimeters, which is demonstrated on the metal-metal oxide catalysts (3, 27, 35). These elaborate Pt-Ni catalysts demonstrate superior turnover rates compared with single-metal Pt or Ni catalysts for CO oxidation (14, 33); a similar trend is also measured in the batch reactor system using model catalysts (fig. S8).
To gain further insight into the energy requirements of the CO oxidation reaction on the interfacial Pt-NiO1−x nanostructure, DFT calculations were carried out on the confined model structures of the NiO1−x/Pt-skin/Pt3Ni and NiO/Pt3Ni, as visualized in fig. S9. For these calculations, we assumed that the each reaction follows either the Langmuir-Hinshelwood (LH) [CO* + O* → CO2(g)] or Eley-Rideal (ER) [CO(g) + O* → CO2(g)] mechanism, which are generally accepted reaction pathways in confined metal-metal oxide model structures (27). The adsorption energies of the CO, O2, and O on the confined model surfaces are summarized in Table 1.
Table 1. Calculated molecular adsorption energies and activation energy barriers.
DFT calculation results for the molecular adsorption of CO, O2, and O on the surface of each theoretically designed model and their apparent activation energy barriers.
| Eads(CO), eV | Eads(O2), eV | Eads(O), eV | Ea, eV/mol | |
| Interfacial Pt-NiO1−x | −1.30 | — | −0.88 | +0.37 |
| NiO/Pt3Ni | Unbound | Unbound | — | +1.08 |
| Pt(111) | −1.41 | −0.31 | −0.53 | +0.86 |
In Fig. 4A, a CO molecule preferentially adsorbs on an atop site of Pt (Eads = −1.30 eV) next to the NiO1−x island (Eads = −0.80 eV) on the interfacial Pt-NiO1−x nanostructure surface. The adsorbed intermediate CO* then interacts with a neighboring oxygen species (O*) from the NiO1−x to form the COO* intermediate. After the stabilization step, the COO* intermediate is promptly desorbed as a CO2(g) molecule. The calculated activation energy barrier is +0.37 eV at the rate-determining step, which is much lower than that on the Pt(111) surface for CO oxidation via the LH mechanism (+0.86 eV) in fig. S10 or that on the fully oxidized NiO on the Pt3Ni surface via the ER mechanism (+1.08 eV) in Fig. 4B. Because a CO molecule does not bind to the NiO/Pt3Ni structure when the Ni surface is fully oxidized, the CO can only react with an exposed terminal oxygen on the NiO surface. This ER mechanism, however, is an unfavorable reaction pathway compared with the steps proposed in the LH mechanism for the interfacial Pt-NiO1−x nanostructure. In addition, a possible origin of the low activation energy barrier for the LH mechanism would be the relatively weaker bond of the CO* (Table 1) on the Pt-NiO1−x nanostructure because of the modified electronic structure of the Pt3Ni(111) and the atop O* binding configuration at the associated interface (fig. S11). Notably, spontaneous dissociative adsorption of an O2 molecule also takes place at the confined interfacial atop sites with an adsorption energy of −0.88 eV per O. This barrier-less O2 dissociation on the interfacial Pt-NiO1−x nanostructure can be compared with the dissociation energy barrier of +0.59 eV in the case of the 2O* configuration on the hollow site for Pt(111) (fig. S12); thus, this barrier-less source of dissociated oxygen at the interfacial sites of the Pt-NiO1−x nanostructure is responsible, in part, for the experimentally observed enhanced catalytic activity for CO oxidation.
Fig. 4. DFT calculation results of the chemical reaction pathway for CO oxidation.
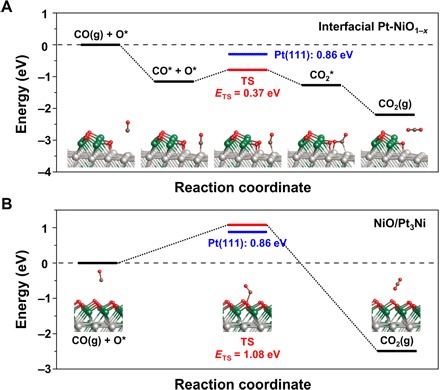
Energy profiles for (A) the interfacial Pt-NiO1−x and (B) the fully oxidized NiO on the stoichiometric Pt3Ni structure. The optimized minimum and transition state (TS) structures are shown, and their relative energies are summarized by the solid black and red lines, respectively. For the energy barrier (ETS) of the rate-determining step, the results for Pt(111) are also shown (blue) for comparison. The adsorbed species are denoted with asterisks (*). The colors for each atom in the model: Pt (light gray), Ni (green), O (red), and C (brown).
DISCUSSION
Our findings demonstrate improved chemical interactions between adsorbate molecules and the interfacial Pt-NiO1−x nanostructure on the bimetallic Pt3Ni(111) surface during CO oxidation. The enhancement of catalytic activity on the bimetallic catalyst surface originates from the thermodynamically efficient reaction pathways at the metal-metal oxide interface, which demonstrates a straightforward process for the SMSI effect. By using an alloy where the metals have highly mismatched lattices, we show the segregation of NiO islands on top of the Pt-skin, which creates active sites at the metal-metal oxide interfaces under chemical reaction environments at ambient pressure. There is a clear difference between the Pt-Ni bimetal surface and single-component Pt or Ni surfaces, which provides reasonable evidence for the enhanced catalytic activity caused by the SMSI effect at the metal-metal oxide interface. The evolution of a bimetal surface during the catalytic reaction demonstrated in this study can be applicable for other bimetal systems and catalytic reactions that exhibit the SMSI effect. The formation of these interfacial metal-metal oxide nanostructures increases catalytic activity while providing a thermodynamically efficient reaction pathway by lowering the heat of reaction on the surface. This model system was used to gain knowledge of the role of the metal-metal oxide interface and active sites on the surface; it also establishes a strategy for improving catalytic activity for catalytic reactions in industrial chemical reactors.
MATERIALS AND METHODS
Materials
Commercially available high-purity (99.99%) Pt(111), Ni(111), and Pt3Ni(111) single crystals were purchased from MaTeck GmbH. The polished samples were provided using a high-accuracy cutting angle within 0.1°.
Sample preparation
The atomically flat Pt-skin covering the layered Pt-Ni surface was prepared using several cycles of Ar+ ion bombardment sputtering at 1 × 10−5 torr (measured sample current: 13.3 μA) for 20 min, followed by vacuum annealing at 1100 K for 5 min in an UHV chamber. The sample cleaning process was repeated until a well-defined clean surface was obtained with no contamination (for example, carbon or oxygen). The experimental measurements were then taken using STM and XPS.
AP-STM observations
The AP-STM observations were performed using a small-volume (~15 ml) reaction cell integrated into a scanning probe microscope (SPM) instrument (SPECS) in a UHV chamber at room temperature. The inside of the reaction cell was separated from the outside by two tightly sealed O-rings. The topographic STM images were acquired using constant current mode with a chemically etched tungsten tip that was calibrated to standard measurements for each length in dimensions x, y, and z that were obtained from an atomically flat clean Au(111) surface. To obtain the in situ observations at ambient pressure, gas molecules were introduced to the reaction cell of the SPM instrument using a precision leak valve before measurements were taken under the various catalytic reaction environments. The pressure inside the reaction cell was measured using a full-range gauge (Pfeiffer Vacuum). The tunneling parameters of each AP-STM image are denoted as Vs for sample bias and It for tunneling current, respectively.
Synchrotron-based AP-XPS analysis
AP-XPS spectra were obtained at the TEMPO beamline at the SOLEIL synchrotron in France and at the BL13 beamline at the Photon Factory at the High Energy Accelerator Research Organization (PF-KEK) in Japan. The selected photon energies were 1050 to 1200 eV for Ni 2p, 650 eV for O 1s, and 180 eV for Pt 4f, respectively. Detailed descriptions of each beamline facility can be found elsewhere (36, 37).
Catalytic activity measurements
The catalytic activity measurements of the Pt(111), Ni(111), and Pt3Ni(111) single crystals for CO oxidation were performed in a batch reactor under 40 torr of CO and 100 torr of O2 as the reactant gases, and 620 torr of He as the balancing gas in the system. The mixed gas was continuously circulated at a rate of 2 liters/min and was analyzed using a gas chromatography instrument equipped with a thermal conductivity detector; a molecular sieve 5A column was used to separate each gas component when at the reaction equilibrium temperature. The measured CO conversion rate for each sample was used to calculate the turnover number in the batch reactor.
DFT calculation
Spin-polarized DFT calculations were performed using the Vienna ab initio simulation package (38, 39) with the revised Perdew-Burke-Ernzerhof exchange-correlation functional (40, 41). The potentials of the atoms were described by the projector-augmented wave (42). Throughout this study, we used a cutoff energy of 400 eV, and all atoms were relaxed using a conjugate gradient algorithm until the total force on each atom was less than 0.05 eV/Å. The interfacial Pt-NiO1−x structure on the stoichiometric Pt3Ni slab was modeled on the basis of the ordered fcc structure (43) because Pt3Ni alloy materials have a closed-packed fcc bulk structure. A (4 × 6) supercell [that is, two Pt3Ni bottom layers and one Pt(111) topmost layer] was used, and a NiO1−x (Ni8O4) ribbon cluster was placed on the stoichiometric Pt3Ni substrate to construct the interfacial nanostructure. The Brillouin zone was sampled using a 2 × 2 × 1 Monkhorst-Pack mesh for geometric optimization. To create the Pt(111) model, three layers of a (2 × 2) Pt(111) slab and 4 × 4 × 1 k-points were used. For both models, the bottom two layers were fixed in their bulk positions, and a vacuum space of 15 Å was used in the z direction. The climbing image nudged elastic band (CI-NEB) method (44) was used to calculate the reaction barriers for CO oxidation.
Supplementary Material
Acknowledgments
We acknowledge M. Salmeron and N. M. Markovic for their fruitful discussions. We thank the beamline scientists at the TEMPO beamline at the SOLEIL synchrotron in France and at the BL13 beamline at the PF-KEK in Japan for helping with the AP-XPS experiments. Funding: This work was supported by Institute for Basic Science (IBS) (IBS-R004). Financial support was provided by National Research Foundation of Korea (NRF-2015R1A5A1009962, NRF-2015R1A2A2A01004084, NRF-2015K1A3A1A14021261, NRF-2016M3D1A1021147, NRF-2017K1A3A7A09016316, and NRF-2017R1A2B3010176) and the GIST Research Institute Grant funded by Gwangju Institute of Science and Technology (GIST) 2018. The AP-XPS experiments were performed under the approval of the Photon Factory Program Advisory Committee (PF PAC- 2016G128) and SOLEIL Program Advisory Committee (project 20170132). Author contributions: J.K. and J.Y.P. planned and designed the project. J.K., W.H.D., and M.C.N. performed the AP-STM experiments. W.H.P. and Y.J. carried out the DFT calculations. S.W.L. measured the catalytic activity of the single-crystal samples. J.-J.G., F.B., H.K., K.M., and B.S.M. acquired the AP-XPS spectra. J.K., Y.J., B.S.M., and J.Y.P. prepared the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/7/eaat3151/DC1
Supplementary Text
Fig. S1. Formation of the bimetallic domain structures.
Fig. S2. Oxygen-induced surface restructuring.
Fig. S3. Statistical analysis plots for NiO1−x clusters.
Fig. S4. Direct observation of the Pt3Ni(111) after CO/O2 gas evacuation.
Fig. S5. AP-XPS analysis at 100 mtorr of O2.
Fig. S6. Tracking the mass profiles at room temperature.
Fig. S7. AP-XPS analysis in mixed CO/O2 gas at elevated temperature.
Fig. S8. Arrhenius plots for catalytic activity measurements.
Fig. S9. Model structures of the interfacial Pt-NiO1−x and NiO/Pt3Ni.
Fig. S10. Energy profile for CO oxidation on the Pt(111).
Fig. S11. Activation barriers for the CO oxidation reaction (CO* + O* → CO2) for different O* configurations on the model surfaces.
Fig. S12. Energy profile for O2 dissociation on the Pt(111) surface and on the interfacial Pt-NiO1−x nanostructure.
Movie S1. Time-lapse AP-STM movie on the Pt3Ni(111) surface under mixed CO/O2 (1:5 ratio) gas at 300 K.
REFERENCES AND NOTES
- 1.Schwab G.-M., Electronics of supported catalysts. Adv. Catal. 27, 1–22 (1979). [Google Scholar]
- 2.Haruta M., Kobayashi T., Sano H., Yamada N., Novel gold catalysts for the oxidation of carbon monoxide at a temperature far below 0°C. Chem. Lett. 16, 405–408 (1987). [Google Scholar]
- 3.Green I. X., Tang W., Neurock M., Yates J. T. Jr, Spectroscopic observation of dual catalytic sites during oxidation of CO on a Au/TiO2 catalyst. Science 333, 736–739 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Tao F., Grass M. E., Zhang Y., Butcher D. R., Renzas J. R., Liu Z., Chung J. Y., Mun B. S., Salmeron M., Somorjai G. A., Reaction-driven restructuring of Rh-Pd and Pt-Pd core-shell nanoparticles. Science 322, 932–934 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Yu W., Porosoff M. D., Chen J. G., Review of Pt-based bimetallic catalysis: From model surfaces to supported catalysts. Chem. Rev. 112, 5780–5817 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Stamenkovic V. R., Strmcnik D., Lopes P. P., Markovic N. M., Energy and fuels from electrochemical interfaces. Nat. Mater. 16, 57–69 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Zeng Z., Chang K.-C., Kubal J., Markovic N. M., Greeley J., Stabilization of ultrathin (hydroxy)oxide films on transition metal substrates for electrochemical energy conversion. Nat. Energy 2, 17070 (2017). [Google Scholar]
- 8.Stamenkovic V. R., Mun B. S., Arenz M., Mayrhofer K. J. J., Lucas C. A., Wang G., Ross P. N., Markovic N. M., Trends in electrocatalysis on extended and nanoscale Pt-bimetallic alloy surfaces. Nat. Mater. 6, 241–247 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Gan L., Cui C., Heggen M., Dionigi F., Rudi S., Strasser P., Element-specific anisotropic growth of shaped platinum alloy nanocrystals. Science 346, 1502–1506 (2014). [DOI] [PubMed] [Google Scholar]
- 10.van der Vliet D. F., Wang C., Li D., Paulikas A. P., Greeley J., Rankin R. B., Strmcnik D., Tripkovic D., Markovic N. M., Stamenkovic V. R., Unique electrochemical adsorption properties of Pt-skin surfaces. Angew. Chem. 124, 3193–3196 (2012). [DOI] [PubMed] [Google Scholar]
- 11.van der Vliet D. F., Wang C., Tripkovic D., Strmcnik D., Zhang X. F., Debe M. K., Atanasoski R. T., Markovic N. M., Stamenkovic V. R., Mesostructured thin films as electrocatalysts with tunable composition and surface morphology. Nat. Mater. 11, 1051–1058 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Porter N. S., Wu H., Quan Z., Fang J., Shape-control and electrocatalytic activity-enhancement of Pt-based bimetallic nanocrystals. Acc. Chem. Res. 46, 1867–1877 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Chen C., Kang Y., Huo Z., Zhu Z., Huang W., Xin H. L., Snyder J. D., Li D., Herron J. A., Mavrikakis M., Chi M., More K. L., Li Y., Markovic N. M., Somorjai G. A., Yang P., Stamenkovic V. R., Highly crystalline multimetallic nanoframes with three-dimensional electrocatalytic surfaces. Science 343, 1339–1343 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Mu R., Fu Q., Xu H., Zhang H., Huang Y., Jiang Z., Zhang S., Tan D., Bao X., Synergetic effect of surface and subsurface Ni species at Pt−Ni bimetallic catalysts for CO oxidation. J. Am. Chem. Soc. 133, 1978–1986 (2011). [DOI] [PubMed] [Google Scholar]
- 15.Kim Y. S., Jeon S. H., Bostwick A., Rotenberg E., Ross P. N., Stamenkovic V. R., Markovic N. M., Noh T. W., Han S., Mun B. S., Role of transition metal in fast oxidation reaction on the Pt3TM (111) (TM = Ni, Co) surfaces. Adv. Energy Mater. 3, 1257–1261 (2013). [Google Scholar]
- 16.Xin H. L., Alayoglu S., Tao R., Genc A., Wan C.-M., Kovarik L., Stach E. A., Wang L.-W., Salmeron M., Somorjai G. A., Zheng H., Revealing the atomic restructuring of Pt–Co nanoparticles. Nano Lett. 14, 3203–3207 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Lee H. C., Kim B. M., Jeong C. K., Toyoshima R., Kondoh H., Shimada T., Mase K., Mao B., Liu Z., Lee H., Huang C.-Q., Li W. X., Ross P. N., Mun B. S., Surface segregation and oxidation of Pt3Ni(1 1 1) alloys under oxygen environment. Catal. Today 260, 3–7 (2016). [Google Scholar]
- 18.Su H.-Y., Bao X.-H., Li W.-X., Modulating the reactivity of Ni-containing Pt(111)-skin catalysts by density functional theory calculations. J. Chem. Phys. 128, 194707 (2008). [DOI] [PubMed] [Google Scholar]
- 19.Longwitz S. R., Schnadt J., Vestergaard E. K., Vang R. T., , Stensgaard I., Brune H., Besenbacher F., High-coverage structures of carbon monoxide adsorbed on Pt(111) studied by high-pressure scanning tunneling microscopy. J. Phys. Chem. B 108, 14497–14502 (2004). [Google Scholar]
- 20.Salmeron M., Schlögl R., Ambient pressure photoelectron spectroscopy: A new tool for surface science and nanotechnology. Surf. Sci. Rep. 63, 169–199 (2008). [Google Scholar]
- 21.Kim J., Noh M. C., Doh W. H., Park J. Y., Thermal evolution and instability of CO-induced platinum clusters on the Pt(557) surface at ambient pressure. J. Am. Chem. Soc. 138, 1110–1113 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Somorjai G. A., Park J. Y., Molecular surface chemistry by metal single crystals and nanoparticles from vacuum to high pressure. Chem. Soc. Rev. 37, 2155–2162 (2008). [DOI] [PubMed] [Google Scholar]
- 23.Tao F., Crozier P. A., Atomic-scale observations of catalyst structures under reaction conditions and during catalysis. Chem. Rev. 116, 3487–3539 (2016). [DOI] [PubMed] [Google Scholar]
- 24.van Spronsen M. A., Frenken J. W. M., Groot I. M. N., Surface science under reaction conditions: CO oxidation on Pt and Pd model catalysts. Chem. Soc. Rev. 46, 4347–4374 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Toyoshima R., Yoshida M., Monya Y., Suzuki K., Amemiya K., Mase K., Mun B. S., Kondoh H., A high-pressure-induced dense CO overlayer on a Pt(111) surface: A chemical analysis using in situ near ambient pressure XPS. Phys. Chem. Chem. Phys. 16, 23564–23567 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Miller D. J., Öberg H., Kaya S., Casalongue H. S., Friebel D., Anniyev T., Ogasawara H., Bluhm H., Pettersson L. G. M., Nilsson A., Oxidation of Pt(111) under near-ambient conditions. Phys. Rev. Lett. 107, 195502 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Fu Q., Li W.-X., Yao Y., Liu H., Su H.-Y., Ma D., Gu X.-K., Chen L., Wang Z., Zhang H., Wang B., Bao X., Interface-confined ferrous centers for catalytic oxidation. Science 328, 1141–1144 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Park J. Y., Baker L. R., Somorjai G. A., Role of hot electrons and metal–oxide interfaces in surface chemistry and catalytic reactions. Chem. Rev. 115, 2781–2817 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Calderón S. K., Grabau M., Óvári L., Kress B., Steinrück H.-P., Papp C., CO oxidation on Pt(111) at near ambient pressures. J. Chem. Phys. 144, 044706 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Kim J., Noh M. C., Doh W. H., Park J. Y., In situ observation of competitive CO and O2 adsorption on the Pt(111) surface using near-ambient pressure scanning tunneling microscopy. J. Phys. Chem. C 122, 6246–6254 (2018). [Google Scholar]
- 31.Noh M. C., Kim J., Doh W. H., Kim K.-J., Park J. Y., Reversible oxygen-driven nickel oxide structural transition on the nickel(1 1 1) surface at near-ambient pressure. ChemCatChem 10, 2046–2050 (2018). [Google Scholar]
- 32.Kuhlenbeck H., Odörfer G., Jaeger R., Illing G., Menges M., Mull T., Freund H.-J., Pöhlchen M., Staemmler V., Witzel S., Scharfschwerdt C., Wennemann K., Liedtke T., and Neumann M., Molecular adsorption on oxide surfaces: Electronic structure and orientation of NO on NiO(100)/Ni(100) and on NiO(100) as determined from electron spectroscopies and ab initio cluster calculations. Phys. Rev. B Condens. Matter 43, 1969–1986 (1991). [DOI] [PubMed] [Google Scholar]
- 33.Hwang S. Y., Yurchekfrodl E., Zhang C., Peng Z., Low-temperature preferential oxidation of carbon monoxide on Pt3Ni alloy nanoparticle catalyst with engineered surface. ChemCatChem 8, 97–101 (2016). [Google Scholar]
- 34.Menning C. A., Chen J. G., Regenerating Pt–3d–Pt model electrocatalysts through oxidation–reduction cycles monitored at atmospheric pressure. J. Power Sources 195, 3140–3144 (2010). [Google Scholar]
- 35.Kattel S., Ramírez P. J., Chen J. G., Rodriguez J. A., Liu P., Active sites for CO2 hydrogenation to methanol on Cu/ZnO catalysts. Science 355, 1296–1299 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Naitabdi A., Fagiewicz R., Boucly A., Olivieri G., Bournel F., Tissot H., Xu Y., Benbalagh R., Silly M. G., Sirotti F., Gallet J.-J., Rochet F., Oxidation of small supported platinum-based nanoparticles under near-ambient pressure exposure to oxygen. Top. Catal. 59, 550–563 (2016). [Google Scholar]
- 37.Toyoshima R., Yoshida M., Monya Y., Kousa Y., Suzuki K., Abe H., Mun B. S., Mase K., Amemiya K., Kondoh H., In situ ambient pressure XPS study of CO oxidation reaction on Pd(111) surfaces. J. Phys. Chem. C 116, 18691–18697 (2012). [Google Scholar]
- 38.Kresse G., Furthmüller J., Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996). [DOI] [PubMed] [Google Scholar]
- 39.Kresse G., Joubert D., From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999). [Google Scholar]
- 40.Hammer B., Hansen L. B., Nørskov J. K., Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B 59, 7413–7421 (1999). [Google Scholar]
- 41.Perdew J. P., Burke K., Ernzerhof M., Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996). [DOI] [PubMed] [Google Scholar]
- 42.Blöchl P. E., Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994). [DOI] [PubMed] [Google Scholar]
- 43.Ma Y., Balbuena P. B., Pt surface segregation in bimetallic Pt3M alloys: A density functional theory study. Surf. Sci. 602, 107–113 (2008). [Google Scholar]
- 44.Henkelman G., Uberuaga B. P., Jónsson H., A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/7/eaat3151/DC1
Supplementary Text
Fig. S1. Formation of the bimetallic domain structures.
Fig. S2. Oxygen-induced surface restructuring.
Fig. S3. Statistical analysis plots for NiO1−x clusters.
Fig. S4. Direct observation of the Pt3Ni(111) after CO/O2 gas evacuation.
Fig. S5. AP-XPS analysis at 100 mtorr of O2.
Fig. S6. Tracking the mass profiles at room temperature.
Fig. S7. AP-XPS analysis in mixed CO/O2 gas at elevated temperature.
Fig. S8. Arrhenius plots for catalytic activity measurements.
Fig. S9. Model structures of the interfacial Pt-NiO1−x and NiO/Pt3Ni.
Fig. S10. Energy profile for CO oxidation on the Pt(111).
Fig. S11. Activation barriers for the CO oxidation reaction (CO* + O* → CO2) for different O* configurations on the model surfaces.
Fig. S12. Energy profile for O2 dissociation on the Pt(111) surface and on the interfacial Pt-NiO1−x nanostructure.
Movie S1. Time-lapse AP-STM movie on the Pt3Ni(111) surface under mixed CO/O2 (1:5 ratio) gas at 300 K.