

Abstract
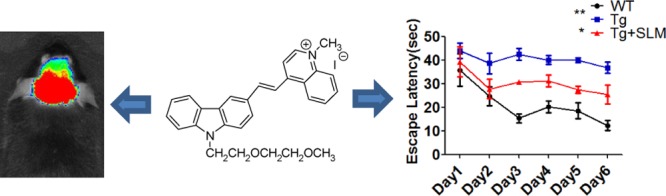
We report herein an investigation of carbazole-based cyanine, (E)-4-(2-(9-(2-(2-methoxyethoxy)ethyl)-9H-carbazol-3-yl)-vinyl)-1-methyl-quinolin-1-iumiodide (SLM), as an effective theranostic agent for Alzheimer’s disease (AD). This cyanine exhibited desirable multifunctional and biological properties, including amyloid-β (Aβ)-oligomerization inhibition, blood–brain barrier permeability, low neurotoxicity, neuroprotective effect against Aβ-induced toxicities, high selectivity and strong binding interactions with Aβ peptide/species, good biostability, as well as strong fluorescence enhancement upon binding to Aβ species for diagnosis and therapy of AD. This cyanine has been successfully applied to perform near-infrared in vivo imaging of Aβ species in transgenic AD mouse model. The triple transgenic AD mice intraperitoneally treated with SLM showed significant recovery of cognitive deficits. Furthermore, those SLM-treated mice exhibited a substantial decrease in both of oligomeric Aβ contents and tau proteins in their brain, which was attributed to the induction of autophagic flux. These findings demonstrated for the first time that SLM is an effective theranostic agent with in vivo efficacy for diagnosis and treatment of AD in mouse models.
Introduction
Alzheimer’s disease (AD) is the most prevalent neurodegenerative dementia, which leads to an impairment of various cognitive functions and memory loss, affecting 47 million people worldwide in 2015.1 Since AD is still incurable, it does place a huge burden to both the medical field and the society. Two of the pathological hallmarks of AD include the presence of extracellular amyloid-β (Aβ) plaques, primarily composed of misfolded Aβ(1–40) and Aβ(1–42) aggregates and intracellular tau neurofibrillary tangles, due to the accumulation of hyperphosphorylated tau proteins.2−4 Since Aβ peptides and tau proteins are widely believed to be involved in the pathogenesis of AD, they represent the primary drug targets for the development of disease-modifying therapy in AD.5,6 Among various drug development strategies, targeting the overproduction and inefficient clearance of the Aβ peptides has constituted one of the primary focuses.7,8 Increasing evidence suggests that soluble Aβ oligomers are more neurotoxic than monomers and aggregates.9,10 Hence, the recent trials of the treatment approach for AD is to reduce the level and inhibit the formation of soluble toxic Aβ oligomers, which are believed to impair intracellular membranes leading to mitochondrial and lysosomal dysfunctions and further resulting in neuronal and synaptic damage, as implicated by the modified amyloid cascade hypothesis. The studies also showed that soluble Aβ oligomers would facilitate the tau pathology by influencing the signaling pathways that mediate the phosphorylation of the tau protein, thereby triggering AD.11 Studies have indicated that the deposition of tau-associated neurofibrillary tangles correlates well with the disease progression of AD and thus it also becomes an important therapeutic target of AD.12,13
Meanwhile, Aβ oligomers can be used as predictive disease biomarkers for monitoring of progression and diagnosis of AD. Among various detection techniques, noninvasive near-infrared (NIR) fluorescence imaging is the most intriguing, particularly for early diagnosis of AD. A variety of fluorescence probes have been developed for detecting Aβ peptides; nonetheless, most of them are still not suitable for in vivo NIR imaging for their limited blood–brain barrier (BBB) permeability, intrinsic neurotoxicity, poor binding affinity toward Aβ, and interference by autofluorescence.
Molecules that exhibit theranostic functions of Aβ oligomerization/aggregation inhibition and imaging capability of Aβ species have recently been reported;14−18 however, their respective in vivo efficacy for cognitive improvement and simultaneously performing imaging of Aβ species in vivo in AD mouse model have not been explored and demonstrated. It is attributed to the diverse and strict criteria of an effective theranostic agent for therapy and diagnosis of AD, which include good BBB permeability, low neurotoxicity, potent Aβ aggregation inhibition, neuroprotective effect against Aβ- induced toxicity, high selectivity and targeting to Aβ peptide/species, good biostability as well as strong fluorescence enhancement upon binding to Aβ peptide/species.19−21 Cyanine fluorophores are biocompatible and versatile as a fluorescence sensing probe for in vitro and in vivo bioimaging.22−25 It was previously shown that carbazole-based quinolinium cyanines not only exhibited selective binding to Aβ peptides rendering the potential for direct imaging of Aβ species but also exerted strong inhibition on Aβ peptide aggregation, offering promise as a therapeutic agent for AD.24 We herein report that the carbazole-based cyanine, (E)-4-(2-(9-(2-(2-methoxyethoxy)ethyl)-9H-carbazol-3-yl)-vinyl)-1-methyl-quinolin-1-iumiodide (SLM) (Figure 1) has been successfully used as an in vivo imaging agent for Aβ species in mouse model. In addition, administration of Aβ-aggregation inhibition and neuroprotective SLM significantly protects triple transgenic (3 × Tg-AD) mice from cognitive decline, concomitant with a substantial decrease in contents of oligomeric Aβ and tau proteins in the cerebral hippocampal region. To the best of our knowledge, this is the first time that an effective theranostic agent shows integrated functions of (i) in vivo NIR imaging of Aβ species in AD mouse models as well as (ii) potent in vivo efficacy of improving cognitive deficit and reduction of Aβ plaques and tau content.
Figure 1.

Molecular structure of carbazole-based cyanine, SLM.
Results and Discussion
NIR imaging can be a useful and promising tool for early AD diagnosis, as it can be applied to visualize and monitor the formation of Aβ aggregates and to evaluate the efficacy of the AD treatment in the animal model. Taking advantage of its strong binding to Aβ species (Kd(Aβ1–40) of fibril = 13.1 μM, Kd(Aβ1–40) of monomer = 96.6 μM; Kd(Aβ1–42) of fibril = 11.4 μM, Kd(Aβ1–42) of monomer = 40.2 μM) that results in strong fluorescence enhancement (Table S1 and Figure S1), SLM shows great potential as in vivo Aβ NIR imaging agent. We demonstrated the application of SLM in detecting and imaging of Aβ species in a 9-month-old amyloid precursor proteins (APP)/PS1 transgenic (Tg) mouse in vivo. Figure 2a displays the fluorescence images of Tg and wild-type (WT) mice at different time points before and after intravenous injection of SLM at a dosage of 5 mg/kg. As clearly seen, the fluorescence intensity from the brain of Tg mouse is significantly higher than that of WT at all time points recorded. The fluorescence signal also retained significantly longer in the Tg mouse when compared with that of the WT, which was attributed to the strong binding of SLM to the Aβ species in the brain of the Tg mouse. This is consistent with the results obtained from the semiquantitative kinetic clearance profile analysis of fluorescence images of the brains of Tg and WT mice (Figure S4). The longer clearance time further confirmed the strong binding interactions of SLM with Aβ species. Nonetheless, accurate quantification of SLM for its permeability and pharmacokinetics by liquid chromatography–mass spectrometry study would be definitely important for further clinical studies. To ascertain the targeting property of SLM toward Aβ species, the brain slices of Tg mouse (9 months old) after tail-vein injection with SLM was ex vivo co-stained with Aβ plaque-specific thioflavin (ThT) dye. As shown in Figure 2b, the fluorescence of SLM overlapped well with that of ThT in the central core of the cluster plaques, indicating that SLM is highly specific to Aβ plaques. In addition to staining the Aβ plaques/fibrils, the weaker fluorescence of SLM was observed in the peripheral of the clusters, which could not be labeled by ThT. It is due to the fact that ThT only labels aggregated Aβ but SLM can also bind strongly with Aβ oligomers.
Figure 2.

(a) Fluorescence images of Tg (9 months old) and wild-type mice at different time points before and after intravenous injection of SLM (5 mg/kg). λex = 535 nm, λem = 600–640 nm. (b) Histological staining of the brain slices from 9-month transgenic mice stained with SLM via tail-vein injection (λex = 488 nm, λem = 640–680 nm) and co-stained with ThT (λex = 405 nm, λem = 460–490 nm). The images are 40× magnification of the selected regions.
SLM also exhibits inhibition effect on the oligomerization of Aβ(1–42), as evaluated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) analysis (Figure S2), where it showed a smaller amount of dimeric Aβ(1–42) formation in the presence of SLM. Besides, LC50 of SLM was determined to be 20 μM using SH-SY5Y cells by MTT assay (Figure S3). Neuroprotective effect of inhibitor compounds is one of the important screening parameters for pharmaceutical potential in AD treatment. The neuroprotective effect of SLM against the more neurotoxic Aβ(1–42) on primary hippocampal cells was then studied. The results, summarized in Figure 3, indicated that SLM showed significantly higher survival rate and, thus, beneficial to the neuronal cells that were incubated with Aβ(1–42) over a period of 24 h, highlighting tremendous promise of this cyanine for preventive and therapeutic treatment of AD.
Figure 3.

(a) Neuroprotective effect of the cyanine compounds against the toxicity induced by Aβ(1–42) on primary hippocampal cells. SLM was found to be neuroprotective, (b) the neuroprotective cyanine attenuated the Aβ(1–42)-induced reactive oxygen species (ROS) level on primary hippocampal cells. The relative percentage represented the cell death and ROS level of the Aβ(1–42)-treated cells, respectively, in the presence and absence of SLM.
Given that elevation in oxidative stress constitutes parts of the causative factors for Aβ-induced neurotoxicity,26,27 we investigated the neuroprotective effect of SLM against reactive oxygen species (ROS) in neuronal primary cells. A ROS fluorescent dye, carboxyl-2′,7′-dichlorofluorescein diacetate (carboxyl-DCFDA), was employed to monitor the ROS level in primary hippocampal cells that were exposed to Aβ(1–42) in the presence and absence of the neuroprotective SLM. Indeed, a significant elevation of ROS level in the control cells was induced by the addition of 10 μM Aβ(1–42) in 24 h. The ROS level was decreased by about 20%, as illustrated in Figure 3, when the neuronal cells were co-incubated with neuroprotective SLM. These results unambiguously indicated that SLM could protect the health of the neuronal cells by attenuating the ROS level.
In view that SLM possessed all of the desired functional and biological properties for potential therapeutics for AD, we then examined the in vivo efficacy of SLM against cognitive impairment using triple transgenic AD model mice (3 × Tg-AD) expressing human gene mutants APPswe, PS1M146V, and tauP301L. These 3 × Tg-AD mice showed intracellular Aβ at 3–6 months of age and cognitive impairment at 6 months of age. The 8-month-old 3 × Tg-AD mice received the cyanine inhibitors, SLM, for 45 days (2.25 mg/kg/day) intraperitoneally. No obvious differences in body weight or changes in physical behavior (swimming speed) were observed among the groups during the 45 days administration (Figures S5 and S6). Morris water maze tests were performed after the 45 days SLM treatment. As shown in Figure 4a,b, the SLM-treated 3 × Tg-AD mice exhibited a significantly shorter latency to escape onto the hidden platform, as compared to the control Tg mice on days 3, 4, and 5 (*P < 0.05, n = 19; **P < 0.001, n = 19; ***P < 0.0001, n = 19). Moreover, the SLM-treated 3 × Tg-AD mice traveled to the previously learned platform quadrant more frequently than the control ones, as assessed by the probe test (Figure 4c,d). The time spent in the previously learned platform quadrant was also longer for the treated 3 × Tg-AD mice after 24 and 72 h of the hidden platform test. All these results consistently support that the SLM-treated 3 × Tg-AD mice showed a significant cognitive improvement, not only enhancing learning skills in the hidden platform test but also improving the spatial memory in the probe trial.
Figure 4.
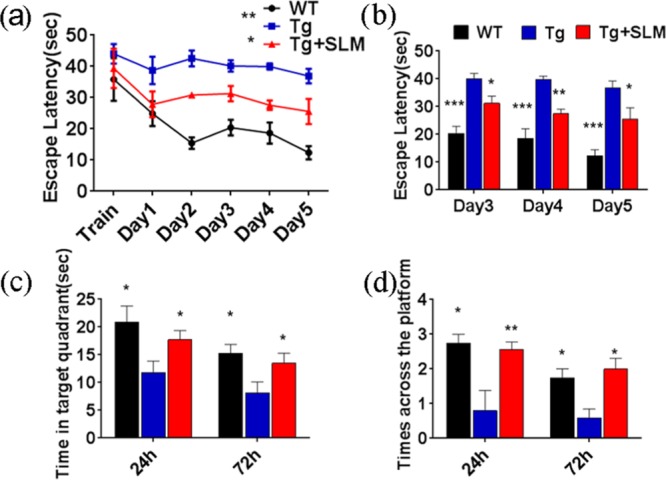
(a, b) Results of hidden platform tests in the Morris water maze experiment for the SLM-treated 3 × Tg-AD mice, the control, and the WT, on training day and days 1–5 (*P < 0.05, n = 19; **P < 0.001, n = 19; ***P < 0.0001, n = 19). (c, d) Results of the probe trial and the time spent in the previously learned platform quadrant for the WT, control, and the treated mice after 24 and 72 h of the hidden platform test. The data were analyzed by two-way analysis of variance (ANOVA).
We also examined the Aβ and tau contents in the brain of the SLM-treated Tg mice via the western blot analysis and immunofluorescence after the cognitive test. Remarkably, the Aβ content substantially decreased, particularly those of the toxic Aβ oligomers with 8, 12, 16, 25, 40, and 55 kDa as the total tau (tau-5) and phosphorylated tau (ps422) proteins in hippocampal regions, as compared with those of the control (see Figure 5a–c,e–g), suggesting that the cyanine inhibitor can ameliorate both Aβ and tau pathologies in the 3 × Tg-AD mouse model. The presence of Aβ and tau, which was visualized by immunostaining in the brain slices of the treated 3 × Tg-AD mice, was obviously reduced compared to those of the untreated 3 × Tg-AD mice (Figure 5d,h). The activity of glycogen synthase kinase-3 (GSK3) can be inhibited by Akt-mediated phosphorylation at Ser9 of GSK3β. The level of inactive phosphorylated glycogen synthase kinase-3β (p-GSK3β) was significantly higher in the SLM-treated Tg mice, as well as the increase in the ratio of p-GSK3β/GSK3β, indicating the reduction of GSK3β activity, which provides a direct insight into the decrease of the phosphorylated tau content (Figure S7c,d). These results also indicate that the reduction of the toxic Aβ oligomers level alleviates the tau dysfunction in the SLM-treated Tg mice.
Figure 5.

Levels of Aβ and tau in the hippocampal and cortical region of SLM-treated 3 × Tg-AD mice were analyzed with the western blot and immunofluorescence. The protein contents of the brain lysates were analyzed with the western blot ((a–c) for oligomeric Aβ, (e–g) for tau, and pS422). Brain slices of the hippocampus and the cortex were immunostained for Aβ (d) and tau (h).
Increasing evidence implies that dysfunctional autophagy plays a crucial role in the AD pathogenesis, such as the failure of Aβ clearance.28,29 Thus, we have investigated whether SLM would modulate autophagic dysfunction. It was found that the levels of amyloid precursor proteins (APPs) and the β-site APP-cleaving enzyme 1 (Bace 1) in the SLM-treated Tg mice were similar to those of the control, suggesting that the Aβ production was not disrupted or suppressed (Figure S7a,b). On the other hand, SLM treatment yielded a significantly reduced level of a key autophagy gatekeeper, phosphorylated form of mammalian target of rapamycin (mTOR), which was concomitant with substantially attenuated phosphorylation of Akt, an upstream effector of mTOR, that modulates its activation as well as a significant increase in a proautophagic protein, the mammalian orthologue of yeast Atg6 (Beclin 1) level, as compared with that of the control (see Figure S8), strongly suggesting autophagic induction.30−32 Furthermore, both the levels of microtubule-associated protein light chain 3-II (LC3-II), which was degraded by lysosomal hydrolases in autophagolysosomes and the LC3-associated p62 protein (sequestosome 1), a marker of autophagic flux, were found to be markedly decreased. The level of the lysosomal protease, cathepsin D (CatD), that mediates the degradation in autophagolysosomes was found to be significantly increased. All these results unanimously support the fact that autophagic pathways were significantly modulated in the SLM-treated 3 × Tg-AD mice relative to the untreated counterparts, resulting in the reduction of Aβ deposits and tau contents as well as recovery of cognitive deficits in the AD mouse model.
Conclusions
A systematic evaluation of carbazole-based cyanine, SLM, including Aβ binding behavior, Aβ1–42 oligomerization inhibition, cytotoxicity, neuroprotective effect, BBB permeability as well as in vivo imaging and efficacy as a novel and effective theranostic agent for diagnosis and treatment of AD was investigated. Remarkably, SLM possessing all of the desirable properties as an in vivo imaging agent can perform NIR imaging of Aβ species in vivo. Furthermore, the SLM-treated 3 × Tg-AD mice showed substantial improvement in cognitive functions, including spatial learning and memory. SLM also exhibited the capability to significantly decrease the oligomeric Aβ contents, as well as reduce t-tau (tau-5) and p-tau protein (ps422) levels in the brain of the 3 × Tg-AD mice, which was attributed to the modulation of autophagic flux. The present study represents the first in vivo evidence that SLM can not only conduct the imaging of Aβ species in vivo but also ameliorate both Aβ and tau pathologies concomitant with a reduction in the levels of toxic Aβ oligomers and p-tau protein. SLM also represents the first effective theranostic agent for the therapy and diagnosis of AD. Our findings open up a new vista to develop more potent theranostic agents for clinical applications in AD.
Materials and Methods
Sample Preparation of Aβ(1–40) Fibril
Stock monomeric Aβ(1–40) (Invitrogen) solution was prepared by dissolving 1 mg of Aβ(1–40) powder in 400 μL of 0.02% ammonia solution without any purification and stored at −20 °C until use. Aβ(1–40) fibril was prepared by diluting the stock monomeric Aβ(1–40) to 50 mM with filtered phosphate buffer solution and incubated at 37 °C for 24 h.
Sample Preparation of Aβ(1–42) Aggregates
Stock monomeric Aβ(1–42) (rPeptide) solution was prepared by dissolving 1 mg of Aβ(1–42) powder in 400 μL of 1% ammonium solution without any purification and stored at −20 °C until use. The aggregate was prepared by diluting the stock monomeric solution to 200 μM with 10 mM filtered HCl and incubated at 37 °C for 6 days.
Fluorescence Titration
SLM solution of 400 and 50 μM Aβ(1–40) monomer/fibril and Aβ(1–42) monomer/aggregate solution were prepared in phosphate buffer. By mixing a corresponding amount of cyanine solution, buffer, and a constant amount of Aβ solution, 20 μL of mixture solution with different SLM-to-Aβ ratios were prepared. Emission measurement was obtained on PTI Time Master C-720.
SDS–PAGE
Monomeric Aβ(1–42) was first treated with 100% hexafluoroisopropanol (HFIP). The residual HFIP was removed under vacuum, and the resulting peptide was stored at −20 °C. The oligomeric Aβ(1–42) was prepared by resuspending the treated monomeric Aβ(1–42) in anhydrous dimethyl sulfoxide (DMSO) with a final concentration of 5 mM. The peptide solution was diluted to a final concentration of 100 μM with F-12 (without phenol red) culture medium and co-incubated with an appropriate amount of SLM solution at 4 °C for 24 h. All samples were then treated with PICUP and followed by SDS–PAGE and Coomassie Blue staining.
Cell Assay Procedures
Cell Culture and Medium
All cell culture reagents were purchased from Gibco. Human neuroblastoma SH-SY5Y cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM)/Ham’s F-12 medium, supplemented with 10% fetal bovine serum. Cells were grown in a humidified cell incubator at 37 °C under a 5% CO2 atmosphere.
Primary Cell Isolation
All animal experiments were performed under the guidelines of Animals (Control of Experiments) Ordinance, approved by the Committee on the Use of Human and Animal Subjects in Teaching and Research of the Hong Kong Baptist University and conformed to The Principles of Laboratory Animal Care (NIH publication no. 86-23, revised 1985). Sprague–Dawley rats were obtained from the Chinese University of Hong Kong. Hippocampus neuronal cells were isolated from 1-day-old Sprague–Dawley rats. Briefly, the brains were placed in cold Hank’s balanced salt solution (HBSS; Gibco), and the hippocampus cells were extracted. The cells were resuspended in DMEM/F-12 medium (Gibco) with 10% fetal bovine serum (Gibco) and papain solution (Sigma-Aldrich) in 2 μg/mL was added into the system. The mixture was incubated at 37 °C for 20 min under gentle shaking. The solution was purified by a 0.4 μm nylon filter (BD falcon), and the filtrate was further centrifuged at 1000 rpm for 5 min. The pellet containing the dissociated neurons was resuspended in neurobasal medium (Gibco) with supplements 2% B-27 (B-27 Supplement 50; Gibco), 0.2% penicillin (PSN; Gibco), and 0.25% glutamax (GlutaMAX-I 100X; Gibco). The centrifugation steps were repeated twice. The cells were plated into a poly-d-lysine (Sigma-Aldrich)-coated plate and incubated in a humidified incubator at 37 °C with 5% CO2 for 7 days before treatment.
Cell Viability Assay
Exponentially growing SH-SY5Y cells were plated onto 96-well plates in cell culture medium at a density of 5 × 105 cells/mL/well. The day after plating, the culture medium was replaced with 0.2% serum containing 24 μL of SLM at a final concentration ranging from 700 nM to 800 μM for determining LC50. After 24 h incubation at 37 °C, the culture medium of each well was replaced with 100 μL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution (0.25 mg/mL; Sigma-Aldrich) and further incubated at 37 °C for 3 h. The MTT solution was then aspirated off, followed by the addition of 100 μL of DMSO (Sigma-Aldrich) to dissolve the formazan crystals. The plates were agitated on a plate shaker for 15 min, and the absorbance at 540 nm with a reference absorbance set to 690 nm was recorded by the microplate reader (BioTek). Wells without cells were referred to as blanks and were subtracted as background from each sample. For the neuroprotective studies on the primary hippocampal cells, SLM at a final concentration of 10 μM was applied together with monomeric Aβ(1–42) at a concentration of 10 μM. Results were expressed as the relative percentage of control. The relative percentage of cytotoxicity was calculated as ((MTTcontrol – MTTbg) – (MTT SLM – MTTbg))/(MTTcontrol – MTTbg) × 100%. Three independent trials were analyzed, and the results were expressed as the mean ± standard error of mean (SEM).
ROS Measurement
Primary hippocampal cells were plated onto a 96 black plate at a density of 5 × 105 cells/mL in cell culture medium per well. The day after plating, the culture medium was replaced with 0.2% serum, containing 100 μM 5-(and-6)-carboxy-2′,7′-dichlorofluorescein diacetate (carboxy-DCFDA) purchased from Invitrogen (Camarillo, CA). The medium was then incubated in dark in a humidified cell incubator at 37 °C under a 5% CO2 atmosphere for 30 min. The medium was removed, and the wells were washed twice with phosphate-buffered saline (PBS). The cells were then resuspended in fresh culture medium and incubated for 30 min. Afterward, the medium was replaced by SLM together with monomeric Aβ(1–42) in 1:1 ratio in HBSS and incubated in dark at 37 °C for 24 h. The fluorescence intensity of cells was measured by a microplate reader (Tecan Schweiz AG, Switzerland) with excitation/emission at 485/530 nm. The values were expressed as fluorescence intensity ratio relative to control (treated with monomeric Aβ(1–42) only).
In Vivo and ex Vivo Study on Blood–Brain Barrier Permeability
First, 9-month-old APP/PS1 Tg mice with age-matching wild-type mice as control were anesthetized under isoflurane gas (2.0 mL/min), supplemented with oxygen (1.0 mL/min) until motionless, then followed by the injection of 100 μL of SLM (5 mg/kg) via the tail vein. Fluorescence imaging of the brain section was captured by an IVIS imaging system at different postinjection time points. All of the images were acquired using an excitation filter at ∼535 nm and an emission filter at ∼600–640 nm, with an exposure time set at 1 s. The resultant images were further analyzed using Living Image software. The normalized fluorescence intensity of brain sections was obtained by dividing the radiant efficiency by the background fluorescence of each mouse [i.e., F(t)/F(pre)] at the selected region of interest. F(t) and F(pre) referred to the fluorescence intensity of the time point of interest and the background fluorescence signal, respectively. For the ThT co-staining assay, Tg mice were sacrificed and the brain tissues of Tg mice were collected after 1 h postinjection of SLM. The brain was then embedded in optimum cutting temperature compound and sliced into 20 μm in thickness for micrography. The brain slices were immersed in a solution cocktail, containing 1.0 μM ThT solution and 10% DMSO for 5 min, then washed with 40% ethanol for 2 min, and rinsed with water for 30 s. Finally, the fluorescence images of the brain slices were recorded by laser scanning confocal microscope, 40× oil-immersion type objective.
Animals and Experimental Procedure
The protocol that we adopted was approved by the Animal Ethical and Welfare Committee of Shenzhen University (Permit Number: AEWC-20140615-002) of China. All operations were performed under ether anhydrous inhalation of anesthesia to minimize suffering.
Animal Model
3 × Tg-AD mice, expressing human gene mutants APPswe, PS1M146V, and tauP301L and wild-type (WT) mice of same genetic background, were purchased from the Jackson laboratory (BarHarbor, ME) for breeding. Male and female mice were kept in a separated cage with ad libitum food and water supply with 12 h light/dark cycle prior to treatment.
Drug Administration
For the drug treatment, the mice were divided into two groups and each group contained ten 8-month-old male mice and nine 8-month- old female mice. Group 1 was the control 3 × Tg mice; they received daily intraperitoneal injection of 100 μL PB, whereas group 2 (3 × Tg mice) received daily intraperitoneal injection of 100 μL SLM at 2.25 mg/kg for 45 days. All procedures followed institutional guidelines and were approved by the local state authorities for animal welfare.
Morris Water Maze Test
After treatment, the spatial reference memory of the mice was examined by Morris water maze. The test generally consisted of three parts: (i) 1 day training, where the mice were placed in a 26–29 °C circular aluminum tank (1.5 m in diameter). The tank was virtually divided into four quadrants (north, east, south, and west) and a visible platform (10 cm in diameter), which is 1 cm higher than the water surface. The mice were placed in one of the quadrants. During the training, the mice could find and escape onto the platform. If the mouse failed to find the platform within 60 s, it was manually guided to the platform and allowed to stay on the platform for 10 s. (ii) After the training, the mice were then placed in the same circular platform to perform a 5-day hidden platform test. (iii) Probe trials were performed on day 7 (24 h) and day 9 (72 h). The platform was removed, and the mice were placed in the quadrant of the pool, which was located opposite to the previous platform quadrant. All of the behavior of the mice, including the distance traveled to the target quadrant and the number of times they passed the platform, was recorded by automated video tracking system, and the data were analyzed by two-way ANOVA.
Protein Extraction from the Brain
After the behavioral study, the mice were killed under anesthesia. Half of the brains were immediately removed, and hippocampus was separated from cortex carefully and the tissues were frozen on ice, stored at −80 °C prior to western blot analysis. To analyze the protein content in the cortex and hippocampus region, the brain sections were first lysed by ultrasonic fragmentation for 1 min in buffer solution (cell lysis buffer for western blot and IP; Beyotime; Shanghai, China) with protease (1:200, PMSF; Beyotime, Shanghai, China) and phosphatase (1:100, PhosSTOP; Roche, Basel, Switzerland) inhibitors. Tissue extracts were centrifuged at 12 000 rpm for 30 min, and the supernatants were collected for further analysis.
Western Blot
The protein content in the brain lysates were determined by the BCA protein assay kit (Beyotime, China), and 30 μg of proteins were resolved in a 12% SDS–PAGE gel, then transferred to a nitrocellulose membrane in the range of 45–90 min, according to molecular weight of the protein. For antibody staining, the membrane was first blocked with 5% skim milk to reduce nonspecific binding, followed by the overnight incubation with the corresponding diluted primary antibodies (see Table S1) at 4 °C. Afterward, the membrane was washed thrice and further incubated with corresponding secondary 1:5000 diluted antibodies, horseradish peroxidase (HRP)-conjugated immunoglobulin (IgG) antimouse (1:5000; Neobioscience, China), and antirabbit (1:5000; Neobioscience, China). The supersignal chemiluminescent substrate (Thermo) was added to visualize HRP activity on Fuji films (GE Healthcare, U.K.). The protein concentrations in the immunoblots were determined by software Quantity One. The statistical significance of the test was performed by using t-test analysis function in GraphPad Prism 5.0. Data were presented as the mean + standard error of mean (SEM) with 95% confidence interval (Table 1).
Table 1. Information of Purchased Antibodies.
| antibody | catalog # | source |
|---|---|---|
| full-length APP | ab2072 | Abcam |
| Bace 1 | 5606 | CSTa |
| Aβ | 9888 | CST |
| tau-5 | MN1000 | Life Technologies |
| ps422 | 2612-1 | Epitomics |
| GSK3β | 9832 | CST |
| p-GSK3β | 5558 | CST |
| PP2A | 2259 | CST |
| mTOR | 2983 | CST |
| p-mTOR | 2971 | CST |
| p70S6k | 2708 | CST |
| p-p70S6k | 9205 | CST |
| Beclin 1 | 3495 | CST |
| LC3 | GTX116080 | GeneTex |
| P62 | GTX1006856 | GeneTex |
| cathepsin D | GTX62063 | GeneTex |
CST = Cell Signaling Technology.
Immunohistochemistry and Immunofluorescence
The brains obtained in the previous section (Materials and Methods section) were bisected in the mid-sagittal plane. The left hemisphere was fixed in 4% formaldehyde for 24–36 h and soaked in different concentrations of ethanol (30, 50, 75, 85, 95, and 100%). Tissues were moved to xylene and were then paraffin-embedded. Immunohistochemistry was performed on 5 μm sagittal paraffin sections. The sections were then treated with 0.3% H2O2 in 0.01 M PBS to block endogenous peroxidases and incubated with 70% formic acid for 3 min. The sections were then treated with fetal calf serum for 15 min at room temperature to block any nonspecific binding, followed by the immunostaining of mouse polyclonal antibodies against Aβ(1–42) (1:300; Cell Signaling Technology) overnight at 4 °C. After incubation, the slices were rinsed thrice with PBS. Subsequently, tissues were incubated with Alexa Fluor 488-conjugated Fab fragments of goat antimouse immunoglobulin (IgG) (H_L) for 2 h (1:500; Invitrogen). After several washes, tissues were mounted using an antifading mounting medium and air-dried away from light for confocal imaging. For immune-fluorescent staining, slices were incubated with tau-5 specific primary antibody (1:300, Abcam) overnight at 4 °C, followed by high-temperature heating to repair antigen. After incubation, the slices were rinsed thrice with PBS. Tissues were then incubated with Alexa Fluor 488-conjugated Fab fragments of goat antimouse immunoglobulin (IgG) (H_L) for 2 h (1:500; Invitrogen). After several washes, tissues were mounted using an antifading mounting medium and air-dried for confocal imaging.
Acknowledgments
This work was supported by the Health and Medical Research Fund (HMRF 01122066) from the Hong Kong Government, and Collaborative Research Fund of Hong Kong Research Grant Council (C2012-15G). We are also thankful for the support of the National Natural Sciences Foundation of China (nos 81400847 and 21675135).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b00475.
Synthesis of SLM; physical properties of SLM; fluorescence titration; oligomerization inhibition; MTT assay of SLM; retention of SLM in brain; body weights of mice after treatment; swimming speed of mice; expression level of p-GSK3β, GSK3β, APP, and Bace 1; expression of autophagy-related proteins; NMR spectra of SLM (PDF)
Author Contributions
∥ Y.L., C.C., and D.X. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Qian X.; Hamad B.; Dias-Lalcaca G. The Alzheimer disease market. Nat. Rev. Drug Discovery 2015, 14, 675–676. 10.1038/nrd4749. [DOI] [PubMed] [Google Scholar]
- Wood H. Alzheimer disease: Evidence for trans-synaptic and exo-synaptic tau propagation in Alzheimer disease. Nat. Rev. Neurol. 2015, 665. 10.1038/nrneurol.2015.205. [DOI] [PubMed] [Google Scholar]
- Serrano-Pozo A.; Hyman B. T. Alzheimer Disease: Alzheimer dementia with sparse amyloid[mdash]AD mimic or variant?. Nat. Rev. Neurol. 2015, 674. 10.1038/nrneurol.2015.221. [DOI] [PubMed] [Google Scholar]
- Ittner L. M.; Götz J. Amyloid-β and tau — a toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 2011, 12, 67–72. 10.1038/nrn2967. [DOI] [PubMed] [Google Scholar]
- Hampel H.; Mitchell A.; Blennow K.; Frank R. A.; Brettschneider S.; Weller L.; Moller H. J. Core biological marker candidates of Alzheimer’s disease - perspectives for diagnosis, prediction of outcome and reflection of biological activity. J. Neural Transm. 2004, 111, 247–272. 10.1007/s00702-003-0065-z. [DOI] [PubMed] [Google Scholar]
- Panza F.; Frisardi V.; Solfrizzi V.; Imbimbo B. P.; Logroscino G.; Santamato A.; Greco A.; Seripa D.; Pilotto A. Immunotherapy for Alzheimer’s disease: from anti-beta-amyloid to tau-based immunization strategies. Immunotherapy 2012, 4, 213–238. 10.2217/imt.11.170. [DOI] [PubMed] [Google Scholar]
- Bates K. A.; Verdile G.; Li Q. X.; Ames D.; Hudson P.; Masters C. L.; Martins R. N. Clearance mechanisms of Alzheimer’s amyloid-[beta] peptide: implications for therapeutic design and diagnostic tests. Mol. Psychiatry 2009, 14, 469–486. 10.1038/mp.2008.96. [DOI] [PubMed] [Google Scholar]
- Mandavilli A. The amyloid code. Nat. Med. 2006, 12, 747–751. 10.1038/nm0706-747. [DOI] [PubMed] [Google Scholar]
- Ladiwala A. R. A.; Litt J.; Kane R. S.; Aucoin D. S.; Smith S. O.; Ranjan S.; Davis J.; Van Nostrand W. E.; Tessier P. M. Conformational Differences between Two Amyloid beta Oligomers of Similar Size and Dissimilar Toxicity. J. Biol. Chem. 2012, 287, 24765–24773. 10.1074/jbc.M111.329763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani M. Structural features and cytotoxicity of amyloid oligomers: Implications in Alzheimer’s disease and other diseases with amyloid deposits. Prog. Neurobiol. 2012, 99, 226–245. 10.1016/j.pneurobio.2012.03.002. [DOI] [PubMed] [Google Scholar]
- Musiek E. S.; Holtzman D. M. Three dimensions of the amyloid hypothesis: time, space and ‘wingmen’. Nat. Neurosci. 2015, 800–806. 10.1038/nn.4018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller E. C.; Teravskis P. J.; Dummer B. W.; Zhao X. H.; Huganir R. L.; Liao D. Z. Tau phosphorylation and tau mislocalization mediate soluble A beta oligomer-induced AMPA glutamate receptor signaling deficits. Eur. J. Neurosci. 2014, 39, 1214–1224. 10.1111/ejn.12507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacobini E.; Gold G. Alzheimer disease therapy[mdash]moving from amyloid-[beta] to tau. Nat. Rev. Neurol. 2013, 9, 677–686. 10.1038/nrneurol.2013.223. [DOI] [PubMed] [Google Scholar]
- Rajasekhar K.; Narayanaswamy N.; Murugan N. A.; Kuang G. L.; Agren H.; Govindaraju T. A High Affinity Red Fluorescence and Colorimetric Probe for Amyloid beta Aggregates. Sci. Rep. 2016, 6, 23668 10.1038/srep23668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasekhar K.; Narayanaswamy N.; Murugan N. A.; Viccaro K.; Lee H. G.; Shah K.; Govindaraju T. A beta plaque-selective NIR fluorescence probe to differentiate Alzheimer’s disease from tauopathies. Biosens. Bioelectron 2017, 98, 54–61. 10.1016/j.bios.2017.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasekhar K.; Suresh S. N.; Manjithaya R.; Govindaraju T. Rationally Designed Peptidomimetic Modulators of A beta Toxicity in Alzheimer’s Disease. Sci. Rep. 2015, 5, 8139 10.1038/srep08139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasekhar K.; Narayanaswamy N.; Mishra P.; Suresh S. N.; Manjithaya R.; Govindaraju T. Synthesis of Hybrid Cyclic Peptoids and Identification of Autophagy Enhancer. ChemPlusChem 2014, 79, 25–30. 10.1002/cplu.201300343. [DOI] [PubMed] [Google Scholar]
- Rajasekhar K.; Chakrabarti M.; Govindaraju T. Function and toxicity of amyloid beta and recent therapeutic interventions targeting amyloid beta in Alzheimer’s disease. Chem. Commun. 2015, 51, 13434–13450. 10.1039/C5CC05264E. [DOI] [PubMed] [Google Scholar]
- Citron M. Strategies for disease modification in Alzheimer’s disease. Nat. Rev. Neurosci. 2004, 5, 677–685. 10.1038/nrn1495. [DOI] [PubMed] [Google Scholar]
- Sheridan C. J&J’s billion dollar punt on anti-amyloid antibody. Nat. Biotechnol. 2009, 27, 679–681. 10.1038/nbt0809-679. [DOI] [PubMed] [Google Scholar]
- Wang Q. M.; Yu X.; Li L. Y.; Zheng J. Inhibition of Amyloid-beta Aggregation in Alzheimer’s Disease. Curr. Pharm. Des. 2014, 20, 1223–1243. 10.2174/13816128113199990068. [DOI] [PubMed] [Google Scholar]
- Yang W.; Wong Y.; Ng O. T. W.; Bai L.-P.; Kwong D. W. J.; Ke Y.; Jiang Z.-H.; Li H.-W.; Yung K. K. L.; Wong M. S. Inhibition of Beta-Amyloid Peptide Aggregation by Multifunctional Carbazole-Based Fluorophores. Angew. Chem., Int. Ed. 2012, 51, 1804–1810. 10.1002/anie.201104150. [DOI] [PubMed] [Google Scholar]
- Guo L.; Chan M. S.; Xu D.; Tam D. Y.; Bolze F.; Lo P. K.; Wong M. S. Indole-based Cyanine as a Nuclear RNA-Selective Two-Photon Fluorescent Probe for Live Cell Imaging. ACS Chem. Biol. 2015, 10, 1171–1175. 10.1021/cb500927r. [DOI] [PubMed] [Google Scholar]
- Yang W.; Chan P. S.; Chan M. S.; Li K. F.; Lo P. K.; Mak N. K.; Cheah K. W.; Wong M. S. Two-photon fluorescence probes for imaging of mitochondria and lysosomes. Chem. Commun. 2013, 49, 3428–3430. 10.1039/c3cc41240g. [DOI] [PubMed] [Google Scholar]
- Li Y.; Xu D.; Ho S.-L.; Li H.-W.; Yang R.; Wong M. S. A theranostic agent for in vivo near-infrared imaging of β-amyloid species and inhibition of β-amyloid aggregation. Biomaterials 2016, 94, 84–92. 10.1016/j.biomaterials.2016.03.047. [DOI] [PubMed] [Google Scholar]
- Barnham K. J.; Masters C. L.; Bush A. I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discovery 2004, 3, 205–214. 10.1038/nrd1330. [DOI] [PubMed] [Google Scholar]
- LaFerla F. M. Calcium dyshomeostasis and intracellular signalling in alzheimer’s disease. Nat. Rev. Neurosci. 2002, 3, 862–872. 10.1038/nrn960. [DOI] [PubMed] [Google Scholar]
- Ghavami S.; Shojaeid S.; Yeganeh B.; Ande S. R.; Jangamreddy J. R.; Mehrpour M.; Christoffersson J.; Chaabane W.; Moghadam A. R.; Kashani H. H.; Hashemi M.; Owji A. A.; Los M. J. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol. 2014, 112, 24–49. 10.1016/j.pneurobio.2013.10.004. [DOI] [PubMed] [Google Scholar]
- Nixon R. A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- Boland B.; Kumar A.; Lee S.; Platt F. M.; Wegiel J.; Yu W. H.; Nixon R. A. Autophagy induction and autophagosome clearance in neurons: Relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 2008, 28, 6926–6937. 10.1523/JNEUROSCI.0800-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita M.; Young A. R. J.; Arakawa S.; Samarajiwa S. A.; Nakashima T.; Yoshida S.; Hong S.; Berry L. S.; Reichelt S.; Ferreira M.; Tavare S.; Inoki K.; Shimizu S.; Narita M. Spatial Coupling of mTOR and Autophagy Augments Secretory Phenotypes. Science 2011, 332, 966–970. 10.1126/science.1205407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q.; Fan W.; Chen K.; Ding X.; Chen S.; Zhong Q. Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 19211–19216. 10.1073/pnas.0810452105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.