

Abstract

ω-Transaminases are attractive biocatalysts for the production of chiral amines. These enzymes usually have a broad substrate range. Their substrates include hydrophobic amines as well as amino acids, a feature referred to as dual-substrate recognition. In the present study, the reaction mechanism for the half-transamination of l-alanine to pyruvate in (S)-selective Chromobacterium violaceum ω-transaminase is investigated using density functional theory calculations. The role of a flexible arginine residue, Arg416, in the dual-substrate recognition is investigated by employing two active-site models, one including this residue and one lacking it. The results of this study are compared to those of the mechanism of the conversion of (S)-1-phenylethylamine to acetophenone. The calculations suggest that the deaminations of amino acids and hydrophobic amines follow essentially the same mechanism, but the energetics of the reactions differ significantly. It is shown that the amine is kinetically favored in the half-transamination of l-alanine/pyruvate, whereas the ketone is kinetically favored in the half-transamination of (S)-1-phenylethylamine/acetophenone. The calculations further support the proposal that the arginine residue facilitates the dual-substrate recognition by functioning as an arginine switch, where the side chain is positioned inside or outside of the active site depending on the substrate. Arg416 participates in the binding of l-alanine by forming a salt bridge to the carboxylate moiety, whereas the conversion of (S)-1-phenylethylamine is feasible in the absence of Arg416, which here represents the case in which the side chain of Arg416 is positioned outside of the active site.
I. Introduction
Transaminases (TAs) catalyze the reversible interchange of amino and keto groups, by the use of the coenzyme pyridoxal-5′-phosphate (PLP).1 There are two types of TAs, α-transaminases, which convert α-amino and α-keto acids, and ω-transaminases (ωTAs), which also accept amino and keto acids in which the amino or keto group is in a non-α position relative to the carboxyl group, called ω-amino or ω-keto acids, respectively.2−4 Some ωTAs additionally have the ability to enantioselectively convert a variety of different amines and ketones without a carboxyl group.2−7 Consequently, they are also called amine transaminases in the literature.3,4,8,9 ωTAs have received much attention in the last decade as they can be used as biocatalysts for the production of chiral amines.10−18 One example of such a biocatalytically useful ωTA is the (S)-selective ωTA ((S)-ωTA) from Chromobacterium violaceum (Cv-ωTA). Cv-ωTA has a broad substrate range, converting both hydrophobic amines and amino acids, such as (S)-1-phenylethylamine ((S)-1-PEA) and l-alanine (l-Ala), respectively,19 and has been applied successfully for the production of optically pure amines.20
Many PLP-dependent enzymes, including TAs, require the formation of a so-called internal aldimine (E-PLP), before the main reaction. In this process, the PLP coenzyme becomes covalently bound to an active-site lysine residue as a protonated Schiff base, which constitutes the active form of the enzyme.3,10 The transamination reaction consists of two half-transamination equilibria, as shown in Scheme 1. An amino donor is converted to the corresponding ketone or aldehyde, resulting in the conversion of E-PLP to pyridoxamine-5′-phosphate (PMP). The E-PLP is then regenerated by the reaction of another ketone or aldehyde, which is then converted to an amine.21,22
Scheme 1. Complete Reaction Cycle of the ωTA-Catalyzed Transamination of (S)-1-PEA to Acetophenone and Pyruvate to l-Ala.

The box represents stereoselective synthesis, where (S)-1-PEA is produced by the use of l-Ala as amino donor and the prochiral acetophenone as amino acceptor. In kinetic resolution, (R)-1-PEA can be isolated from racemic 1-PEA as the (S)-enantiomer is consumed.
In biocatalytic applications, chiral amines may be kinetically resolved by applying an ωTA and a keto acid, for example, pyruvate.12,14,18 In cases where the enantiospecificity is high, essentially only one enantiomer of the amino substrate is consumed to produce the corresponding ketone and alanine, and the other enantiomer of the amino substrate can then be isolated in an enantiopure form. For stereoselective synthesis, alanine, for example, can be used as an amino donor for a prochiral ketone, to produce an enantiopure target amine.11,12,14,18 The kinetic resolution option is limited to a maximum theoretical conversion of 50%, whereas the stereoselective synthesis may give a full conversion but then usually requires equilibrium displacement.11−14,18
In TAs, two structurally and chemically different substrates (e.g., a hydrophobic and a hydrophilic substrate) are converted in a subsequent manner, as shown in Scheme 1. The feature of recognizing two different substrates while discriminating against other substrates using the same active site is called dual-substrate recognition or dual specificity.4,23−25 Various proposals have been put forward as to how this can be achieved in different TAs.4,23−25 For (S)-ωTAs, like Cv-ωTA, it has been suggested that a flexible arginine residue is responsible for the binding of acidic substrates having a carboxylate functional group, and forms thus the structural basis for the dual-substrate recognition.9 The side chain of this arginine, located near the channel to the active site, is found to be oriented in different directions in the crystal structures of several known (S)-ωTAs, indicating that it is highly flexible (see Figure 1).
Figure 1.
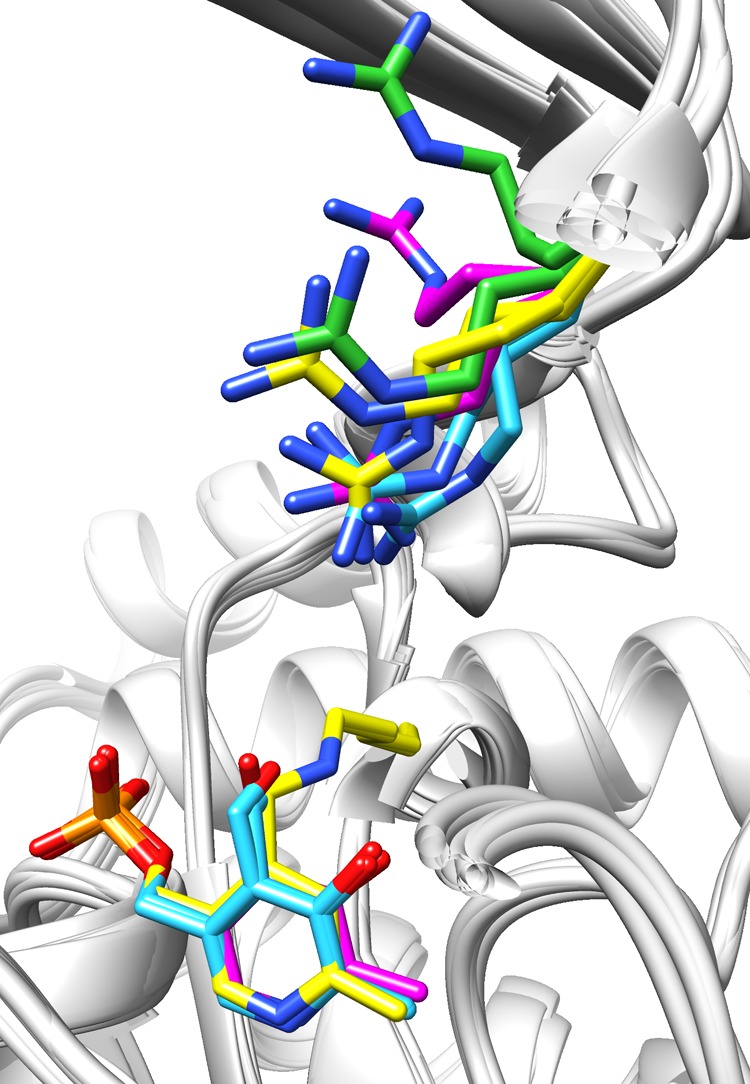
Superposition of crystal structures of several (S)-ωTAs with the flexible arginine residue (Arg416 in Cv-ωTA) and the PLP coenzyme (PLP covalently bound to Lys288 in Cv-ωTA) in stick representation. In PDB 4A6T/Cv-ωTA (yellow), the side chain of the arginine residue has two different orientations in the two monomers (chains A and B); the same is observed in PDB 3HMU/Silicibacter pomeroyi (green) and PDB 3I5T/Rhodobacter sphaeroides KD131 (cyan). In PDB 3FCR/Silicibacter sp. TM1040 (magenta), the side chain of the arginine residue has two different orientations in the same monomer.
By applying molecular docking and molecular dynamics simulations to the crystal structure of (S)-ωTA from S. pomeroyi (PDB 3HMU), Steffen-Munsberg et al. showed that the arginine residue (Arg417) may form a salt bridge with the carboxylate group of α- and ω-amino acids, such as l-Ala and γ-aminobutyrate, respectively, if the side chain of the arginine points toward the PLP.9 On the other hand, hydrophobic amines, such as (S)-1-PEA, may be accommodated in the active site if the side chain of the arginine is oriented away from the PLP.9 Similar results were obtained in a molecular docking study using the crystal structure of (S)-ωTA from Paracoccus denitrificans (PDB 4GRX).26 Furthermore, in the crystal structure of Cv-ωTA with suicide inhibitor gabaculine (PDB 4BA5), it is seen that the flexible arginine residue (Arg416) forms a salt bridge with the carboxylate group of the inhibitor.27
The idea of an arginine switch for dual-substrate recognition has also been proposed for other TAs, for example, aromatic amino acid TAs,28 aspartate TAs,29−31 and recently also for (R)-selective ωTAs.32−34 These enzymes have different active-site architectures as compared to those of (S)-ωTAs, and consequently the proposed flexible arginine is in different positions.9,25,32
Recently, we have described by density functional theory (DFT) calculations the detailed mechanism of the half-transamination reaction for the conversion of (S)-1-PEA to acetophenone in Cv-ωTA.35 On the basis of a crystal structure of the holo enzyme containing the PLP coenzyme covalently bound to an active-site lysine residue (E-PLP), a large active-site model was designed, and the involved transition states and intermediates were characterized. The calculations showed that the amino substrate becomes covalently bound to PLP, replacing the lysine residue, through geminal diamine intermediates, to form the so-called external aldimine. The exchange of the amino group of the substrate to a keto group then proceeds through the deprotonation of the external aldimine by the lysine residue, forming a planar quinonoid intermediate. Subsequent proton transfers yield a ketimine, to which water is added to form a hemiaminal. In the final steps, proton rearrangements and cleavage of the carbon–nitrogen bond yield the half-transamination products, ketone and PMP, which are no longer covalently bound to each other. These steps are shown in Scheme 2, for the l-alanine substrate, which is the subject of the current study.
Scheme 2. Reaction Mechanism for the Half-Transamination of l-Ala to Pyruvate in Cv-ωTA.

In the present study, we use DFT calculations to investigate the reaction mechanism for the half-transamination of l-Ala to pyruvate in Cv-ωTA. Together with the previous study,35 the complete transamination cycle of (S)-1-PEA to acetophenone and pyruvate to l-Ala is thus described. On the basis of the geometries of the intermediates and transition states, and the energy profiles of the reaction mechanism obtained in the current and previous works, we examine and discuss the differences and similarities in the conversions of hydrophobic amines and amino acids in ωTAs. We also examine the role of the flexible arginine residue in the dual-substrate recognition by employing two active-site models, one including this residue and one lacking it. The calculated energy profiles are evaluated, and the effect of the flexible arginine residue on the energetics of the reaction is assessed.
II. Results and Discussion
II.I. Active-Site Models
In this work, two active-site models of Cv-ωTA are employed, one including the flexible Arg416 residue and one lacking it. The models were generated from the crystal structure of the holo enzyme (PDB 4A6T(36)) and include the l-Ala and water substrates, the amino acids that make up the binding site (Phe22, Leu59, Trp60, Phe88′, Tyr153, Ile262, and Thr321′, and Gly230 and Ala231 and the backbone between them), a part of the PLP coenzyme covalently bound to the active-site lysine residue Lys288 (E-PLP), and Asp259 that forms a hydrogen bond to the nitrogen of PLP. In the model that includes Arg416, this residue was taken from the chain A monomer of the enzyme, and the guanidinium group was manually turned to face the active site, thus forming a salt bridge to the carboxylate group of the l-Ala substrate (see Figure 2c). The model without the Arg416 residue (Figure 2b) is thus identical to the one used in the previous study (Figure 2a),35 but with l-Ala as a substrate instead of (S)-1-PEA.
Figure 2.
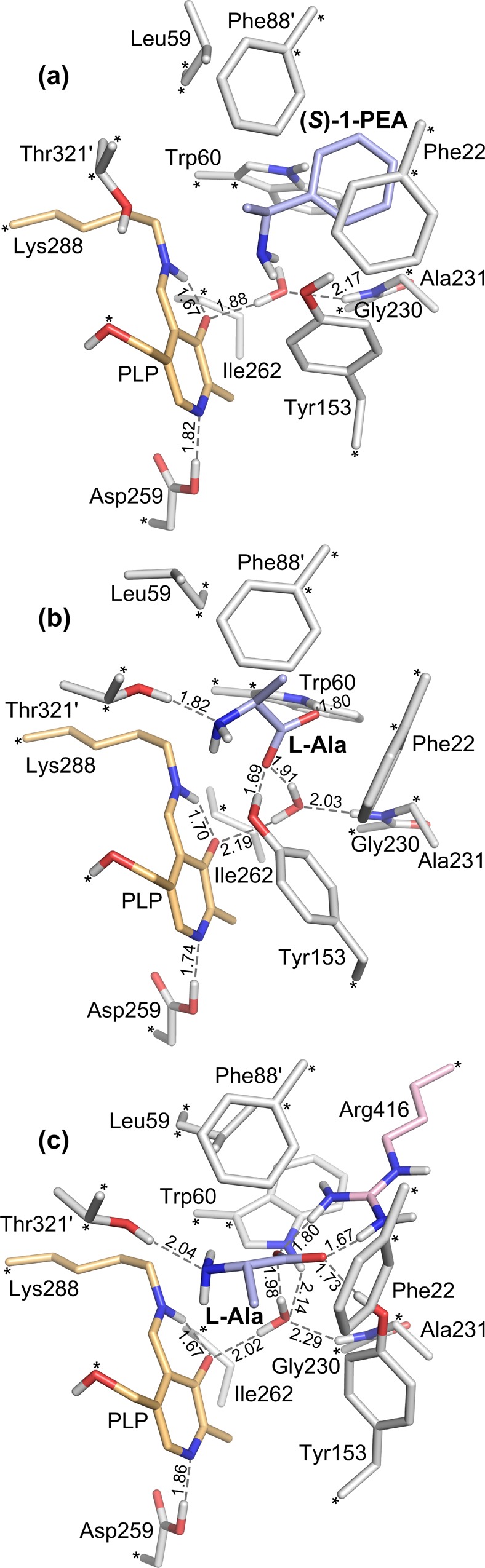
Optimized structures of the Cv-ωTA active-site models (ES) with (a) the (S)-1-PEA substrate, (b) the l-Ala substrate, and (c) the l-Ala substrate and additionally the flexible arginine residue, Arg416. In ES, the PLP coenzyme is covalently bound to Lys288, referred to as the internal aldimine (E-PLP). Asterisks indicate atoms that were kept fixed at their crystallographic positions during the geometry optimizations, and primes indicate residues from one of the two subunits in the homodimer. Color scheme for the atoms is as follows: light blue for carbons of the substrate, orange for carbons of the PLP-Lys288 moiety, pink for carbons of Arg416, red for oxygens, blue for nitrogen, and white for hydrogens. Nonpolar hydrogen atoms have been omitted. Distances are given in angström.
The various residues were truncated at either the α- or β-carbons, as shown in Figure 2, and all hydrogen atoms were added manually. The phosphate group of PLP was not included, apart from the bridging oxygen, saturated with a hydrogen atom. The alanine substrate in solution is mainly in the zwitterionic form. However, because the reaction starts with the amino group of the substrate making a nucleophilic attack on the internal aldimine (E-PLP), l-Ala has to lose a proton on entry into the active site such that the transamination reaction can take place. Therefore, the l-Ala substrate was modeled in the deprotonated state in the active site, that is, with a neutral amino group and a negatively charged carboxylate group.
A number of atoms were kept fixed to their crystallographic positions during the geometry optimizations to preserve the overall structure of the active site. These atoms are indicated by asterisks in Figure 2. The atom fixation is a standard procedure in the cluster approach. The fixed atoms were chosen on the basis of initial preliminary geometry optimizations of one of the active-site models, to avoid large artificial movements as compared to the crystal structure. The residues of the active-site models are granted enough flexibility to adapt to the specific substrate studied, be it (S)-1-PEA or l-Ala. This is evident from the optimized geometries of the enzyme–substrate complexes shown in Figure 2, where the residues (e.g., Tyr153, Trp60, Thr321, and Leu59) move to accommodate the substrates in a favorable manner.
The active-site models with and without Arg416 consist of 211 and 189 atoms, respectively, and have total charges of 0 and −1, respectively.
II.II. Mechanism Including Arg416
The reaction mechanism of the Cv-ωTA-catalyzed half-transamination of l-Ala to pyruvate obtained on the basis of the present calculations is presented in Scheme 2. The calculated energy profiles of this reaction using the two active-site models, that is, with and without the flexible arginine residue (see above), are shown in Figure 3 together with the results from the previous study, describing the half-transamination of (S)-1-PEA to acetophenone.35 Optimized structures of selected intermediates and transition states are shown in Figure 4. First, we focus on examining the mechanism of the conversion of l-Ala to pyruvate compared to the previously described mechanism of the conversion of (S)-1-PEA to acetophenone.35 As it turned out, the active-site model that includes the flexible arginine residue, Arg416, resulted in the mechanism with the most plausible energies (Scheme 2). In this model, Arg416 forms a salt bridge to the carboxylate group of l-Ala. Hence, Arg416 participates in the binding of acidic substrates, that is, substrates having a carboxylate functional group. The role of Arg416 in the dual-substrate recognition, evaluated on the basis of the energetics of the two active-site models (Figure 3), will be discussed in the next subsection.
Figure 3.

Calculated energy profiles of the Cv-ωTA-catalyzed half-transamination reaction of l-Ala to pyruvate, employing two active-site models, with Arg416 (blue) and without Arg416 (red), along with the previously published results for (S)-1-PEA to acetophenone (black) using the active-site model without Arg416.
Figure 4.

Optimized structures of selected stationary points along the reaction pathway in the Cv-ωTA active-site model that includes Arg416. Some residues and most nonpolar hydrogens have been omitted for clarity. The nonpolar hydrogens on the carbons adjacent to the substrate nitrogen are shown when relevant.
The obtained detailed reaction mechanism, presented in Scheme 2, reveals that the deamination of the amino acid, l-Ala, follows essentially the same mechanism as the deamination of the hydrophobic amine, (S)-1-PEA, with some minor changes that are indicated below. There are, however, significant differences in the calculated energies of intermediates and transition states, which will have implications on the kinetics of the reaction.
The first difference in the reaction of l-Ala compared to that of (S)-1-PEA is already seen in the enzyme–substrate complex (ES). By comparing the geometries of ES for l-Ala (Figure 2c) and (S)-1-PEA (Figure 2a), it can be noted that there are differences in the hydrogen-bonding network. Because of the existence of a carboxylate moiety in l-Ala, several hydrogen bonds to the surrounding residues and co-substrate water arise. The l-Ala substrate is positioned such that the carboxylate group forms a salt bridge with the guanidinium group of Arg416 as well as hydrogen bonds to Tyr153 and the water molecule. This water molecule is further hydrogen-bonded to Trp60, the oxygen of PLP, and the backbone nitrogen of Ala231. The amino group of l-Ala is hydrogen-bonded to Thr321. The salt bridge to Arg416 and the hydrogen bond to Tyr153 are maintained throughout the entire reaction pathway.
As described in Introduction, in the initial steps of the transamination reaction, the active-site lysine residue, Lys288, which is covalently bound to the PLP coenzyme as a protonated Schiff base, is replaced by the amino substrate, which becomes covalently bound to PLP, generating a new Schiff base (see Scheme 2). The former is called the internal aldimine (E-PLP), and the latter is called the external aldimine. From the calculated energy profiles in Figure 3, it can be concluded that the initial steps of the reaction, from the internal aldimine (ES) to the formation of the external aldimine at TS3, proceed through transition states of low energies (≤10.6 kcal/mol relative to ES) in the reactions with both l-Ala and (S)-1-PEA.
In the reaction with l-Ala, the initial nucleophilic attack by the amino group of the substrate on the internal aldimine (ES → Int1a) and the proton transfer between the amino group of the substrate and the oxygen of PLP (Int1a → Int1b) can take place as either two separate steps or a single step accompanied by a concerted proton shuttling by the water molecule. The energy difference between the two scenarios is very small, with the stepwise case (via TS1a and TS1b) being only 0.5 kcal/mol lower than the concerted case. Similarly, in the reaction with (S)-1-PEA, two alternative concerted transition states were located for the transformation ES → Int1b.35 In that case, the proton transfer can occur directly or with the assistance of the water molecule in the same manner as for l-Ala. The latter path was found to be 1.4 kcal/mol lower in energy. Thus, for both l-Ala and (S)-1-PEA, the energy differences are very small, and the two cases are basically indistinguishable. However, for consistency, in Figure 3 we report the transition state with the slightly lower energy for each substrate.
In the external aldimine (Int3) for l-Ala, the active-site lysine residue, Lys288, and the water substrate are positioned in a similar manner to that in Int3 for (S)-1-PEA. In the newly created hydrogen-bonding network, Lys288 is hydrogen-bonded to the oxygen of PLP and the water molecule, which, in turn, is hydrogen-bonded to Thr321 and the carboxylate group of the substrate (see Figure 4). The latter hydrogen bond is missing in the reaction with (S)-1-PEA as it has a benzene ring instead of a carboxylate group. This hydrogen-bonding network is essentially maintained in the following reaction steps until the water molecule is consumed in the formation of the hemiaminal at TS7. For example, in the ketimine (Int6), the same hydrogen-bonding network as in Int3 is observed, whereas in the hemiaminal (Int7), Lys288 and Thr321 are hydrogen-bonded to the newly formed hydroxyl group (see Figure 4).
Interestingly, there is a difference in the relative energies of Int3 and Int6 in the two half-transamination reactions (see Figure 3). In the reaction with l-Ala, Int3 is calculated to be 2.8 kcal/mol lower in energy than ES, that is, the external aldimine, Int3, is more stable than the internal aldimine, ES. On the other hand, in the reaction with (S)-1-PEA, they are of equal energy. At the ketimine Int6, the opposite trend is observed. In the reaction with (S)-1-PEA, Int6 is 2.1 kcal/mol lower in energy than ES, whereas they are of equal energy in the reaction with l-Ala. How this difference in the relative energies of Int3 and Int6 influences the kinetics of the reactions will be discussed below.
The exchange of the amino group of the substrate to a keto group in the half-transamination product is initiated at Int3 and proceeds through several transition states of relatively high-energy barriers as compared to those in the initial steps. In both reactions, with l-Ala and (S)-1-PEA, the three TSs (TS4, TS6, and TS7) are of similar heights (see Figure 3). TS4 is the deprotonation of the external aldimine by Lys288 with the assistance of the water molecule as a proton shuttle, TS6 is the protonation of the planar quinonoid intermediate by Lys288, and TS7 is the addition of water to the ketimine intermediate with the assistance of Lys288 as a proton shuttle (see Scheme 2 and Figure 4).
In the reaction with (S)-1-PEA, the calculated energies of TS4 and TS6 are 19.3 and 15.3 kcal/mol, respectively, relative to Int3, whereas TS7 has a calculated energy of 14.6 kcal/mol, relative to Int6. Hence, TS4 was concluded to be the rate-determining step in this reaction.35 For l-Ala, the calculated energies of TS4, TS6, and TS7 relative to Int3 are 21.3, 21.7, and 20.2 kcal/mol, respectively. The difference in energy between these TSs is thus very small, certainly within the expected error of the adopted methodology. It is therefore not possible on the basis of the current calculations to determine which one of these TSs is rate-limiting, or whether one, two, or all three of them contribute to the observed rate of the reaction. A rather small change in the energy of any of these three TSs would change the conclusion regarding this issue.
As seen in Figure 3, the final steps of the reaction, which yield the ketone product and PMP (E:PMP), also proceed through transition states of low energies (≤7.6 kcal/mol relative to ES), similarly to the initial steps of the reaction. In the reaction with l-Ala, the proton rearrangements with the assistance of Lys288 as a proton shuttle and the cleavage of the carbon–nitrogen bond in the hemiaminal were found to occur concertedly in TS8, as shown in Scheme 2. In the reaction with (S)-1-PEA, this transformation was previously found to be a stepwise process.35
As mentioned above, there is a difference in the relative energies of Int3 and Int6 in the two half-transamination reactions, which influences the kinetics of the reactions. In the reaction with (S)-1-PEA, the lowest energy point was found to be the ketimine Int6, at −2.1 kcal/mol relative to ES, which gives an overall energy barrier of 21.1 kcal/mol for the reverse reaction (Int6 → TS4). Thus, in the half-transamination of (S)-1-PEA/acetophenone, the ketone is kinetically favored, as the energy barrier of the forward reaction is 1.8 kcal/mol lower than that of the reverse reaction.35 In contrast, for the half-transamination of l-Ala/pyruvate, the opposite is observed, that is, the amine is kinetically favored. Here, the lowest energy point is the external aldimine, Int3, at −2.8 kcal/mol relative to ES, which results in an overall energy barrier of 18.7 kcal/mol for the reverse reaction (Int6 → TS6), that is, 3 kcal/mol lower than the energy barrier of the forward reaction (21.7 kcal/mol). These observations are consistent with experimental findings. Thus, it has been shown that ωTAs are suitable for the kinetic resolution of racemic 1-PEA using pyruvate as amino acceptor because of a high deamination reaction rate from (S)-1-PEA and a high amination reaction rate to pyruvate.37 That is, acetophenone is the kinetically favored product in the first half-transamination reaction, and l-Ala is favored in the second half, which effectively regenerates the holo enzyme (E-PLP).
II.III. Mechanism without Arg416
In the above-presented mechanism for the reaction with l-Ala (Scheme 2), Arg416 forms a salt bridge to the carboxylate group of l-Ala and thus participates in the binding of the substrate. To evaluate the role of Arg416 as an arginine switch, we employed an active-site model without this residue and calculated the energy profile for the entire reaction. The effect of Arg416 on the energetics of the reaction is assessed by analyzing the energy profiles of the two active-site models, with and without Arg416, as presented in Figure 3. It is found that the deamination of l-Ala follows the same mechanism with and without Arg416, the only exception being the step Int7 → TS8 → E:PMP. In the reaction of l-Ala without Arg416, the proton transfer from the oxygen of PLP to the nitrogen of the hemiaminal occurs as a separate step before TS8, similarly to the reaction with (S)-1-PEA.35 However, the calculations show that the overall energy barrier without Arg416 is 25.2 kcal/mol (ES → TS6), which is 3.5 kcal/mol higher than the overall calculated energy barrier for l-Ala with Arg416 (Int3 → TS6). This corresponds to a more than 100-fold slower reaction.
The high overall energy barrier in the reaction of l-Ala without Arg416 stems from the fact that the enzyme–substrate complex (ES) is the lowest energy point, that is, 3.0 kcal/mol lower in energy than the external aldimine, Int3. As seen in Figure 2b, when Arg416 is not present to bind to the carboxylate moiety of the substrate, l-Ala is positioned such that the carboxylate group is oriented toward PLP and the water molecule, rather than toward the opening of the active site. l-Ala is further hydrogen-bonded to Thr321, Trp60, Tyr153, and the water molecule. This energetically favorable conformation found for ES without Arg416 represents a nonproductive binding mode, as a repositioning of the substrate and a movement of Tyr153 are required before the first reaction step, which is the nucleophilic attack by the amino group of the substrate on the internal aldimine. An alternative conformation of ES, in which the l-Ala substrate is more suitably positioned for the first reaction step, similarly to that in ES with Arg416 (see optimized structure in the Supporting Information), was found to be 6.6 kcal/mol higher in energy than ES, that is, 3.6 kcal/mol higher in energy than Int3.
The results above are consistent with the mutagenesis experiments on (S)-ωTAs from R. sphaeroides KD131 and Silicibacter sp. TM1040.9 It has thus been shown that mutating the flexible arginine residue (Arg418 and Arg420, respectively) to alanine results in a significantly decreased transamination activity toward (S)-1-PEA in combination with acidic amino acceptors (pyruvate or succinate semialdehyde), and a hardly affected transamination activity when using an aliphatic amino acceptor (butyraldehyde).9 As mentioned above, it is proposed that the side chain of the arginine residue adopts a stretched conformation to form a salt bridge to the carboxylate moiety of acidic substrates, whereas it is oriented away from the active site when substrates lacking a carboxylate group are accommodated in the enzyme.9 Hence, mutating this arginine to alanine affects the binding of acidic substrates by eliminating the possibility of a salt bridge but does not significantly affect the binding of hydrophobic substrates. The calculations performed in the current study and the previous study35 support these findings. As seen in Figure 3, the half-transamination of l-Ala to pyruvate is less feasible without Arg416 because of the high overall energy barrier of 25.2 kcal/mol (ES → TS6), whereas the half-transamination of (S)-1-PEA to acetophenone is attainable because of the lower barrier of 19.3 kcal/mol (Int3 → TS4). In the latter reaction, the absence of Arg416 in the active-site model can effectively be considered to represent both the mutant reaction and the wild-type reaction (in which the side chain of the arginine residue is positioned outside of the active site). Thus, it is reasonable that the transamination of (S)-1-PEA in combination with an aliphatic amino acceptor is barely affected by the mutation, as both substrates lack a carboxylate group. Further, it was observed that the alanine mutants had some residual activity for the transamination of (S)-1-PEA in combination with acidic amino acceptors.9 To explain this, it was proposed that a tryptophan/tyrosine residue (Trp61 and Tyr59, respectively, equivalent to Trp60 in Cv-ωTA) is also involved in the substrate binding.9 This is also supported by our calculations, which indicate that both Trp60 and Tyr153 are hydrogen-bonded to the carboxylate group of l-Ala without Arg416 (see Figure 2b). These hydrogen bonds are maintained throughout the reaction pathway.
III. Conclusions
In this study, we have investigated the reaction mechanism for the half-transamination of l-Ala to pyruvate in the (S)-ωTA from C. violaceum using DFT calculations. We have also examined the role of a flexible arginine residue, Arg416, in the dual-substrate recognition, that is, the ability of an enzyme to recognize and convert two different substrates. For this, two active-site models were employed, one including Arg416 and one lacking it. The obtained reaction mechanism is presented in Scheme 2, and the associated energy profiles are shown in Figure 3. The mechanism of the conversion of l-Ala to pyruvate has been evaluated compared to that of the conversion of (S)-1-PEA to acetophenone, and the effect of the flexible arginine residue on the energetics of the reaction has been assessed by analyzing the energy profiles of the two active-site models, with and without this residue.
The calculations suggest that the deamination of l-Ala and (S)-1-PEA follows essentially the same mechanism, with some minor differences. However, the energetics of the reactions are shown to differ significantly. The calculations are not conclusive on the nature of the rate-determining step in the reaction with l-Ala, as TS4, TS6, and TS7 are calculated to have very similar energies. For (S)-1-PEA, the rate-determining step was previously found to be TS4, which is the deprotonation of the external aldimine.35 In the half-transamination of (S)-1-PEA/acetophenone, the ketimine Int6 is the lowest energy point; therefore, the ketone is kinetically favored.35 In the half-transamination of l-Ala/pyruvate, the external aldimine, Int3, is the lowest energy point; therefore, the amine is kinetically favored. These observations are consistent with experimental results, which demonstrate that ωTAs are suitable for the kinetic resolution of racemic 1-PEA when using pyruvate as the amino acceptor.37
The calculations further support the notion that the dual-substrate recognition in (S)-ωTAs is governed by the flexible arginine residue functioning as an arginine switch, which, depending on the substrate, positions its side chain inside or outside of the active site. It is shown that the half-transamination of l-Ala to pyruvate is feasible in the presence of Arg416, as the arginine binds l-Ala by forming a salt bridge to the carboxylate moiety. Without Arg416, the conversion of l-Ala is calculated to have a higher overall energy barrier. The conversion of (S)-1-PEA to acetophenone was previously shown to have a reasonable energy barrier in line with experimental data without Arg416,35 which here represents the case in which the side chain of the arginine residue is positioned outside of the active site.
The high overall energy barrier in the reaction of l-Ala without Arg416 stems from the fact that the nonproductive binding mode of the internal aldimine, ES, is the lowest energy point. However, this binding mode might not be as energetically favorable with larger acidic substrates, such as ω-amino acids, for example, γ-aminobutyrate, which would then alter the overall energy barrier in the reaction without Arg416. This might indicate that the conclusion regarding the role of the flexible arginine could be substrate-dependent.
IV. Computational Details
In the current study, we use the same computational protocol as in the previous work.35 That is, all calculations were performed using DFT with the B3LYP functional,38,39 as implemented in the Gaussian09 software package.40 Geometry optimizations were conducted using the 6-31G(d,p) basis set, while more accurate energies were obtained by performing single-point calculations on the optimized geometries using the larger 6-311+G(2d,2p) basis set. The 6-31G(d,p) basis set was also used for frequency calculations to obtain zero-point energy (ZPE) corrections. The solvation effect of the protein surrounding was estimated by single-point calculations using the CPCM conductor-like polarizable continuum model,41,42 the 6-31G(d,p) basis set, and the standard value of ε = 4 for the dielectric constant. Empirical dispersion corrections were added using the B3LYP-D2 method.43,44 Thus, the final energies are those calculated with the large basis set, corrected for ZPE, solvation, and dispersion. The enzyme–substrate complex (ES), that is, the optimized active-site model, is defined as the zero on the energy scale (0 kcal/mol), and the energies of intermediates and transition states are reported relative to this in the energy graph of Figure 3.
Performing the geometry optimizations in the gas phase with a medium-sized basis set and then adding the corrections (large basis set, ZPE, solvation, and dispersion) on the basis of those geometries result in some low-energy transition states having the final calculated energy lower than one of its connecting intermediates (e.g., TS2 and TS5). This is an artifact of the adopted methodology and has been observed also in the previous study.35 In this case, the energy of the connecting intermediate can effectively be considered as the barrier for that step, and the transition-state energy is therefore not indicated in the energy graph (Figure 3).
Acknowledgments
The authors acknowledge the financial support from the Swedish Research Council, the Göran Gustafsson Foundation, and the Knut and Alice Wallenberg Foundation. Computer time was generously granted by the Swedish National Infrastructure for Computing.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.6b00376.
Additional figure of an alternative conformation of the internal aldimine; Cartesian coordinates of all optimized stationary points along the reaction pathway for the half-transamination of l-Ala/pyruvate, with and without Arg416 (PDF)
Author Present Address
† EnginZyme AB, Teknikringen 38A, SE-114 28 Stockholm, Sweden (K.E.C.).
The authors declare no competing financial interest.
Supplementary Material
References
- Christen P., Metzler D. E., Eds. Transaminases; John Wiley and Sons: New York, 1985. [Google Scholar]
- Malik M. S.; Park E.-S.; Shin J.-S. Appl. Microbiol. Biotechnol. 2012, 94, 1163. 10.1007/s00253-012-4103-3. [DOI] [PubMed] [Google Scholar]
- Höhne M.; Bornscheuer U. T.. Application of Transaminases. In Enzyme Catalysis in Organic Synthesis, 3rd ed.; Drauz K., Gröger H., May O., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012; pp 779–820. [Google Scholar]
- Steffen-Munsberg F.; Vickers C.; Kohls H.; Land H.; Mallin H.; Nobili A.; Skalden L.; van den Bergh T.; Joosten H.-J.; Berglund P.; Höhne M.; Bornscheuer U. T. Biotechnol. Adv. 2015, 33, 566. 10.1016/j.biotechadv.2014.12.012. [DOI] [PubMed] [Google Scholar]
- Shin J.-S.; Kim B.-G. Biotechnol. Bioeng. 1997, 55, 348.. [DOI] [PubMed] [Google Scholar]
- Shin J.-S.; Kim B.-G. Biotechnol. Bioeng. 1999, 65, 206.. [DOI] [PubMed] [Google Scholar]
- Berglund P.; Humble M. S.; Branneby C. In Comprehensive Chirality; Carreira E. M., Yamamoto H., Eds.; Elsevier: Amsterdam, 2012; pp 390–401. [Google Scholar]
- Steffen-Munsberg F.; Vickers C.; Thontowi A.; Schätzle S.; Tumlirsch T.; Humble M. S.; Land H.; Berglund P.; Bornscheuer U. T.; Höhne M. ChemCatChem 2013, 5, 150. 10.1002/cctc.201200544. [DOI] [Google Scholar]
- Steffen-Munsberg F.; Vickers C.; Thontowi A.; Schätzle S.; Meinhardt T.; Humble M. S.; Land H.; Berglund P.; Bornscheuer U. T.; Höhne M. ChemCatChem 2013, 5, 154. 10.1002/cctc.201200545. [DOI] [Google Scholar]
- Hwang B.-Y.; Cho B.-K.; Yun H.; Koteshwar K.; Kim B.-G. J. Mol. Catal. B: Enzym. 2005, 37, 47. 10.1016/j.molcatb.2005.09.004. [DOI] [Google Scholar]
- Höhne M.; Bornscheuer U. T. ChemCatChem 2009, 1, 42. 10.1002/cctc.200900110. [DOI] [Google Scholar]
- Koszelewski D.; Tauber K.; Faber K.; Kroutil W. Trends Biotechnol. 2010, 28, 324. 10.1016/j.tibtech.2010.03.003. [DOI] [PubMed] [Google Scholar]
- Tufvesson P.; Lima-Ramos J.; Jensen J. S.; Al-Haque N.; Neto W.; Woodley J. M. Biotechnol. Bioeng. 2011, 108, 1479. 10.1002/bit.23154. [DOI] [PubMed] [Google Scholar]
- Mathew S.; Yun H. ACS Catal. 2012, 2, 993. 10.1021/cs300116n. [DOI] [Google Scholar]
- Huisman G. W.; Collier S. J. Curr. Opin. Chem. Biol. 2013, 17, 284. 10.1016/j.cbpa.2013.01.017. [DOI] [PubMed] [Google Scholar]
- Kohls H.; Steffen-Munsberg F.; Höhne M. Curr. Opin. Chem. Biol. 2014, 19, 180. 10.1016/j.cbpa.2014.02.021. [DOI] [PubMed] [Google Scholar]
- Ghislieri D.; Turner N. J. Top. Catal. 2014, 57, 284. 10.1007/s11244-013-0184-1. [DOI] [Google Scholar]
- Fuchs M.; Farnberger J. E.; Kroutil W. Eur. J. Org. Chem. 2015, 32, 6965. 10.1002/ejoc.201500852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaulmann U.; Smithies K.; Smith M. E. B.; Hailes H. C.; Ward J. M. Enzyme Microb. Technol. 2007, 41, 628. 10.1016/j.enzmictec.2007.05.011. [DOI] [Google Scholar]
- Koszelewski D.; Göritzer M.; Clay D.; Seisser B.; Kroutil W. ChemCatChem 2010, 2, 73. 10.1002/cctc.200900220. [DOI] [Google Scholar]
- Kirsch J. F.; Eichele G.; Ford G. C.; Vincent M. G.; Jansonius J. N.; Gehring H.; Christen P. J. Mol. Biol. 1984, 174, 497. 10.1016/0022-2836(84)90333-4. [DOI] [PubMed] [Google Scholar]
- Silverman R. B.The Organic Chemistry of Enzyme-Catalyzed Reactions; Academic Press, 2000. [Google Scholar]
- Eliot A. C.; Kirsch J. F. Annu. Rev. Biochem. 2004, 73, 383. 10.1146/annurev.biochem.73.011303.074021. [DOI] [PubMed] [Google Scholar]
- Hirotsu K.; Goto M.; Okamoto A.; Miyahara I. Chem. Rec. 2005, 5, 160. 10.1002/tcr.20042. [DOI] [PubMed] [Google Scholar]
- Schiroli D.; Peracchi A. Biochim. Biophys. Acta 2015, 1854, 1200. 10.1016/j.bbapap.2015.02.023. [DOI] [PubMed] [Google Scholar]
- Park E.-S.; Park S.-R.; Han S.-W.; Dong J.-Y.; Shin J.-S. Adv. Synth. Catal. 2014, 356, 212. 10.1002/adsc.201300786. [DOI] [Google Scholar]
- Sayer C.; Isupov M. N.; Westlake A.; Littlechild J. A. Acta Crystallogr., Sect. D: Struct. Biol. Cryst. 2013, 69, 564. 10.1107/S0907444912051670. [DOI] [Google Scholar]
- Okamoto A.; Nakai Y.; Hayashi H.; Hirotsu K.; Kagamiyama H. J. Mol. Biol. 1998, 280, 443. 10.1006/jmbi.1998.1869. [DOI] [PubMed] [Google Scholar]
- Malashkevich V. N.; Onuffer J. J.; Kirsch J. F.; Jansonius J. N. Nat. Struct. Biol. 1995, 2, 548. 10.1038/nsb0795-548. [DOI] [PubMed] [Google Scholar]
- Oue S.; Okamoto A.; Yano T.; Kagamiyama H. J. Biol. Chem. 1999, 274, 2344. 10.1074/jbc.274.4.2344. [DOI] [PubMed] [Google Scholar]
- Oue S.; Okamoto A.; Yano T.; Kagamiyama H. J. Biochem. 2000, 127, 337. 10.1093/oxfordjournals.jbchem.a022612. [DOI] [PubMed] [Google Scholar]
- Łyskowski A.; Gruber C.; Steinkellner G.; Schürmann M.; Schwab H.; Gruber K.; Steiner K. PLoS One 2014, 9, e87350 10.1371/journal.pone.0087350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen M.; Skalden L.; Palm G. J.; Höhne M.; Bornscheuer U. T.; Hinrichs W. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2014, 70, 1086. 10.1107/S1399004714001084. [DOI] [PubMed] [Google Scholar]
- Skalden L.; Thomsen M.; Höhne M.; Bornscheuer U. T.; Hinrichs W. FEBS J. 2015, 282, 407. 10.1111/febs.13149. [DOI] [PubMed] [Google Scholar]
- Cassimjee K. E.; Manta B.; Himo F. Org. Biomol. Chem. 2015, 13, 8453. 10.1039/C5OB00690B. [DOI] [PubMed] [Google Scholar]
- Humble M. S.; Cassimjee K. E.; Håkansson M.; Kimbung Y. R.; Walse B.; Abedi V.; Federsel H.-J.; Berglund P.; Logan D. T. FEBS J. 2012, 279, 779. 10.1111/j.1742-4658.2012.08468.x. [DOI] [PubMed] [Google Scholar]
- Seo J.-H.; Kyung D.; Joo K.; Lee J.; Kim B.-G. Biotechnol. Bioeng. 2011, 108, 253. 10.1002/bit.22930. [DOI] [PubMed] [Google Scholar]
- Becke A. D. J. Chem. Phys. 1993, 98, 5648. 10.1063/1.464913. [DOI] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Phys. Rev. B 1988, 37, 785. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas O.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian09, revision A.02; Gaussian, Inc.: Wallingford, CT, 2009.
- Barone V.; Cossi M. J. Phys. Chem. A 1998, 102, 1995. 10.1021/jp9716997. [DOI] [Google Scholar]
- Cossi M.; Rega N.; Scalmani G.; Barone V. J. Comput. Chem. 2003, 24, 669. 10.1002/jcc.10189. [DOI] [PubMed] [Google Scholar]
- Grimme S. J. Comput. Chem. 2006, 27, 1787. 10.1002/jcc.20495. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. J. Chem. Phys. 2010, 132, 154104 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.