

Abstract
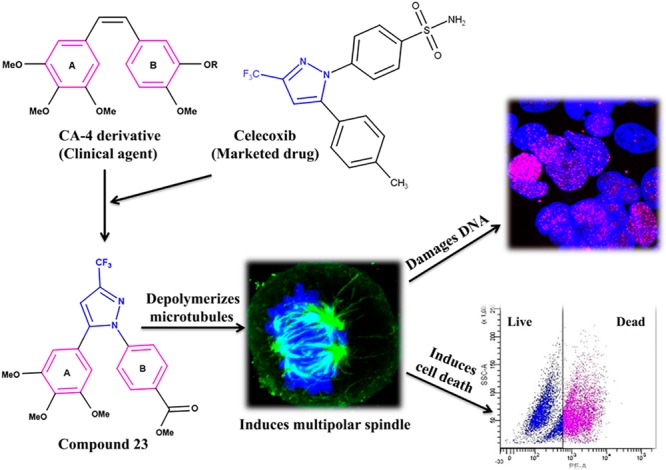
Twenty-three combretastatin A-4 (CA-4) analogues were synthesized by judiciously incorporating a functional N-heterocyclic motif present in Celecoxib (a marketed drug) while retaining essential pharmacophoric features of CA-4. Combretastatin-(trifluoromethyl)pyrazole hybrid analogues, i.e., 5-trimethoxyphenyl-3-(trifluoromethyl)pyrazoles with a variety of relevantly substituted aryls and heteroaryls at 1-position were considered as potential tubulin polymerization inhibitors. The cytotoxicity of the compounds was evaluated using MCF-7 cells. Analog 23 (C-23) was found to be the most active among the tested compounds. It showed pronounced cytotoxicity against HeLa, B16F10, and multidrug-resistant mammary tumor cells EMT6/AR1. Interestingly, C-23 displayed significantly lower toxicity toward noncancerous cells, MCF10A and L929, than their cancerous counterparts, MCF-7 and B16F10, respectively. C-23 depolymerized interphase microtubules, disrupted mitotic spindle formation, and arrested MCF-7 cells at mitosis, leading to cell death. C-23 inhibited the assembly of tubulin in vitro. C-23 bound to tubulin at the colchicine binding site and altered the secondary structures of tubulin. The data revealed the importance of (trimethoxyphenyl)(trifluoromethyl)pyrazole as a cis-restricted double bond-alternative bridging motif, and carboxymethyl-substituted phenyl as ring B for activities and interaction with tubulin. The results indicated that the combretastatin-(trifluoromethyl)pyrazole hybrid class of analogues has the potential for further development as anticancer agents.
Introduction
Microtubules are dynamic polymers that form the structural framework of eukaryotic cells. The dynamic microtubules form a spindle during mitosis, which guides the sister chromatids to the equatorial plane.1 A perturbation of the dynamicity of microtubules halts chromosome separation and causes the cells to arrest at mitosis leading to apoptosis. Because microtubules play essential roles during the cell division, they are considered as drug targets for the treatment of various diseases.2 Presently, several antitubulin agents are being used for the treatment of several types of cancers, neurological diseases, and fungal and parasitic infections. Antitubulin agents such as paclitaxel, vinblastine, and epothilones are being successfully used as anticancer agents.3,4 In addition to being antimitotic, some of the antitubulin agents also possess vascular disrupting5,6 and antiangiogenic properties.7 These antivascular agents can disrupt the existing tumor vasculature and inhibit blood vessel formation thereby rendering the tumor devoid of nutrition. One such remarkable microtubule-targeting agent is combretastatin A-4 (CA-4).8
CA-4 is a stilbenoid phenol, isolated from the African bushwillow tree, Combretum caffrum(9) It binds to tubulin and induces microtubule depolymerization. It acts as a potent antimitotic agent and also exhibits vascular disrupting property.10,11 CA-4 is reported to be poorly soluble in water.12 To overcome this limitation, many water-soluble prodrug forms of CA-4, such as 3-O-phosphate derivative (CA-4P, Zybrestat)13 and a serine amino acid conjugate (AVE8062)14 have been developed. At present, combretastatin A4 phosphate (CA-4P),13,15 the phosphate form of the prodrug, in combination with carboplatin/paclitaxel is in phase III clinical trials for anaplastic thyroid cancer (ClinicalTrials.gov; Identifier NCT00507429). Further, several phase I and phase II clinical trials (ClinicalTrials.gov; Identifiers NCT01423149, NCT00968916, NCT00977210, NCT00960557, NCT01560325) were completed for CA-4P and its analogues against different types of tumors.16−19 In addition, a synthetic phosphorylated prodrug of combretastatin A-1, Oxi450317,18 is presently undergoing phase I/II trials in combination with Cytarabine for acute myelogenous leukemia and myelodysplastic syndromes (ClinicalTrials.gov; Identifier NCT02576301).
Owing to its important pharmacological features and simple structure, CA-4 serves as a potential lead molecule for the generation of new anticancer compounds. The structure–activity relationship studies of CA-4 and its analogues has revealed the importance of the syn-orientation of rings A and B and 3,4,5-trimethoxyphenyl as ring A for antitubulin activity/cytotoxicity.20,12 Although several combretastatin derivatives are under clinical investigations, their stability is a major concern. CA-4 undergoes isomerization of the double bond of stilbene from cis-isomer (active) to trans-isomer (inactive) in physiological conditions and in the presence of heat, light, or protic media.21,22 The binding of trans-CA-4 to tubulin leads to structural distortions of the bound molecule, which can be the most likely cause of its reduced activity in comparison with its cis-form.23 To circumvent the issue of cis-to-trans-isomerization, various CA-4 analogues have been explored via the incorporation of heterocyclic,24,25 carbocyclic,26,27 and functional bridging groups28−31 as a replacement for the double bond. We have reported one such analog, compound 12,32 which was found to show more potent antiproliferative activity than CA-4.
Nowadays, the preparation of hybrid compounds that possess important skeletons present in drugs/clinical agents has become an important research area for medicinal chemists.33,34 This can be an effective approach to possibly avoid physicochemical/pharmacokinetic/toxicity problems that appear in the later stages of development.35,36 Celecoxib is a cyclooxygenase-2 (COX-2) inhibiting anti-inflammatory drug. Currently, USFDA has approved it for the treatment of breast, colon, and urinary cancers. Celecoxib inhibited the growth of human endometrial, gastric, and prostate carcinomas37,38 and was also found to display chemopreventive effect against lung cancer in former smokers.39 These unique features of celecoxib incited us to suitably blend its skeleton with CA-4 to generate a new molecular motif as a potential antitubulin agent (Figure 1). The assembly of a trifluoromethyl substitution, a pharmacologically and physicochemically important functionality,40 and a pyrazole motif41 can provide additional hydrogen bond acceptors and donors (via water molecules) for interaction with tubulin. In the investigated 1,5-diaryl-3-(trifluoromethyl)pyrazole series of compounds, the 3,4,5-trimethoxyphenyl motif in ring A and a variety of relevant aryls and heteroaryls as ring B were considered. In addition, a compound that mimics the diaryls of CA-4 was also synthesized.
Figure 1.

Design of novel 1,5-diaryl-3-(trifluoromethyl)pyrazoles.
In this work, we have evaluated the cytotoxicity of the synthesized compounds against MCF-7 cells. Among the compounds tested, C-23 displayed most potent antiproliferative activity against MCF-7 cells. The cytotoxic effect of C-23 against different types of cancer cells including the multidrug resistant cancer cells as well as on noncancerous cells was determined. Further, we have characterized the antiproliferative mechanism of C-23. In addition, the structural features of the compounds along with their antiproliferative activity have been discussed.
Results and Discussion
Chemistry
A 1,5-diarylpyrazole scaffold is usually constructed via cyclocondensation of 1,3-dicarbonyl with hydrazine.42 Utilizing this method, preparation of our designed compounds, 5-(3,4,5-trimethoxyphenyl)-3-(trifluoromethyl)pyrazoles with various aryl and heteroaryls at 1-position require (hetero) arylhydrazines as starting substrates. However, these hydrazines are difficult to prepare43 and are limited in commercial availability. Moreover, in our investigation, this approach42 involving the cyclocondensation reaction of (Z)-4,4,4-trifluoro-3-hydroxy-1-(3,4,5-trimethoxyphenyl)but-2-en-1-one with 4-methoxyphenylhydrazine provided the desired 1-(4-methoxyphenyl)-3-(trifluoromethyl)-5-(3,4,5-trimethoxyphenyl)-1H-pyrazole in low yield (38%). Moreover, an undesired product, regioisomeric N-arylated pyrazole, 1-(4-methoxyphenyl)-5-(trifluoromethyl)-3-(3,4,5-trimethoxyphenyl)-1H-pyrazole, was also obtained in considerable quantity (15% yield). Therefore, this approach was considered to be nonconvenient. 1,5-Diarylpyrazoles were also previously prepared via transition-metal (Pd,44 Cu,45,46 Fe47,48)-mediated N-arylation of preformed 1H-pyrazoles. For the preparation of targeted compounds, 5-(3,4,5-trimethoxyphenyl)-3-(trifluoromethyl)pyrazole (IV) as the precursor for N-arylation was considered (Figure 2). The reaction of hydrazine hydrate with enol intermediate (III) prepared by a reported method49 of Claisen–Schmidt transformation produced pyrazole IV.42
Figure 2.

Synthesis of investigated compounds. Substrates, reagents, and conditions: compound IV (1 mmol), RBr (2equiv), CuI (5 mol %), DMEDA (20 mol %), K2CO3 (2.1 equiv), 1,4-dioxane (anhyd, 2 mL), 110 °C. Yield for maximum conversion in optimum time.
In this approach, pyrazole IV was obtained in one pot with an overall 60% yield. A method for cupric acetate–mediated N-arylation of pyrazoles with arylboronic acid is known in the literature.50 The reaction for N-arylation of 5-(3,4,5-trimethoxyphenyl)-3-(trifluoromethyl)pyrazole (IV) with 4-methoxyphenylboronic acid using this method was performed. However, incomplete conversion, poor regioselectivity (N1- vs N2-arylation), and low yield were observed. Investigating varied reaction conditions did not improve the conversion and yield (Table S1).We then investigated N-arylation of pyrazole IV by the Ullmann–Golderg coupling method.45 The reaction with 4-bromoanisole by CuI catalysis was performed to prepare the targeted diarylpyrazole. The desired 1,5-diaryl regioisomer was formed in 15% yield. An optimization study was then carried out (Tables S2 and S3). Various ligands known for N-arylation reactions were evaluated. The bidentate ligands proved to be superior to monodentate ligands in terms of conversion, regioselectivity, and yield of desired product. Next, copper catalysts were examined. In general, Cu (I) catalysts (Cl, Br, I, OTf) were found slightly better than other tested Cu(II) catalysts. CuI was found to be best. With DMEDA ligand and CuI catalyst, gratifyingly, the desired 1,5-regioisomer was obtained in 93% yield. Moreover, the optimized conditions enabled us to reduce the production of undesired regioisomeric N-arylated product (1,3-diaryl) in trace quantity (4% yield only). With the optimized protocol in hand (Figure 2), a series of investigated compounds (1–23) with relevant substitutions were synthesized. Several substituted aryls and heteroaryls important for displaying potential tubulin polymerization inhibitory activity were considered in ring B. Compound 3 that exactly mimics the diaryls in CA-4 was synthesized from compound 21 by Pd–C catalyzed hydrogenative debenzylation. All the products were identified by 1H and 13C NMR, IR and HRMS spectroscopies. Single crystal X-ray crystallographic analysis of compound 1, as a representative example, confirmed the 1,5-regioisomer, a desired cis-restricted pyrazole analog of combretastatin (Figure S1). HPLC study of all synthesized compounds indicated the purity >95%.
Biological Studies
Screening Antiproliferative Activity of Combretastatin Analogues in MCF-7 Cells
Using sulforhodamine assay,51 the synthesized combretastatin analogues (1–23) were screened for their antiproliferative activity in MCF-7 (human breast cancer) cells (Figure 3). Compound 23 (C-23) was found to be the most potent antiproliferative agent among the tested compounds. Hence, the antiproliferative activity of C-23 was further characterized and its mechanism of action was elucidated.
Figure 3.

Screening of the antiproliferative activity of combretastatin analogues in MCF-7 cells. The percentage inhibition of MCF-7 cell proliferation by 1 μM CA-4 analogues was determined. The assay was performed four times. Error bar represents standard deviation.
Effects of C-23 on the Proliferation of Different Types of Cancer Cells
The antiproliferative activity of C-23 was tested against various cancer cell lines. C-23 inhibited the proliferation of MCF-7, B16F10 (mouse skin cancer), HeLa (human cervical cancer), and EMT6/AR1 (multidrug drug-resistant mouse mammary cancer) cells with a half-maximal inhibitory concentration (IC50) of 1.3 ± 0.8, 6 ± 0.6, 5.5 ± 0.6, and 14.7 ± 0.3 μM, respectively (Table 1 and Figure S50).
Table 1. Half-Maximal Inhibitory Concentration (IC50) of C-23 in MCF-7, MCF10A, B16F10, L929, HeLa, and EMT6/AR1 Cellsa.
| cells | IC50 values (μM) |
|---|---|
| MCF-7 | 1.3 ± 0.8 |
| MCF10A | 23.2 ± 0.8 |
| B16F10 | 6 ± 0.6 |
| L929 | 14 ± 1 |
| HeLa | 5.5 ± 0.6 |
| EMT6/AR1 | 14.7 ± 0.3 |
The data represent an average ± SD IC50 value from three independent experiments.
Effects of C-23 on Noncancerous Cells
To determine the cytotoxic potential of C-23 in noncancerous cells, the effect of C-23 was evaluated against normal epithelial human breast cells (MCF10A) and normal fibroblast mouse skin cells (L929). The half-maximal inhibitory concentration (IC50) of C-23 against MCF10A was determined to be 23.2 ± 0.8 μM, which is ∼18 times more than its IC50 value in MCF-7 (1.3 ± 0.8 μM) (Table 1 and Figure S50). Also, the IC50 of C-23 against L929 was 14 ± 1 μM, ∼2 times more than its IC50 value in B16F10 (6 ± 0.6 μM) (Table 1). The results indicated that C-23 displayed low toxicity in noncancerous cells compared to the cancer cells used in the study. Though the low toxicity of the investigated compound against noncancerous cells is encouraging, tubulin-targeting agents are known to show several side effects including neurotoxicity.
Effects of C-23 on Microtubule Depolymerization in MCF-7 Cells
Perturbation of cellular microtubules is one of the characteristic morphological changes associated with antitubulin compounds. Control interphase cells displayed an intact organization of microtubules with distinct filaments (Figure 4a) whereas C-23 treatment depolymerized interphase microtubules in MCF-7 cells (Figure 4a). Further, C-23 depolymerized spindle microtubules and disrupted the chromosome organization (Figure 4b). Vehicle treated MCF-7 cells exhibited regular bipolar spindles required for proper chromosome segregation during metaphase. C-23 exposure disrupted the bipolar astral formation of spindles leading to the formation of either one or many astral bodies at the spindle poles (Figure 4b). In Figure 4b, the upper panel shows that the chromosomes are properly aligned at the metaphase plate in the control cells. However, in the C-23 treated cells, the chromosomes were pulled closer to the centrosomes toward each pole (Figure 4b, middle panel). In C-23 treated monopolar cells, the metaphasic plate was not properly formed and the chromosomes were centered at the pole near the centrosome (Figure 4b, lower panel). The results indicated that C-23 caused spindle defects in cells, leading to missegregation and misalignment of chromosomes at the metaphase plate. In addition, we quantified the level of polymer/soluble tubulin in C-23 treated MCF-7 cells. The polymer to soluble tubulin levels was decreased by 52 and 62% in the presence of 3 and 6 μM C23 indicating that C-23 depolymerized microtubules in cells (Figure 4c,d).
Figure 4.

C-23 depolymerized microtubules in MCF-7 cells. (a) C-23 depolymerized interphase microtubules in cells. Cells were incubated with vehicle (0.1% DMSO) and 3 and 6 μM C-23 for 36 h, fixed, and processed for immunostaining using α-tubulin IgG. The scale bar is 10 μm. (b) C-23 (3 and 6 μM) perturbed microtubule spindle formation in MCF-7 cells. DNA was stained with Hoechst 33258 (blue). The scale bar is 10 μm. (c) C-23 treatment leading to a decrease in the ratio of polymeric/soluble tubulin in MCF-7 cells. Cells were treated with vehicle (lane 1) and with 3 μM (lane 2) and 6 μM (lane 3) of C-23 for 36 h. Fifteen nanomolar vinblastine (lane 4) was used as a positive control. The experiment was performed four times. Shown is a representative blot. (d) Polymer and soluble level of tubulin quantified using ImageJ software and the ratio of polymeric/soluble tubulin was plotted. Error bar represents standard deviation. **p < 0.01 indicates statistical significance of the data.
C-23 Caused Mitotic Block and Induced DNA Damage in MCF-7 Cells
The perturbation of mitotic microtubules and improper formation of metaphase plate led us to hypothesize that the antiproliferative properties of C-23 were due to its ability to induce mitotic arrest. To test this, the effect of C-23 on cell cycle progression of MCF-7 cells was determined by flow cytometry (Figure 5a and Table 2). The flow cytometric analysis revealed that 10 ± 2, 31 ± 9, and 46 ± 8% of the cells were accumulated at the G2/M phase when treated with either vehicle or 3 and 6 μM C-23, respectively (Table 2). The results indicated that C-23 arrested cells at the G2/M phase. Further, the mitotic index (percentage of cells in mitosis) was determined to be 5 ± 2, 23 ± 4, and 43 ± 5 in the absence and presence of 3 and 6 μM C-23, respectively, showing that C-23 blocks the cells at mitosis (Figure 5b,c and Table 2).
Figure 5.

C-23 blocked MCF-7 cells at mitosis. (a) Flow cytograms showing DNA distribution profiles of vehicle and C-23 (3 and 6 μM) treated MCF-7 cells in different phases of the cell cycle. (b) Effects of C-23 on mitotic progression. MCF-7 cells treated with vehicle and 3 and 6 μM C-23 for 36 h were fixed and DNA was stained with Hoechst 33258 (blue). The experiment was performed three times. (c) C-23 treatment increasing the mitotic index in MCF-7 cells. The experiment was performed three times and 500 cells were scored in each case. The error bar represents standard deviation. **p < 0.01 indicates statistical significance of the data.
Table 2. Effects of C-23 on Cell Cycle Progression of MCF-7 Cellsa.
| % of cells
in different phases of cell cycle |
||||
|---|---|---|---|---|
| samples | G1 | S | G2/M | mitotic index |
| control | 72 ± 3 | 14 ± 1 | 10 ± 2 | 5 ± 2 |
| 3 μM C-23 | 37 ± 6 | 25 ± 9 | 31 ± 9 | 23 ± 4 |
| 6 μM C-23 | 27 ± 16 | 20 ± 9 | 46 ± 8 | 43 ± 5 |
Cell cycle analysis and mitotic indices of MCF-7 cells treated without and with different concentrations of C-23. Data were an average ± SD of three sets of experiments. For mitotic index calculation, 500 cells were scored in each case.
It is well-known that a prolonged arrest in mitosis leads to induction of DNA damage in cells.52 We therefore ought to find out the effect of C-23 on DNA damage in MCF-7 cells. Phosphorylation at Ser139 of H2AX (γ-H2AX) at the site of double-stranded breaks (DSBs) is a typical marker used to examine DNA damage in cells.53,54 DNA damage in C-23 treated cells was determined by evaluating phosphorylation (γ-H2AX) of histone-2AX (H2AX) by immunofluorescence microscopy. MCF-7 cells were treated with 3 and 6 μM C-23 and processed for immunostaining using γ-H2AX antibody. DNA was stained with Hoechst. The level of phosphorylation of H2AX (γ-H2AX intensity) in C-23 treated cells was higher compared to that of the untreated (control) cells, indicating that C-23 produced DNA damage in MCF-7 cells (Figure 6a,b).
Figure 6.

C-23 induced DNA damage in cells. (a) MCF-7 cells were treated with vehicle and 3 and 6 μM C-23 for 36 h and were fixed and processed for immunostaining using γ-H2AX IgG to stain double-stranded DNA breaks (pink). DNA was stained using Hoechst 33258 (blue). The scale bar is 20 μm. (b) The γ-H2AX intensity was calculated using ImageJ software and plotted. The experiment was performed three times and 100 cells were scored for intensity calculation in each case. Error bar represents standard deviation. **p < 0.01 indicates statistical significance of the data.
C-23 Induced PARP Cleavage and Apoptosis in MCF-7 Cells
To determine whether C-23 could induce cell death in MCF-7 cells, we performed a live and dead assay using flow cytometry. MCF-7 cells were treated without and with 3 and 6 μM C-23 for 48 h and the cells were processed for flow cytometry after incubating with propidium iodide (PI). As shown in Figure 7a,b, 2 ± 1, 52 ± 2, 78 ± 5, and 83 ± 6% of the total cells were found to be dead/apoptotic when treated with vehicle or 3 and 6 μM C-23 and 15 nM vinblastine, respectively (Table 3).
Figure 7.

C-23 treatment caused PARP cleavage and induced cell death in MCF-7 cells. (a) Flow cytograms show live and dead cells after PI staining. MCF-7 cells were incubated without and with C-23 (3 and 6 μM) for 48 h. Fifteen nanomolar vinblastine was used as a positive control. Representative images from three experiments are shown. (b) The percent of live and dead cells was quantified and plotted. The error bar shows standard deviation. **p < 0.01 indicates statistical significance of the data. (c) C-23 cleaves PARP in MCF-7 cells. Cells were treated with vehicle and 3 and 6 μM C-23 for 48 h. Cell lysate for each sample was prepared, and PARP cleavage was determined by Western blot using anti-PARP-1 IgG. Actin was used as a loading control. Fifteen nanomolar vinblastine was used as a positive control. The experiment was performed three times. A representative blot is shown.
Table 3. Percentage of Live and Dead Cells Determined Using Flow Cytometrya.
| live % | dead % | |
|---|---|---|
| control | 98 ± 1 | 2 ± 1 |
| 3 μM C-23 | 49 ± 8 | 52 ± 2 |
| 6 μM C-23 | 23 ± 5 | 78 ± 5 |
| 15 nM Vinblastine | 18 ± 6 | 83 ± 6 |
Data were an average ± SD of three experiments.
The cleavage of poly(ADP-ribose) polymerase (PARP) is a well-known indicator of apoptosis in cells.54,55 Therefore, we determined whether C-23 treatment could induce PARP cleavage in MCF-7 cells. Immunoblot analysis of MCF-7 cells treated without and with 3 and 6 μM C-23 for 48 h indicated that C-23 activated PARP, as evident by the cleavage of PARP (Figure 7c). Vinblastine (15 nM) was used as positive control. The results indicated that C-23 treatment induced apoptosis in MCF-7 cells.
C-23 Inhibited Polymerization of Purified Tubulin
The assembly kinetics of tubulin is known to be perturbed by antitubilin agents. As C-23 depolymerized the cellular microtubules, we ought to find out its effect on in vitro tubulin polymerization. Purified tubulin was incubated in the absence and presence of different concentrations of C-23 and the effect of the compound on the polymerization of tubulin was monitored by turbidimetry. C-23 inhibited tubulin assembly in a concentration-dependent manner with a half-maximal inhibitory concentration of 39 ± 3 μM (Figure 8a).
Figure 8.

C-23 bound to purified tubulin and inhibited its polymerization. (a) Tubulin (13 μM) was polymerized in the presence of vehicle (DMSO) (■) and 10 (●), 20 (▲), 40 (▼), 60 (◀), and 75 (▶) μM C-23. The kinetics of tubulin assembly was monitored at 350 nm. The experiment was performed three times. One of the independent sets is shown. (b) Electron micrographs of DMSO-induced tubulin polymers polymerized without (control) and with 20 μM C-23 are shown. The scale bar is 0.5 μm. (c)The elution profile of tubulin (20 μM) (□) and C-23 (60 μM) (Δ) when loaded individually onto the column are shown. Tubulin (20 μM) was incubated with C-23 (60 μM) in 25 mM PIPES at 25 °C for 30 min and then eluted through the same column. The elution profile of tubulin (■) and C-23 (▲) of the tubulin-C-23 complex is shown. The experiment was performed two times.
In addition, the electron micrographs of tubulin polymers were obtained by polymerizing tubulin in the presence of only vehicle DMSO (control) or with 20 μM C-23 (Figure 8b). The electron micrographs showed that C-23 perturbed the formation of tubulin polymers. In the absence of C-23, several polymers were observed per microscopic field, most of which were long and straight. In the presence of 20 μM C-23, few short polymers were observed per microscopic field supporting the turbidimetry data that C-23 inhibits tubulin assembly in vitro.
C-23 Bound to Purified Tubulin in Vitro
To check if C-23 interacts with pure tubulin, we performed size exclusion chromatography. Tubulin (100 kDa), C-23 (436.4 Da), and a mixture of tubulin and C-23 was separately loaded onto a P4 resin column. Tubulin and C-23 when loaded individually eluted at 1.5 and 7.5 mL of elution volume, respectively (Figure 8c). When the mixture of tubulin and C-23 was loaded onto the same column, tubulin was eluted at 1.5 mL as a single peak and C-23 was eluted in two different elution volumes, one peak at 1.5 mL and another peak at 7.5 mL, suggesting that C-23 coeluted with tubulin. The result indicated that C-23 can form a complex with purified tubulin.
C-23 Perturbed the Secondary Structure of Tubulin
The effect of C-23 on the secondary structure of tubulin was monitored by far-UV CD spectroscopy. An analysis of the CD spectra of tubulin indicated that tubulin contains 49 ± 2% helix, 21 ± 2% sheets, and 28 ± 3% turns and random coils (Figure 9a,b). The observed secondary structure content was found to be similar to the reported crystal structure of tubulin (PDB ID: 5LYJ). A significant decrease in the α-helical content of tubulin was observed when incubated with C-23 (Figure 9a). The α-helical content decreased from 49 ± 2% to 47 ± 2%, and 44 ± 2% when treated with 6 and 10 μM C-23, respectively. Also, a significant increase in the random coil structure was observed as it increased from 13 ± 2% to 16 ± 1% and 18 ± 2% in the presence of 6 and 10 μM C-23, respectively indicating that the binding of C-23 disrupts the secondary structures of tubulin (Figure 9b).
Figure 9.

C-23 disrupted the secondary structure of purified tubulin. (a) Tubulin (1 μM) was incubated with vehicle (DMSO) (■) and 6 (●) and 10 (▲) μM C-23. The far-UV CD spectra were recorded. One of the three independent sets is shown. (b) The percentage of helix, sheet, turn, and random coil of tubulin when incubated without and with C-23 was determined by CDPro software and was plotted. The error bar indicates standard deviation. *p < 0.05 indicates statistical significance of the data.
C-23 Bound to Tubulin at the Colchicine Binding Site
Antitubulin agents perturb microtubule assembly dynamics by binding to tubulin at three different sites, namely, the vinca binding site,56 colchicine binding site,57 and taxol binding site.58 Combretastatins are known to interact with tubulin by binding at the interface of the tubulin dimer specifically at the colchicine site.59 As C-23 is an analog of CA-4, we hypothesized that it will also bind at the colchicine site on tubulin. To test this hypothesis, a competitive assay of C-23 with colchicine was carried out. When excited at 350 nm, colchicine exhibits low fluorescence in aqueous solution whereas a significant increase in the fluorescence intensity with a fluorescence maxima at 440 nm is observed when it is bound to tubulin.60 The preincubation of C-23 with tubulin led to a reduction in the development of fluorescence of tubulin–colchicine complex in a concentration-dependent manner (Figure 10a). This indicated that C-23 impedes the binding of colchicine to tubulin. The inhibitory constant (Ki) of C-23 for colchicine was determined to be 2.2 ± 0.5 μM (Figure 10b).
Figure 10.

C-23 bound at the colchicine binding site on tubulin. (a) Tubulin (5 μM) was incubated in the absence (■) and presence of 2 (●), 5 (▲), 7 (▼), 10 (◀), 12 (▶), 15 (◆), and 20 (⬟) μM C-23. Then the mixtures were incubated with 5 μM colchicine for 45 min at 37 °C. The fluorescence spectra (410–500 nm) were monitored using 350 nm as the excitation wavelength. (b) C-23 reduced the fluorescence intensity of tubulin–colchicine complex. One of the three independent sets is shown. (c) Tubulin (5 μM) was incubated in the absence (■) and presence of 5 (●), 10 (▲), 20 (▼), 30 (◀), 40 (▶), and 50 (◆) μM C-23. The mixtures were incubated with 5 μM C-12 for 10 min at 37 °C, and the fluorescence spectra (410–550 nm) were monitored using excitation wavelength 350 nm. One of the three independent sets is shown. (d) The percentage inhibition of the tubulin-C-12 fluorescence was plotted against C-23 concentration. The error bar indicates standard deviation.
Similarly, a competitive assay of C-23 with C-12 was performed. C-12 is an analog of CA-4 and is known to bind at the colchicine site in tubulin.32 On binding to tubulin, C-12 fluoresces with maximum fluorescence intensity at 450 nm. C-23 decreased the binding of C-12 to tubulin in a concentration-dependent manner (Figure 10c,d), indicating that C-23 binds at the combretastatin binding pocket as that of C-12.32 The results together suggested that C-23 binds to tubulin at the colchicine site.
Analysis of C-23 Binding Site on Tubulin by Molecular Docking
Using molecular docking, a putative binding site for C-23 in tubulin dimer was investigated. To validate the docking results, DAMA-colchicine and CA-4 (Figure 11a,d) were docked on tubulin dimer (PDB ID: 5LYJ). The docked conformations of DAMA-colchicine and CA-4 were found to be at the interface of the tubulin dimer (Figure 11b,e). The docked conformation of DAMA-colchicine and CA-4 was then compared with the PDB coordinates of their respective X-ray crystallographically determined structures, 1SA057 and 5LYJ,23 respectively. The RMSD (root-mean-square deviation) between the docked conformation and the crystal structure of the molecules was estimated to be 1.6 and 1.1 Å for DAMA-colchicine and CA-4, respectively (Figure 11c,f). A RMSD value less than 2–3 Å indicates an appropriate docking.61 Thus, a similar docking protocol was used to elucidate the binding site of C-23 in tubulin.
Figure 11.
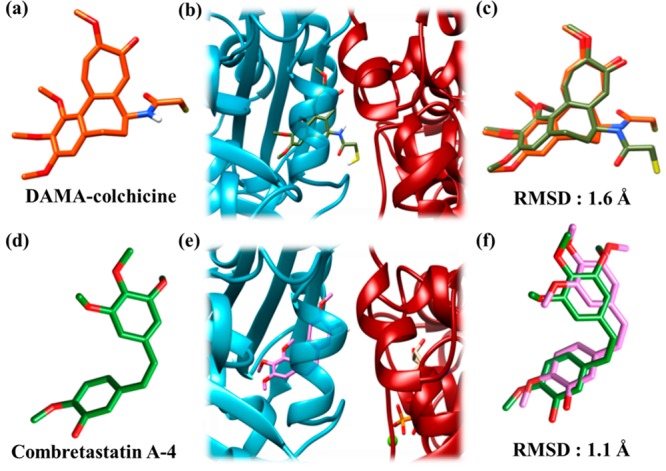
DAMA-colchicine and CA-4 docked at the interface of the tubulin dimer. The color scheme for α-tubulin is dark red, for β-tubulin is cyan, for DAMA-colchicine (crystal structure) is orange red, for DAMA-colchicine (docked) is olive green, for CA-4 (crystal structure) is green, and for CA-4 (docked) is orchid pink. The compounds and tubulin are in stick and ribbon representations, respectively. Sulfur, hydrogen, oxygen, and nitrogen atoms are depicted as yellow, white, red, and dark blue sticks, respectively. (a) Crystal structure of DAMA-colchicine in sticks. (b) Docked conformation of DAMA-colchicine at the interface of the tubulin dimer. The conformation with lowest binding energy was found to be docked at the interface of the dimer. (c) RMS deviation between the crystal structure and docked conformation of DAMA-colchicine. The coordinates of the docked DAMA-colchicine (olive green) were superimposed over the X-ray crystallographically determined coordinates (orange red). (d) Crystal structure of CA-4 in sticks. (e) Docked conformation of CA-4 at the interface of tubulin dimer. The conformation of CA-4 with the lowest binding energy was found to be docked at the interface of the dimer. (f) RMS deviation between the docked and crystal structure of CA-4 obtained by superimposing the docked CA-4 (orchid pink) over the crystallographically determined coordinates (green).
A docking analysis of C-23 (Figure 12a) suggested that C-23 binds to tubulin at the colchicine-binding pocket (Figure 12b). On superimposing the docked conformations of C-23, CA-4, and DAMA-colchicine, it was observed that both CA-4 and C-23 overlapped with DAMA-colchicine, indicating that they bind to the colchicine binding pocket (Figure 12c and d).
Figure 12.
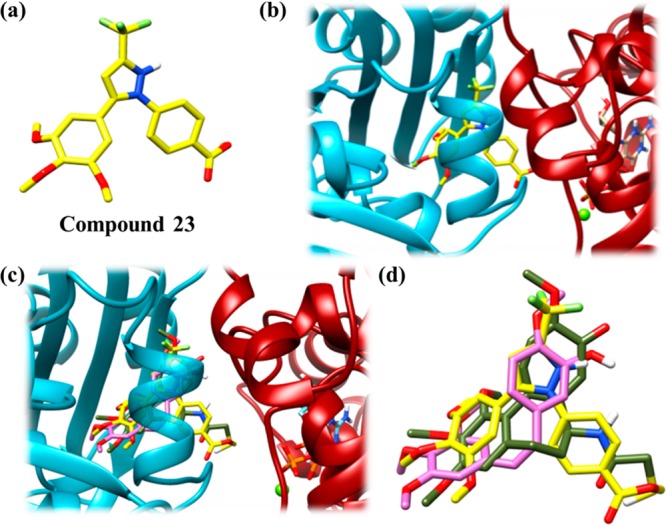
C-23 docked at the colchicine binding pocket on the tubulin dimer. The color scheme for α-tubulin is dark red, for β-tubulin is cyan, for C-23 is yellow, for DAMA-colchicine (docked) is olive green and for CA-4 (docked) is orchid pink. The compounds and tubulin are in stick and ribbon representations, respectively. Hydrogen, oxygen, nitrogen, sulfur, and fluorine atoms are depicted as white, red, dark blue, yellow, and light green sticks, respectively. (a) Structure of C-23 in sticks. (b) Docked conformation of C-23 at the interface of tubulin dimer. The conformation of C-23 with the lowest binding energy was found to be docked at the interface of the dimer (c) Docked conformation of C-23 overlapped with docked conformations of DAMA-colchicine and CA-4. All the three compounds were shown to occupy the same binding pocket. (d) Zoomed view of (c) showing overlapped C-23, CA-4, and DAMA-colchicine.
The colchicine-binding pocket mostly consists of hydrophobic amino acids, and therefore, the site is hydrophobic in nature. An analysis of the residues lying within a 4 Å distance from the docked conformation of C-23 indicated that the binding pocket is mostly constituted of hydrophobic residues (Figure 13). Most of the residues present in the binding pocket of C-23 were also found to be present in the binding pocket of CA-4 and DAMA-colchicine. Further analysis indicated that several common hydrophobic residues were present in the vicinity of all the three compounds and thus may provide possible hydrophobic interactions (Table 4).
Figure 13.
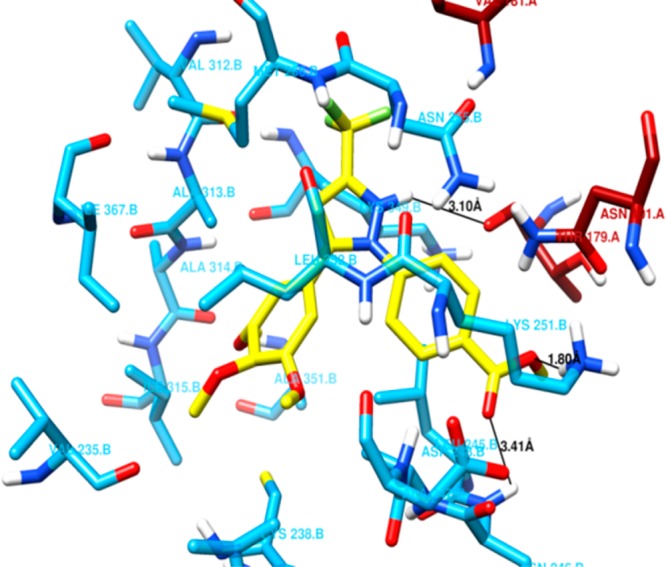
Amino acid residues of the tubulin dimer present within 4 Å distance of C-23. Residues of α-tubulin and β-tubulin are shown in dark red and cyan sticks, respectively. C-23 is in yellow stick representation. The color scheme for the atoms is same as in Figure 12. The black line represents the possibility of a hydrogen bond between C-23 and the amino acid present in its binding pocket. C-23 was found to have possible hydrogen bonds with Thr179A (3.10 Å), Asp248B (3.41 Å), and Lys251B (1.80 Å).
Table 4. Tubulin Residues Present around the Docked Compounds within a Distance of 4 Åa.
| docked compound | α-tubulin residues | β-tubulin residues |
|---|---|---|
| DAMA-colchicine | Asn (101), Thr (179), Ala (180), Val (181) | Val (235), Cys (238), Leu (239), Leu (245), Asn (246), Ala (247), Lys (251), Leu (252), Asn (255), Met (256), Val (312), Ala (313), Ile (315), Asn (347), Lys (349), Ile (367) |
| CA-4 | Thr (179), Ala (180), Val (181) | Gly (234), Cys (238), Leu (239), Leu (245), Ala (247), Asp (248), Lys (251), Leu (252),Asn (255), Met (256), Val (312), Ala (313), Ala (314), Ile (315), Asn (346), Asn (347), Ala (351), Ile (367) |
| C-23 | Asn (101), Thr (179), Val (181) | Val (235), Cys (238), Leu (239), Leu (245), Asn (246), Ala (247), Asp (248), Lys (251), Leu (252), Asn (255), Met (256), Val (312), Ala (313), Ala (314), Ile (315), Lys (349), Ala (351), Ile (367) |
Residues in bold are the common residues found in the binding pocket of DAMA-colchicine, CA-4, and C-23.
In addition, a close look at the binding pocket of docked C-23 revealed possible hydrogen bonding interactions. The distance between the ester-methoxy oxygen of C-23 to the amide hydrogen of Lys251B was measured to be 1.80 Å (Figure 13), indicating a hydrogen bond between them. Another possible hydrogen bond was observed between the hydrogen of pyrazole moiety of C-23 and carbonyl oxygen of Thr179A as the distance between them was 3.10 Å (Figure 13). Further, a weak hydrogen bond of bond length 3.41 Å (Figure 13) was observed between the oxygen atom of carboxyl ester group of C-23 and amide hydrogen of Asp248B. These hydrogen bonding interactions are possibly involved in stabilizing C-23 in the binding pocket along with other hydrophobic interactions. Similarly, possible hydrogen bonds of DAMA-colchicine and CA-4 with tubulin were also measured and analyzed (Table 5).
Table 5. Binding Energy and Hydrogen Bonding Interactions of the Three Compounds Docked on Tubulin Dimer Individuallya.
| hydrogen bonding interactions |
|||
|---|---|---|---|
| ligand | binding energy (kcal/mol) | residues involved | distance (Å) |
| DAMA-colchicine | –10.43 | Thr179A | 1.98 |
| Val181A | 2.75 | ||
| Cys238B | 3.86 | ||
| CA-4 | –7.50 | Thr179A | 2.03 |
| C-23 | –8.49 | Thr179A | 3.10 |
| Asp248B | 3.41 | ||
| Lys251B | 1.80 | ||
The binding energies were estimated by Autodock 4.2 and the lengths of the hydrogen bonds were measured using UCSF Chimera version 1.11.
Using the Autodock software, the binding energy for C-23, CA-4, and DAMA-colchicine interaction with tubulin was estimated to be −8.5, −7.50, and −10.43 kcal/mol, respectively, indicating that C-23 may bind to tubulin with a greater affinity than CA-4 (Table 5). The biochemical competitive binding data and the docking analysis together strongly suggested that C-23 binds to tubulin at the colchicine-binding site.
Structure–Activity Relationship
The present series of compounds involves the 3-(trifluoromethyl)pyrazole as bridging scaffold, trimethoxyphenyl motif as ring A, and varied substituted aryls and heteroaryls as ring B. The IC50 values (Table 1) and antiproliferative activity of C-23 indicated important structure–activity relationship. The presence of methoxy, trifluoromethoxy, benzyloxy, dimethoxy, dioxomethylene, methoxy-benzyloxy, and methoxy-hydroxyl in the aryls (ring B) exhibited low to moderate antiproliferative activities. The presence of quinoline or 2-methylquinoline as ring B in molecules displayed low and moderate antiproliferative activity, respectively. This observation is in contrast to our earlier finding of combretastatin-2-aminoimidazole analog that possesses quinoline as ring B and exhibited potent antiproliferative activity. The ring B motifs such as the aryls containing chloro, fluoro, and acetyl, and the heteroaryls including thiophene, pyridine, and indole displayed good activities. (Trifluoromethyl)pyrazole derivative (C-23) that possesses carboxyl ester-substituted phenyl as ring B was found to be most active and showed potent antiproliferative activity against various cancer cells, MCF-7, HeLa, B16F10, and EMT6/AR1.The ester-methoxy oxygen and oxygen of carboxyl ester provides important hydrogen bonding interactions with Lys251B and Asp 248B, respectively. The presence of carboxyl ester functionality in the aryl ring B (which is known to be amenable) of this most active and new combretastatin-(trifluoromethyl)pyrazole analog is interesting and useful.
Conclusion
In an attempt toward overcoming disadvantages including instability and trans-isomerization susceptibility of the Z-double bond of CA-4 and analogues/derivatives, a new series of heterocyclic compounds was designed. It involved a blend of the structural features of the clinical agents and a marketed drug celecoxib, recently approved for treatment of various cancers. The compounds consisted of 3-(trifluoromethyl)pyrazole as the bridging motif replacing the double bond of CA-4, 3,4,5-trimethoxyphenyl group in ring A and various relevant (hetero)aryls in ring B. For the synthesis of these compounds, a convenient method with high regioselectivity and yield was developed. Twenty-three compounds with relevantly substituted aryls and heteroaryls were prepared. All the compounds were tested against MCF-7 cells and C-23 was found to be the most effective antiproliferative agent. C-23 also showed significant inhibitory activity against several other cancer cells including the drug-resistant EMT6/AR1cells. Interestingly, C-23 showed substantially weaker cytotoxicity toward noncancerous cells than the cancer cells indicating the anticancer potential of the compound. Although several of the tubulin targeting agents have been successfully used in cancer chemotherapy, toxicity, and development of resistance limit their applications. Altered isotype compositions, mutations in the drug binding site and drug efflux are thought to be the major causes of the development of drug resistance.2−4In vitro experiments showed that C-23 binds to tubulin at the colchicine site and the binding of the compound disrupts the secondary structure of tubulin. It inhibited tubulin polymerization both in cells and in vitro and interfered with the cell cycle progression by arresting the cells at mitosis leading to cell death by apoptosis.
Experimental Section
Materials
Vinblastine sulfate, propidium iodide, sulforhodamine B (SRB), mouse monoclonal anti-α tubulin IgG, mouse monoclonal anti-β actin IgG, fluorescein isothiocyanate (FITC)-conjugated antimouse IgG, Hoechst 33258, and bovine serum albumin (BSA) were purchased from Sigma (St. Louis, MO, USA). Alexa flour 555 conjugated goat antirabbit, alexa flour 594 conjugated goat antirabbit IgG, and fetal bovine serum (FBS) were purchased from Molecular probes, Invitrogen (Eugene, OR, USA). Rabbit polyclonal anti-PARP-1 IgG was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Horseradish peroxidase conjugated horse antimouse IgG and rabbit monoclonal anti-γ-H2AX IgG were purchased from Cell Signaling Technologies. Horseradish peroxidase conjugated goat antirabbit IgG was purchased from Biorad (USA). Supersignal west picochemiluminescent substrate was purchased from Thermofisher Scientific. All other reagents were of analytical grade and were purchased from HiMedia (Mumbai, India) and Sigma.
Cell Culture
EMT6/AR1 cells were procured from Sigma and were cultured in Eagle’s minimal essential medium (MEM) (HiMedia). MCF-7, HeLa, A549, B16F10, and L929 cells were obtained from National Centre for Cell Science, Pune. The cell lines were authenticated using STR analysis and they were tested free from mycoplasma. MCF-7, HeLa, B16F10, and L929 cells were cultured in Dulbecco’s modified eagle’s medium (DMEM) (HiMedia). A549 cells were cultured in F-12k medium (Kaighn’s Modification of Ham’s F-12 Medium) (HiMedia). MCF10A cells were cultured in 1:1 mixture of DMEM/F12 medium supplemented with 0.5 μg/mL hydrocortisone, 20 ng/mL EGF, and 10 μg/mL insulin. The cells were maintained as described earlier.62
Methods
Screening of Combretastatin Analogues in MCF-7 Cells
Twenty-three combretastatin analogues (C-1–C-23) were dissolved in 100% DMSO. For the screening, MCF-7 cells were grown in a 96-well cell culture plates at a density of 1 × 104 cells/well.54 Then, the cells were grown in the presence of 1 μM concentration of all the CA-4 analogues for 48 h. The inhibition of cell proliferation by CA-4 analogues was determined by sulforhodamine B assay.63,64
Cell Proliferation Assay
Inhibition of cell proliferation by most potent compound C-23 in HeLa, B16F10, L929, MCF10A, and EMT6/AR1was determined using sulforhodamine B assay as mentioned above. The half-maximal inhibitory concentration was determined as described earlier.54
Determination of PARP Cleavage by Western Blot
MCF-7 cells (0.8 × 106) were grown in 60 mm cell culture dishes and incubated with vehicle and 3 and 6 μM C-23 or 15 nM vinblastine for 48 h. After the required incubation, cells were collected and immunoblotted using specific antibodies for PARP-1 and β-actin as described earlier.62 The blots were developed by chemiluminescence using HRP conjugated secondary IgG. Band intensities were measured using ImageJ software.
Immunofluorescence Microscopy
MCF-7 cells were grown on glass coverslip in a 24 well cell culture plate at a density of 5 × 104 cells/mL, and then, the cells were incubated without or with 3 and 6 μM C-23 for 36 h. The cells were processed and immunostaining was performed using specific antibodies as described earlier.62 For microtubule staining, anti-α tubulin antibody was used and DNA was stained with Hoechst 33258 dye. To determine DNA damage, cells were stained with anti-γ-H2AX antibody. Intensity was calculated by ImageJ. A total of 500 cells were scored in each case. For the quantification of mitotic index, the cells were scored on the basis of DNA morphology. A total of 1000 cells were scored in each case. Vinblastine (15 nM) was taken as a positive control. Immunofluorescence imaging was performed using iPlan-apochromat 63×/1.4 NA oil immersion or iPlan-apochromat 40×/1.3 NA oil immersion objective in a Confocal laser scanning microscope and images were processed by Image-Pro Plus software (Media Cybernetics, Silver Spring, MD).
Cell Cycle Analysis and Live and Dead Assay by Flow Cytometry
MCF-7 cells (0.8 × 106) were grown in 60 mm cell culture dishes and incubated with either vehicle or 3 and 6 μM C-23 for 36 h for cell cycle analysis and 48 h for live–dead assay. After the incubation, cells were processed for flow cytometry using PI.
Isolation of Polymer/Soluble Mass of Tubulin
The polymer and soluble levels of tubulin in cells was quantified using an assay modified from Giannakakou et al.65 In brief, MCF-7 cells were treated with 3 and 6 μM C-23 for 36 h and then lysed in a hypotonic buffer solution of 1 mM MgCl2, 2 mM EGTA, 1% Nonidet P-40, 50 mM Tris–HCl (pH 6.8), and 10 μL/mL protease inhibitor (Thermofisher scientific). The supernatant containing soluble tubulin and the pellet containing polymerized tubulin were separated by centrifugation at 14000g for 15 min. The pellet fraction was lysed with GST-cell lysis buffer (20 mM Tris, 200 mM NaCl, 0.1% Triton-X 100, 1 mM DTT, pH 7.6) and centrifuged to collect the polymeric fraction of tubulin. The protein concentration of the soluble and polymeric fractions were measured by the Bradford method.66 Immunoblotting was performed using the antibody specific for α-tubulin, and the blot was developed as described above. The tubulin band intensities were measured using ImageJ software.
In Vitro Tubulin Polymerization Assay
Purification of tubulin from goat brain was done by two cycles of polymerization and depolymerization using 1 M glutamate.67 The purified tubulin was checked for its purity using Coomassie Brilliant Blue stained SDS-PAGE and its concentration was determined by Bradford method.66
Tubulin (13 μM) was suspended in PEM buffer [50 mM piperazine–N,N′-bisethanesulfonic acid (PIPES) of pH 6.8, 3 mM MgCl2, 1 mM EGTA] and incubated without and with 10, 20, 40, 60, and 75 μM C-23 for 15 min on ice. DMSO (10% v/v) was added to the reaction mixtures, followed by the addition of 1 mM GTP (guanosine-5′-triphosphate). The reaction mixtures were then immediately transferred to a Multiplate reader Spectramax M2 preheated at 37 °C and the kinetics of tubulin assembly was monitored at 37 °C by measuring turbidity at 350 nm for 45 min. Reaction mixtures containing tubulin and different concentrations of C-23 in the absence of DMSO and GTP were used as a control. The final spectra were calculated by subtracting the spectra of control from the respective reaction mixtures.
Electron Microscopy Analysis
Tubulin (13 μM) was polymerized in the absence and presence of 20 μM C-23 in PEM buffer containing 10% DMSO and 1 mM GTP for 15 min at 37 °C as described above. The protein polymers formed were then loaded on Formvar carbon-coated copper grids (300 mesh) for 45 s and blot dried. The grids were then washed with Milli-Q water and stained with 2% uranylacetate68 for 45 s, dried, and observed under an electron microscope (JEM 2100 ultra HRTEM) at 200 kV.68
Gel Filtration Chromatography
Tubulin (20 μM) was incubated with C-23 (60 μM) in 25 mM PIPES (pH 6.8) at 25 °C for 30 min. Tubulin, C-23, and a mixture of tubulin and C-23 were individually loaded onto a P4 resin column pre-equilibrated with 25 mM PIPES. The protein was eluted using 25 mM PIPES and elution fractions of 250 μL each were collected. The presence of tubulin in the fractions was determined by Bradford method and C-23 was detected by monitoring its absorbance at 400 nm.
Circular Dichroism (CD) Spectroscopy
Tubulin (1 μM) was incubated without or with (6 and 10 μM) C-23 in 1 mM phosphate buffer (pH 7.2) at 25 °C for 30 min. Far-UV CD spectra (195–260 nm) were measured in a CD spectrophotometer (JASCO J-1500) using a quartz cuvette of 0.1 cm path length.69 Each spectrum was an average of three continuously measured spectra. The data were analyzed by CDPro software and CONTINLL, CDSSTR, and SELCON3 programs were used to predict the content of secondary structures.70
Inhibition of Colchicine Binding to Tubulin by C-23
Tubulin (5 μM) was incubated without or with 2, 5, 7, 10, 12, 15, and 20 μM C-23 in 25 mM PIPES (pH 6.8) for 30 min at 37 °C. Colchicine (5 μM) was then added to the reaction mixtures and incubated for further 45 min at 37 °C. The fluorescence spectra (410–500 nm) were recorded by exciting the mixtures at 350 nm in a 0.3 cm path length cuvette using a spectrofluorometer (FP-6500 JASCO, Tokyo, Japan). The fluorescence spectra of colchicine without and with different concentrations of C-23 were measured and subtracted from the respective spectra of the test samples. Under the experimental conditions, C-23 displayed extremely low fluorescence and it did not change in the presence of tubulin. The inner filter effect correction of the observed fluorescence intensities was performed as described earlier.69
The change in the fluorescence intensity of the tubulin–colchicine complex in the presence of C-23 was calculated and Ki value was determined using71
where Ki is the half-inhibitory concentration of C-23, the concentration of C-23 required to inhibit the binding of colchicine by 50%, EC50 is the value at which the fluorescence intensity is reduced by 50% in the presence of C-23, [L] is the concentration of C-23, and Kd is the dissociation constant of binding of colchicine to tubulin. The value of Ki was determined by using Graph Pad Prism 6 software.
Inhibition of C-12 Binding to Tubulin by C-23
Compound 12 (C-12) binds to tubulin at the colchicine binding pocket.32 Tubulin (5 μM) was incubated without or with 5, 10, 20, 30, 40, and 50 μM C- 23 in 25 mM PIPES (pH 6.8) for 30 min at 37 °C. Subsequently, 5 μM C-12 was added to the reaction mixtures and incubated for an additional 10 min at 37 °C. Fluorescence spectra from 410 to 550 nm were then recorded by exciting the mixtures at 350 nm.32 The fluorescence spectra of C-12 without and with above concentrations of C-23 were measured and subtracted from the respective reaction sets. Inner filter effect correction of the observed fluorescence intensities was done.69
Molecular Docking
To examine the binding site of C-23 on tubulin, C-23 was docked on tubulin crystal structure using molecular docking software Autodock 4.2.72 The crystal structure of tubulin (PDB ID: 5LYJ)23 was used as a template and was modified prior to docking. The coordinates of chain A, B, stathmin-like domain, tubulin–tyrosine ligase, CA-4, 2-(N-morpholino)ethanesulfonic acid, AMP-PCP, and glycerol were deleted from the crystal structure using PyMOL73 resulting in αβ tubulin dimer along with GTP, GDP, two magnesium atoms, and one calcium atom. PDB coordinates for C-23 were obtained from the PRODRG server.74 To verify the docking parameters, docking of DAMA-colchicine and CA-4 on the tubulin dimer was individually performed. The PDB coordinates of DAMA-colchicine and CA-4 were obtained from their PDB structures 1SA0 and 5LYJ, respectively. The resulting docked conformation of DAMA-colchicine and CA-4 was then superimposed with the PDB coordinates of their respective crystal structures to measure RMSD. As the docking parameters were verified by docking of known ligands, a similar approach was used for the docking of C-23 on the tubulin dimer. At first, a blind docking75 of C-23 on tubulin dimer was performed by covering the entire protein surface in a grid box of 126 × 126 × 126 Å with spacing kept at 0.785 Å. Ten independent docking jobs were run keeping tubulin as a rigid and C-23 as a flexible molecule. Docking was carried out using default parameters of the Lamarckian genetic algorithm. A total of 100 runs were carried out keeping the maximum number of energy evaluation as 2 500 000, i.e., medium. Ten cycles of 100 runs yielded 1000 conformations, which were then clustered into clusters of RMSD 4.0 Å. The maximum number of conformations was found to be at the interface of the dimer. So, local docking was carried out at the dimer interface.
For the local docking, a grid box of 98 × 92 × 78 Å was made with spacing kept at 0.375 Å, which covered the interface of the αβ tubulin dimer. A total of 50 independent docking jobs were carried out using the Lamarckian genetic algorithm. A total of 50 cycles of 100 runs resulted in 5000 conformations, which were clustered in clusters of 4.0 Å.76 All the clusters were analyzed and compared on the basis of the cluster size and the binding energy, which was calculated by Autodock 4.2 scoring function. The docked conformation with the least binding energy was considered to be a probable binding conformation. The interaction of the compounds with tubulin was further analyzed using UCSF Chimera version 1.11.77
Acknowledgments
The work is supported by Tata Innovation Fellowship from Department of Biotechnology, Government of India to D.P., and a grant 02(0192)/13/EMR-II from CSIR, Government of India, to S.K.G. N.H. is supported by DST-INSPIRE fellowship, DST, New Delhi, India. We gratefully acknowledge the Confocal laser scanning microscope, FACS, and Cryo-HRTEM central facilities of IIT Bombay.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b01784.
Optimization studies in synthesis, experimental synthetic procedure, spectral data, X-ray crystallographic data and ORTEP diagram, spectra (1H and 13C NMR) of compounds, and antiproliferative activity (PDF)
Author Present Address
# Hospira Healthcare India Pvt. Ltd., a Pfizer Company, Kancheepuram, Tamil Nadu 602117, India.
Author Contributions
∥ N.H. and A.N. contributed equally to the work. N.H., A.N., and S.S.P. designed and performed experiments, analyzed data, and contributed to manuscript preparation. D.P. and S.K.G. designed the strategy of study and experiments, analyzed data, and contributed to manuscript preparation.
The authors declare no competing financial interest.
Supplementary Material
References
- Jordan M. A.; Wilson L. Microtubules as a Target for Anticancer Drugs. Nat. Rev. Cancer 2004, 4 (4), 253–265. 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- Field J. J.; Kanakkanthara A.; Miller J. H. Microtubule-Targeting Agents Are Clinically Successful due to Both Mitotic and Interphase Impairment of Microtubule Function. Bioorg. Med. Chem. 2014, 22 (18), 5050–5059. 10.1016/j.bmc.2014.02.035. [DOI] [PubMed] [Google Scholar]
- Singh P.; Rathinasamy K.; Mohan R.; Panda D. Microtubule Assembly Dynamics: An Attractive Target for Anticancer Drugs. IUBMB Life 2008, 60 (6), 368–375. 10.1002/iub.42. [DOI] [PubMed] [Google Scholar]
- Dumontet C.; Jordan M. A. Microtubule-Binding Agents: A Dynamic Field of Cancer Therapeutics. Nat. Rev. Drug Discovery 2010, 9 (10), 790–803. 10.1038/nrd3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanthou C.; Tozer G. M. Microtubule Depolymerizing Vascular Disrupting Agents: Novel Therapeutic Agents for Oncology and Other Pathologies. Int. J. Exp. Pathol. 2009, 90 (3), 284–294. 10.1111/j.1365-2613.2009.00651.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaiah G.; Remick S. C. Combretastatin A4 Phosphate: A Novel Vascular Disrupting Agent. Future Oncol. 2010, 6 (8), 1219–1228. 10.2217/fon.10.90. [DOI] [PubMed] [Google Scholar]
- Bijman M. N. A.; van Nieuw Amerongen G. P.; Laurens N.; van Hinsbergh V. W. M.; Boven E. Microtubule-Targeting Agents Inhibit Angiogenesis at Subtoxic Concentrations, a Process Associated with Inhibition of Rac1 and Cdc42 Activity and Changes in the Endothelial Cytoskeleton. Mol. Cancer Ther. 2006, 5 (9), 2348–2357. 10.1158/1535-7163.MCT-06-0242. [DOI] [PubMed] [Google Scholar]
- Lin C. M.; Ho H. H.; Pettit G. R.; Hamel E. Antimitotic Natural-Products Combretastatin-a-4 and Combretastatin-a-2 - Studies on the Mechanism of Their Inhibition of the Binding of Colchicine to Tubulin. Biochemistry 1989, 28 (17), 6984–6991. 10.1021/bi00443a031. [DOI] [PubMed] [Google Scholar]
- Pettit G. R.; Singh S. B.; Hamel E.; Lin C. M.; Alberts D. S.; Garcia-Kendall D. Isolation and Structure of the Strong Cell Growth and Tubulin Inhibitor Combretastatin A-4. Experientia 1989, 45 (2), 209–211. 10.1007/BF01954881. [DOI] [PubMed] [Google Scholar]
- Lin C. M.; Singh S. B.; Chu P. S.; Dempcy R. O.; Schmidt J. M.; Pettit G. R.; Hamel E. Interactions of Tubulin with Potent Natural and Synthetic Analogs of the Antimitotic Agent Combretastatin: A Structure-Activity Study. Mol. Pharmacol. 1988, 34 (2), 200–208. [PubMed] [Google Scholar]
- Dark G. G.; Hill S. A.; Prise V. E.; Tozer G. M.; Pettit G. R.; Chaplin D. J. Combretastatin A-4, an Agent That Displays Potent and Selective Toxicity toward Tumor Vasculature. Cancer Res. 1997, 57 (10), 1829–1834. [PubMed] [Google Scholar]
- Tron G. C.; Pirali T.; Sorba G.; Pagliai F.; Busacca S.; Genazzani A. A. Medicinal Chemistry of Combretastatin A4: Present and Future Directions. J. Med. Chem. 2006, 49 (11), 3033–3044. 10.1021/jm0512903. [DOI] [PubMed] [Google Scholar]
- Young S. L.; Chaplin D. J. Combretastatin A4 Phosphate: Background and Current Clinical Status. Expert Opin. Invest. Drugs 2004, 13 (9), 1171–1182. 10.1517/13543784.13.9.1171. [DOI] [PubMed] [Google Scholar]
- Hori K.; Saito S. Microvascular Mechanisms by Which the Combretastatin A-4 Derivative AC7700 (AVE8062) Induces Tumour Blood Flow Stasis. Br. J. Cancer 2003, 89 (7), 1334–1344. 10.1038/sj.bjc.6601261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooney C. J.; Nagaiah G.; Fu P.; Wasman J. K.; Cooney M. M.; Savvides P. S.; Bokar J. A.; Dowlati A.; Wang D.; Agarwala S. S.; Flick S. M.; Hartman P. H.; Ortiz J. D.; Lavertu P. N.; Remick S. C. A Phase II Trial of Fosbretabulin in Advanced Anaplastic Thyroid Carcinoma and Correlation of Baseline Serum-Soluble Intracellular Adhesion Molecule-1 with Outcome. Thyroid 2009, 19 (3), 233–240. 10.1089/thy.2008.0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessa C.; Lorusso P.; Tolcher A.; Farace F.; Lassau N.; Delmonte A.; Braghetti A.; Bahleda R.; Cohen P.; Hospitel M.; Veyrat-Follet C.; Soria J.-C. Phase I Safety, Pharmacokinetic and Pharmacodynamic Evaluation of the Vascular Disrupting Agent Ombrabulin (AVE8062) in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2013, 19 (17), 4832–4842. 10.1158/1078-0432.CCR-13-0427. [DOI] [PubMed] [Google Scholar]
- Cummings J.; Zweifel M.; Smith N.; Ross P.; Peters J.; Rustin G.; Price P.; Middleton M. R.; Ward T.; Dive C. Evaluation of Cell Death Mechanisms Induced by the Vascular Disrupting Agent OXi4503 during a Phase I Clinical Trial. Br. J. Cancer 2012, 106 (11), 1766–1771. 10.1038/bjc.2012.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson D. M.; Zweifel M.; Middleton M. R.; Price P. M.; Folkes L. K.; Stratford M. R. L.; Ross P.; Halford S.; Peters J.; Balkissoon J.; Chaplin D. J.; Padhani A. R.; Rustin G. J. S. Phase I Clinical and Pharmacokinetic Evaluation of the Vascular-Disrupting Agent OXi4503 in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2012, 18 (5), 1415–1425. 10.1158/1078-0432.CCR-11-2414. [DOI] [PubMed] [Google Scholar]
- Kim K. W.; Lee J. M.; Jeon Y. S.; Lee I. J.; Choi Y.; Park J.; Kiefer B.; Kim C.; Han J. K.; Choi B. I. Vascular Disrupting Effect of CKD-516: Preclinical Study Using DCE-MRI. Invest. New Drugs 2013, 31 (5), 1097–1106. 10.1007/s10637-012-9915-6. [DOI] [PubMed] [Google Scholar]
- Nam N. H. Combretastatin A-4 Analogues as Antimitotic Antitumor Agents. Curr. Med. Chem. 2003, 10 (17), 1697–1722. 10.2174/0929867033457151. [DOI] [PubMed] [Google Scholar]
- Aprile S.; Del Grosso E.; Tron G. C.; Grosa G. In Vitro Metabolism Study of Combretastatin A-4 in Rat and Human Liver Microsomes. Drug Metab. Dispos. 2007, 35 (12), 2252–2261. 10.1124/dmd.107.016998. [DOI] [PubMed] [Google Scholar]
- Cushman M.; Nagarathnam D.; Gopal D.; He H. M.; Lin C. M.; Hamel E. Synthesis and Evaluation of Analogues of (Z)-1-(4-Methoxyphenyl)-2-(3,4,5-Trimethoxyphenyl)ethene as Potential Cytotoxic and Antimitotic Agents. J. Med. Chem. 1992, 35 (12), 2293–2306. 10.1021/jm00090a021. [DOI] [PubMed] [Google Scholar]
- Gaspari R.; Prota A. E.; Bargsten K.; Cavalli A.; Steinmetz M. O. Structural Basis of Cis- and Trans-Combretastatin Binding to Tubulin. Chem. 2017, 2 (1), 102–113. 10.1016/j.chempr.2016.12.005. [DOI] [Google Scholar]
- Simoni D.; Grisolia G.; Giannini G.; Roberti M.; Rondanin R.; Piccagli L.; Baruchello R.; Rossi M.; Romagnoli R.; Invidiata F. P.; Grimaudo S.; Jung M. K.; Hamel E.; Gebbia N.; Crosta L.; Abbadessa V.; Di Cristina A.; Dusonchet L.; Meli M.; Tolomeo M. Heterocyclic and Phenyl Double-Bond-Locked Combretastatin Analogues Possessing Potent Apoptosis-Inducing Activity in HL60 and in MDR Cell Lines. J. Med. Chem. 2005, 48 (3), 723–736. 10.1021/jm049622b. [DOI] [PubMed] [Google Scholar]
- Beale T. M.; Allwood D. M.; Bender A.; Bond P. J.; Brenton J. D.; Charnock-Jones D. S.; Ley S. V.; Myers R. M.; Shearman J. W.; Temple J.; Unger J.; Watts C. A.; Xian J. A-Ring Dihalogenation Increases the Cellular Activity of Combretastatin-Templated Tetrazoles. ACS Med. Chem. Lett. 2012, 3 (3), 177–181. 10.1021/ml200149g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.; Li Y.; Sheng C.; Lv Z.; Dong G.; Wang T.; Liu J.; Zhang M.; Li L.; Zhang T.; Geng D.; Niu C.; Li K. Design and Synthesis of Cyclopropylamide Analogues of Combretastatin-A4 as Novel Microtubule-Stabilizing Agents. J. Med. Chem. 2013, 56 (3), 685–699. 10.1021/jm301864s. [DOI] [PubMed] [Google Scholar]
- Jonnalagadda S. S.; ter Haar E.; Hamel E.; Lin C. M.; Magarian Ra.; Day B. W. Synthesis and Biological Evaluation of 1,1-Dichloro-2,3-Diarylcyclopropanes as Antitubulin and Anti-Breast Cancer Agents. Bioorg. Med. Chem. 1997, 5 (4), 715–22. 10.1016/S0968-0896(97)00014-X. [DOI] [PubMed] [Google Scholar]
- Liou J. P.; Chang C. W.; Song J. S.; Yang Y. N.; Yeh C. F.; Tseng H. Y.; Lo Y. K.; Chang Y. L.; Chang C. M.; Hsieh H. P. Synthesis and Structure-Activity Relationship of 2-Aminobenzophenone Derivatives as Antimitotic Agents. J. Med. Chem. 2002, 45 (12), 2556–2562. 10.1021/jm010365+. [DOI] [PubMed] [Google Scholar]
- La Regina G.; Edler M. C.; Brancale A.; Kandil S.; Coluccia A.; Piscitelli F.; Hamel E.; De Martino G.; Matesanz R.; Diaz J. F.; Scovassi A. I.; Prosperi E.; Lavecchia A.; Novellino E.; Artico M.; Silvestri R. Arylthioindole Inhibitors of Tubulin Polymerization. 3. Biological Evaluation, Structure-Activity Relationships and Molecular Modeling Studies. J. Med. Chem. 2007, 50 (12), 2865–2874. 10.1021/jm061479u. [DOI] [PubMed] [Google Scholar]
- Ohsumi K.; Nakagawa R.; Fukuda Y.; Hatanaka T.; Morinaga Y.; Nihei Y.; Ohishi K.; Suga Y.; Akiyama Y.; Tsuji T. Novel Combretastatin Analogues Effective against Murine Solid Tumors: Design and Structure-Activity Relationships. J. Med. Chem. 1998, 41 (16), 3022–3032. 10.1021/jm980101w. [DOI] [PubMed] [Google Scholar]
- Lee H. Y.; Chang J. Y.; Nien C. Y.; Kuo C. C.; Shih K. H.; Wu C. H.; Chang C. Y.; Lai W. Y.; Liou J. P. 5-Amino-2-Aroylquinolines as Highly Potent Tubulin Polymerization Inhibitors. Part 2. The Impact of Bridging Groups at Position C-2. J. Med. Chem. 2011, 54 (24), 8517–8525. 10.1021/jm201031f. [DOI] [PubMed] [Google Scholar]
- Chaudhary V.; Venghateri J. B.; Dhaked H. P. S.; Bhoyar A. S.; Guchhait S. K.; Panda D. Novel Combretastatin-2-Aminoimidazole Analogues as Potent Tubulin Assembly Inhibitors: Exploration of Unique Pharmacophoric Impact of Bridging Skeleton and Aryl Moiety. J. Med. Chem. 2016, 59 (7), 3439–3451. 10.1021/acs.jmedchem.6b00101. [DOI] [PubMed] [Google Scholar]
- Hu Y.; Stumpfe D.; Bajorath J. Recent Advances in Scaffold Hopping. J. Med. Chem. 2017, 60 (4), 1238–1246. 10.1021/acs.jmedchem.6b01437. [DOI] [PubMed] [Google Scholar]
- Guchhait S. K.; Hura N.; Sinha K.; Panda D. Pyridine C3-Arylation of Nicotinic Acids Accessible via a Multicomponent Reaction: An Entry to All-Substituted-3,4-Diarylated Pyridines. RSC Adv. 2017, 7 (14), 8323–8331. 10.1039/C6RA28299G. [DOI] [Google Scholar]
- Morgan P.; Van Der Graaf P. H.; Arrowsmith J.; Feltner D. E.; Drummond K. S.; Wegner C. D.; Street S. D. A. Can. the Flow of Medicines Be Improved? Fundamental Pharmacokinetic and Pharmacological Principles toward Improving Phase II Survival. Drug Discovery Today 2012, 17, 419–424. 10.1016/j.drudis.2011.12.020. [DOI] [PubMed] [Google Scholar]
- van der Graaf P. H.; Benson N. Systems Pharmacology: Bridging Systems Biology and Pharmacokinetics-Pharmacodynamics (PKPD) in Drug Discovery and Development. Pharm. Res. 2011, 28 (7), 1460–1464. 10.1007/s11095-011-0467-9. [DOI] [PubMed] [Google Scholar]
- Mehar A.; Macanas-Pirard P.; Mizokami A.; Takahashi Y.; Kass G. E. N.; Coley H. M. The Effects of Cyclooxygenase-2 Expression in Prostate Cancer Cells: Modulation of Response to Cytotoxic Agents. J. Pharmacol. Exp. Ther. 2008, 324 (3), 1181–1187. 10.1124/jpet.107.131383. [DOI] [PubMed] [Google Scholar]
- Cho S. J.; Kim N.; Kim J. S.; Jung H. C.; Song I. S. The Anti-Cancer Effect of COX-2 Inhibitors on Gastric Cancer Cells. Dig. Dis. Sci. 2007, 52 (7), 1713–1721. 10.1007/s10620-007-9787-3. [DOI] [PubMed] [Google Scholar]
- Mao J. T.; Roth M. D.; Fishbein M. C.; Aberle D. R.; Zhang Z. F.; Rao J. Y.; Tashkin D. P.; Goodglick L.; Holmes E. C.; Cameron R. B.; Dubinett S. M.; Elashoff R.; Szabo E.; Elashoff D. Lung Cancer Chemoprevention with Celecoxib in Former Smokers. Cancer Prev. Res. 2011, 4 (7), 984–993. 10.1158/1940-6207.CAPR-11-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yale H. L. The Trifluoromethyl Group in Medicinal Chemistry. J. Med. Pharm. Chem. 1959, 1 (2), 121–133. 10.1021/jm50003a001. [DOI] [PubMed] [Google Scholar]
- Sawyer J. S.; Anderson B. D.; Beight D. W.; Campbell R. M.; Jones M. L.; Herron D. K.; Lampe J. W.; McCowan J. R.; McMillen W. T.; Mort N.; Parsons S.; Smith E. C. R.; Vieth M.; Wier L. C.; Yan L.; Zhang F. M.; Yingling J. M. Synthesis and Activity of New Aryl- and Heteroaryl- Substituted Pyrazole Inhibitors of the Transforming Growth Factor-B Type I Receptor Kinase Domain. J. Med. Chem. 2003, 46 (19), 3953–3956. 10.1021/jm0205705. [DOI] [PubMed] [Google Scholar]
- Chowdhury M. A.; Abdellatif K. R. A.; Dong Y.; Das D.; Suresh M. R.; Knaus E. E. Synthesis of Celecoxib Analogues Possessing a N-Difluoromethyl-1,2-Dihydropyrid-2-One 5-Lipoxygenase Pharmacophore: Biological Evaluation as Dual Inhibitors of Cyclooxygenases and 5-Lipoxygenase with Anti-Inflammatory Activity. J. Med. Chem. 2009, 52 (6), 1525–1529. 10.1021/jm8015188. [DOI] [PubMed] [Google Scholar]
- Begtrup M.; Rasmussen L. K. Arylhydrazines. Sci. Synth. 2007, 1773–1826. [Google Scholar]
- Anderson K. W.; Tundel R. E.; Ikawa T.; Altman R. A.; Buchwald S. L. Monodentate Phosphines Provide Highly Active Catalysts for Pd-Catalyzed C-N Bond-Forming Reactions of Heteroaromatic Halides/amines and (H)N-Heterocycles. Angew. Chem., Int. Ed. 2006, 45, 6523–6527. 10.1002/anie.200601612. [DOI] [PubMed] [Google Scholar]
- Antilla J. C.; Baskin Jm.; Barder Te.; Buchwald S. L. Copper-Diamine-Catalyzed N-Arylation of Pyrroles, Pyrazoles, Indazoles, Imidazoles, and Triazoles. J. Org. Chem. 2004, 69 (17), 5578–5587. 10.1021/jo049658b. [DOI] [PubMed] [Google Scholar]
- Antilla J. C.; Klapars A.; Buchwald S. L.; Soc J. A. C. The Copper-Catalyzed N-Arylation of Indoles. J. Am. Chem. Soc. 2002, 124 (39), 11684–11688. 10.1021/ja027433h. [DOI] [PubMed] [Google Scholar]
- Correa A.; Bolm C. Iron-Catalyzed N-Arylation of Nitrogen Nucleophiles. Angew. Chem., Int. Ed. 2007, 46 (46), 8862–8865. 10.1002/anie.200703299. [DOI] [PubMed] [Google Scholar]
- Taillefer M.; Xia N.; Ouali A. Efficient Iron/copper Co-Catalyzed Arylation of Nitrogen Nucleophiles. Angew. Chem., Int. Ed. 2007, 46 (6), 934–936. 10.1002/anie.200603173. [DOI] [PubMed] [Google Scholar]
- Yang D. M.; Zhou Y. H.; Xue N.; Qu J. P. Synthesis of Trifluoromethyl Ketones via Tandem Claisen Condensation and Retro-Claisen C-C Bond-Cleavage Reaction. J. Org. Chem. 2013, 78 (8), 4171–4176. 10.1021/jo400280p. [DOI] [PubMed] [Google Scholar]
- Lam P. Y. S.; Clark C. G.; Saubern S.; Adams J.; Averill K. M.; Chan D. M. T.; Combs A. Copper Promoted Aryl/saturated Heterocyclic C-N Bond Cross-Coupling with Arylboronic Acid and Arylstannane. Synlett 2000, 5, 674–676. [Google Scholar]
- Skehan P.; Storeng R.; Scudiero D.; Monks A.; McMahon J.; Vistica D.; Warren J. T.; Bokesch H.; Kenney S.; Boyd M. R. New Colorimetric Cytotoxicity Assay for Anticancer-Drug Screening. J. Natl. Cancer Inst. 1990, 82 (13), 1107–1112. 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- Ganem N. J.; Pellman D. Linking Abnormal Mitosis to the Acquisition of DNA Damage. J. Cell Biol. 2012, 199 (6), 871–881. 10.1083/jcb.201210040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariotti L. G.; Pirovano G.; Savage K. I.; Ghita M.; Ottolenghi A.; Prise K. M.; Schettino G. Use of the γ-H2AX Assay to Investigate DNA Repair Dynamics Following Multiple Radiation Exposures. PLoS One 2013, 8 (11), e79541. 10.1371/journal.pone.0079541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A.; Naaz A.; Prakasham A. P.; Gangwar M. K.; Butcher R. J.; Panda D.; Ghosh P. Potent Anticancer Activity with High Selectivity of a Chiral Palladium N-Heterocyclic Carbene Complex. ACS Omega 2017, 2 (8), 4632–4646. 10.1021/acsomega.7b00688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldani C.; Scovassi A. I. Poly(ADP-Ribose) Polymerase-1 Cleavage during Apoptosis: An Update. Apoptosis 2002, 7 (4), 321–328. 10.1023/A:1016119328968. [DOI] [PubMed] [Google Scholar]
- Gigant B.; Wang C.; Ravelli R. B. G.; Roussi F.; Steinmetz M. O.; Curmi P. A.; Sobel A.; Knossow M. Structural Basis for the Regulation of Tubulin by Vinblastine. Nature 2005, 435 (7041), 519–522. 10.1038/nature03566. [DOI] [PubMed] [Google Scholar]
- Ravelli R. B. G.; Gigant B.; Curmi P. A.; Jourdain I.; Lachkar S.; Sobel A.; Knossow M. Insight into Tubulin Regulation from a Complex with Colchicine and a Stathmin-like Domain. Nature 2004, 428 (6979), 198–202. 10.1038/nature02393. [DOI] [PubMed] [Google Scholar]
- Nogales E.; Grayer Wolf S.; Khan I. A.; Luduena R. F.; Downing K. H. Structure of Tubulin at 6.5A and Location of the Taxol-Binding Site. Nature 1995, 375 (6530), 424–427. 10.1038/375424a0. [DOI] [PubMed] [Google Scholar]
- Sackett D. L. Podophyllotoxin, Steganacin and Combretastatin: Natural Products That Bind at the Colchicine Site of Tubulin. Pharmacol. Ther. 1993, 59 (2), 163–228. 10.1016/0163-7258(93)90044-E. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya B.; Wolff J. Promotion of Fluorescence Upon Binding of Colchicine to Tubulin. Proc. Natl. Acad. Sci. U. S. A. 1974, 71 (7), 2627–2631. 10.1073/pnas.71.7.2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai A.; Gupta T. K.; Kini S.; Kunwar A.; Surolia A.; Panda D. CXI-Benzo-84 Reversibly Binds to Tubulin at Colchicine Site and Induces Apoptosis in Cancer Cells. Biochem. Pharmacol. 2013, 86 (3), 378–391. 10.1016/j.bcp.2013.05.024. [DOI] [PubMed] [Google Scholar]
- Rai A.; Kapoor S.; Naaz A.; Kumar Santra M.; Panda D. Enhanced Stability of Microtubules Contributes in the Development of Colchicine Resistance in MCF-7 Cells. Biochem. Pharmacol. 2017, 132, 38–47. 10.1016/j.bcp.2017.02.018. [DOI] [PubMed] [Google Scholar]
- Skehan P.; Storeng R.; Scudiero D.; Monks A.; McMahon J.; Vistica D.; Warren J. T.; Bokesch H.; Kenney S.; Boyd M. R. New Colorimetric Cytotoxicity Assay for Anticancer-Drug Screening. J. Natl. Cancer Inst. 1990, 82 (13), 1107–1112. 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- Mohan R.; Panda D. Kinetic Stabilization of Microtubule Dynamics by Estramustine Is Associated with Tubulin Acetylation, Spindle Abnormalities, and Mitotic Arrest. Cancer Res. 2008, 68 (15), 6181–6189. 10.1158/0008-5472.CAN-08-0584. [DOI] [PubMed] [Google Scholar]
- Giannakakou P.; Sackett D. L.; Kang Y. K.; Zhan Z.; Buters J. T.; Fojo T.; Poruchynsky M. S. Paclitaxel-Resistant Human Ovarian Cancer Cells Have Mutant Beta-Tubulins That Exhibit Impaired Paclitaxel-Driven Polymerization. J. Biol. Chem. 1997, 272 (27), 17118–17125. 10.1074/jbc.272.27.17118. [DOI] [PubMed] [Google Scholar]
- Bradford M. M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Hamel E.; Lin C. M. Glutamate-Induced Polymerization of Tubulin - Characteristics of the Reaction and Application to the Large-Scale Purification of Tubulin. Arch. Biochem. Biophys. 1981, 209 (1), 29–40. 10.1016/0003-9861(81)90253-8. [DOI] [PubMed] [Google Scholar]
- Hire R. R.; Srivastava S.; Davis M. B.; Kumar Konreddy A.; Panda D. Antiproliferative Activity of Crocin Involves Targeting of Microtubules in Breast Cancer Cells. Sci. Rep. 2017, 7 (March), 44984. 10.1038/srep44984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S.; Mishra S.; Surolia A.; Panda D. C1, a Highly Potent Novel Curcumin Derivative, Binds to Tubulin, Disrupts Microtubule Network and Induces Apoptosis. Biosci. Rep. 2016, 36, e00323. 10.1042/BSR20160039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreerama N.; Woody R. W. Estimation of Protein Secondary Structure from Circular Dichroism Spectra: Comparison of CONTIN, SELCON, and CDSSTR Methods with an Expanded Reference Set. Anal. Biochem. 2000, 287 (2), 252–260. 10.1006/abio.2000.4880. [DOI] [PubMed] [Google Scholar]
- Cheng Y.; Prusoff W. H. Relationship between the Inhibition Constant (K1) and the Concentration of Inhibitor Which Causes 50 per Cent Inhibition (I50) of an Enzymatic Reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner Mf.; Belew Rk.; Goodsell Ds.; Olson A. J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30 (16), 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano W. L. The PyMOL Molecular Graphics System, Version 1.1; Schrodinger LLC, 2002. [Google Scholar]
- Schuttelkopf A. W.; van Aalten D. M. F. PRODRG: A Tool for High-Throughput Crystallography of Protein-Ligand Complexes. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 1355–1363. 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- Hetenyi C.; van der Spoel D. Efficient Docking of Peptides to Proteins without Prior Knowledge of the Binding Site. Protein Sci. 2002, 11 (7), 1729–1737. 10.1110/ps.0202302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumbhar B. V.; Borogaon A.; Panda D.; Kunwar A. Exploring the Origin of Differential Binding Affinities of Human Tubulin Isotypes Alpha Beta II, Alpha Beta III and Alpha Beta IV for DAMA-Colchicine Using Homology Modelling, Molecular Docking and Molecular Dynamics Simulations. PLoS One 2016, 11 (5), e0156048. 10.1371/journal.pone.0156048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera - A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25 (13), 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.