

Abstract
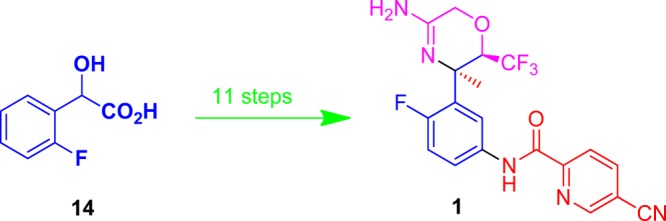
Development of a scalable synthesis of an oxazine class of β-secretase inhibitor is described. Trifluoromethylated acyloin synthesis by the reaction of a mandelic acid with trifluoroacetic anhydride in the presence of pyridine (Dakin–West reaction) was used as an efficient strategy to install the key trifluoromethyl substituent on the oxazine ring. Diastereoselective addition of methyl magnesium bromide to a cyclic sulfamidate imine and trimethylsilyl trifluoromethanesulfonate catalyzed intramolecular amidine formation to yield oxazine-3-amine are some of the significant, novel synthetic methods developed in this synthesis. These critical transformations allowed a concise 11-step route to the target compound with excellent overall yields.
Introduction
The amyloid oligomer protein Aβ42 has now been widely accepted as the principal culprit behind the formation of fibrils and eventually their deposition as plaques in the brain, a key, measurable, causative factor suspected in the pathogenesis of Alzheimer’s disease (AD) and age-related dementia.1 Many amyloid-lowering strategies have thus been under investigation as potential interventional therapies to arrest the progression of AD and related neurodegenerative diseases.2 Prominent among the small molecule drug targets actively being pursued in this approach is the enzyme β-secretase 1 (BACE 1), which cleaves the amyloid precursor protein (APP) to a soluble 99 amino acid peptide (C 99), the rate-determining step in the pathway for fibrilogenic Aβ42 formation.3 Work from our laboratories has led to a series of potent BACE 1 inhibitors.4 Among the oxazine class of BACE 1 inhibitors, compound 1 was identified as one of the most promising and was selected for further pharmacological evaluation. Toward this purpose, multigram quantities of active pharmaceutical ingredient (API) were needed. Herein, we describe development of a new synthetic route to 1. Our primary focus has been to replace the discovery route that suffers from poor overall yield (<2%), use of hazardous and expensive reagents, and numerous chromatographic purifications with a practical and step-economic route suitable for large-scale synthesis for preclinical development (Figure 1).
Figure 1.

Structure of 1.
Results and Discussion
The discovery synthesis, as outlined in Scheme 1, starts from 2-fluoro-5-bromoacetophenone, which was converted to the amino acid, 3, using the classical Strecker synthesis. The amino acid was then converted to the morpholinedione, 4, and then resolved by chiral chromatography. The required trifluoromethyl group was then introduced by addition of the Ruppert–Prakash reagent, followed by halogenation and reduction in a three-step sequence with 38% overall yield to give the morpholin-3-one, 5, as a mixture of diastereoisomers in 4:1 ratio. Conversion to amino-oxazine 6 was accomplished by a two-step sequence by first converting morpholin-3-one 5 to the corresponding thione, followed by substitution with ammonia. Introduction of the amine at the 5-position was accomplished by reaction with sodium azide with stoichiometric copper at high temperature. Standard amide coupling with 5-cyano-pyridine-1-carboxylic acid furnished the final product.
Scheme 1. Discovery Synthesis of 7 Starting from 2.

Reagents and conditions: (a) TMSCN, NH4Cl, 7 N NH3/MeOH, room temperature (rt), 4 days; (b) 6 N HCl, reflux, 16 h; (c) (i) chloroacetyl chloride, 1 M NaOH, 1,4-dioxane, 2 h, (ii) dimethylformamide (DMF), NaHCO3, 80 °C, 3 h; (d) TMSCF3, TBAT, tetrahydrofuran (THF), 0 °C to rt, 20 min; (e) SOCl2, DCM, 0 °C, 30 min, then add pyridine, 0 °C, 30 min; (f) Zn, AcOH, 80 °C, 3 h; (g) P2S5, THF, 70 °C, 3 h; (h) 33% NH3 (aq), 60 °C, 18 h; (i) NaN3, CuI, DMEDA, Na2CO3, dimethyl sulfoxide (DMSO), 110 °C, 4 h; (j) 5-cyanopicolinic acid, DMTMM, MeOH, 0 °C, 6 h.
The discovery synthesis as outlined above, although quite adequate for preparation of small amounts of the final product, presented several problems for large-scale synthesis. The main issues are (1) low throughput in the majority of steps, (2) poor diastereoselectivity in the formation of trifluoromethyl morpholinone 5, (3) high-temperature azide substitution reaction to introduce the amine toward the end of the synthesis, and (4) chromatography required for nearly all of the intermediates.
Bearing in mind these issues, we focused our attention on the penultimate intermediate, 8, as our target for new route development (Scheme 2). At the very outset, we decided that a nitration–reduction sequence from 10 would be the most suitable approach to install the desired amine substituent. It was anticipated that the directing influence of the fluorine in the electrophilic nitration would result in the desired para substitution. We envisaged synthesis of amidine 10 from the stereochemically well-defined amino alcohol, 11.
Scheme 2. Retrosynthesis.

While considering a synthetic route for the amino alcohol, 11, we recognized that an efficient approach to the tertiary amine stereo center would be through addition of a methyl carbon nucleophile to a suitably elaborated imine functionality. We were particularly attracted by the elegant work of Lee and Chang5 on the addition of Grignard reagents to sulfamidate imines for the synthesis of quaternary sulfamidate building blocks. Following their work, we envisaged synthesis of 11, as outlined in Scheme 3, with the sulfamate playing a remarkable role in the route from 15. It simultaneously delivers the required nitrogen atom into the molecule, acts as a protecting group for the alcohol, activates the imine for Grignard addition, and locks the conformation of the protected hydroxyimine, 13, such that Grignard addition should be stereoselective. Lam and co-workers6 have shown that the sulfamidates like 12 can be readily cleaved by lithium aluminum hydride (LAH) to yield the desired amino alcohol, 11.
Scheme 3. Cyclic Sulfamate Imine Route.

We now only needed ready access to trifluoromethyl acyloin, 15, which we found in an interesting variant of the Dakin–West reaction reported by Kawase and co-workers.7 We were delighted to find that 15 could be obtained in good yields by starting with fluoromandelic acid 14 and using trifluoroacetic anhydride (TFAA) under the conditions prescribed by Kawase without modification (Scheme 4). This reaction was subsequently scaled to 200 g without the need for optimization studies; however, an exotherm from 40 to 62 °C during addition of TFAA was observed and should be taken into account for any larger scale development of this transformation. The scaled reaction product, 15, was a crystalline solid8a with sufficient purity to carry forward to the next step without additional purification.
Scheme 4. Synthesis of 15.

This opening transformation established the carbon framework of the chiral portion of the molecule in a single step from a readily available starting material. In addition, we have introduced the trifluoromethyl group with a relatively inexpensive reagent, TFAA, whereas the medicinal chemistry route made use of the more expensive Ruppert–Prakash reagent (TMSCF3) as the source of the trifluoromethyl group.8
With sufficient quantities of trifluoromethyl acyloin 15 in hand, we proceeded to examine the forward synthesis of amino alcohol 11 as outlined in Scheme 5.
Scheme 5. Forward Synthesis of Amino Alcohol 11.

Generation of cyclic sulfamate imine 13 from acyloin 15 was performed on a larger scale using in situ-generated sulfamoyl chloride (Scheme 5).9 Rather than combining chlorosulfonyl isocyanate and formic acid neat, the reagent formation was carried out in acetonitrile (ACN), and the solution was used as is, allowing sufficient time for reagent generation. The initial sulfamoylation of the hydroxyl group was carried out following literature precedent using dimethylacetamide (DMA) as the solvent.10 In the absence of DMA (e.g., using ACN as the sole solvent), no reaction was observed until DMA was added. DMA appears to serve as a nucleophilic catalyst to promote the sulfonylation event. After consumption of 15 was complete, the reaction mixture contained mostly sulfamoylated ketone 16 with a small amount of imine 13. Upon heating the reaction to 55 °C, 16 was smoothly converted to 13. Sulfamoyl chloride (2 equiv) is used in the reaction, with the second equivalent presumably acting as a water scavenger to drive imine formation. With less than 2 equiv, the yield of 13 is decreased.
Addition of methyl Grignard to 13 was expected to have a high probability of success based on literature precedent.5 We envisaged that the addition would take place from the least hindered face, forming diastereomer with RR/SS relative stereochemistry rac-12. However, the reaction presented difficulties with reproducibility until it was found that using excess Grignard reagent resulted in consistent, very clean, high-yielding (>95%) reactions in small-scale screens (∼50–100 mg). It was also found necessary to add the Grignard reagent at temperatures below −40 °C (−60 °C preferable) to avoid significant impurity formation. Using less than 1 equiv of Grignard or adding reagent at too high a temperature resulted in dark-colored reactions that always showed numerous impurities.
Upon addition of aqueous HCl to quench the reaction, the clean, high-yielding reactions always bubbled off methane gas from the quench of unreacted methyl Grignard, as expected. The poor-quality, dark-colored reaction mixtures uniformly did not outgas when quenched indicating that the excess methyl Grignard had been consumed in some manner, which presumably plays a role in the multiple side products observed in the poor reactions.
The diastereoselectivity of the Grignard addition was always very high, however. NOE experiments of the product confirmed the expected RR/SS relative stereochemistry.11 The other possible diastereomer was never observed in any reactions from which adduct rac-12 was isolated. Analysis by high-performance liquid chromatography (HPLC) of the unpurified product consistently showed a single product peak for 12, and 1H NMR spectra of the unpurified product always showed the presence of only one diastereomer.
On a 5 g scale reaction, rac-12 was isolated in 95% yield. The reaction was then scaled to 176 g, which produced rac-12 in only 67% yield. This large-scale reaction produced the dark red-brown colored reaction mixture during the addition of Grignard reagent that was characteristic of significant impurity formation. As observed during small-scale reaction screening, the large-scale reaction mixture did not outgas methane when quenched. HPLC analysis showed the production of numerous side products; therefore, a much more extensive purification had to be undertaken and a much lower yield was observed than expected. It is believed that the problem may lie with the extended time (3.5 h) required to add all of the methyl Grignard reagent on scale, while keeping the temperature below −40 °C. Impurity production may be the result of the anionic product reacting with unreacted starting material in some manner during the long addition.
If the interaction of the product and starting material is the cause of impurity formation, using an inverse addition protocol deserves further scrutiny. One small-scale reaction using inverse addition in tert-butyl methyl ether (TBME) produced rac-12 cleanly. TBME itself deserves further investigation as some small-scale reactions run in TBME may indicate that TBME could be superior to THF. Project timelines prevented a deeper exploration of reaction parameters, such as solvent and order of addition, to optimize this Grignard addition on scale, but we feel that the reaction could be improved significantly with further study.
We determined that sulfamate rac-12 was a good candidate for separation of enantiomers by chiral stationary-phase HPLC. Excellent resolution of the enantiomers was achieved using the CHIRALPAK-AS stationary phase with heptane/ethanol as the eluent. Rac-12 (100 g) was separated with near-quantitative recovery (48%) of the desired (R,R) enantiomer.12
Desulfuration would now complete the preparation of the key intermediate amino alcohol 11. The cleavage of the sulfoxy group from the molecule was accomplished by reduction with LAH based on literature precedent.6,11 A moderate exotherm (50–63 °C) at the initiation of the reaction should be noted, and precautions should be taken for larger scale applications. After reaction quench by the “Fieser” method,1211 was isolated in very high yield from the resultant aluminum salts by simple filtration.
Scheme 6 illustrates the completion of the synthesis in seven steps commencing with the selective O-alkylation of 11 to give cyanomethyl ether 17. We recognized this alkylation as a potentially difficult task. We expected that the steric hindrance around the nitrogen center may attenuate the rate of N-alkylation, thereby favoring O-alkylation. On the other hand, the inductive withdrawal effect of the α-trifluoromethyl group would attenuate oxygen nucleophilicity bringing the balance of reactivity between O- and N-alkylation into question. Furthermore, base-mediated elimination of hydrogen fluoride might also shunt the reaction to unproductive pathways.
Scheme 6. Complete Synthesis of 1 Starting from 11.

Ultimately, the alkylation was accomplished on multigram scale using lithium ethoxide and bromoacetonitrile in THF at rt. Adduct 17 was obtained in 98% unpurified yield with bromoacetonitrile as a major contaminant. Nevertheless, this material could be taken directly into the next step without need for purification. The conditions for this alkylation were the result of an extensive screening of combinations of base, alkylating agent, and solvent (Table 1). The combination of lithium counterion with bromoacetonitrile and THF singularly produced the proper balance of nucleophile and electrophile reactivity, which delivered high-yielding O-alkylation with minimal side products. Remarkably, alteration of any one of these parameters resulted in much poorer reaction performance. For example, the use of sodium or potassium alkoxides resulted in the formation of numerous unidentified products, whereas pairing chloroacetonitrile with a lithium alkoxide produced no reaction at all at rt. All attempts with the weaker inorganic bases in Table 1 gave either no reaction or just a trace of reaction. Whereas lithium ethoxide was selected for use in our scaled-up reactions, lithium tert-butoxide behaved similarly in smaller scale reactions. Among the other solvents screened, DMF promoted the formation of numerous side products, whereas reactions in methanol and ethanol showed no conversion of the starting material.
Table 1. Selective O-Alkylation Reaction Screen.

| screening results | bases | solvents | alkylating agents |
|---|---|---|---|
| no conversion | NaOR | MeOH, EtOH | ClCH2CN |
| BrCH2CN | |||
| low conversion | K2CO3, Cs2CO3 | ACN, DMF | ClCH2CN |
| KF, CsF, K3PO4 | BrCH2CN | ||
| product formed with numerous unidentified side products | NaH, NaOMe, KH | ACN, DMF, THF | ClCH2CN |
| NaOt-Bu, KOt-Bu | |||
| LiHMDS, KHMDS | BrCH2CN | ||
| generally clean reactions with THF as solvent using bromoacetonitrile | LiH, LiOt-Bu, LiOEt | THF | ClCH2CN |
| BrCH2CN |
The cyclization of 17 to amidine 10 was promoted on scale by trimethylsilyl trifluoromethanesulfonate (TMSOTf) with excellent results. A 99% yield of unpurified 10 was obtained, and the material was again of sufficient purity to be carried forward without further manipulation. These novel reaction conditions were also born from intensive screening. We had first examined copper(I) salts to promote the cyclic amidine formation.13−15 We found stoichiometric quantities of CuCl to be effective giving a 79% isolated yield of amidine in a small-scale reaction; however, we did not feel that using a full equivalent of a copper salt was optimum for scale-up. In particular, there appeared to be difficulty with releasing the amidine from copper coordination during work-up.
To find an alternative, we initiated a screen of Brønsted acid and Lewis acid activating agents using microwave heating for convenience. The results of the screen are summarized in Table 2. Mineral acids at a higher temperature (100 °C) produced decomposition or undesired products. At a moderate temperature (60 °C), protic acids did not decompose the starting material but also did not promote the cyclization to amidine with good conversion. Trimethysilyl chloride was observed to give a clean reaction but conversion was limited. Simple thermal conditions in o-dichlorobenzene and isopropanol (IPA) were also not successful. Interestingly, heating in hexafluoroisopropanol yielded significantly more 10 than IPA. In addition to CuCl, zinc salts and Cu(OTf)2 were found to give significant conversion to amidine, and TMSOTf also showed significant, clean conversion to amidine during screening.
Table 2. Amidine Formation Reaction Screen.

| screening result | conditions | |
|---|---|---|
| decomposition or other products | conc. H2SO4, tol, 100 °C | |
| HCI, MeOH, 100 °C | ||
| clean but low or no conversion | HCI/dioxane, tol, 60 °C | TMSCI (1.0), tol, 100 °C |
| TFA, tol, 60 °C | o-dichlorobenzene, 200 °C | |
| TfOH, tol, 100 °C | IPA, 150 °C | |
| moderate to excellent conversion | CuCl(1.0), EtOH, 100 °C | Zn(OTf)2 (1.0), IPA, 120 °C |
| CuCl(1.0), IPA, 100 °C | ZnCl2(1.0), IPA, 120 °C | |
| CuCl(1.0), DMSO, 100 °C | TMSOTf (1.0), tol, 100 °C | |
| hexafluoro-IPA, 120 °C | Cu(OTf)2 (1.0), IPA, 100 °C | |
We decided to further examine Zn(OTf)2 and TMSOTf (Table 3). We found that Zn(OTf)2 could be used in catalytic amounts (5 mol %) in IPA, and gave a 79% yield of the amidine, but the reaction was complicated by formation of the dimeric side product, 18. The cyclization using TMSOTf, however, was extremely clean giving a 98% yield of 10 with no further purification required. Work-up and isolation was very simple, requiring only a quench with aqueous Na2CO3, extraction, and concentration of the organic layer. One equivalent of TMSOTf was found to be required. Using less reagent resulted in reduced conversion. We also determined upon further scale-up that heating was not required. Reasonable reaction rate was observed at rt.
Table 3. Comparison of Zn(OTf)2 and TMSOTf for Cyclic Amidine Formation.

We now had in place a concise six-step synthesis of advanced intermediate 10 from 2-fluoromandelic acid that notably establishes the vicinal chiral centers of our target with 100% diastereoselectivity. Most steps have been developed to high-yielding transformations, and significant purification effort was required only for the Grignard reaction adduct, rac-12, a problem that we feel is solvable with further development. For expediency, we opted for chiral HPLC for resolution during our work, but we note that the basic amine and amidine intermediates through compound 10 may be candidates for resolution by scalable crystallization processes.
Continuing the route, the nitration of 10 was straightforward in 90% fuming nitric acid, giving a high yield of desired product 9 in a 97:3 mixture with the ortho nitration isomer. To avoid nitration of residual toluene from the previous reaction, toluene was removed from 10 by twice concentrating the crude material from acetic acid (AcOH). The AcOH solution of 10 was then fed directly into the nitration. It was found advantageous to minimize the amount of AcOH (or any other co-solvent) in this reaction, as the rate of nitration slowed considerably with dilution of the nitric acid. For example, the usual concentrated nitric acid (∼60% HNO3 in water) did not nitrate 10 at rt and was slow to nitrate even at 100 °C. Reactions with no co-solvent in which solid 10 was added directly to 90% fuming nitric acid proceeded to completion more rapidly than reactions in which AcOH solutions of 10 were used; nevertheless, working with 10 in AcOH solution was preferable on larger scale.
The mixture of nitration isomers was carried forward into the Boc protection step, which occurred smoothly in acetonitrile. It was important to eliminate all traces of AcOH from 9, as N-acetyl amidine 22 was observed as an impurity in test reactions on initial samples of crude 9 (Scheme 7).
Scheme 7. Impurity 22 Arising from Boc Protection of 9.

Attempts at nitro group reduction on the unpurified mixture of 19 under typical Pd/C catalytic hydrogenation conditions (rt, 1 atm H2) were not successful. The reactions stalled at partial conversion from apparent catalyst poisoning. Therefore, the decision was made to purify 19 by silica gel chromatography. The benefit of the chromatography was that the minor nitration regioisomer, 19b, could be removed at this stage.
With purified 19a, hydrogenation of the nitro group occurred without incident in very high yield. The nature of the catalyst poisoning with impure 19 was not studied further nor were alternative catalysts examined in our work.
Aniline 20 was efficiently coupled to picolinic acid 21 using the peptide coupling agent EDCI in DMF. The reaction was initiated with 1.1 equiv each of the acid and EDCI. When the reaction remained incomplete after repeated monitoring, an additional 0.2 equiv of each component was introduced, and the reaction proceeded to complete conversion. Excess reactants and spent peptide coupling by-product were easily removed by aqueous washes, leaving the product in the organic layer in a pure enough state to take directly into the final deprotection.
Final Boc deprotection was accomplished in a straightforward manner using trifluoroacetic acid (TFA) in dichloromethane. In small-scale experimentation, 1 was obtained in an 89:11 ratio with tert-butyl amide 24 arising from the Ritter reaction with the liberated tert-butyl cation (Scheme 8). To solve this problem, anisole (10 equiv) was included in the reaction to scavenge the rogue cation, thereby completely suppressing the Ritter by-product. Final purification of 1 was accomplished by trituration with hot 3:1 ethanol/water, followed by partial recrystallization from hot toluene with 43 g of 1 being delivered for in vivo profiling.
Scheme 8. Boc Deprotection of 23, Leading to 1 and 24.

In conclusion, choice of synthetic routes selected by chemists in the pharmaceutical industry is largely purpose driven. Early discovery synthesis for example is invariably focused on routes that go through key advanced intermediates to allow maximum flexibility for exploration of structure–activity relationships rapidly with less emphasis on the overall synthetic efficiency. However, as the discovery program matures to lead candidate selection stage, the chemistry focus quickly shifts to designing efficient, concise routes to allow preparation of select candidate APIs on larger scale quickly for in vivo profiling. This dichotomy in synthetic objectives is especially pronounced in the case of complex drug molecules, which take a large number of steps and considerable time to synthesize, as illustrated in the above example. To summarize, a concise, 11-step synthesis of the candidate compound has been developed. The cumulative yield for all chemical transformations was 29%, which gives an average chemical yield per step of 89.4%. Including the resolution step, the overall yield was 14% starting from mandelic acid 14.
Experimental Section
General Experimental Methods
Reagents were purchased from commercial suppliers and used without purification unless otherwise noted. 1H NMR and 13C NMR (101, 126, 150 MHz) spectra were acquired on 400, 500, or 600 MHz NMR spectrometers. Chemical shifts (δ) are reported in parts per million (ppm) downfield from an internal tetramethylsilane standard. Spin multiplets are given as s (singlet), d (doublet), t (triplet), q (quartet), and m (multiplet). Coupling constants (J) are given in hertz (Hz). Mass spectra were recorded using electrospray ionization (ESI) or using electron impact ionization (EI) in either positive or negative mode as indicated. HPLC conditions: 35 °C, 0.75 mL/min; Eclipse XDB-C8, 5 μm, 4.6 × 150 mm2; gradient: 5–99% CH3CN (0.05% TFA) in H2O (0.05% TFA) 3.8 min; then 99% CH3CN (0.05% TFA) in H2O (0.05% TFA) 0.6 min; then 99–5% CH3CN (0.05% TFA) in H2O (0.05% TFA) 0.6 min.
3,3,3-Trifluoro-1-(2-fluorophenyl)-2-hydroxypropan-1-one (15)8a
To a 20 L jacketed reactor was added toluene (2.7 L), mandelic acid 14 (200 g, 1.18 mol), and pyridine (570 mL, 7.05 mol). The mixture was heated to 40 °C, and TFAA (654 mL, 4.70 mol) was added resulting in gas evolution (CO2) and an exotherm to 62 °C. The reaction was heated to 80 °C and stirred for 3 h. Analysis by HPLC indicated complete consumption of the mandelic acid and generation of a single product peak. The reaction was cooled to rt and quenched by careful addition of 2 M HCl (2.7 L). The mixture was heated to 60 °C and stirred for 30 min. The layers were separated, and the organic layer was washed with saturated aqueous NaHCO3 (2 L), dried (Na2SO4), and concentrated to give the desired product as a tan solid. The product was dried in a vacuum oven under house vacuum (∼10 Torr) at 60 °C overnight. Some sublimation of the product occurred in the vacuum oven. The desired keto alcohol 15 (212 g, 81%) was isolated as a slightly impure crystalline solid. The initial acidic aqueous layer (∼3.5 L) was found to contain the product. The acidic aqueous layer was extracted 3 times with toluene (1:5 v/v toluene/aq. layer). The combined toluene extracts were washed with saturated NaHCO3 (equal volume), dried (Na2SO4), and concentrated to give more 15 (16 g, 6%). Total isolated yield = 228 g (87%). The keto alcohol was used without further purification. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.92 (ddd, J = 7.7, 7.3, 1.8 Hz, 1H), 7.72–7.62 (m, 1H), 7.32 (ddd, J = 7.8, 7.8, 1.0 Hz, 1H), 7.22 (ddd, J = 11.4, 8.4, 0.9 Hz, 1H), 5.48 (dq, J = 8.0, 6.8 Hz, 1H), 4.24 (dd, J = 8.2, 1.2 Hz, 1H).
4-(2-Fluorophenyl)-5-(trifluoromethyl)-5H-1,2,3-oxathiazole 2,2-dioxide (13)
To a 1 L round-bottom flask purged with N2 was added ACN (176 mL) and chlorosulfonyl isocyanate (138 mL, 1.59 mol). The mixture was stirred with cooling in an ice bath, while neat formic acid (60 mL, 1.59 mol) was added slowly via syringe over 2 h, maintaining the internal temperature below 11 °C. Gas evolution was observed. The mixture was stirred for 1 h in the ice bath. Then, the bath was removed and the reaction was allowed to warm to rt and stir for 21 h. The solution of sulfamoyl chloride precipitated a small amount of white solid generating a slightly cloudy suspension. Very slow gas evolution was still observable. The solution of sulfamoyl chloride was used as is. Keto alcohol 15 (176.44 g, 794 mmol) and DMA (440 mL) were combined in a 5 L jacketed reactor purged with N2. The mixture was cooled to 0 °C, and the solution of sulfamoyl chloride in ACN was added via cannula over 1 h, maintaining the internal temperature below 5 °C. The reaction was allowed to warm to rt and stir for 11 h. Analysis by HPLC indicated a trace of keto alcohol remaining with the bulk of material converted to uncyclized keto sulfamate. There was minor conversion to the cyclic sulfamate imine final product. To complete the cyclization to 13, the reactor was heated to 55 °C, and the reaction was allowed to stir at this temperature for 25 h. Analysis by HPLC showed excellent conversion to the desired cyclic imine with just a trace of open-chain keto sulfamate visible. The reaction was cooled to −6 °C, and water (3.1 L, 5-fold the volume of ACN/DMA) was added to quench remaining sulfamoyl chloride and precipitate the product. An exotherm to 23 °C was observed. The mixture was stirred at rt for 1.5 h to complete the precipitation. The product was collected by vacuum filtration and washed with excess water. The solid was dried in a vacuum oven under house vacuum (∼10 Torr) at 50 °C overnight to provide 185.71 g (82%) of crude 13 as a tan solid. To purify the crude product, the material was redissolved in hot 15% toluene/heptane (10 mL/g crude product) in a 5 L jacketed reactor (solution temperature, 95 °C) leaving an undissolved dark red-brown gummy residue on the walls of the flask. The hot solution was drained from the reactor into two 2 L Erlenmeyer flasks. The solutions were agitated with magnetic stirring and allowed to slowly cool and crystallize. The product was collected by vacuum filtration, washed with excess 15% toluene/heptane, and dried under house vacuum at 50 °C overnight to provide 163 g (72%) of 13 as an off-white crystalline solid. The mother liquor was concentrated and purified by flash chromatography (330 silica gel cartridge, 5–70% EtOAc/hexanes gradient, 40 mL/min). Fractions containing the product were combined and concentrated to give an impure product, which was recrystallized from hot 15% toluene/heptane as before to provide a further 12.3 g (5%) of the desired imine. Total isolated yield = 175.3 g (77%). 1H NMR (400 MHz, CDCl3): δ (ppm) 8.08 (ddd, J = 8.0, 7.2, 1.8 Hz, 1H), 7.84–7.73 (m, 1H), 7.42 (ddd, J = 7.8, 7.8, 1.0 Hz, 1H), 7.30 (ddd, J = 11.4, 8.4, 0.9 Hz, 1H), 6.26 (dq, J = 5.9, 1.5 Hz, 1H). 13C NMR (150 MHz, CDCl3): δ (ppm) 168.3, 162.4 (d, JC–F = 256.1 Hz), 138.4 (d, JC–F = 9.7 Hz), 131.9 (d, JC–F = 1.0 Hz), 125.9 (d, JC–F = 3.1 Hz), 120.2 (q, JC–F = 282.4 Hz), 117.1 (d, JC–F = 21.8 Hz), 115.4 (d, JC–F = 10.6 Hz), 83.4 (dq, JC–F = 35.2, 11.3 Hz). Anal. Calcd for C9H5F4NO3S: C, 38.17%; H, 1.78%; N, 4.95%. Found: C, 38.33%; H, 1.67%; N, 4.92%.
Racemic 4-(2-Fluorophenyl)-4-methyl-5-(trifluoromethyl)-1,2,3-oxathiazolidine 2,2-dioxide (rac-12)
A 3 L three-necked round-bottom flask was fitted with mechanical stirring, purged with N2 (g), and charged with sulfamate imine 13 (178.77 g, 631.2 mmol) and THF (1.26 L). The solution was cooled to −67 °C in a dry ice/IPA bath and treated dropwise via cannula with MeMgBr (295 mL of a 3.0 M solution in Et2O, 883.7 mmol) over 3.5 h, while keeping the internal temperature below −60 °C. The mixture took on a dark red coloration during the addition. The reaction was allowed to warm to −10 °C over 4 h, then was quenched by addition of 2 M HCl (500 mL). No gas evolution was observed. Layers were separated, and the aqueous layer was extracted twice with TBME (2 × 200 mL). The combined organic layers were dried (MgSO4) and concentrated to give the crude product as a dark orange semisolid. The crude product was dissolved in hot 40% IPA/heptane (400 mL) and allowed to cool and crystallize overnight, producing large, translucent crystals infused with a dark green color. The crystals were broken up, collected by vacuum filtration, and washed with 60% IPA/heptane. The product was dried in a vacuum oven under house vacuum (∼10 Torr) at 60 °C for 2 days. Some sublimation of the product was observed. The initial product (95 g) was obtained as large dark brown to black colored crystals. HPLC and 1H NMR analysis indicated excellent purity. To decolorize the product, the crystals were dissolved in warm TBME (21 mL/g) and treated with activated charcoal (10 wt %). The charcoal was removed by filtration through a pad of Celite, and the filter cake was washed with excess TBME. The solvent was removed in vacuo to give the first crop of rac-12 (93.83 g, 50%) as a white solid. The mother liquor from the first crystallization was partially concentrated at elevated temperature and allowed to cool and crystallize. The crystals were slurried with additional 60% IPA/heptane, collected by suction filtration, washed with 60% IPA/heptane, and dried to give a second crop of the product (∼13 g). A third crop (∼7 g) was obtained from the mother liquor in the same manner as above. The second and third crops were combined and treated with activated charcoal in the same manner as described above to provide the product (20.6 g, 11%) as a white to off-white solid. The remaining mother liquor was concentrated and passed through a 2.5 in. silica gel plug in a 2 L fritted funnel eluting with a gradient of 10% EtOAc/hexanes to 50% EtOAc/hexanes. Product-containing fractions were combined and concentrated to give a thick red-orange oil, which precipitated more product upon cooling. The viscous mother liquor was decanted off, and the solid was slurried in 40% IPA/heptane, collected by vacuum filtration, and air dried to give a fourth crop (∼11 g). The mother liquors from crop 4 were combined, concentrated, and chromatographed (330 g silica gel cartridge, 5–50% EtOAc/hexanes gradient, 60 mL/min). Product-containing fractions were partially concentrated and allowed to crystallize. The crystals were slurried in 40% IPA/heptane and collected by vacuum filtration to give a fifth crop of the product (∼5 g). Crops 4 and 5 were combined and treated with activated charcoal as above to provide a final batch of the product (15.7 g, 8%). Total combined yield = 130.13 g (69%), mp 147.2–148.2 °C. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.49–7.41 (m, 1H), 7.31–7.23 (m, 2H), 7.21–7.13 (m, 1H), 5.26 (dq, J = 6.6, 3.0 Hz, 1H), 5.14 (bs, 1 H), 2.04 (s, 3H). 13C NMR (150 MHz, CDCl3): δ (ppm) 159.5 (d, JC–F = 245.3 Hz), 131.5 (d, JC–F = 9.0 Hz), 125.3 (d, JC–F = 3.1 Hz), 125.2 (d, JC–F = 3.2 Hz), 123.7 (d, JC–F = 12.7 Hz), 121.7 (q, JC–F = 282.6 Hz), 116.6 (d, JC–F = 22.2 Hz), 85.5 (dq, JC–F = 31.1, 4.6 Hz), 64.9 (d, JC–F = 2.0 Hz), 27.4 (d, JC–F = 3.0 Hz).
(4R,5R)-4-(2-Fluorophenyl)-4-methyl-5-(trifluoromethyl)-1,2,3-oxathiazolidine 2,2-dioxide (12)
The racemic mixture (100 g) was separated by preparative chiral stationary-phase chromatography using a 110 mm × 40 cm column packed with 2 kg of CHIRALPAK-AS 20 μm stationary phase eluting with heptane/ethanol (85:15) at a flow rate of 450 mL/min. First eluting enantiomer: (S,S) stereochemistry. Yield = 45 g (45%). [α]D20 = +83.0° (c = 1.0, MeOH). Second eluting enantiomer, 12: (R,R) stereochemistry. Yield = 48 g (48%). [α]D = −82.2° (c = 1.0, MeOH). 1H NMR (400 MHz, CDCl3): δ (ppm) 7.49–7.41 (m, 1H), 7.31–7.23 (m, 2H), 7.21–7.13 (m, 1H), 5.26 (dq, J = 6.6, 3.0 Hz, 1H), 5.14 (bs, 1 H), 2.04 (s, 3H).
(2R,3R)-3-Amino-1,1,1-trifluoro-3-(2-fluorophenyl)butan-2-ol (11)
To a 2 L round-bottom three-necked flask fitted with mechanical stirring was added cyclic sulfamate 12 (48.0 g, 160.4 mmol) and THF (450 mL). The homogeneous mixture was heated to 50 °C, and crushed pellets of LAH (10.07 g, 265.3 mmol) were added in portions. Vigorous gas evolution and an exotherm to 63 °C were observed. Analysis by HPLC after 30 min indicated unreacted sulfamate. Additional solid LAH (1.83 g, 48.1 mmol) was added. Analysis by HPLC after an additional 30 min showed complete consumption of the sulfamate. The mixture was cooled in an ice bath to 5 °C and quenched by careful addition of water (12 mL), while maintaining the internal temperature below 20 °C. Vigorous gas evolution was observed during the initial quench. The mixture was stirred for 30 min during which time the reaction became thick and gelled. The ice bath was removed, and more THF (50 mL) was added. Aqueous 15% NaOH solution (12 mL) was added, and the gel began to break down. After stirring for 30 min, water (36 mL) was added in one portion, and the mixture was stirred overnight, resulting in a gray suspension. Celite (30 g) was added and allowed to mix well with the suspension to aid filtration. After several minutes of stirring, the suspension was filtered through a pad of Celite. The filter cake was washed with excess THF, and the solvent was removed in vacuo on a rotary evaporator. After the bulk of the solvent was removed, the product was dried at 67 °C on the rotary evaporator for 30 min, then was placed under high vacuum for 3 h, while cooling to rt. Yield = 37.3 g (98%), pale yellow solid. The amino alcohol was used without further purification. [α]D20 = −17.6° (c = 1.0, MeOH). 1H NMR (400 MHz, CDCl3): δ (ppm) 7.44 (ddd, J = 8.0, 8.0, 1.7 Hz, 1H), 7.34–7.27 (m, 1H), 7.18 (ddd, J = 7.7, 7.5, 1.3 Hz, 1H), 7.05 (ddd, J = 12.2, 8.1, 1.2 Hz, 1H), 4.70 (bd, J = 10.1 Hz, 1H), 4.46 (broad quint, J = 8.2 Hz, 1H), 1.71 (bs, 2H), 1.58 (d, J = 0.4 Hz, 3H). 13C NMR (150 MHz, CDCl3): δ (ppm) 160.5 (d, JC–F = 243.8 Hz), 131.4 (d, JC–F = 14.0 Hz), 129.5 (d, JC–F = 8.7 Hz), 125.6 (d, JC–F = 4.9 Hz), 125.2 (q, JC–F = 283.6 Hz), 124.3 (d, JC–F = 3.2 Hz), 116.1 (d, JC–F = 23.9 Hz), 72.1 (dq, JC–F = 27.1, 7.0 Hz), 56.8 (d, JC–F = 3.9 Hz), 27.5 (d, JC–F = 3.0 Hz). LRMS (ESI) m/z calcd for C10H12F4NO, 238.1 [M + H]+; found, 238.0. Anal. calcd for C10H11F4NO: C, 50.64%; H, 4.67%; N, 5.91%. Found: C, 50.77%; H, 4.66%; N, 5.86%.
2-(2R,3R)-3-Amino-1,1,1-trifluoro-3-(2-fluorophenyl)butan-2-yl)oxy)acetonitrile (17)
To a solution of the unpurified amino alcohol 11 (36.81 g, 155.2 mmol) in THF (155 mL) at rt was added LiOEt (200 mL of 1 M solution in THF, 200 mmol, and 4.1 g of solid LiOEt, 79 mmol). Bromoacetonitrile (16.5 mL, 248.3 mmol) was added immediately in one portion. The mixture was stirred at rt overnight. Analysis by HPLC after 16 h indicated incomplete conversion of 11. More solid LiOEt (0.81 g, 15.5 mmol) and bromoacetonitrile (1.0 mL, 15.5 mmol) were added. Analysis by HPLC after 2 h showed further conversion of the amino alcohol, but unreacted 11 was still present. A third aliquot of solid LiOEt (0.81 g, 15.5 mmol) and bromoacetonitrile (1.0 mL, 15.5 mmol) was added. Analysis by HPLC after 2 h indicated complete consumption of 11. The reaction was quenched by addition of saturated NH4Cl (1 L). The biphasic mixture was stirred overnight. The mixture was partially concentrated to remove the bulk of the THF, and the remaining aqueous layer was extracted three times with TBME (500 mL, then 2 × 300 mL). The combined organic layers were dried (Na2SO4), and the drying agent was removed by filtration. The dark orange organic layer (1.2 L) was treated with activated charcoal (4.3 g, 10 wt %) and heated at 40 °C, while spinning on a rotary evaporator for 30 min. The mixture was cooled to rt, and the charcoal was removed by filtration through a pad of Celite. The filter cake was washed with excess TBME. The coloration of the filtrate was significantly lighter after the charcoal treatment. The TBME was removed in vacuo leaving the crude product as a dark orange liquid. 1H NMR indicated the product contained bromoacetonitrile (11.7 mol %, 5.4 wt %). To reduce bromoacetonitrile contamination, the product was allowed to stand under high vacuum at rt for 2 d. 1H NMR showed that the bromoacetonitrile content was 4.8 mol %, 2.2 wt %. Mass recovery = 43.15 g. Yield of 17 corrected for bromoacetonitrile content = 42.2 g (98%). This material was used without further purification. [α]D20 = +37.3° (c = 1.0, MeOH). 1H NMR (400 MHz, CDCl3): δ (ppm) 7.61 (ddd, J = 8.1, 8.1, 1.7 Hz, 1H), 7.34–7.27 (m, 1H), 7.18 (ddd, J = 7.8, 7.6, 1.2 Hz, 1H), 7.05 (ddd, J = 12.8, 6.9, 1.2 Hz, 1H), 4.59 (d, J = 16.2 Hz, 1H), 4.46 (d, J = 16.2 Hz, 1H), 4.45 (dq, J = 6.8, 0.9 Hz, 1H), 1.78 (bs, 2H), 1.64 (s, 3H). 13C NMR (150 MHz, CDCl3): δ (ppm) 160.4 (d, JC–F = 242.9 Hz), 129.8 (d, JC–F = 11.6 Hz), 129.6 (d, JC–F = 9.0 Hz), 127.4 (d, JC–F = 4.5 Hz), 124.7 (q, JC–F = 284.6 Hz), 124.6 (d, JC–F = 3.0 Hz), 116.0 (d, JC–F = 24.0 Hz), 114.6, 81.5 (dq, JC–F = 25.8, 6.5 Hz), 58.2 (d, JC–F = 1.4 Hz), 56.3 (d, JC–F = 3.4 Hz), 26.9. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C12H13F4N2O, 277.0959; found, 277.0967.
(5R,6R)-5-(2-Fluorophenyl)-5-methyl-6-(trifluoromethyl)-5,6-dihydro-2H-1,4-oxazin-3-amine (10)
To a stirred solution of the unpurified amino cyanomethyl ether 17 (41.8 g, 151.3 mmol) in toluene (756 mL) at rt was added TMSOTf (27.4 mL, 151.3 mmol) in one portion. A very mild exotherm to 26 °C was observed within 5 min of addition. A drying tube was placed on the flask, and the reaction was stirred at rt for 1.5 h. Analysis by HPLC indicated complete conversion of 17. The reaction was quenched by addition of 10% aqueous Na2CO3 (450 mL), and the biphasic mixture was stirred overnight at rt. Layers were separated, and the aqueous layer was extracted twice with toluene (2 × 200 mL). The combined organic layers were dried (Na2SO4) and concentrated in vacuo at 60 °C to give crude amidine 10 as a viscous dark orange oil, which was placed under high vacuum for several minutes to remove solvent. 1H NMR indicated that the toluene content was 3.6 wt %. Yield of 10 corrected for toluene content = 41.4 g (99%). The amidine was used without further purification. [α]D20 = −104.2° (c = 1.0, MeOH). 1H NMR (400 MHz, CDCl3): δ (ppm) 7.84 (ddd, J = 8.0, 8.0, 1.8 Hz, 1H), 7.29–7.22 (m, 1H), 7.15 (ddd, J = 7.8, 7.4, 1.2 Hz, 1H), 7.00 (ddd, J = 12.5, 8.1, 1.2 Hz, 1H), 4.60 (q, J = 8.3 Hz, 1H), 4.25 (bs, 2H), 4.22 (s, 2H), 1.68 (d, J = 1.0 Hz, 3H). 13C NMR (150 MHz, CDCl3): δ (ppm) 160.9 (d, JC–F = 244.0 Hz), 154.2, 129.6 (d, JC–F = 12.1 Hz), 129.2 (d, JC–F = 4.5 Hz), 129.0 (d, JC–F = 8.6 Hz), 124.1 (q, JC–F = 285.7 Hz), 124.0 (d, JC–F = 3.1 Hz), 115.5 (d, JC–F = 23.7 Hz), 73.0 (dq, JC–F = 27.2, 5.1 Hz), 58.7, 55.3 (d, JC–F = 3.4 Hz), 28.5 (d, JC–F = 4.0 Hz). HRMS (ESI-TOF) m/z: [M + H]+ calcd for C12H13F4N2O, 277.0959; found, 277.0962.
(5R,6R)-5-(2-Fluoro-5-nitrophenyl)-5-methyl-6-(trifluoromethyl)-5,6-dihydro-2H-1,4-oxazin-3-amine (9)
To eliminate toluene from 10 before nitration, the amidine (40.9 g, 148 mmol) was dissolved in glacial acetic acid (AcOH) (100 mL) and concentrated in vacuo at 60 °C. Another aliquot of AcOH (100 mL) was added, and the solution was partially concentrated to a volume of approximately 45 mL. The resulting solution of amidine 10 in AcOH was free of toluene, as indicated by 1H NMR analysis. The solution of 10 in a 2 L round-bottom flask was cooled to 0 °C in an ice bath. Fuming nitric acid (90% HNO3) (200 mL) was cooled to −20 °C and then added in portions to the ice cold solution of 10 in AcOH. The reaction gave a moderate exotherm to 34 °C during addition. The final ratio of HNO3 to AcOH was approximately 4.4:1 (v/v). When addition was complete, the reaction was stirred for 5 min in the ice bath. The ice bath was then removed, and the reaction was allowed to warm to rt and stirred overnight. Analysis by HPLC and MS indicated complete consumption of 10 with clean formation of the nitrated products. The reaction was diluted with ice (200 g) and water (200 mL), causing precipitation of a solid (presumably the nitric acid salt of 10). EtOAc (500 mL) was added, and the mixture was cooled on ice as the aqueous layer was basified with NaOH. Initially, solid NaOH was added until the pH reached 5–6, and then aqueous NaOH solution was added until pH 8. The layers were separated, and the aqueous layer was further basified to pH 10 and extracted twice with EtOAc (2 × 200 mL). The combined organic layers (dark orange) were dried (Na2SO4) and concentrated to give the product as a foam. This crude material was found to contain significant quantities of AcOH; therefore, the foam was redissolved in TBME (1 L) and washed 4 times with 0.5 M NaOH (4 × 200 mL). The organic layer was dried (Na2SO4) and concentrated to give 53.74 g of the mixture of nitration isomers as a dark orange viscous oil with TBME (15 wt %) and EtOAc (0.6 wt %) contamination as indicted by 1H NMR analysis. Yield corrected for solvent contamination = 45.36 g (95%). This material was used without further purification. Spectral data for 9: [α]D20 = −99.3° (c = 1.0, MeOH). 1H NMR (400 MHz, CDCl3): δ (ppm) 8.85 (dd, J = 6.6, 3.0 Hz, 1H), 8.19 (ddd, J = 8.9, 4.1, 3.0 Hz, 1H), 7.16 (dd, J = 10.8, 8.9 Hz, 1H), 4.62 (dq, J = 8.2, 1.1 Hz, 1H), 4.35 (bs, 2H), 4.26, 4.23 (ABq, JAB = 15.7 Hz, 2H), 1.67 (d, J = 1.1 Hz, 3H). 13C NMR (150 MHz, CDCl3): δ (ppm) 164.3 (d, JC–F = 255.4 Hz), 154.5, 144.5, 132.2 (d, JC–F = 14.9 Hz), 126.1 (d, JC–F = 6.9 Hz), 125.0 (d, JC–F = 10.8 Hz), 123.9 (q, JC–F = 285.9 Hz), 116.5 (d, JC–F = 26.8 Hz), 72.4 (dq, JC–F = 27.6, 5.5 Hz), 58.5, 55.2 (d, JC–F = 3.6 Hz), 28.3. LRMS (ESI) m/z: [M + H]+ calcd for C12H12F4N3O3, 322.1; found, 321.9. Anal. calcd for C12H11F4N3O3: C, 44.87%; H, 3.45%; N, 13.08%. Found: C, 45.27%; H, 3.49%; N, 13.05%.
tert-Butyl ((5R,6R)-5-(2-Fluoro-5-nitrophenyl)-5-methyl-6-(trifluoromethyl)-5,6-dihydro-2H-1,4-oxazin-3-yl)carbamate (19a)
To a stirred solution of the mixture of nitrated amidines (44.85 g, 139.6 mmol) in ACN (280 mL) was added Boc anhydride (Boc2O) (45.71 g, 209.4 mmol). The reaction was heated to 50 °C and allowed to stir until HPLC analysis indicated complete consumption of amidine (15.5 h). To scavenge unreacted Boc2O, the reaction was cooled in an ice bath and treated with N-methylpiperazine (10.8 mL, 98 mmol). A mild exotherm and gas evolution were observed. The reaction was allowed to warm to rt and stir for 2 h. Most of the ACN was removed in vacuo, and the residue was dissolved in TBME (500 mL) and washed 3 times with 2 M HCl (3 × 50 mL), once with 1:1 saturated NaHCO3/water (100 mL) and once with brine (100 mL). The organic layer was dried (MgSO4) and concentrated to give the mixture of Boc-amidines as a viscous orange oil, which was purified by flash chromatography (750 g silica gel cartridge, hexanes to 40% EtOAc/hexanes gradient) to provide 57.56 g of the desired product, 19a, as a colorless solid foam after drying under high vacuum for 1 h. 1H NMR analysis indicated that the product contained 7 wt % TBME. Yield of 19a corrected for TBME contamination = 53.53 g (91%). Spectral data for 19a: [α]D20 = −89.3° (c = 1.0, MeOH). 1H NMR (400 MHz, CDCl3): δ (ppm) 8.72 (dd, J = 6.6, 3.0 Hz, 1H), 8.20 (ddd, J = 8.9, 4.1, 3.0 Hz, 1H), 7.18 (dd, J = 10.7, 8.9 Hz, 1H), 7.15 (bs, 1H), 4.93 (d, J = 18.4 Hz, 1H), 4.68 (d, J = 18.4 Hz, 1H), 4.68 (q, J = 9.6 Hz, 1H), 1.67 (s, 3H), 1.51 (s, 9H). 13C NMR (150 MHz, CDCl3): δ (ppm) 164.1 (d, JC–F = 255.5 Hz), 153.3, 151.6, 144.5, 131.4 (d, JC–F = 14.8 Hz), 125.5 (d, JC–F = 6.9 Hz), 125.2 (d, JC–F = 10.9 Hz), 123.8 (q, JC–F = 285.9 Hz), 116.7 (d, JC–F = 26.4 Hz), 82.3, 72.1 (dq, JC–F = 28.3, 5.6 Hz), 60.3, 55.7 (d, JC–F = 3.7 Hz), 28.1, 28.0 (d, JC–F = 3.4 Hz). LRMS (ESI) m/z: [M + H]+ calcd for C17H20F4N3O5, 422.1; found, 366.0 [M – tBu + 2H]+. Anal. calcd for C17H19F4N3O5: C, 48.46%; H, 4.55%; N, 9.97%. Found: C, 48.55%; H, 4.50%; N, 9.89%.
tert-Butyl ((5R,6R)-5-(5-Amino-2-fluorophenyl)-5-methyl-6-(trifluoromethyl)-5,6-dihydro-2H-1,4-oxazin-3-yl)carbamate (20)
To a 2 L round-bottom flask were added 19a (53.03 g, 125.8 mmol), 10% Pd/C (Aldrich, dry) (5.3 g), and THF (265 mL, 5 mL/g nitro compound). The reaction was stirred under 1 atm of H2 (g) overnight. Analysis by HPLC showed that the reaction was approximately 50% complete. The reaction contents were transferred to a 2 L Parr bottle and placed on a shaker under 40 psi of H2 (g) for 4 h. Analysis by HPLC indicated complete consumption of 19a with formation of a single product. The Pd catalyst was removed by filtration through a pad of Celite, and the filter cake was washed with excess THF to give a solution of the product that was pale lavender in color. The solution was dried (MgSO4) and then filtered through a 1 in. plug of silica gel eluting with excess THF to remove the color. The filtrate was concentrated to give 53.29 g of the desired aniline, 20, as a colorless solid foam. 1H NMR analysis indicated 8 wt % THF content. Yield of 20 corrected for THF contamination = 49.03 g (99%). This material was used without further purification. [α]D20 = −85.0° (c = 1.0, MeOH). 1H NMR (400 MHz, CDCl3): δ (ppm) rotameric broadening 7.12–7.02 (bm, 2H), 6.81 (bdd, J = 11.7, 8.6 Hz, 1H), 6.60–6.50 (bm, 1H), 4.88 (d, J = 18.4 Hz, 1H), 4.70–4.59 (m, 2H), 3.54 (bs, 2H), 1.64 (s, 3H), 1.48 (s, 9H). 13C NMR (150 MHz, CDCl3): δ (ppm) 154.3 (d, JC–F = 234.7 Hz), 152.6, 151.8, 142.4, 129.4 (d, JC–F = 13.5 Hz), 124.0 (q, JC–F = 286.5 Hz), 116.0 (d, JC–F = 25.0 Hz), 115.2 (d, JC–F = 3.1 Hz), 115.0 (d, JC–F = 8.1 Hz), 81.9, 72.4 (q, JC–F = 27.1 Hz), 60.3, 55.8 (d, JC–F = 3.7 Hz), 28.2 (d, JC–F = 4.2 Hz), 28.1. LRMS (ESI) m/z: [M + H]+ calcd for C17H22F4N3O3, 392.2; found 392.0. Anal. calcd for C17H21F4N3O3: C, 52.17%; H, 5.41%; N, 10.74%. Found: C, 52.06%; H, 5.26%; N, 10.64%.
tert-Butyl (5R,6R)-5-(5-(5-Cyanopicolinamido)-2-fluorophenyl)-5-methyl-6-(trifluoromethyl)-5,6-dihydro-2H-1,4-oxazin-3-yl)carbamate (23)
1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDCI) (23.72 g, 123.8 mmol) was added in one portion to a rt solution of 20 (44.03 g, 112.5 mmol) and 5-cyanopicolinic acid (21) (18.33 g, 123.8 mmol) in DMF (240 mL). Analysis by HPLC after 1 h showed significant conversion, but the reaction was not complete. After 7 h, there was no further conversion. Second portions of EDCI (4.31 g, 22.5 mmol) and picolinic acid 21 (3.33 g, 22.5 mmol) were added, and the reaction was stirred for 1 h. Analysis by HPLC indicated complete conversion of 20. The DMF was removed in vacuo with the rotary evaporator bath temperature at 40 °C. The bright orange residue was partitioned between TBME (500 mL) and 0.5 M HCl (200 mL). Layers were separated. The organic layer was washed a second time with 0.5 M HCl causing a precipitate, which was determined to be the product, to form in the separatory funnel. Ethyl acetate (300 mL) was added to bring the product into solution. The organic layer was washed once more with 0.5 M HCl (200 mL), then was washed twice with 5% Na2CO3 (2 × 100 mL) and once with brine (300 mL). The organic layer was dried (MgSO4) and concentrated. The off-white solid was dried under high vacuum for 2 h at rt to give 62.13 g of the coupled product. 1H NMR analysis indicated contamination by EtOAc (0.6 wt %) and TBME (0.1 wt %). Yield corrected for solvent contamination = 61.70 g (94%). This material was used without further purification. [α]D20 = −19.8° (c = 1.0, MeOH). 1H NMR (400 MHz, CDCl3): δ (ppm) 9.87 (bs, 1H), 8.91 (dd, J = 1.9, 0.7 Hz, 1H), 8.44 (d, J = 8.1 Hz, 1H), 8.22 (dd, J = 8.2, 2.0 Hz, 1H), 7.99–7.89 (m, 2H), 7.14 (bs, 1H), 7.08 (dd, J = 11.4, 8.8 Hz, 1H), 4.92 (d, J = 18.8 Hz, 1H), 4.71 (q, J = 8.4 Hz, 1H), 4.68 (d, J = 18.2 Hz, 1H), 1.68 (s, 3H), 1.51 (s, 9H). 13C NMR (150 MHz, CDCl3): δ (ppm) 159.9, 157.6 (d, JC–F = 243.1 Hz), 152.9, 152.3, 151.7, 150.7, 141.3, 133.2, 130.1 (d, JC–F = 14.3 Hz), 124.0 (q, JC–F = 286.0 Hz), 122.4, 120.6 (d, JC–F = 8.7 Hz), 120.4 (d, JC–F = 4.7 Hz), 116.3 (d, JC–F = 25.0 Hz), 115.9, 112.5, 82.1, 72.3 (dq, JC–F = 26.0, 3.9 Hz), 60.3, 55.8 (d, JC–F = 3.7 Hz), 28.2 (d, JC–F = 3.0 Hz), 28.1. LRMS (ESI) m/z: [M + H]+ calcd for C24H24F4N5O4, 522.2; found 422.1 [M – Boc + H]+. Anal. calcd for C24H23F4N5O4: C, 55.28%; H, 4.45%; N, 13.43%. Found: C, 55.36%; H, 4.42%; N, 13.32%.
N-(3-((2R,3R)-5-Amino-3-methyl-2-(trifluoromethyl)-3,6-dihydro-2H-1,4-oxazin-3-yl)-4-fluorophenyl)-5-cyanopicolinamide (1)
To a rt solution of Boc-amidine (61.73 g, 118.4 mmol) and anisole (129 mL, 1.18 mol) in dichloromethane (CH2Cl2) (800 mL) was added trifluoroacetic acid (200 mL). Analysis by HPLC after 4 h indicated complete removal of the Boc group. Volatiles were removed in vacuo by rotary evaporation with the water bath set at 40 °C. The thick orange residue was taken up in ethyl acetate (EtOAc) (500 mL) and washed 3 times with 10% Na2CO3 (3 × 300 mL) and once with brine (200 mL). The organic layer was dried (Na2SO4) and concentrated to give a moist off-white solid that contained significant anisole. The material was dried further in vacuo under rotary evaporation with the water bath set at 60 °C. The resulting crude solid was triturated with hot 3:1 ethanol/water (100 mL) for 20 min by spinning on the rotary evaporator with the water bath set at 92 °C. The suspension was cooled to 7 °C, and the product was collected by vacuum filtration and washed with cold quantities of 3:1 ethanol/water. The solid was dried in a vacuum oven under house vacuum (∼10 Torr) at 70 °C overnight. 1H NMR analysis indicated 4.5 wt % ethanol content. The product was then triturated and partially recrystallized from hot toluene, as described. Toluene (approximately 700 mL) was added to the solid, and the mixture was heated to 100 °C with rapid stirring. The solid was partially dissolved after stirring for 20 min. The mixture was removed from heat and allowed to cool and crystallize. When the temperature of the solution had reached rt, the flask was placed in the freezer and cooled to −5 °C. The product was collected by vacuum filtration and washed first with cold toluene, then with warm heptane. The finely divided white solid was dried under high vacuum (∼50 mTorr) at 70 °C overnight to provide 43.55 g (87%) of 1. Mp 242 °C (decomp). 1H NMR analysis indicated the toluene content to be 0.3 wt %. [α]D20 = −11.2° (c = 1.0, DMF). 1H NMR (400 MHz, CDCl3): δ (ppm) 10.77 (s, 1H), 9.20 (dd, J = 2.0, 0.8 Hz, 1H), 8.58 (dd, J = 8.2, 2.0 Hz, 1H), 8.28 (dd, J = 8.2, 0.8 Hz, 1H), 8.18 (dd, J = 7.2, 2.8 Hz, 1H), 7.86–7.79 (m, 1H), 7.13 (dd, J = 12.0, 8.8 Hz, 1H), 5.86 (s, 2H), 4.54 (q, J = 8.1 Hz, 1H), 4.23 (d, J = 15.9 Hz, 1H), 4.10 (d, J = 15.9 Hz, 1H), 1.56 (s, 3H). 13C NMR (150 MHz, CDCl3): δ (ppm) 159.9, 157.7 (d, JC–F = 242.7 Hz), 154.6, 152.4, 150.7, 141.3, 133.2 (d, JC–F = 2.3 Hz), 130.8 (d, JC–F = 14.0 Hz), 124.1 (q, JC–F = 286.1 Hz), 122.3, 120.9 (d, JC–F = 4.7 Hz), 120.4 (d, JC–F = 8.7 Hz), 116.2 (d, JC–F = 25.1 Hz), 116.0, 112.4, 72.5 (dq, JC–F = 27.3, 5.6 Hz), 58.5, 55.2 (d, JC–F = 3.6 Hz), 28.6 (d, JC–F = 3.8 Hz). LRMS (ESI) m/z: [M + H]+ calcd for C19H16F4N5O2, 422.1; found, 422.1. Anal. calcd for C19H15F4N5O2: C, 54.16%; H, 3.59%; N, 16.62%. Found: C, 54.23%; H, 3.36%; N, 16.54%.
Acknowledgments
We thank our colleagues Drs. Daniel Oehlrich, Nicholas Carruthers, Harry Gijsen, and Gregor Macdonald for helpful discussions and support throughout the course of this project. We thank Hilde Vanbaelen and Jozef Proost for their excellent work in chiral separations and Dr. Jiejun Wu and Heather McAllister for analytical support.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.6b00362.
Proton and carbon NMR spectra of all compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Farris W.; Schutz S. G.; Cirrito J. R.; Shankar G. M.; Sun X.; George A.; Leissring M. A.; Walsh D. M.; Qiu W. Q.; Holtzman D. M.; Selkoe D. J. Am. J. Pathol. 2007, 171, 241–251. 10.2353/ajpath.2007.070105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimmer T.; Henriksen G.; Wester H.-J.; Foerstl H.; Klunk W. E.; Mathis C. A.; Kurz A.; Drzezga A. Neurobiol. Aging 2009, 30, 1902–1909. 10.1016/j.neurobiolaging.2008.01.016. [DOI] [PubMed] [Google Scholar]
- Evin G. BioDrugs 2016, 30, 173–194. 10.1007/s40259-016-0168-3. [DOI] [PubMed] [Google Scholar]; May P. C.; Willis B. A.; Lowe S. L.; Dean R. A.; Monk S. A.; Cocke P. J.; Audia J. E.; Boggs L. N.; Borders A. R.; Brier R. A.; Calligaro D. O.; Day T. A.; Ereshefsky L.; Erickson J. A.; Gevorkyan H.; Gonzales C. R.; James D. E.; Jhee S. S.; Komjathy S. F.; Li L.; Lindstrom T. D.; Mathes B. M.; Martenyi F.; Sheehan S. M.; Stout S. L.; Timm D. E.; vaught G. M.; Watson B. M.; Winneroski L. L.; Yang Z.; Mergott D. J. J. Neurosci. 2015, 35, 1199–1210. 10.1523/JNEUROSCI.4129-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rombouts F. J. R.; Tresadern G.; Delgado O.; Martinez-Lamenca C.; Van Gool M.; Garcia-Molina A.; Alonso de Diego S. A.; Oehlrich D.; Prokopcova H.; Alonso J. M.; Austin N.; Borghys H.; Van Brandt S.; Surkyn M.; De Cleyn M.; Vos A.; Alexander R.; Macdonald G.; Moechars D.; Gijsen H.; Trabanco A. A. J. Med. Chem. 2015, 58, 8216–8235. 10.1021/acs.jmedchem.5b01101. [DOI] [PubMed] [Google Scholar]
- Chang S.; Lee E. E. Synthesis 2010, 2361–2366. 10.1055/s-0029-1218778. [DOI] [Google Scholar]
- Hepburn H. B.; Chotsaeng N.; Luo Y.; Lam H. W. Synthesis 2013, 45, 2649–2661. 10.1055/s-0033-1339499. [DOI] [Google Scholar]
- Kawase M.; Saito S.; Kurihara T. Chem. Pharm. Bull. 2000, 48, 1338–1343. 10.1248/cpb.48.1338. [DOI] [PubMed] [Google Scholar]
- a Ramanjaneyulu B. T.; Mahesh S.; Anand R. V. Org. Lett. 2015, 17, 6–9. 10.1021/ol502581b. [DOI] [PubMed] [Google Scholar]; b Ruppert I.; Schlich K.; Volbach W. Tetrahedron Lett. 1984, 25, 2195–2198. 10.1016/S0040-4039(01)80208-2. [DOI] [Google Scholar]; c Prakash G. K. S.; Krishnamurti R.; Olah G. A. J. Am. Chem. Soc. 1989, 111, 393–395. 10.1021/ja00183a073. [DOI] [Google Scholar]; d Prakash G. K. S.; Mandal M. J. Am. Chem. Soc. 2002, 124, 6538–6539. 10.1021/ja020482+. [DOI] [PubMed] [Google Scholar]
- Appel R.; Berger G. Chem. Ber. 1958, 91, 1339–1341. 10.1002/cber.19580910633. [DOI] [Google Scholar]
- Okada M.; Iwashita S.; Koizumi N. Tetrahedron Lett. 2000, 41, 7047–7051. 10.1016/S0040-4039(00)01130-8. [DOI] [Google Scholar]
- See the Supporting Information for NOE experiments to determine relative stereochemistry.
- Absolute stereochemistry was assigned based on a comparison of HPLC and optical rotations of compounds of known configuration determined by VCD; see ref (4).
- Wang Y.-Q.; Yu C.-B.; Wang D.-W.; Wang X.-B.; Zhou Y.-G. Org. Lett. 2008, 10, 2071–2074. 10.1021/ol800591u. [DOI] [PubMed] [Google Scholar]
- Lithium Aluminum Hydride. In Reagents for Organic Synthesis; Fieser L. F., Fieser M., Eds.; John Wiley & Sons, Inc., 1967; p 581. [Google Scholar]
- Rousselet G.; Capdevielle P.; Maumy M. Tetrahedron Lett. 1993, 34, 6395–6398. 10.1016/0040-4039(93)85054-Z. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.