

Abstract
Little is known about the biological and structural features that govern the isoform selectivity for class I histone deacetylases (HDACs) over HDAC6. In addition to that for known inhibitors, like benzamides, psammaplin A, and cyclodepsipeptide-derived thiols, selectivity was also observed for naturally occurring cyclopeptide HDAC inhibitors with an aliphatic flexible linker and ketonelike zinc-binding group (ZBG). The present study reports that this isoform selectivity is mainly due to the linker and ZBG, as replacement of the cyclopeptide cap region by a simple aniline retained class I HDAC isoform selectivity toward HDAC6 in enzymatic assays. The best cyclopeptide-free analogues preserved efficacy against Plasmodium falciparum and cancer cell lines. Molecular modeling provided hypotheses to explain this selectivity and suggests different behaviors of the flexible linker on HDAC1 and HDAC6 pockets, which may influence, on the basis of the strength of the ZBG, its coordination with the zinc ion.
Introduction
Histone deacetylases (HDACs) have been extensively investigated for many years as targets to combat cancer,1 neurological disorders, as well as inflammatory, viral,2 and, more recently, parasitic diseases.3 In recent years, four HDAC inhibitors (HDACi’s) have been approved by the FDA for use in cancer treatment.4 HDACs and histone acetyltransferases (HATs) form two families of enzyme with antagonist actions on the acetylation status of histone. Together with other epigenetic marks (e.g., DNA methylation, histone methylation, phosphorylation, and ubiquitination), histone acetylation is involved in gene regulation via cross-talk leading to chromatin remodeling and accessibility.5 HDACs are responsible for the removal of acetyl groups from lysine residues in the core histones. In humans, 18 HDAC isoforms have been identified and split among four classes. Eleven HDACs contain a conserved deacetylase domain that involves a zinc-dependent catalytic site. They are divided into class I (HDACs 1, 2, 3, and 8), class IIa (HDACs 4, 5, 7, and 9), class IIb (HDACs 6 and 10), and class IV (HDAC 11). The class III isoforms, named sirtuins (sirtuins 1–7), require nicotine adenine dinucleotide as a cofactor and their catalytic activity is independent of Zn2+. Class I HDACs are ubiquitous in all tissues and are key players in epigenetic processes. They are highly expressed in various cancer cell lines and sustain malignant growth, making tumor cells more sensitive to HDACi treatment compared with normal cells.6 With regard to emerging therapeutic approaches, anti-latency therapy for persistent human immunodeficiency virus type 1 (HIV-1) infection used selective inhibitors of class I HDACs to induce reactivation of HIV-1 transcription in the 2D10 cell line.7 Recent studies on apicomplexan intracellular parasites, such as Plasmodium falciparum and Toxoplasma gondii, which are the causative agents of malaria and toxoplasmosis, respectively, have demonstrated the central role of class I HDACs in parasite survival and proliferation in the host cell.8 By targeting HDAC3, FR235222, in nanomolar concentrations (Figure 1), inhibited the proliferative and cystic forms of T. gondii.8,9 Class IIb HDAC6 is another prominent HDAC member found mainly in the cytoplasm and hence not strictly involved in epigenetic functions.10 Thus, HDAC6 deacetylates α-tubulin and chaperones, playing a role in cell trafficking, motility, and transcriptional regulation.11
Figure 1.

Representative structures of natural and synthetic selective class I HDAC inhibitors and the pan-HDAC inhibitor SAHA.
From the perspective of deciphering specific epigenetic pathways linked to single HDAC isoforms, potent and isoform-selective HDACi’s are required. The search for inhibitors having isoform selectivity is also attractive from a therapeutic point of view to reduce off-target activities and toxicity.12
To design better HDACi’s, it is essential to understand the structural regions necessary for class selectivity.13 The triad of cap/linker/zinc-binding group (ZBG) is typically found in all potent HDACi’s, such as SAHA (Figure 1). The cap region interacts with the rim of the enzyme, whereas the linker enters the deep hydrophobic tunnel to properly position the ZBG in the catalytic site.
Four main families of class I-selective HDACi’s have been identified on the basis of their ZBGs: benzamides (MS-275); psammaplin A; and two groups of naturally occurring cyclopeptide compounds,14 a cyclodepsipeptide with a thiol ZBG (romidepsin, largazole) and a cyclotetrapeptide with a ketonelike ZBG (FR235222, trapoxin, apicidin). MS-275 has been reported to be a selective inhibitor of class I HDAC1–3, without an effect on class I HDAC8.15 Optimization of the benzamide ZBG has led to improved potency and high selectivity.16 The natural product psammaplin A and its thiol derivatives showed high selectivity for class I HDACs (HDAC1–3 and HDAC8) versus that for HDAC6.17 Trapoxin B is known to be a highly potent inhibitor of class I HDACs, with no effect on the activities of HDAC6 in cellulo.18 Apicidin also showed marked isoform selectivity for class I HDACs over other classes19 and has inspired the development of new ethyl ketone ZBG derivatives for selective class I inhibition, such as 1(20) and 2.21 In these cases, efforts were focused on modulating the cap region to increase the potency for class I HDACs.
In the course of our attempts to synthesize new HDACi analogues inspired by FR235222,22 we found evidence that modulating the ZBG could also increase the potency. Keeping the cyclotetrapeptide part unchanged (with R1, R2 = H; Figure 1), compound 3, having the (R)-hydroxyl ketone ZBG, was shown to be more efficient in inhibiting the T. gondii RH strain (IC50 = 28.6 nM) than the corresponding analogue with an ethyl ketone group (IC50 = 172 nM).22 However, why the ketonelike ZBG inhibitors are selective for class I HDACs and do not affect HDAC6 remains unclear and has been little explored.
Here, we report the identification and study of the structural regions in FR235222 necessary for class I versus class IIb selectivity. The combination of (R)-hydroxyl ketone, as a medium-range zinc chelator, with a flexible linker, on its own, is sufficient to provide selectivity. This simple pharmacophore combined with minimal caps can maintain HDAC isoform selectivity and inhibitory activities against P. falciparum and human cancer cell lines. Finally, docking calculations followed by molecular dynamic simulations were performed to gain insights into HDAC structure/selectivity relationships.
Results and Discussion
Synthesis
The analogue cyclotetrapeptide 3 showed cellular effects similar to those observed with FR23522222 and was used as the basis for structural deconvolution. The goal of this study was to replace the cyclotetrapeptide part by a simple cap, like the aniline amide group found on SAHA. Given that the ZBG seems to be necessary for the activity and on the basis of previous studies,22 various ZBGs could easily be grafted onto any cap group having a linker ending with a terminal olefin. Aniline amides with various linker lengths, 6a–c, were synthesized from aniline 4 (Scheme 1) to determine the optimal linker length. Derivatives 6a–c were condensed with (R)-hydroxyl ketone xanthate 7(22) to give the corresponding xanthate products, 10a–c, according to Zard’s reaction conditions.23 The hypophosphorus acid condition24 was used to remove the xanthate group. During this step, an unexpected concomitant silyl deprotection occurred, providing us with direct access to the desired products, 11a–c. Using the best linker length determined with 11b, (S)-hydroxyl ketone 8 and ethyl ketone 9 xanthates22 were used to make analogues 11f and 11g, respectively. Their potency in comparison to that of the (R)-hydroxyl ketone ZBG was determined. As the acetylated version of the (R)-hydroxyl ketone ZBG with cyclotetrapeptide 13 showed promising activity in cellular assays,22 the simplified analogue, 12b, was made by acylation of 11b. 3-Ethynyl aniline 5 was later used as the starting material to obtain 3-phenyl-triazol-aniline as the cap group (6e). This choice was influenced by the work of Kozikowski,25 who reported that similar motifs significantly increased HDAC3-selective inhibition. The 3-phenyl-triazol-aniline cap analogue, 11e, was synthesized with the optimal linker length and the ZBG found in the natural product FR235222.
Scheme 1. Synthesis of Aniline Amides with Various Linker Lengths 6a–c from Aniline 4.

Reagents and conditions: (i) N,N’-dicyclohexylcarbodiimide (DCC), 4-dimethylaminopyridine (DMAP), CH2Cl2, room temperature (rt), overnight; (ii) Et3N, CH2Cl2, rt, overnight; (iii) aniline, tBuONO, TMSN3, CH3CN, 2 h, then 6d, Amberlyst-A21 CuI cat., rt, overnight; (iv) xanthate, dilauroyl peroxide (DLP), ClCH2CH2Cl, reflux, overnight; (v) H3PO2/Et3N, azobisisobutyronitrile (AIBN), dioxane, reflux, 2 h; (vi) Ac2O, Et3N, DMAP, 1 h at 0 °C, 2 h at rt.
Biological Evaluation
Our previous results22 indicated that cyclotetrapeptides 3 and 13 strongly inhibited the proliferation of P. falciparum and human cancer cell lines. The HDAC inhibitory activity of compounds 3, 11a–c, 11e–g, 12b, 13, and 14 was initially evaluated using a HeLa nuclear extract containing a mixture of nuclear HDAC isoforms (mainly HDAC1, HDAC2, and HDAC3); the activity under these conditions was compared to that with HDAC6 (Table 1). All compounds were initially tested at 100 μM on HeLa nuclear extracts to detect class I HDACs inhibitory compounds. A 50% inhibition cutoff was applied to select compounds for class I IC50 determination. Compounds 3 and 13 exhibited a good inhibition potency against class I HDACs, whereas no effect was observed with HDAC6 (IC50 > 300 μM). The high selectivity is remarkable for 3 and is comparable to results obtained with apicidin.19 The next step was to determine whether the cyclotetrapeptide plays a critical role in HDAC inhibition or selectivity. The cyclotetrapeptide region without ZBG 14(22) was evaluated using HDAC enzymatic assays, and the observations were in line with the demonstrations by Ghadiri26 and Olsen27 that a cyclodepsipeptide lacking the ZBG could efficiently inhibit HDAC activity. The cyclotetrapeptide without ZBG 14 had no inhibitory effect on class I HDACs or HDAC6 (IC50 > 300 μM), suggesting that the linker and ZBG are necessary for its biological activity. The cyclotetrapeptide region was then replaced by the aniline amide cap found on SAHA. The HDAC inhibitory activities of compounds 11a–c were compared to define the optimal linker length. Compound 11b showed the best activity and, notably, has the same linker length as SAHA. Interestingly, this truncated structure showed good selectivity for class I HDACs (IC50 = 8.9 μM) versus that for HDAC6 (IC50 > 300 μM), whereas the shorter-linker-length analogue, 11a, displayed marginal HDAC6 activity. Using the optimal linker length, the impact of substituting the FR235222 (R)-hydroxyl ketone ZBG with the (S)-hydroxyl ketone (11f) or ethyl ketone (11g) found on apicidin was studied. With the same cap (simple aniline), ketone hydroxyl groups 11b and 11f were more efficient than ethyl ketone group 11g, and the (R)-hydroxyl configuration, 11b, remained the best. Finally, substitution of aniline with ortho-phenyl triazole aniline (11e) efficiently increased the potency of the compound for class I HDACs while maintaining the selectivity.
Table 1. HDAC Inhibition Assays (IC50) Using a Nuclear Extract and Class IIb HDAC6a,b.

Results are presented as mean ± SD of three independent experiments; N.D., not determined.
Not active (inhibition < 50% at 100 μM); HeLa nuclear extract (IC50 cutoff; 20 μM).
IC50 values lower than 20 μM were determined for the displayed compounds 3, 13, 11b, and 11e; therefore, these compounds were further tested on a panel of recombinant class I HDACs (HDAC1, HDAC3/NCOR2, HDAC8) and class IIa HDAC4 to determine dose responses (starting at 100 μM). These products showed a lack of activity on class IIa HDAC4 extending the isoform selectivity of the hydroxyl ketone ZBG to this class of HDACs. Inhibition of class I HDAC8 was observed with the two most potent inhibitors (3 and 13), underlining the positive contribution of the cyclotetrapeptide to ligand efficiency. In class I inhibition, compound 11b showed a noticeable preference for HDAC1 over HDAC3. Modulation of 11b to produce 11e in turn improved the selectivity for HDAC3 compared to that for HDAC1. The results from comparing the performances of recombinant HDACs, which are purified enzymes, and HeLa nuclear extracts, which contain a mixture of nuclear HDAC isoforms in a multienzyme complex, should be interpreted with caution and may explain the lower IC50 values determined in recombinant HDAC assays (Table 2) compared to those determined with HeLa nuclear extracts (Table 1). However, the possibility that these products inhibit another HDACs cannot be ruled out.
Table 2. HDAC Inhibition Assays (IC50) Using Class I and Class IIa HDAC Isoforms.
| IC50 (μM) | ||||
|---|---|---|---|---|
| molecule | HDAC1 | HDAC3/NCOR2 | HDAC4 | HDAC8 |
| 3 | 0.96 ± 0.6 | 0.33 ± 0.01 | >100 | 7.02 ± 1.53 |
| 13 | 0.39 ± 0.02 | 0.35 ± 0.003 | >100 | 1.87 ± 0.42 |
| 11b | 7.9 ± 0.8 | 50.2 ± 4.7 | >100 | >100 |
| 11e | 5.04 ± 0.07 | 1.97 ± 0.03 | >100 | >100 |
| SAHAa | 0.123 ± 0.003 | 0.957 ± 0.07 | 0.101b | 1.52b |
| trichostatin A (TSA)a | 0.624 | |||
| TMP269a | 0.275 | |||
Assay-positive controls according to relevant isoform.
Data from ref (28).
Next, the most enzymatically active compounds were evaluated for their antiproliferative activity against P. falciparum and a panel of human cell lines (Table 3). The two cyclotetrapeptides, 3 and 13, were known to be good inhibitors of cell proliferation, with GI50 values against P. falciparum of 17.4 and 22.2 nM,22 respectively, and with GI50 ranging from 6 to 170 nM when used with the Jurkat, K562, HeLa, or HEK293 cell lines. The simplified analogues remained bioactive on cells but at much lower levels. Compound 11e appeared to be the most promising simplified analogue. An additional assay on normal B lymphocytes was performed and showed that 11e had GI50 > 50 μM. However, these inhibitors showed little selectivity in terms of the cytotoxicity between P. falciparum and the human cell lines.
Table 3. Antiproliferative Activity (GI50) Assays with P. falciparum and a Panel of Human Cell Linesa.
| GI50 (μM) | |||||
|---|---|---|---|---|---|
| molecule | P. falciparum | Jurkat | K562 | HeLa | HEK293 |
| 3 | 0.0174 ± 0.002b | 0.048 ± 0.001b | 0.018 ± 0.002b | 0.170 ± 0.010 | 0.04 ± 0.020 |
| 13 | 0.0221 ± 0.001b | 0.127 ± 0.011b | 0.061 ± 0.002b | 0.119 ± 0.008 | 0.006 ± 0.001 |
| 11b | 37.9 ± 3.8 | 92 ± 1.0 | 62.3 ± 5.8 | >50 | 35.6 ± 1.6 |
| 12b | 40.1 ± 4.3 | ≥100 | ≥100 | >50 | 36.3 ± 1.7 |
| 11e | 4.85 ± 1.3 | 2.84 ± 0.7 | 7.12 ± 0.9 | 115.1 ± 25.3 | 15.9 ± 1.13 |
| SAHA | N.D. | N.D. | N.D. | 2.84 ± 0.5 | 0.080 ± 0.006 |
Results are shown as mean ± SD of three independent experiments; N.D.: not determined.
Data from ref (22).
Molecular Modeling and Docking Calculations
Compounds 11b and 11e (Tables 1 and 2) are good examples of HDACi’s that selectively inhibit class I HDACs over class IIb HDAC6 and class IIa HDAC4. Compound 11b was selected for the computational study as it only differs from SAHA in its ZBG (same cap and same linker length) and therefore offers the opportunity to directly compare the (R)-hydroxy ketone ZBG with a classic hydroxamate ZBG. Thus, its structure is very similar to that of SAHA, which in addition to class I HDACs also inhibits HDAC6 and HDAC4. To understand this difference, docking studies were performed for SAHA and 11b on HDAC1, HDAC6, and HDAC4. The X-ray structures available for HDAC1 and HDAC4 were used (PDB: 4BKX, 5A2S). For HDAC6, all studies presented below were performed with a HDAC6 homology model, as described in the Experimental Methods section, as no structure was available at the time the experiments were performed. However, the main result was confirmed by the very recently published HDAC6 X-ray structure (PDB: 5EDU).29
Docking Calculations
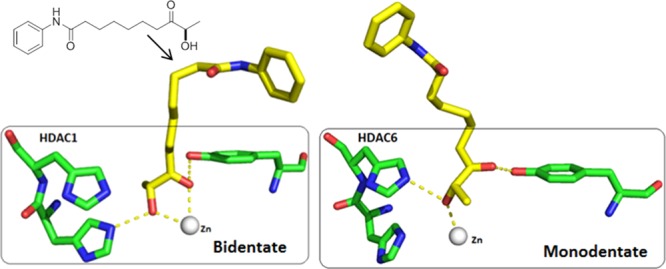
For HDAC1, the docking positions of SAHA and 11b showed that their ZBGs bound in a similar manner, with bidentate coordination (Figure 2A). For HDAC4, different docking patterns were obtained for SAHA and 11b. For SAHA, as expected, a bidentate binding mode was observed (Figure 2C), whereas the 11b ligand penetrates into the catalytic site from the other side (aniline cap) to form a π–cationic interaction with an arginine and its phenyl group (Figure 2D). This π–cationic interaction has been described in trisubstituted cyclopropane-bearing hydroxamate ZBGs designed as selective class IIa inhibitors.30 In the case of 11b, the π–cationic interaction alone is weak, which could explain the lack of activity observed for HDAC4. Finally, for HDAC6, monodentate coordination was observed for both SAHA and 11b. Docking of 11b with a deprotonated ZBG was also performed, and a similar position was observed (Figure S2). The aliphatic linker in the two compounds appears to adopt a curled conformation and penetrates less deeply into the catalytic site (Figure 2B). Compared with HDAC1, HDAC6 is known to have a broader catalytic cavity,31 which may account for this difference in the binding mode. However, this result remains insufficient to explain the difference in affinity observed experimentally for SAHA and 11b in HDAC6. To better understand this selectivity, molecular dynamics simulations were performed on the docked position obtained for both ligands in HDAC1 and HDAC6.
Figure 2.

Best-ranked docked positions obtained for SAHA (cyan stick atoms) and 11b (yellow stick atoms) in HDAC1 (A), HDAC6 (B), and HDAC4 (C, D).
Molecular Dynamics Simulations
To further extend our understanding of the differences between compounds, molecular dynamics simulations were performed. In HDAC1, the bidentate coordination with the zinc atom remained the same for both SAHA and 11b (Figures 3 and S3). Ligands formed H-bonds with His140 and 141 and Tyr303 for SAHA and with His140 and Tyr303 for 11b.
Figure 3.

Main interactions of SAHA (cyan stick atoms, A) and 11b (yellow stick atoms, B) with the characteristic residues of the ZBG in HDAC1, determined by molecular dynamics simulations. The average distances between O1 and Zn and O2 and Zn are, respectively, 2.40 and 2.32 Å for SAHA and 2.36 and 2.40 Å for 11b.
However, a different pattern was obtained for the binding of SAHA and 11b to HDAC6. As SAHA has a strong ZBG, the monodentate coordination obtained from docking studies became bidentate as observed for HDAC1 (Figures 4A and S4). This strong affinity of the hydroxamate ZBG should explain why SAHA inhibits all HDAC isoforms (Tables 1 and 2). In contrast, compound 11b seems to have a weaker ZBG, and the monodentate coordination remained stable in HDAC6 after molecular dynamics simulations (Figures 4B and S3). This binding mode compared with the bidentate coordination of 11b in HDAC1 might explain the experimental data showing isoform selectivity of the keto-hydroxyl ZBG for class I HDACs over HDAC6. Similarly, 11g can only show monodentate coordination through its carbonyl group, which explains its lack of activity on both HDAC1 and HDAC6. The weaker affinity of the keto-hydroxyl ZBG for the zinc atom is also revealed by experimental data, as a simple change in configuration from the hydroxyl group (11f) led to a significant decrease in HDAC inhibition.
Figure 4.

Main interactions of SAHA (cyan stick atoms, A) and 11b (yellow stick atoms, B) with the residues characterizing the zinc-binding domain of HDAC6, determined by molecular dynamics simulations. The average distances between O1 and Zn and O2 and Zn are, respectively, 2.51 and 2.44 Å for SAHA, 2.37 and 3.91 Å for 11b.
Docking and dynamic simulations performed with 11b on the very recent crystallographic structure of HDAC6 (PDB: 5EDU) confirmed the monodentate binding of the keto-hydroxyl ZBG (Figures S5 and S6). These results support the robustness of our model, in agreement with the superposition of its backbone with the HDAC6 RX (Figure S7). The positions of the residues involved in the catalytic mechanism are well conserved. The position determined for SAHA in HDAC6 from molecular dynamics simulations was also compared to the binding mode described for SAHA in Danio rerio HDAC6 (PDB: 5EDU), and the binding mode described for SAHA and the zinc atom was confirmed (Figure S8).
Conclusions
The reasons for isoform selectivity among HDACs are often difficult to rationalize. From the naturally occurring cyclotetrapeptide HDACi, which has a ketonelike ZBG, we were able to shed light on a determining factor that may trigger the selective inhibition of class I HDACs (HDAC1) versus class IIb HDAC6. Reducing the structure of these natural products to simple SAHA analogues (an aniline cap, a flexible linker, and a ketonelike ZBG) led to the identification of two compounds, 11b and 11e, that showed significant activity (in micromolar range) toward class I HDACs in enzymatic assays and were also efficient inhibitors in cellular assays with P. falciparum and cancer cell lines, apparently due to their ability to inhibit class I HDACs. Although benzamide and thiol ZBG derivatives showed higher class I/class II HDACs selectivity and potency, modulation by simple aniline caps is possible without affecting the HDAC isoform selectivity of the hydroxyl ketone ZBG. However, all simplified aniline analogues remain less efficient than the cyclotetrapeptide cap, suggesting that this cap optimizes ligand potency. Compound 11b and SAHA only differ in their ZBGs (same cap and linker length) and offer the opportunity to directly compare the (R)-hydroxy ketone ZBG with a classic hydroxamate ZBG. Docking studies suggested that the ZBG of 11b could not coordinate in HDAC4. The comparison between class I HDACs and HDAC6 based on docking studies followed by molecular dynamics simulations revealed that the ZBG of 11b binds with bidentate and monodentate coordination to HDAC1 and HDAC6, respectively. These differences might explain the isoform selectivity of the keto-hydroxyl ZBG for class I HDACs over HDAC6, while also providing a new perspective for the design of selective HDACi’s. It has been established that a key aspect may be related to the subtle association of a flexible linker with a medium-range ZBG, such as (R)-hydroxyl ketone.
Experimental Methods
Synthetic Chemistry General Comments
All starting materials were obtained from commercial sources and were used without further purification. Air-sensitive reactions were performed under argon conditions. Anilines 6a–c were obtained according to the literature procedures.32,33 Cyclotetrapeptides 3, 13, and 14 were synthesized according to our previous work.22 All intermediates were purified by flash chromatography. All final compounds had purities greater than 95% on the basis of HPLC analysis (Agilent 1100 series using a diode array detector and a C18 reversed-phase column (5 μm particle size, 125 mm × 3 mm, Nucleosil C18; Macherey-Nagel) at 25 °C, with a mobile phase composed of A = water + 0.1% TFA and B = MeOH + 0.1% TFA with a 90:10 to 0:100 A/B gradient over 15 min, 1 mL/min, 10 μL injection, detection at 254 nm). NMR spectra were recorded on a Bruker Advance spectrometer at 400 MHz for 1H NMR spectra and 100 MHz for 13C NMR spectra. Chemical shifts are reported in ppm (δ) relative to tetramethylsilane (TMS) as an internal standard or to the solvents: 13C NMR (CDCl3) δ = 77.23 ppm. The asterisk symbol (*) in this section refers to duplicate NMR signals due to diastereoisomerism of the mixture. Accurate mass spectra were recorded on a time-of-flight spectrometer.
N-(3-Ethynylphenyl)hex-5-enamide 6d
A solution of 5-hexenoic acid (974 mg, 8.53 mmol, 1.1 equiv) at 0 °C was slowly added to a solution of DCC (1.93 g, 9.39 mmol, 1.1 equiv) in anhydrous CH2Cl2 (60 mL), followed by the addition of 3-aminophenylacetylene 5 (1.0 g, 8.53 mmol, 1 equiv) and DMAP (104 mg, 10 mol %). The mixture was allowed to reach rt and was stirred vigorously overnight. After the reaction was completed, the solid was filtered and the organic layer was washed with 1 N HCl, 5% NaHCO3, and brine. The organic layer was then dried over MgSO4 and filtered, and the solvent was evaporated. The resulting residue was purified by column chromatography on silica gel (CH2Cl2/cyclohexane, 8:2) to give compound 6d in 67% yield (1.22 g, 5.72 mmol). Rf = 0.2 (CH2Cl2/cyclohexane, 8:2); IR νmax (film, cm–1): 3289, 2927, 1660, 1607, 1586, 1543, 1485, 1426, 1405, 1288; 1H NMR (400 MHz, CDCl3) δ (ppm): 1.81 (m, 2H), 2.13 (dt, J = 7.0, 7.0 Hz, 2H), 2.35 (t, J = 7.5 Hz, 2H), 3.05 (s, 1H), 5.00 (d, J = 10.1 Hz, 1H), 5.04 (d, J = 16.9, 1H), 5.79 (ddt, J = 16.9, 10.1, 6.7 Hz, 1H), 7.14–7.33 (m, 2H, H2), 7.54 (d, J = 7.7 Hz, 1H), 7.63 (s, 1H); 13C NMR (100 MHz, CDCl3) δ (ppm): 24.7 (CH2), 33.2 (CH2), 36.9 (CH2), 83.3 (C, CH), 115.7 (CH2), 120.6 (CH), 123.0 (C), 123.5 (CH), 128.1 (CH), 129.1 (CH), 137.9 (C), 138.1 (CH), 171.6 (C); HRMS (ESI+) m/z: [M + Na]+ calcd for C14H15NONa, 236.1051; found, 236.1051.
N-(3-(1-Phenyl-1H-1,2,3-triazol-4-yl)phenyl)hex-5-enamide 6e
To a solution of aniline (86.6 mg, 0.93 mmol) in CH3CN (10 mL) was added tBuONO (149 mg, 1.39 mmol, 1.5 equiv) at 0 °C. A solution of TMSN3 (128 mg, 1.11 mmol, 1.2 equiv) was added dropwise, and the mixture was stirred over 2 h. Then, compound 6d (200 mg, 0.93 mmol, 1 equiv) and polymer-supported Amberlyst-A21/CuI catalyst (0.5 g)34 were added to the mixture. The mixture was stirred overnight at rt. After the reaction was completed, the polymer was filtered and washed several times with EtOAc. The combined organic layers were evaporated. The resulting residue was purified by column chromatography on silica gel (EtOAc/cyclohexane, 2:8) to give compound 6e as a white solid in 58% yield (180.3 mg, 0.54 mmol). Rf = 0.2 (EtOAc/cyclohexane, 2:8); IR νmax (film, cm–1): 3279, 2935, 1651, 1593, 1530, 1509, 1440, 1408; 1H NMR (400 MHz, CDCl3) δ (ppm): 1.85 (m, 2H), 2.15 (dt, J = 7.1, 7.0 Hz, 2H), 2.39 (t, J = 7.5 Hz, 2H), 5.01 (d, J = 10.1, 1H), 5.05 (d, J = 16.9, 1H), 5.80 (ddt, J = 16.9, 10.1, 6.7 Hz, 1H), 7.38 (m, 1H), 7.45 (m, 1H), 7.50–7.57 (m, 3H, H3), 7.67 (d, J = 7.7 Hz, 1H), 7.77 (m, 2H), 8.12 (s, 1H), 8.21 (s, 1H); 13C NMR (100 MHz, CDCl3) δ (ppm): 24.7 (CH2), 33.2 (CH2), 37.0 (CH2), 115.7 (CH2), 117.2 (CH), 118.2 (CH), 119.8 (CH), 120.7 (2 × CH), 121.7 (CH), 129.0 (CH), 129.9 (3 × CH), 130.0 (C), 137.1 (C), 137.9 (CH), 138.8 (C), 148.1 (C), 171.6 (C); HRMS (ESI+) m/z: [M + Na]+ calcd for C20H20N4ONa, 355.1534; found, 355.1535.
S-((8R)-8-((tert-Butyldimethylsilyl)oxy)-1,7-dioxo-1-(phenylamino)nonan-4-yl) O-Ethyl Carbonodithioate 10a
A mixture of compound 6a (80 mg, 0.45 mmol, 1 equiv) and xanthate 7 (148 mg, 0.45 mmol, 1 equiv) in 1,2-dicholoroethane (1 mL) was stirred at 165 °C. Over 15 min, DLP (130 mg, 0.326 mmol, 0.7 equiv) was added in three portions every 3 h. The reaction mixture was then stirred overnight at reflux. The 1,2-dicholoroethane was evaporated, and compound 10a was obtained after purification by column chromatography on silica gel (CH2Cl2/cyclohexane/MeOH, 68:30:2) as an oil in 70% yield (153.7 mg, 0.31 mmol). Rf = 0.2 (CH2Cl2/cyclohexane/MeOH, 68:30:2); IR νmax (film, cm–1): 3692, 3317, 2937, 1721, 1662, 1598, 1545, 1443, 1213, 1116, 1047; 1H NMR (400 MHz, CDCl3) δ (ppm): 0.07 (s, 6H), 0.90 (s, 9H), 1.26 (m, 3H), 1.37 (t, J = 7.1 Hz, 3H), 1.68–1.88 (m, 2H), 1.97–2.15 (m, 2H), 2.53, 2.55* (t, J = 7.1 Hz, 2H), 2.79 (m, 2H), 3.80 (m, 1H), 4.13 (m, 1H), 4.60 (q, J = 7.1 Hz, 2H), 7.09 (t, J = 7.9 Hz, 1H), 7.31 (t, J = 7.9 Hz, 2H), 7.51 (d, J = 7.9 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ (ppm): −4.8 (CH3), 13.9 (CH3), 20.0, 20.1* (CH3), 25.9 (3 × CH3), 30.5 (2 × CH2), 34.8, 34.9 (2 × CH2), 50.7, 50.8* (CH), 70.6 (CH2), 72.9, 73.0* (CH), 120.0 (2 × CH), 124.5 (CH), 129.2 (2 × CH), 138.0 (C), 170.4 (C), 212.0 (C), 214.1 (C); HRMS (ESI+) m/z: [M + H]+ calcd for C24H40NO4SiS2, 498.2168; found, 498.2151.
S-((9R)-9-((tert-Butyldimethylsilyl)oxy)-1,8-dioxo-1-(phenylamino)decan-5-yl) O-Ethyl Carbonodithioate 10b
On the basis of the synthesis of compound 10a and starting from compound 6b (0.1 g, 0.53 mmol), xanthate 7 (171 mg, 0.53 mmol), and DLP (148 mg, 0.37 mmol), compound 10b was obtained after purification by column chromatography on silica gel (CH2Cl2/cyclohexane/MeOH, 68:30:2) as an oil in 77% yield (209.3 mg, 0.41 mmol). Rf = 0.2 (CH2Cl2/cyclohexane/MeOH, 68:30:2); IR νmax (film, cm–1): 3676, 3310, 2938, 1714, 1666, 1602, 1549, 1501, 1442, 1373, 1309, 1208; 1H NMR (400 MHz, CDCl3) δ (ppm): 0.07 (s, 3H), 0.91 (s, 9H), 1.27, 1.28* (d, J = 6.8 Hz, 3H), 1.41 (t, J = 7.6 Hz, 3H), 1.70–1.84 (m, 3H), 1.91 (m, 2H), 2.09 (m, 1H), 2.41 (t, J = 7.6 Hz, 2H), 2.77 (m, 2H), 3.81 (m, 1H), 4.14, 4.15* (q, J = 6.8 Hz, 1H), 4.62, 4.63* (q, J = 7.1 Hz, 2H), 7.09 (t, J = 7.9 Hz, 1H), 7.31 (t, J = 7.9 Hz, 2H), 7.53 (d, J = 7.9 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ (ppm): −4.8 (CH3), 14.0, 14.3* (CH3), 18.2 (C, C20), 21.0 21.1* (CH3), 23.0 (CH2), 25.9 (3 × CH3), 27.5 (CH2), 34.4, 34.5* (CH2), 34.6, 34.7* (CH2), 37.2 (CH2), 50.8 (CH), 70.2 (CH2), 75.0 (CH), 119.9 (2 × CH), 124.3 (CH), 129.1 (2 × CH), 138.2 (C), 171.0 (C), 214.0 (C), 214.1 (C); HRMS (ESI+) m/z: [M + Na]+ calcd for C25H41NO4SiS2Na, 534.2144; found, 534.2143.
S-((10R)-10-((tert-Butyldimethylsilyl)oxy)-1,9-dioxo-1-(phenylamino)undecan-6-yl) O-Ethyl Carbonodithioate 10c
On the basis of the synthesis of compound 10a and starting from compound 6c (137 mg, 0.62 mmol), xanthate 7 (202 mg, 0.62 mmol), and DLP (173 mg, 0.43 mmol), compound 10c was obtained after purification by column chromatography on silica gel (CH2Cl2/cyclohexane/MeOH, 68:30:2) as an oil in 40% yield (126.8 mg, 0.241 mmol). Rf = 0.2 (CH2Cl2/cyclohexane/MeOH, 68:30:2); IR νmax (film, cm–1): 3686, 3317, 2926, 2862, 1716, 1673, 1603, 1550, 1502, 1443, 1250, 1116, 1052; 1H NMR (400 MHz, CDCl3) δ (ppm): 0.07 (s, 6H), 0.91 (m, 9H), 1.27 (m, 3H), 1.40 (m, 3H), 1.46–1.60 (m, 2H), 1.65–1.84 (m, 4H), 1.95–2.12 (m, 2H), 2.35 (t, J = 7.5 Hz, 2H), 2.65–2.85 (m, 2H), 3.77 (m, 1H), 4.13 (qd, J = 6.8, 2.7 Hz, 1H), 4.61 (m, 2H), 7.09 (t, J = 7.9 Hz, 1H), 7.30 (t, J = 7.9 Hz, 2H), 7.52 (d, J = 7.9 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ (ppm): −4.8, −4.5* (2 × CH3), 13.9 (CH3), 18.2 (CH3), 21.0 (CH2), 25.4 (CH2), 25.9 (3 × CH3), 26.4 (CH2), 27.6 (CH2), 34.4 (CH2), 37.6 (CH2), 51.1 (CH), 70.1 (CH2), 75.0 (CH), 120.0 (2 × CH), 124.3 (CH), 129.1 (2 × CH), 138.1 (C), 171.3 (C), 213.8 (C), 214.7 (C); HRMS (ESI+) m/z: [M + H]+ calcd for C26H44NO4SiS2, 526.2481; found, 526.2462.
S-((9S)-9-((tert-Butyldimethylsilyl)oxy)-1,8-dioxo-1-((3-(1-phenyl-1H-1,2,3-triazol-4-yl)phenyl)amino)decan-5-yl) O-Ethyl Carbonodithioate 10e
On the basis of the synthesis of compound 10a and starting from compound 6e (80 mg, 0.24 mmol), xanthate 7 (78 mg, 0.85 mmol), and DLP (67 mg, 0.168 mmol), compound 10e was obtained after purification by column chromatography on silica gel (CH2Cl2/cyclohexane/MeOH, 68:30:2) as an oil in 60% yield (94.4 mg, 0.14 mmol). Rf = 0.15 (CH2Cl2/cyclohexane/MeOH, 68:30:2); IR νmax (film, cm–1): 3322, 2926, 1721, 1666, 1616, 1600, 1566, 1533, 1508, 1433, 1213, 1116, 1047; 1H NMR (400 MHz, CDCl3) δ (ppm): 0.07 (s, 3H), 0.91 (s, 9H), 1.28 (m, 3H), 1.41 (m, 3H), 1.67–1.85 (m, 3H), 1.92 (m, 2H), 2.09 (m, 1H), 2.44 (t, J = 7.6 Hz, 2H), 2.77 (m, 2H), 3.82 (m, 1H), 4.15 (qd, J = 6.8, 4.4 Hz, 1H), 4.62 (m, 2H), 7.39 (m, 1H), 7.45 (m, 1H), 7.50–7.57 (m, 3H, H2), 7.70 (d, J = 7.7 Hz, 1H), 7.78 (m, 2H), 8.15 (s, 1H), 8.24 (s, 1H); 13C NMR (100 MHz, CDCl3) δ (ppm): −4.8 (2 × CH3), 14.0 (CH3), 18.2 (CH3), 21.1 (C), 23.0 (CH2), 25.9 (3 × CH3), 27.6 (CH2), 34.5 (2 × CH2), 37.2 (CH2), 50.9 (CH), 70.2 (CH2), 75.0 (CH), 117.2 (CH), 118.3 (CH), 119.9 (CH), 120.7 (2 × CH), 121.7 (CH), 129.0 (CH), 129.9 (3 × CH), 130.9 (C), 137.1 (C), 138.9 (C), 148.1 (C), 171.3 (C), 214.0 (C), 214.9 (C); HRMS (ESI+) m/z: [M + H]+ calcd for C33H47N4O4SiS2, 655.2808; found, 655.2807.
S-((9S)-9-((tert-Butyldimethylsilyl)oxy)-1,8-dioxo-1-(phenylamino)decan-5-yl) O-Ethyl Carbonodithioate 10f
On the basis of the synthesis of compound 10a and starting from compound 6b (150 mg, 0.79 mmol), xanthate 8 (282 mg, 0.87 mmol), and DLP (222 mg, 0.55 mmol), compound 10f was obtained after purification by column chromatography on silica gel (CH2Cl2/cyclohexane/MeOH, 68:30:2) as an oil in 91% yield (368.2 mg, 0.72 mmol). Rf = 0.2 (CH2Cl2/cyclohexane/MeOH, 68:30:2); IR νmax (film, cm–1): 3676, 3310, 2938, 1714, 1666, 1602, 1549, 1501, 1442, 1373, 1309, 1208; 1H NMR (400 MHz, CDCl3) δ (ppm): 0.07 (s, 3H), 0.91 (s, 9H), 1.27, 1.28* (d, J = 6.8 Hz, 3H), 1.41 (t, J = 7.6 Hz, 3H), 1.70–1.84 (m, 3H), 1.91 (m, 2H), 2.09 (m, 1H), 2.41 (t, J = 7.6 Hz, 2H), 2.77 (m, 2H), 3.81 (m, 1H), 4.14, 4.15* (q, J = 6.8 Hz, 1H), 4.62, 4.63* (q, J = 7.1 Hz, 2H), 7.09 (t, J = 7.9 Hz, 1H), 7.31 (t, J = 7.9 Hz, 2H), 7.53 (d, J = 7.9 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ (ppm): −4.8, −4.4* (2 × CH3), 14.3 (CH3), 18.2 (C), 21.1 (CH3), 22.8 (CH2), 25.7 (3 × CH3), 27.4 (CH2), 34.5 (2 × CH2), 37.2 (CH2), 50.8 (CH), 70.2 (CH2), 75.0 (CH), 119.9 (2 × CH), 124.3 (CH), 129.1 (2 × CH), 138.2 (C), 171.0 (C), 214.0 (C), 214.1 (C); HRMS (ESI+) m/z: [M + H]+ calcd for C25H42NO4SiS2, 512.2324; found, 512.2311.
S-(1,8-Dioxo-1-(phenylamino)decan-5-yl) O-Ethyl Carbonodithioate 10g
On the basis of the synthesis of compound 10a and starting from compound 6b (116 mg, 0.615 mmol), xanthate 9 (118 mg, 0.615 mmol), and DLP (172 mg, 0.43 mmol), compound 10g was obtained after purification by column chromatography on silica gel (CH2Cl2/cyclohexane/MeOH, 68:30:2) as an oil in 49% yield (113.4 mg, 0.3 mmol). Rf = 0.4 (CH2Cl2/cyclohexane/MeOH, 68:30:2); IR νmax (film, cm–1): 3670, 3306, 2921, 1716, 1668, 1598, 1545, 1502, 1443, 1213, 1116, 1047; 1H NMR (400 MHz, CDCl3) δ (ppm): 1.06 (t, J = 7.3 Hz, 3H), 1.41 (t, J = 7.1 Hz, 3H), 1.71–1.84 (m, 3H), 1.84–1.97 (m, 2H), 2.11 (m, 1H), 2.37–2.48 (m, 4H), 2.50–2.69 (m, 2H), 3.76 (m, 1H), 4.62, 4.63* (q, J = 7.1 Hz, 2H), 7.09 (t, J = 7.9 Hz, 1H), 7.31 (t, J = 7.9 Hz, 2H), 7.54 (d, J = 7.9 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ (ppm): 8.0 (CH3), 14.3 (CH3), 22.8 (CH2), 28.0 (CH2), 34.3 (CH2), 36.2 (CH2), 37.1 (CH2), 39.4 (CH2), 50.7 (CH), 70.3 (CH2), 119.9 (2 × CH), 124.3 (CH), 129.1 (2 × CH), 138.2 (C), 171.1 (C), 211.2 (C), 215.0 (C); HRMS (ESI+) m/z: [M + Na]+ calcd for C19H27NO3S2Na, 404.1330; found, 404.1332.
(R)-8-Hydroxy-7-oxo-N-phenylnonanamide 11a
A mixture of compound 10a (24 mg, 0.084 mmol) and phosphonic acid/Et3N (1 M) (0.42 mL, 5 equiv) in dioxane (1 mL) was stirred at 145 °C. Over 15 min, AIBN (7 mg) was added dropwise, and the reaction mixture was stirred over 2 h at 145 °C. Dioxane was evaporated, and compound 11a was obtained after purification by column chromatography on silica gel (EtOAc/cyclohexane, 4:6) as white solid in 45% yield (10 mg, 0.04 mmol). [α]D20 – 7.8 (c = 1, chloroform); Rf = 0.2 (EtOAc/cyclohexane, 4:6); IR νmax (film, cm–1): 3308, 2939, 1718, 1662, 1599, 1543, 1500, 1444, 1313, 1246, 1103, 1032; 1H NMR (400 MHz, CDCl3) δ (ppm): 1.30–1.48 (m, 5H), 1.60–1.84 (m, 4H), 2.36 (t, J = 7.4 Hz, 2H), 2.50 (m, 2H), 4.23 (q, J = 7.1 Hz, 1H), 7.10 (t, J = 7.9 Hz, 1H), 7.32 (t, J = 7.9 Hz, 2H), 7.51 (d, J = 7.9 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ (ppm): 20.0 (CH3), 23.3 (CH2), 25.3 (CH2), 28.8 (CH2), 37.4 (CH2), 37.5 (CH2), 72.8 (CH), 119.9 (2 × CH), 124.5 (CH), 129.2 (2 × CH), 138.1 (C), 171.2 (C), 212.8 (C); HRMS (ESI+) m/z: [M + Na]+ calcd for C15H21NO3Na, 286.1419; found, 286.1418.
(R)-9-Hydroxy-8-oxo-N-phenyldecanamide 11b
On the basis of the synthesis of compound 11a and starting from compound 10b (0.1 g, 0.19 mmol), phosphonic acid/Et3N (1 M) (0.98 mL), and AIBN (10 mg), compound 11b was obtained after purification by column chromatography on silica gel (EtOAc/cyclohexane, 1:9) as white solid in 61% yield (32.4 mg, 0.11 mmol). [α]D20 – 2.8 (c = 1, chloroform); Rf = 0.2 (EtOAc/cyclohexane 3:7); IR νmax (film, cm–1): 3312, 2927, 1718, 1662, 1603, 1528, 1500, 1444, 1329, 1246, 1103, 1044; 1H NMR (400 MHz, CDCl3) δ (ppm): 1.30–1.46 (m, 7H), 1.65 (m, 2H), 1.74 (m, 2H), 2.35 (t, J = 7.5 Hz, 2H), 2.48 (m, 2H), 4.24 (q, J = 7.1 Hz, 1H), 7.1 (t, J = 7.9 Hz, 1H), 7.32 (t, J = 7.9 Hz, 2H), 7.51 (d, J = 7.9 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ (ppm): 20.0 (CH3), 23.5 (CH2), 25.4 (CH2), 29.0 (2 × CH2), 37.5 (CH2), 37.7 (CH2), 72.8 (CH), 119.9 (2 × CH), 124.6 (CH), 129.2 (2 × CH), 138.4 (C), 171.4 (C), 212.8 (C); HRMS (ESI+) m/z: calcd for C16H24NO3, 278.1756; found, 278.1763.
(R)-10-Hydroxy-9-oxo-N-phenylundecanamide 11c
On the basis of the synthesis of compound 11a and starting from compound 10c (80 mg, 0.15 mmol), phosphonic acid/Et3N (1 M) (0.76 mL), and AIBN (13 mg), compound 11c was obtained after purification by column chromatography on silica gel (EtOAc/cyclohexane, 4:6) as an oil in 60% yield (26.2 mg, 0.09 mmol). [α]D20 – 5.3 (c = 1, chloroform); Rf = 0.2 (EtOAc/cyclohexane, 4:8); IR νmax (film, cm–1): 3294, 2938, 1716, 1666, 1602, 1538, 1501, 1442, 1373, 1314, 1251; 1H NMR (400 MHz, CDCl3) δ (ppm): 1.20–1.41 (m, 9H), 1.63 (m, 2H), 1.72 (m, 2H), 2.35 (t, J = 7.5 Hz, 2H), 2.39– 2.57 (m, 2H), 4.23 (q, J = 7.1 Hz, 1H), 7.11 (t, J = 7.9 Hz, 1H), 7.31 (t, J = 7.9 Hz, 2H), 7.51 (d, J = 7.9 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ (ppm): 20.0 (CH3), 23.6 (2 × CH2), 25.6 (CH2), 29.2 (CH2), 29.3 (CH2), 37.6 (CH2), 39.9 (CH2), 72.8 (CH), 119.9 (2 × CH), 124.4 (CH), 129.2 (2 × CH), 138.2 (C), 171.6 (C), 212.9 (C); HRMS (ESI+) m/z: [M + Na]+ calcd for C17H25NO3Na, 314.1732; found, 314.1734.
(S)-9-Hydroxy-8-oxo-N-(3-(1-phenyl-1H-1,2,3-triazol-4-yl)phenyl)decanamide 11e
On the basis of the synthesis of compound 11a and starting from compound 10e (74.5 mg, 0.11 mmol), phosphonic acid/Et3N (1 M) (0.55 mL), and AIBN (10 mg), compound 11e was obtained after purification by column chromatography on silica gel (EtOAc/cyclohexane, 4:6) as white solid in 72% yield (33.4 mg, 0.08 mmol). [α]D20 – 9.9 (c = 1, chloroform); Rf = 0.1 (EtOAc/cyclohexane, 4:6); IR νmax (film, cm–1): 3348, 2927, 1708, 1666, 1613, 1565, 1533, 1490, 1405, 1229; 1H NMR (400 MHz, CDCl3) δ (ppm): 1.27 (d, J = 7.1 Hz, 3H), 1.40 (m, 4H), 1.67 (m, 2H), 1.77 (m, 2H), 2.40 (m, 2H), 2.50 (m, 2H), 4.24 (q, J = 7.1 Hz, 1H), 7.38 (t, J = 7.9 Hz, 1H), 7.45 (m, 1H), 7.54 (m, 3H), 7.67 (d, J = 7.7 Hz, 1H), 7.77 (d, J = 7.5 Hz, 2H), 8.12 (s, 1H), 8.23 (s, 1H); 13C NMR (100 MHz, CDCl3) δ (ppm): 20.0 (CH3), 23.5 (CH2), 25.4 (CH2), 29.0 (2 × CH2), 37.7 (2 × CH2), 72.8 (CH), 117.1 (2 × CH), 119.8 (CH), 120.7 (2 × CH), 121.7 (CH), 129.1 (CH), 129.9 (3 × CH), 131.1 (C), 137.2 (C), 138.9 (C), 148.5 (C), 171.4 (C), 212.9 (C); HRMS (ESI+) m/z: [M + H]+ calcd for C24H29N4O3, 421.2240; found, 421.2241.
(S)-9-Hydroxy-8-oxo-N-phenyldecanamide 11f
On the basis of the synthesis of compound 11a and starting from compound 10f (33 mg, 0.064 mmol), phosphonic acid/Et3N (1 M) (0.32 mL), and AIBN (7 mg), compound 11f was obtained after purification by column chromatography on silica gel (EtOAc/cyclohexane, 4:6) as white solid in 77% yield (13.7 mg, 0.05 mmol). [α]D20 + 2.4 (c = 1, chloroform); Rf = 0.2 (EtOAc/cyclohexane, 4:6); IR νmax (film, cm–1): 3310, 2932, 1719, 1666, 1602, 1538, 1501, 1442, 1373, 1309; 1H NMR (400 MHz, CDCl3) δ (ppm): 1.29–1.43 (m, 7H), 1.64 (m, 2H), 1.73 (m, 2H), 2.35 (t, J = 7.5 Hz, 2H), 2.47 (m, 2H), 4.23 (q, J = 7.1 Hz, 1H), 7.10 (t, J = 7.9 Hz, 1H), 7.31 (t, J = 7.9 Hz, 2H), 7.51 (d, J = 7.9 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ (ppm): 20.0 (CH3), 23.5 (CH2), 25.4 (CH2), 29.0 (CH2), 37.5 (CH2), 72.8 (CH), 119.9 (2 × CH), 124.4 (CH), 129.2 (2 × CH), 138.1 (C), 171.4 (C), 212.8 (C); HRMS (ESI+) m/z: [M + H]+ calcd for C16H24NO3, 278.1756; found, 278.1758.
8-Oxo-N-phenyldecanamide 11g
On the basis of the synthesis of compound 11a and starting from compound 10g (55 mg, 0.144 mmol), phosphonic acid/Et3N (1 M) (0.72 mL), and AIBN (15 mg), compound 11g was obtained after purification by column chromatography on silica gel (EtOAc/cyclohexane, 1:9) as white solid in 96% yield (36.2 mg, 0.14 mmol). Rf = 0.2 (EtOAc/cyclohexane, 1:9); IR νmax (film, cm–1): 3364, 2932, 1687, 1602, 1533, 1501, 1437, 1416, 1373; 1H NMR (400 MHz, CDCl3) δ (ppm): 1.05 (t, J = 7.3 Hz, 3H), 1.20–1.47 (m, 4H), 1.58 (m, 2H), 1.72 (m, 2H), 2.34 (t, J = 7.5 Hz, 2H), 2.41 (m, 4H), 7.09 (t, J = 7.9 Hz, 1H), 7.3 (t, J = 7.9 Hz, 2H), 7.53 (d, J = 7.9 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ (ppm): 8.0 (CH3), 23.7 (CH2), 25.5 (CH2), 29.0 (CH2), 29.1 (CH2), 36.1 (CH2), 37.7 (2 × CH2), 120.0 (2 × CH), 124.3 (CH), 129.1 (2 × CH), 138.2 (C), 171.6 (C), 212.2 (C); HRMS (ESI+) m/z: [M + H]+ calcd for C16H24NO2, 262.1807; found, 262.1814.
(R)-3,10-Dioxo-10-(phenylamino)decan-2-yl Acetate 12b
A mixture of compound 11b (22 mg, 0.079 mmol) in anhydrous CH2Cl2 (2 mL) was cooled to 0 °C and Ac2O (0.011 mL, 0.12 mmol, 1.5 equiv), Et3N (0.022 mL, 0.158 mmol, 2 equiv), and DMAP (19.3 mg, 0.158 mmol, 2 equiv) were added dropwise. The reaction mixture was stirred for 1 h at 0 °C and then for 2 h at rt. After the reaction was completed, EtOAc and a saturated aqueous solution of NH4Cl were added to the mixture. The organic layer was then dried over MgSO4 and filtered, and the solvent was evaporated. Compound 12b was obtained after purification by column chromatography on silica gel (EtOAc/cyclohexane, 3:7) as a white solid in 83% yield (21 mg, 0.066 mmol). [α]D20 + 6.6 (c = 1, chloroform); Rf = 0.25 (EtOAc/cyclohexane, 3:7); IR νmax (film, cm–1): 3326, 2938, 1730, 1714, 1666, 1602, 1533, 1442, 1378, 1240; 1H NMR (400 MHz, CDCl3) δ (ppm): 1.38 (m, 3H), 1.55–1.67 (m, 4H), 1.69–1.79 (m, 4H), 2.14 (s, 3H), 2.35 (m, 2H), 2.47 (m, 2H), 5.08 (q, J = 7.1 Hz, 1H), 7.10 (t, J = 7.9 Hz, 1H), 7.32 (t, J = 7.9 Hz, 2H), 7.52 (d, J = 7.9 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ (ppm): 16.3 (CH3), 21.0 (CH3), 23.1 (CH2), 25.4 (CH2), 28.7 (CH2), 28.8 (CH2), 37.6 (CH2), 38.2 (CH2), 74.9 (CH), 119.9 (CH), 124.3 (CH), 129.2 (CH), 138.2 (C), 170.7 (C), 179.4 (C), 208.1 (C); HRMS (ESI+) m/z: [M + H]+ calcd for C18H26NO4, 320.1862; found, 320.1858.
HDAC Enzymatic Assays
The following compounds were tested for HDAC inhibition of HeLa nuclear extract (Enzo Life Sciences, Lausen, Switzerland): HDAC1, HDAC3/NCOR2, and HDAC6 (Sigma-Aldrich, Buchs, Switzerland) and HDAC4 and HDAC8 (Reaction Biology Corp.). The substrate concentration was 50 μM. The highest concentration used for single isoforms was 300 μM. IC50 values were considered >300 μM when the % inhibition was ≪50% at this concentration. Reactions were carried out in 96-well 1/2 volume microplates. The HeLa nuclear extract or specific isoforms were diluted in assay buffer (50 mM Tris at pH 8.0, adjusted with HCl; 137 mM NaCl; 2.7 mM KCl; and 1 mM MgCl2) and added to each microplate well, except for blanks. The test compounds were diluted in dimethyl sulfoxide (DMSO) and added to the wells (5% DMSO in each well). The reaction was initiated by addition of the MAL substrate (BOC-Ac-Lys-AMC; Sigma-Aldrich) for HeLa nuclear extract, HDAC1, and HDAC6 or the Fluor de Lys SIRT1 deacetylase substrate (BML-KI177; Enzo) for HDAC3. The plate was then incubated at 37 °C for 90 min (HeLa nuclear extract, HDAC1, and HDAC6) or 30 min (HDAC3). The reaction was stopped with 1 μM Trichostatin A. Trypsin from the bovine pancreas, 7500 BAEE units/mg (Sigma-Aldrich), was added 30 min prior to plate reading. HDAC inhibition was calculated by comparing the amount of deacetylated substrate between the control (100% HDAC activity) and test sample. The relative amount of deacetylated substrate was obtained by fluorescence reading with excitation at 360 nm and emission at 460 nm for HeLa nuclear extract, HDAC1, HDAC3, and HDAC6. Dose–response curves were obtained with six different concentrations to determine the IC50 values of the compounds showing more than 50% inhibition at 100 μM using the HeLa nuclear extract. Results are the mean of three independent experiments. SAHA was used as a positive control. HDAC4 and HDAC8 assays were performed by Reaction Biology Corp.
Antiproliferative Activity on Human Cell Lines
XTT assays were conducted as previously described to examine the inhibitory effects of the compounds in various human cell lines.35 Briefly, normal B lymphocytes (15 000 cells/well), HeLa, and HEK cells (10 000 cells/well) were seeded in 96-well plates and treated immediately with increasing concentrations in triplicates. After 72 h of incubation, 50 μL of XTT solution (1 mg/mL in culture medium supplemented with an electron carrier) was added to the cells and incubated for 4 h. The percentage of cell growth was calculated as the absorbance of each test well divided by that of the vehicle control wells, multiplied by 100. The concentration that resulted in a 50% reduction in cell growth (GI50) was determined from at least five different concentrations using GraphPad Prism 5.0 software. With Jurkat and K562, cells were cultured in RPMI 1640 complemented with 10% FCS in 96-well microplates (8000 cells/well) and incubated at 37 °C and 5% CO2 in the presence of various concentration of tested molecules, as above. After 24 h of incubation at 37 °C, [3H]-thymidine was added to each well. After an additional 6 h of incubation at 37 °C, incorporation of [3H]-thymidine in nucleic acids was measured as indicated below.
P. falciparum Assay
In vitro antimalarial activity was measured on microtiter plates, using the [3H]-hypoxanthine incorporation assay with the 3D7 strain of P. falciparum, as previously described.36 Briefly, suspensions of infected erythrocytes at 1.5% final hematocrit and 0.6% parasitemia were cultured in complete medium (RPMI 1640, complemented with 25 mM Hepes, pH 7.4, and 10% AB+ human serum) either in the absence (controls) or in the presence of the compounds within a range of molecular concentrations. The compounds were usually dissolved in DMSO and were diluted in culture medium so that the final DMSO concentration never exceeded 0.25%. After 48 h of incubation at 37 °C, under a gaseous mixture of 5% CO2, 5% O2, and 90% N2, [3H]-hypoxanthine was added to each well for 18 h. Then, the cells were lysed using an automatic cell harvester and the cell macromolecules, including radioactive nucleic acids, were retained onto glass fiber filters and counted for radioactivity.
Molecular Modeling
Molecular docking calculations were conducted using GOLD software (version 5.237) with the preset options for the genetic GOLD algorithm and the ChemPLP scoring function. Zinc atom was used as the centroid, and the size of the docking cavity was 10 Å. One hundred poses were generated for each ligand. To compare all solutions, root-mean-square deviations (rmsd’s) among different structures were computed and clusters of solutions were formed according to their rmsd values. The best-ranked docking poses were then visually inspected in Pymol, version 2006.38
To validate the molecular docking protocol, SAHA was redocked into the catalytic pocket of HDAC2, wherein a co-crystallized structure exists (PDB: 4LXZ). Results are given in the Supporting Information (Figure S1). The same protocol was used to target HDAC1 and HDAC6. HDAC1 was taken from the Protein Data Bank (PDB: 4BKX), whereas a homology model of the HDAC6 catalytic site CD II (from Leu483 to Gly800, Uniprot ID Q9UBN7) was built with MOE 2014.09 (www.chemcomp.com). Despite the higher % of identity with class IIa HDACs (HDAC4 and HDAC7), the crystal structure of HDAC8 (PDB: 1T69) was chosen as the template for building the model, as also recently proposed.39 HDAC8 shares with HDAC6 a catalytic Tyr residue (Tyr306 in HDAC8, Tyr782 in HDAC6) and a similar manner of binding inhibitors, making the design of selective inhibitors toward these two isoforms difficult.40 Alignment of HDAC6 and HDAC8 sequences was carried out with the MOE Align program using default parameters. HDAC6 CD II shares 28% identity and 46% similarity with HDAC8. Comparative model building was then carried out with the MOE Homology Model module. Fifty intermediate models were built, refined, and scored with the GB-IV scoring function, using the AMBER99 force field: A refined and averaged model was finally generated and taken into account. Zinc ion coordinates were transferred from the template to the target. Lateral chains and only the zinc ion were then submitted to 1000 minimization cycles in the Sybyl X 1.3 suite (Tripos Inc., St. Louis, MO) using the AMBER99 force field and Powell algorithm. The quality of the minimized model (coordinates given in the Supporting Information) was checked by monitoring the Ramachandran plot, MOE protein geometry module, Errat, Verify3D, and QMEAN score, and the results are given in the Supporting Information.
Docking was done keeping the proteins fixed, whereas the ligands fully flexible. Ligands were built and their geometries optimized using Gaussian09 software41 at the B3LYP/6-31g* level. Different protonation states were tested for their ZBG groups.
Before docking calculations, short molecular mechanical (MM) dynamics were performed on the structures of the protein (X-ray structure and model) using the AMBER12 software,42 under periodic boundary conditions. For X-ray structures, water and other co-crystallized molecules were also removed. The two neighboring histidines close to the active site (e.g., for HDAC1, His140 and 141) are protonated at the delta position (HID). For other amino acids residues, the tool xleap of Amber12 was used to add the missing protons. The systems were solvated with a TIP3P water rectangular box. For the zinc atom, nonbonded parameters and a charge of two were added. The dimensions of the TIP3P water rectangular box were chosen to be at least 10 Å larger than that of the solute in every direction. Sodium cations were added and long-range electrostatic interactions were computed using the particle-mesh Ewald method, with a cutoff value of 10 Å. The time step was set at 1 fs. Minimization and heating in the NVT ensemble were carried out. Then a molecular dynamics production phase was run in the NPT ensemble at 300 K over 1 ns.
MM simulations were also performed on a few structures of complexes obtained from docking under the same conditions. In the case of the monodentate interaction, 20 ns of simulations was done to check the stability of the structure. For the better known bidentate interaction, only 5 ns of simulations was done. For ligands, parameters were generated using the program Antechamber of Amber with the GAFF force field. Partial charges were obtained by the RESP approach after Gaussian09 calculation of the electrostatic potential in the MK scheme at the HF/6-31g* level.
Acknowledgments
Financial support was received from “Agence Nationale de la Recherche” (French National Research Agency) under the Laboratory of Excellence program (ARCANE project No. ANR-11-LABX-003), “fondation pour le développement de la chimie des substances naturelles et ses applications” Institut de France/Académie des Sciences, and CNRS (French National Center for Scientific Research) is gratefully acknowledged. Financial support from the Swiss National Science Foundation (P300P3 158507) and Pierre Mercier foundation is also acknowledged. The authors are also grateful to CECIC for computer facilities. The P. falciparum 3D7 strain was obtained through the Malaria Research and Reference Reagent Resource Center (MR4; http://www.mr4.org/). ICMG (Institut de Chimie Moléculaire de Grenoble) is acknowledge for analysis and characterization facilities.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.6b00481.
The authors declare no competing financial interest.
Supplementary Material
References
- López J. E.; Sullivan E. D.; Fierke C. A. Metal-dependent deacetylases: cancer and epigenetic regulators. ACS Chem. Biol. 2016, 11, 706–716. 10.1021/acschembio.5b01067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkenberg K. J.; Johnstone R. W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discovery 2014, 13, 673–691. 10.1038/nrd4360. [DOI] [PubMed] [Google Scholar]
- Andrews K. T.; Haque A.; Jones M. K. HDAC inhibitors in parasitic diseases. Immunol. Cell Biol. 2012, 90, 66–77. 10.1038/icb.2011.97. [DOI] [PubMed] [Google Scholar]
- Madhusoodanan M.; Zheng S.; Huang T. L.; Wang G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 2015, 20, 3898–3941. 10.3390/molecules20033898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lee N.; Zhang Y. Chemical answers to epigenetic crosstalk. Nat. Chem. Biol. 2008, 4, 335–337. 10.1038/nchembio0608-335. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Henikoff S.; Greally J. M. Epigenetics, cellular memory and gene regulation. Curr. Biol. 2016, 26, R644–R666. 10.1016/j.cub.2016.06.011. [DOI] [PubMed] [Google Scholar]
- Delcuve G. P.; Khan D. H.; Davie J. R. Targeting class I histone deacetylase in cancer therapy. Expert Opin. Ther. Targets 2013, 17, 29–41. 10.1517/14728222.2013.729042. [DOI] [PubMed] [Google Scholar]
- a Huber K.; Doyon G.; Plaks J.; Fyne E.; Mellors J. W.; Sluis-Cremer N. Inhibitors of histone deacetylases: correlation between isoform specificity and reactivation of HIV Type 1 (HIV-1) from latently infected cells. J. Biol. Chem. 2011, 286, 22211–22218. 10.1074/jbc.M110.180224. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Shirakawa K.; Chavez L.; Hakre S.; Calvanese V.; Verdin E. Reactivation of latent HIV by histone deacetylase inhibitors. Trends Microbiol. 2013, 21, 277–285. 10.1016/j.tim.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Barton K. M.; Archin N. M.; Keedy K. S.; Espeseth A. S.; Zhang Y.-L.; Gale J.; Wagner F. F.; Holson E. B.; Margolis D. M. Selective HDAC inhibition for the disruption of latent HIV-1 infection. PLoS One 2014, 9, e102684 10.1371/journal.pone.0102684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bougdour A.; Maubon D.; Baldacci P.; Ortet P.; Bastien O.; Bouillon A.; Barale J.-C.; Pelloux H.; Menard R.; Hakimi M.-A. Drug inhibition of HDAC3 and epigenetic control of differentiation in Apicomplexa parasites. J. Exp. Med. 2009, 206, 953–966. 10.1084/jem.20082826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maubon D.; Bougdour A.; Wong Y.-S.; Brenier-Pinchart M.-P.; Curt A.; Hakimi M.-A.; Pelloux H. Activity of the histone deacetylase inhibitor FR235222 on Toxoplasma gondii: inhibition of stage conversion of the parasite cyst form and study of new derivative compounds. Antimicrob. Agents Chemother. 2010, 54, 4843–4850. 10.1128/AAC.00462-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallavalle S.; Pisano C.; Zunino F. Development and therapeutic impact of HDAC6-selective inhibitors. Biochem. Pharmacol. 2012, 84, 756–765. 10.1016/j.bcp.2012.06.014. [DOI] [PubMed] [Google Scholar]
- Simões-Pires C.; Zwick V.; Nurisso A.; Shencker E.; Carrupt P. A.; Cuendet M. HDAC6 as a target for neurodegenerative diseases: what makes it different from the other HDACs?. Mol. Neurodegener. 2013, 8, 7 10.1186/1750-1326-8-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand P. Inside HDAC with HDAC inhibitors. Eur. J. Med. Chem. 2010, 45, 2095–2116. 10.1016/j.ejmech.2010.02.030. [DOI] [PubMed] [Google Scholar]
- a Bieliauskas A. V.; Pflum M. K. H. Isoform-selective histone deacetylase inhibitors. Chem. Soc. Rev. 2008, 37, 1402–1413. 10.1039/b703830p. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Butler K. V.; Kozikowski A. P. Chemical origins of isoform selectivity in histone deacetylase inhibitors. Curr. Pharm. Des. 2008, 14, 505–528. 10.2174/138161208783885353. [DOI] [PubMed] [Google Scholar]; c Thaler F.; Mercurio C. Towards selective inhibition of histone deacetylase isoforms: what has been achieved, where we are and what will be next. ChemMedChem 2014, 9, 523–536. 10.1002/cmdc.201300413. [DOI] [PubMed] [Google Scholar]; d Micelli C.; Rastelli G. Histone deacetylases: structural determinants of inhibitor selectivity. Drug Discovery Today 2015, 20, 718–735. 10.1016/j.drudis.2015.01.007. [DOI] [PubMed] [Google Scholar]
- a Newkirk T. L.; Bowers A. A.; Williams R. M. Nat. Prod. Rep. 2009, 26, 1293–1320. 10.1039/b817886k. [DOI] [PubMed] [Google Scholar]; b Cherblanc F. L.; Davidson R. W. M.; Di F. P.; Srimongkolpithak N.; Fuchter M. J. Perspectives on natural product epigenetic modulators in chemical biology and medicine. Nat. Prod. Rep. 2013, 30, 605–624. 10.1039/c3np20097c. [DOI] [PubMed] [Google Scholar]
- Hu E.; Dul E.; Sung C.-M.; Chen Z.; Kirkpatrick R.; Zhang G.-F.; Johanson K.; Liu R.; Lago A.; Hofmann G.; Macarron R.; de los Frailes M.; Perez P.; Krawiec J.; Winkler J.; Jaye M. Identification of novel isoform-selective inhibitors within class I Histone deacetylases. J. Pharmacol. Exp. Ther. 2003, 307, 720–728. 10.1124/jpet.103.055541. [DOI] [PubMed] [Google Scholar]
- a Moradei O. M.; Mallais T. C.; Frechette S.; Paquin I.; Tessier P. E.; Leit S. M.; Fournel M.; Bonfils C.; Trachy-Bourget M. C.; Liu J.; Yan T. P.; Lu A. H.; Rahil J.; Wang J.; Lefebvre S.; Li Z.; Vaisburg A. F.; Besterman J. M. Novel aminophenyl benzamide-type histone deacetylase inhibitors with enhanced potency and selectivity. J. Med. Chem. 2007, 50, 5543–5546. 10.1021/jm701079h. [DOI] [PubMed] [Google Scholar]; b Stolfa D. A.; Stefanachi A.; Gajer J. M.; Nebbioso A.; Altucci L.; Cellamare S.; Jung M.; Carotti A. Design, synthesis, and biological evaluation of 2-aminobenzanilide derivatives as potent and selective HDAC inhibitors. ChemMedChem 2012, 7, 1256–1266. 10.1002/cmdc.201200193. [DOI] [PubMed] [Google Scholar]; c Methot J. L.; Hoffman D. M.; Witter D. J.; Stanton M. G.; Harrington P.; Hamblett C.; Siliphaivanh P.; Wilson K.; Hubbs J.; Heidebrecht R.; Kral A. M.; Ozerova N.; Fleming J. C.; Wang H.; Szewczak A. A.; Middleton R. E.; Hughes B.; Cruz J. C.; Haines B. B.; Chenard M.; Kenific C. M.; Harsch A.; Secrist J. P.; Miller T. A. Delayed and prolonged histone hyperacetylation with a selective HDAC1/HDAC2 inhibitor. ACS Med. Chem. Lett. 2014, 5, 340–345. 10.1021/ml4004233. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Wagner F. F.; Lundh M.; Kaya T.; McCarren P.; Zhang Y.-L.; Chattopadhyay S.; Gale J. P.; Galbo T.; Fisher S. L.; Meier B. C.; Vetere A.; Richardson S.; Morgan N. G.; Christensen D. P.; Gilbert T. J.; Hooker J. M.; Leroy M.; Walpita D.; Mandrup-Poulsen T.; Wagner B. K.; Holson E. B. An isochemogenic set of inhibitors to define the therapeutic potential of histone deacetylases in β-cell protection. ACS Chem. Biol. 2016, 11, 363–374. 10.1021/acschembio.5b00640. [DOI] [PubMed] [Google Scholar]
- a Kim D. H.; Shin J.; Kwon H. J. Psammaplin A is a natural produg that inhibits class I histone deacetylase. Exp. Mol. Med. 2007, 39, 47–55. 10.1038/emm.2007.6. [DOI] [PubMed] [Google Scholar]; b Baud M. G. J.; Leiser T.; Haus P.; Samlal S.; Wong A. C.; Wood R. J.; Petrucci V.; Gunaratnam M.; Hughes S. M.; Buluweda L.; Turlais F.; Neidle S.; Meyer-Almes F.-J.; White A. J. P.; Fuchter M. J. Defining the mechanism of action and enzymatic selectivity of psammaplin A against its epigenetic targets. J. Med. Chem. 2012, 55, 1731–1750. 10.1021/jm2016182. [DOI] [PubMed] [Google Scholar]; c Baud M. G. J.; Haus P.; Leiser T.; Meyer-Almes F.-J.; Fuchter M. J. Highly ligand efficient and selective N-2-(thioethyl)picolinamide histone deacetylase inhibitors inspired by the natural product psammaplin A. ChemMedChem 2013, 8, 149–156. 10.1002/cmdc.201200450. [DOI] [PubMed] [Google Scholar]
- Matsuyama A.; Shimazu T.; Sumida Y.; Saito A.; Yoshimatsu Y.; Seigneurin-Berny D.; Osada H.; Komatsu Y.; Nishino N.; Khochbin S.; Horinouchi S.; Yoshida M. In vivo destabilisation of dynamic microtubules by HDAC6-mediated deacetylation. EMBO J. 2002, 21, 6820–6831. 10.1093/emboj/cdf682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bantscheff M.; Hopf C.; Savitski M. M.; Dittmann A.; Grandi P.; Michon A.-M.; Schlegl J.; Abraham Y.; Becher I.; Bergamini G.; Boesche M.; Delling M.; Duempelfeld B.; Eberhard D.; Huthmacher C.; Mathieson T.; Poeckel D.; Reader V.; Strunk K.; Sweetman G.; Kruse U.; Neubauer G.; Ramsden N. G.; Drewes G. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat. Biotechnol. 2011, 29, 255–265. 10.1038/nbt.1759. [DOI] [PubMed] [Google Scholar]
- Kinzel O.; Llauger-Bufi L.; Pescatore G.; Rowley M.; Schultz-Fademrecht C.; Monteagudo E.; Fonsi M.; Gonzalez Paz O.; Fiore F.; Steinkuhler C.; Jones P. Discovery of a potent class I selective ketone histone deacetylase inhibitor with antitumor activity in vivo and optimized pharmacokinetic properties. J. Med. Chem. 2009, 52, 3453–3456. 10.1021/jm9004303. [DOI] [PubMed] [Google Scholar]
- Olsen C. A.; Ghadiri M. R. Discovery of potent and selective histone deacetylase inhibitors via focused combinatorial libraries of cyclic α3β–tetrapeptides. J. Med. Chem. 2009, 52, 7836–7846. 10.1021/jm900850t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traoré M.; Mietton F.; Maubon D.; Peuchmaur M.; Francisco Hilario F.; Pereira de Freitas R.; Bougdour A.; Curt A.; Maynadier M.; Vial H.; Pelloux H.; Hakimi M.-A.; Wong Y.-S. Flexible synthesis and evaluation of diverse anti-apicomplexa cyclic peptides. J. Org. Chem. 2013, 78, 3655–3675. 10.1021/jo4001492. [DOI] [PubMed] [Google Scholar]
- Quiclet-Sire B.; Zard S. Z. Powerful carbon-carbon bond forming reactions based on a novel radical exchange process. Chem. Eur. J. 2006, 12, 6002–6012. 10.1002/chem.200600510. [DOI] [PubMed] [Google Scholar]
- a Barton D. H. R.; Jang D. O.; Jaszberenyi J. C. Hypophosphorous acid and its salts: new reagents for radical chain deoxygenation, dehalogenation and deamination. Tetrahedron Lett. 1992, 33, 5709–5712. 10.1016/0040-4039(92)89012-2. [DOI] [Google Scholar]; b Boivin J.; Jrad R.; Juge S.; Nguyen V. T. On the reduction of S-alkyl-thionocarbonates (xanthates) with phosphorus compounds. Org. Lett. 2003, 5, 1645–1648. 10.1021/ol0342610. [DOI] [PubMed] [Google Scholar]
- Chen Y.; He R.; Chen Y.; D’Annibale M. A.; Langley B.; Kozikowski A. P. Studies of benzamide- and thiol-based histone deacetylase inhibitors in models of oxidative-stress-induced neuronal death: identification of some HDAC3-selective inhibitors. ChemMedChem 2009, 4, 842–852. 10.1002/cmdc.200800461. [DOI] [PubMed] [Google Scholar]
- Vickers C. J.; Olsen C. A.; Leman L. J.; Ghadiri M. R. Discovery of HDAC inhibitors that lack an active site Zn2+-binding functional group. ACS Med. Chem. Lett. 2012, 3, 505–508. 10.1021/ml300081u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villadsen J. S.; Kitir B.; Wich K.; Friis T.; Madsen A. S.; Olsen C. A. An azumamide C analogue without the zinc-binding functionality. MedChemComm 2014, 5, 1849–1855. 10.1039/C4MD00252K. [DOI] [Google Scholar]
- Khan N.; Jeffers M.; Kumar S.; Hackett C.; Boldog F.; Khramtsov N.; Qian X.; Mills E.; Berghs S. C.; Carey N.; Collins L. S.; Tumber A.; Ritchie J. W.; Jensen P. B.; Lichenstein H. S.; Sehested M.; et al. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem. J. 2008, 409, 581–590. 10.1042/BJ20070779. [DOI] [PubMed] [Google Scholar]
- Hai Y.; Christianson D. W. Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat. Chem. Biol. 2016, 12, 741–747. 10.1038/nchembio.2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bürli R. W.; Luckhurst C. A.; Aziz O.; Matthews K. L.; Yates D.; Lyons K. A.; Beconi M.; McAllister G.; Breccia P.; Stott A. J.; Penrose S. D.; Wall M.; Lamers M.; Leonard P.; Müller I.; Richardson C. M.; Jarvis R.; Stones L.; Hughes S.; Wishart G.; Haughan A. F.; O’Connell C.; Mead T.; McNeil H.; Vann J.; Mangette J.; Maillard M.; Beaumont V.; Munoz-Sanjuan I.; Dominguez C. Design, synthesis, and biological evaluation of potent and selective class IIa histone deacetylase (HDAC) inhibitors as a potential therapy for Huntington’s disease. J. Med. Chem. 2013, 56, 9934–9954. 10.1021/jm4011884. [DOI] [PubMed] [Google Scholar]
- a Di Micco S.; Chini M. G.; Terracciano S.; Bruno I.; Riccio R.; Bifulco G. Structural basis for the design and synthesis of selective HDAC inhibitors. Bioorg. Med. Chem. 2013, 21, 3795–3807. 10.1016/j.bmc.2013.04.036. [DOI] [PubMed] [Google Scholar]; b Miyata Y.; Keusch J. L.; Wang L.; Saito M.; Hess D.; Wang X.; Melancon B. J.; Helquist P.; Gut H.; Matthias P. Structural insights into HDAC6 tubulin deacetylation and its selective inhibition. Nat. Chem. Biol. 2016, 12, 748–754. 10.1038/nchembio.2140. [DOI] [PubMed] [Google Scholar]
- Conti P.; Tamborini L.; Pinto A.; Sola L.; Ettari R.; Mercurio C.; De Micheli C. Design and synthesis of novel isoxazole-based HDAC inhibitors. Eur. J. Med. Chem. 2010, 45, 4331–4338. 10.1016/j.ejmech.2010.06.035. [DOI] [PubMed] [Google Scholar]
- Nicolai S.; Sedigh-Zadeh R.; Waser J. J. Org. Chem. 2013, 78, 3783–3801. 10.1021/jo400254q. [DOI] [PubMed] [Google Scholar]
- Girard C.; Oenen E.; Aufort M.; Beauviere S.; Samson E.; Herscovici J. Reusable polymer-supported catalyst for the [3 + 2] Huisgen cycloaddition in automation protocols. Org. Lett. 2006, 8, 1689–1692. 10.1021/ol060283l. [DOI] [PubMed] [Google Scholar]
- Roehm N. W.; Rodgers G. H.; Hatfield S. M.; Glasebrook A. L. An improved colorimetric assay for cell proliferation and viability utilizing the tetrazolium salt XTT. J. Immunol. Methods 1991, 142, 257–256. 10.1016/0022-1759(91)90114-U. [DOI] [PubMed] [Google Scholar]
- Ancelin M. L.; Calas M.; Bonhoure A.; Herbuté S.; Ringwald P.; Vial H. Potent inhibitors of Plasmodium phospholipid metabolism with a broad spectrum of in vitro antimalarial activities. Antimicrob. Agents Chemother. 2003, 47, 2590–2597. 10.1128/AAC.47.8.2590-2597.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones G.; Willett P.; Glen R. C.; Leach A. R.; Taylor R. J. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- Delano W.The PyMOL Molecular Graphics System; DeLano Scientific: Palo Alto, CA, 2006. [Google Scholar]
- Senger J.; Melesina J.; Marek M.; Romier C.; Oehme I.; Witt O.; Sippl W.; Jung M. Synthesis and biological investigation of oxazole hydroxamates as highly selective histone deacetylase 6 (HDAC6) inhibitors. J. Med. Chem. 2016, 59, 1545–1555. 10.1021/acs.jmedchem.5b01493. [DOI] [PubMed] [Google Scholar]
- Deschamps N.; Simões-Pires C. A.; Carrupt P.-A.; Nurisso A. How the flexibility of human histone deacetylases influences ligand binding: an overview. Drug Discovery Today 2015, 20, 736–742. 10.1016/j.drudis.2015.01.004. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, revision D.01; Gaussian, Inc.: Wallingford, CT, 2009.
- Case D. A.; Darden T. A.; Cheatham T. E. III; Simmerling C. L.; Wang J.; Duke R. E.; Luo R.; Walker R. C.; Zhang W.; Merz K. M.; et al. AMBER 12; University of California: San Francisco, CA, 2012.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.