

Abstract
Bacterial adenosine 5’-diphosphate-ribosylating toxins are encoded by several human pathogens, such as Pseudomonas aeruginosa (exotoxin A (ETA)), Corynebacterium diphtheriae (diphtheria toxin (DT)), and Vibrio cholerae (cholix toxin (CT)). The toxins modify eukaryotic elongation factor 2, an essential human enzyme in protein synthesis, thereby causing cell death. Targeting external virulence factors, such as the above toxins, is a promising alternative for developing new antibiotics, while at the same time avoiding drug resistance. This study aims to establish a reliable computational methodology to find a “silver bullet” able to target all three toxins. Herein, we have undertaken a detailed analysis of the active sites of ETA, DT, and CT, followed by the determination of the most appropriate selection of the size of the docking sphere. Thereafter, we tested two different approaches for normalizing the docking scores and used these to verify the best target (toxin) for each ligand. The results indicate that the methodology is suitable for identifying selective as well as multitoxin inhibitors, further validating the robustness of inverse docking for target-fishing experiments.
Introduction
Antimicrobial resistance is a growing public-health threat,1 making regression to a preantibiotic era in which common infections could kill a very real possibility that we have to address.2−4 Traditional antibacterial agents aim to kill bacteria (bactericidal) or stop their growth (bacteriostatic) and provide an incentive for the bacteria to develop resistance toward them using different mechanisms. Thus, compounds that do not target the genome or metabolic proteins inside the pathogens but act by inhibiting their external virulence factors are an interesting alternative.
The bacterial genus Pseudomonas includes a variety of Gram-negative, rod-shaped, and polar-flagella species. A well-known opportunistic pathogen of this genus, Pseudomonas aeruginosa, most commonly affects immunocompromised patients, such as those with cystic fibrosis, acquired immune deficiency syndrome, and cancer, or burn victims5,6 and is the major cause of infections among hospitalized patients, such as hospital-acquired pneumonia and bloodstream and urinary tract infections. P. aeruginosa is able to produce several toxic proteins that can kill the host cell and is well known for its resistance to many major classes of antibiotics.7−10
In P. aeruginosa, the most toxic factor secreted is exotoxin A (ETA). ETA has an LD50 (50% of the lethal dose) of 0.2 μg/kg upon intraperitoneal injection into mice.11−15 Once ETA has entered a eukaryotic cell through receptor-mediated endocytosis, it catalyzes adenosine 5’-diphosphate (ADP) ribosylation of its target protein, eukaryotic elongation factor 2 (eEF2).16 eEF2 is a GTPase that operates during protein synthesis to facilitate the movement of the peptidyl tRNA–mRNA complex from site A to site P of the ribosome, a process known as translocation.17 eEF2 contains a unique post-translationally modified histidine residue, that is, diphthamide(2-[3-carboxyamido-3-trimethylamonio)propyl]histidine.18 The precise role of diphthamide remains undetermined,19 but its absence has been associated with altered translational fidelity.20−22 Diphthamide is also the unique site of modification of eEF2 by ETA and other NAD+-dependent ADP ribosylase toxins, including Corynebacterium diphtheriae diphtheria toxin (DT) and Vibrio cholerae cholix toxin (CT).18,23,24
The modification involves the transfer of an ADP-ribose moiety from NAD+ to a nitrogen atom of the diphthamide imidazole ring in eEF225−31 (Figure 1).
Figure 1.

Proposed mechanism of diphthamide modification, catalyzed by ETA, DT, and CT.
ADP ribosylation of eEF2 inhibits the translocation step in protein synthesis, irreversibly inactivating eEF2 and leading to cell death.15,25,32−34
DT was discovered in 188835 and is a single-chain enzyme of 58 kDa with 535 amino acid residues. The toxin has two subunits, the active or catalytic (A) domain and the binding (B) domain, which displays both receptor-binding and translocation capabilities.15,36,37 ETA is an AB toxin of 66 kDa, with 613 amino acids, discovered in 1966.14 CT is a 666-residue protein that has an AB domain organization similar to that of ETA,23 and it was discovered as recently as in 2007.38 All three contain a HYYE motif in the active site of the A domain, the latter of these (Glu) being identified as the key catalytic residue, being invariant in all ADP-ribosylating toxins.23,39−45 As proposed for Glu148 in DT, Glu553 in ETA, and Glu581 in CT, the glutamic acid is believed to stabilize the oxacarbenium intermediate after dissociation of nicotinamide by formation of a hydrogen bond with the 2′-OH of the ribose.30,46 The catalytic His is believed to form a hydrogen bond with the adenine ribose of NAD+. Mutation of the His residue considerably reduces the activity of the toxin.43,44,46−48
Finally, the two Tyr residues are part of a hydrophobic pocket that binds the nicotinamide moiety of NAD+ through a π-stacking interaction.42,47,49
Knowing that all three pathogens utilize closely related toxins triggered the idea of developing new potential antibiotics targeting mainly ETA but at the same time displaying activity against DT and CT.
Paul Ehrlich connected chemistry with biology, postulating the existence of specific receptors for binding molecules.50,51 This idea evolves into the “magic bullet” concept, that is, that is the concept of drugs going directly to their predetermined biological target.52 However, this one-compound–one-target picture is a simplification of the reality, where a one-compound–multiple-target model is more appropriate. Thus, the “magic shotgun” or “silver bullet” approach for drug development appears to be more suitable and closer to reality than the magic bullet concept.53−55
To find the “magic shotgun” that is able to target several receptors with one load, inverse-docking approaches have emerged as the in silico prototypical techniques to accomplish that goal. Inverse docking refers to computational docking of a selected small molecule onto a library of receptor structures, originally proposed by Chen and Zhi in 2001.56 Since then, several reports employing inverse-docking approaches for identifying new potential targets have been published.57−61
In this work, we first selected a set of known binders for each of the three bacterial toxins and then performed, for the first time, an inverse-docking study against all three. First, we analyzed the active sites, found the optimal sizes of selected spheres for docking, and “normalized” the binding energies to enable comparisons and detect the best receptor for each ligand. Thereafter, we compared the binding modes and affinities of the known ligands, such as NAD+, to validate the approach.
Results and Discussion
Comparison of Toxins
The prepared models of all three toxins were aligned and superposed in MOE 2015.10,62 and the sequence similarity and identity percentages of DT and CT with respect to those of ETA were calculated (Figure 2).
Figure 2.

Sequence alignment and analysis of ETA (1AER), DT (1DTP), and CT (2Q6M). Sequence similarity and identity were calculated with respect to those of ETA. Positive (blue) bars show similarity and negative (red) bars represent dissimilar residues, as obtained using BLOSUM62 matrix scores. The %-identity was also calculated with respect to that of ETA and is displayed as magenta bars. Identical residues are highlighted in green, whereas red residues show nonidentical amino acids.
The primary structure of CT shows a 39.2% sequence identity with that of ETA for the catalytic domain, whereas DT displays an 18.4% sequence identity. The sequence similarity to that of ETA is also higher for CT (52.8%) as compared to that for DT (31.6%).
Those residues that are described as interacting with the natural substrate (NAD+) and are involved in the catalysis are conserved in all three toxins (HYYE motif, see below) and are highlighted with yellow arrows in Figure 2. Position 42 (arbitrary numbering for alignment) harbors the catalytic His (440 for ETA, 21 for DT, and 460 for CT), which is believed to form a hydrogen bond with the adenine ribose of NAD+. In alignment positions 75 and 86, two almost-parallel Tyr residues are located (470 and 481 for ETA, 54 and 65 for DT, and 493 and 504 for CT, respectively). Finally, the Glu that is located at position 180 is universally conserved among ADP ribosylases (553 for ETA, 148 for DT, and 581 for CT) and has been described as essential for catalytic activity.15,38,39 It forms a hydrogen bond with the 2′-OH of the ribose of NAD+, stabilizing the oxacarbenium intermediate after the first step of the SN1 dissociation of the nicotinamide moiety (see Figure 1).30,40,46
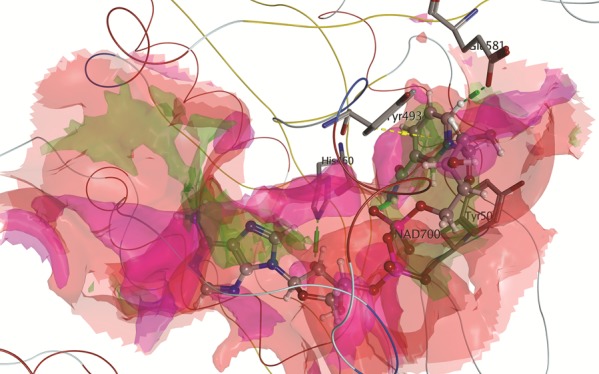
Besides the sequence similarity, the three-dimensional (3D) structures of ADP ribosylases are also similar, as shown in Figure 3. Of particular interest is that the residues belonging to the HYYE motif are not only conserved but also have the same chain conformation among all structures.
Figure 3.

(a) Superposition of the 3D structures of toxins; ETA in magenta, DT in green, and CT in yellow. Residues belonging to the HYYE motif are represented using a ball and stick model. (b) Root-mean-square deviation (RMSD) matrix values of the positions of the Cα atoms for each pair of toxins. 1AER: ETA, 1DTP: DT, 2Q6M: CT.
As evidenced by the RMSD calculation after alignment and superposition, ETA is more similar to CT than to DT. This is also in line with the higher sequence identity and similarity noted for these toxins (cf. Figure 1). It is clear that all three toxins share common features in the active site, which could enable the possibility of finding a potential multitarget inhibitor. Figure 4 shows the molecular surface of the active site of ETA (1AER), calculated using MOE 2015.10.62 An interaction-potential map, as displayed in Figure 4, provides a representation of where a chemical probe has favorable interactions with a particular receptor. In this context, a “probe” is a united-atom representation of a particular functional group. Different probes were employed to calculate the hydrophilic as well as hydrophobic energies at each grid point.63,64 The available probes include a range of different sizes, charges, and hydrogen bond donor/acceptor properties, such as aromatic CH groups, methyl groups, nitrogen atoms with a lone pair, amide NH and NH2 groups, protonated amine groups (both sp2 and sp3), ether and ester oxygen atoms, carbonyl and carboxylic oxygen atoms, halogen atoms, phosphate groups, water, alkaline ions, and hydroxyl groups (phenolic and alcoholic). As can be seen, hydrophobic contacts are formed where the two Tyr residues and His440 aromatic ring are located. Hydrophilic contacts or solvent-exposed regions occur preferably in the proximity of Glu553.
Figure 4.

(a) Active site of ETA showing the spatial arrangement of the most important amino acid residue (HYYE motif). (b) Surface representation of the same image. Blue areas correspond to solvent-exposed regions, magenta corresponds to hydrophilic regions, and green indicates hydrophobic regions.
Optimization of Docking Parameters
It is well known that one of the critical parameters for proper ligand docking is the size of the box used to search for and identify the lowest-energy binding pose of the ligand.65 The ultimate goal of molecular docking is to predict the correct ligand–receptor interactions, that is, the binding pose, and to determine the binding affinity from that pose. When ligands are drug candidates, compound ranking is the most important task. To obtain as accurate a compound ranking and thus screening results and identification of lead candidates as possible, optimum pose prediction is imperative. This in turn requires a correct docking contact sphere size, that is, the actual active site description used in the docking calculations.
Therefore, we first investigated how the size of the grid affects the accuracy of posing a ligand within the search space (the active site). To this end, known ligands with available crystal structures of the corresponding complexes (Table S1 and Figure S1, Supporting Information) were compared to the complexes obtained from docking studies using box sizes of 8 and 6 Å, respectively, and selecting spheres within those radii. First, the molecular surface of the receptor was generated,66 followed by spheres capturing the topology of the surface.67 These overlapping spheres are used to create a negative image of the surface pocket of the target and are selected within some radius, for example, 8 and 6 Å (Figure S2). As the final grid is generated using the spheres, the sphere centers are matched with the ligand atoms during docking to generate the orientations of the ligand in the active site. Too small a search space (very small radius or number of spheres) may give an incomplete set of conformations, and an excessively large search space may produce a large number of inappropriate binding poses. To this end, benchmarking of molecular docking was performed using two different radii for selecting spheres for each known ligand within its receptor, aiming to maximize the docking accuracy.
For NAD+, the crystal structure of the CT complex was employed (3Q9O). When using an 8 Å radius for selecting spheres, the predicted docked pose was flipped compared with the crystal structure, meaning that the nicotinamide moiety was placed in the region of the adenine ring in the crystal structure and vice versa (Figure 5a). The computed RMSD of the heavy atoms for NAD+ with respect to that for the crystal structure was 9.72, calculated using the Schrödinger graphical interface Maestro.68 On the contrary, when using a smaller sphere-selecting radius (6 Å), the predicted binding pose for NAD+ was highly improved (Figure 5b), also evidenced by an RMSD of 1.90.
Figure 5.

Predicted binding poses for NAD+ in CT are displayed in blue, and the crystal structure conformation is represented in red. (a) Sphere-selecting radius of 8 Å and (b) sphere-selecting radius of 6 Å.
The existence of these two possibilities is coherent with the shape and features of NAD+, having aromatic, hydrophobic residues (nicotinamide and adenine) at the termini and hydrophilic groups (ribose rings and phosphates) in between, and makes the reproducibility of experimental information increasingly important in order to serve as a suitable screening setup.44 The interactions for CT and NAD+ are shown in Figure S3 for both the crystal structure and docked conformation. Hence, the pose (conformation and orientation) was accurately reproduced.
The other 10 known complexes for CT, included in Table S1, were also evaluated using sphere-selecting radii of 8 and 6 Å. In all cases, the best pose prediction was obtained for a 6 Å radius, which was thus the selected docking parameter for further studies. The computed RMSD of the heavy atoms for these 10 ligands docked to CT with respect to that of the crystal structure is reported in Table S2.
For ETA, both β-TAD and PJ34 were computationally docked using both sphere radii selections. For β-TAD, the results are shown in Figure 6.
Figure 6.

Predicted binding poses for β-TAD in ETA are displayed in blue, and the crystal structure conformation is represented in red. (a) Sphere-selecting radius of 8 Å and (b) sphere-selecting radius of 6 Å.
Although the pose is not flipped when using an 8 Å selecting radius, as in the above-mentioned case, the predicted docked conformation is rotated (Figure 6a), probably hampering important hydrophobic interactions with the two Tyr residues belonging to the HYYE motif (RMSD 3.28). Again, using a 6 Å radius, the docked pose was significantly improved, also evidenced by a computed RMSD of 1.74 (Figure 6b). For PJ34, using an 8 Å radius again generated a pose with a larger RMSD with respect to that of the crystal structure than that obtained when using a 6 Å radius (6.12 and 3.86, respectively).
Finally for DT, APU was tested as the ligand for binding-pose reproduction, and the results are shown in Figure 7.
Figure 7.

Predicted binding poses for APU in DT are displayed in blue, and the crystal structure conformation is represented in red. (a) Sphere-selecting radius of 8 Å and (b) sphere-selecting radius of 6 Å.
As for β-TAD, using an 8 Å radius generated a rotated pose for APU with respect to that of the crystal structure (RMSD 2.56), whereas the predicted pose when using a 6 Å radius perfectly reproduced the orientation observed in the crystal structure (RMSD 0.42).
These benchmarking calculations using ligands with known crystal structures of the corresponding complexes assisted in defining the optimum docking parameters, aimed at improving the accuracy of the virtual screening protocol. In particular, for ETA, DT, and CT, it was found that on using a 6 Å radius for selecting docking spheres, the docking poses for known ligands are well-reproduced.
Inverse Docking of Known Binders
After benchmarking the systems to reproduce the docking poses with a set of small molecules against the toxin library, the next step was to estimate the relative affinity of those ligands toward each receptor.
The scoring functions used in the docking programs are useful to compare the different ligands for the same receptor (normal or “forward” docking), but they cannot be directly used to compare one ligand interacting with different receptors (inverse docking) because these in general do not have the same binding pocket shape, protein size, or internal energy. These factors can increase or decrease the docking scores across the targets for all ligands, for example, a deeper binding pocket may lead to larger binding affinities for all ligands and hence a wrong conclusion if this then leads to the selection of that particular target as the “best receptor”.69
Therefore, to prioritize targets for a particular ligand, a post-docking analysis in the form of a normalization or standardization to correct for such biases must be performed.
A normalization of the energy values was suggested by Lauro et al.,70,71 using eq 1, where V is the normalized score for a potential ligand on a receptor, V0 is the predicted docking score obtained in the molecular docking calculation, ML is the average binding energy of the ligand across different targets, and MR is the average binding energy of the receptor for all ligands studied (all entries in kcal mol–1) (Table 1).
| 1 |
V is an absolute number that only shows that the higher the value, the more promising is the interaction between a particular ligand and a target from the panel of toxins. To confirm the validity of the approach, a comparison of the results for some ligands with known binding preferences can be performed (bold entries in Table 1). For instance, it has been described that the affinity of ADP-ribosylating toxins for NAD+ follows the trend DT > ETA ≥ CT.44,72 For PJ34, the known affinity for CT is higher than that observed for ETA.38 Finally, for β-TAD a higher affinity for ETA is expected.46
Table 1. DOCK Grid Scores (V0, kcal mol–1) for Known Ligands against Those of All Three Toxinsa.
| receptor |
|||||||
|---|---|---|---|---|---|---|---|
| ETA |
DT |
CT |
|||||
| V0 | V | V0 | V | V0 | V | ML | |
| ligand | |||||||
| APU | –49.18 | 1.03 | –61.88 | 1.30 | –54.41 | 1.14 | –55.16 |
| GPD | –36.16 | 0.92 | –41.98 | 1.07 | –37.56 | 0.96 | –38.57 |
| GPF | –40.53 | 1.03 | –38.14 | 0.97 | –36.73 | 0.93 | –38.47 |
| GPG | –35.52 | 0.94 | –34.32 | 0.90 | –37.06 | 0.98 | –35.63 |
| GPH | –35.78 | 0.98 | –31.24 | 0.85 | –32.73 | 0.89 | –33.25 |
| GPI | –40.14 | 1.00 | –38.87 | 0.97 | –40.96 | 1.02 | –39.99 |
| GPL | –35.97 | 0.94 | –34.36 | 0.90 | –38.42 | 1.01 | –36.25 |
| GPM | –46.19 | 1.07 | –47.55 | 1.10 | –44.77 | 1.04 | –46.17 |
| GPP | –34.48 | 0.93 | –31.50 | 0.85 | –36.93 | 0.99 | –34.30 |
| NAD+ | –51.79 | 1.14 | –53.19 | 1.17 | –48.19 | 1.06 | –51.06 |
| naphthalimide | –32.34 | 0.90 | –30.69 | 0.85 | –32.35 | 0.90 | –31.79 |
| PJ34 | –35.41 | 0.93 | –36.36 | 0.95 | –37.36 | 0.98 | –36.37 |
| β-TAD | –52.94 | 1.21 | –44.56 | 1.01 | –45.49 | 1.04 | –47.66 |
| V30 | –35.25 | 0.91 | –38.66 | 1.00 | –38.33 | 0.99 | –37.41 |
| MR | –40.12 | –40.24 | –40.09 | ||||
From Table 1, it is clear that the normalization approach correctly identified the target of choice for the selected known ligands. However, the span of V values is rather small, thus making it difficult to perform accurate selection. Also, the method is highly dependent on the particular set of molecules included, affecting MR. Therefore, even if the trends are correct using the Lauro normalization, a different approach might lead to more readily interpretable results.
A different correction scheme from that used above is the so-called multiple active site correction (MASC), suggested by Vigers and Rizzi,73 shown in the following equations (eqs 2–4)
| 2 |
| 3 |
| 4 |
where Sij is the original calculated docking score for the ith compound and jth pocket (in kcal mol–1) and Sij′ is the modified score for compound i in active site j. μi and σi are the average and standard deviations of the scores for compound i across all pockets j, respectively (Table 2).
Table 2. DOCK Grid Scores (Sij, kcal mol–1) for Known Ligands against Those of All Three Toxinsa.
| receptor |
||||||||
|---|---|---|---|---|---|---|---|---|
| ETA |
DT |
CT |
||||||
| ligand | Sij | Sij′ | Sij | Sij′ | Sij | Sij′ | μi | (σi)2 |
| APU | –49.18 | 0.94 | –61.88 | –1.05 | –54.41 | 0.12 | –55.16 | 6.38 |
| GPD | –36.16 | 0.79 | –41.98 | –1.12 | –37.56 | 0.33 | –38.57 | 3.04 |
| GPF | –40.53 | –1.07 | –38.14 | 0.17 | –36.73 | 0.91 | –38.47 | 1.92 |
| GPG | –35.52 | 0.08 | –34.32 | 0.96 | –37.06 | –1.04 | –35.63 | 1.37 |
| GPH | –35.78 | –1.09 | –31.24 | 0.87 | –32.73 | 0.22 | –33.25 | 2.31 |
| GPI | –40.14 | –0.15 | –38.87 | 1.06 | –40.96 | –0.92 | –39.99 | 1.05 |
| GPL | –35.97 | 0.14 | –34.36 | 0.92 | –38.42 | –1.06 | –36.25 | 2.04 |
| GPM | –46.19 | –0.02 | –47.55 | –0.99 | –44.77 | 1.01 | –46.17 | 1.39 |
| GPP | –34.48 | –0.06 | –31.50 | 1.03 | –36.93 | –0.97 | –34.30 | 2.72 |
| NAD+ | –51.79 | –0.28 | –53.19 | –0.83 | –48.19 | 1.11 | –51.06 | 2.58 |
| naphthalimide | –32.34 | –0.57 | –30.69 | 1.15 | –32.35 | –0.58 | –31.79 | 0.95 |
| PJ34 | –35.41 | 0.99 | –36.36 | 0.02 | –37.36 | –1.01 | –36.37 | 0.97 |
| β-TAD | –52.94 | –1.15 | –44.56 | 0.68 | –45.49 | 0.47 | –47.66 | 4.59 |
| V30 | –35.25 | 1.15 | –38.66 | –0.66 | –38.33 | –0.49 | –37.41 | 1.88 |
The MASC score is useful in that it includes in the sign information about how far apart a value is from the average and in which direction. In this particular application, if an MASC score is negative, it means that the score for the ligand in that particular receptor is better (more negative) than the average among all targets. Again, the trends for the known binders were accurately reproduced, for example, NAD+ scored negatively for ETA and DT (higher affinity), whereas the MASC score for CT was positive (lower affinity). For PJ34, CT stands out as the best binding target, and for β-TAD, it is ETA, in accord with the experimental data. Thus, our inverse-docking calculations on ADP-ribosylating toxins against known ligands allowed for a clear identification of the target of choice.
The results show that the MASC score works well when calculated for a set of known ligands and suggest that the methodology can be used to predict, on one hand the “best receptor” for a particular potential ligand and, on the other hand a multitarget compound if all receptors obtain similar MASC scores.
Conclusions
This study was undertaken to find the best docking settings to perform an inverse-docking study on three different ADP-ribosylating toxins. First, we compared the active sites of ETA, DT, and CT and found the optimum docking box size for reproducing binding poses of the natural substrate (NAD+) and 13 other ligands with available crystal structures. In addition, we carried out molecular docking experiments against all three toxins and tested two different corrections of the scoring functions aimed at target-fishing the best receptor for each ligand.
We have constructed a ready-for-dock toxin library, prepared to run inverse virtual screening with different databases to identify inhibitor candidates for all toxins. Hence, these results could serve as the starting point for developing potential antibiotics targeting three different toxins from harmful pathogens, that is, P. aeruginosa, C. diphtheriae, and V. cholerae. As the receptors are available and prepared, any small-molecule database can be employed for inverse-docking studies, using the MASC score to detect potential selective or multitarget ligands.
However, we emphasize that the scheme presented herein is completely general. After setting up and benchmarking a set of target receptors, be it a family of related proteins, such as kinases, or a wide set of diverse receptors where binding may result in adverse side effects, screening and MASC normalization will enable the user to identify selective as well as broad-hitting binders.
Methodology
Protein Preparation
The protein crystal structures of ETA (PDB id 1AER(46)), DT (PDB id 1DTP(74)), and CT (PDB id 2Q6M(38)) were retrieved from the Protein Data Bank.75
All molecular modelings were performed using the University of California San Francisco (UCSF) DOCK (version 6.7).76,77 Structure preparation prior to docking was performed using UCSF Chimera.78
Each protein structure was processed according to the usual recommended protocol in DOCK.77 Ligands were removed and the “Dock prep” Chimera tool was employed. Hydrogen atoms were added to generate the protonation states at physiological pH. Charges for standard residues were calculated from Amber ff14SB,79 and histidine side chains were protonated according to their local environment. The molecular surface for each structure was calculated using the chimera tool “DMS”. The corresponding box was created using the program “showbox”, and the final grid was calculated using the “grid” program suite, both included in DOCK. The sizes of the grid and selected spheres were carefully selected in order to reproduce the structures and poses of known crystallized ligands (see the Results and Discussion section).
Ligand Selection and Preparation
The 3D structure of the natural substrate, NAD+, was determined from the CT–NAD+ complex (PDB id 3Q9O(44)), whereas the structure of an analogue of the natural substrate, β-TAD, was obtained from the complex with ETA (PDB id 1AER(46)). Several inhibitors were also retrieved from the crystal structures of their corresponding toxin complexes: PJ34 (PDB id 1XK9(80)), APU (PDB id 1DTP(74)), 1,8-naphthalimide (PDB id 3ESS(81)), V30 (PDB id 3NY6(82)), GPD (PDB id 3KI0(82)), GPF (PDB id 3KI1(82)), GPG (PDB id 3KI2(82)), GPH (PDB id 3KI3(82)), GPP (PDB id 3KI4(82)), GPM (PDB id 3KI5(82)), GPL (PDB id 3KI6(82)), and GPI (PDB id 3KI7(82)) (Table S1 and Figure S1). For all ligands, protonation states at pH 7 were assigned using the chimera tool “AddH” and charges were assigned based on AM1-BCC calculations (“Add Charge” Chimera tool).
Molecular Docking
A grid-based scoring function was used for docking, considering each receptor as rigid and sampling ligand torsion angles during energy-minimization procedures.83,84 A maximum of 500 orientations were sampled for each ligand, they were energy-minimized (score optimization), and the top scored pose was then kept. The scores corresponding to each protein for a single ligand were saved for further analysis. Two normalization or standardization approaches were implemented to enable the comparison among docking scores from different receptors (see the Results and Discussion section).
Acknowledgments
P.S.-M. received funding from the People Program (Marie Curie Actions) of the European Union’s Seventh Framework Program (FP7/2007-2013) under REA grant agreement no 608743. L.A.E. gratefully acknowledges funding from the Swedish Research Council and the Faculty of Science at the University of Gothenburg. We also acknowledge the generous allocation of computing time at the C3SE supercomputing center via a grant from the Swedish National Infrastructure for Computing (SNIC).
Glossary
Abbreviations
- ETA
exotoxin A
- DT
diphtheria toxin
- CT
cholix toxin
- eEF2
eukaryotic elongation factor 2
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b00010.
Known ligands selected (crystal structure references, chemical structures, and RMSD data), selected spheres for ETA, and ligand interactions between CT and NAD+ (PDF)
Author Contributions
P.S.-M., M.E., and L.A.E. conceived and designed the study; P.S.-M. performed the computations and analyzed the data; and P.S.-M., M.E., and L.A.E. contributed to the writing of the paper.
The authors declare no competing financial interest.
Supplementary Material
References
- Antimicrobial Resistance: Global Report on Surveillance; World Health Organization, 2014. [Google Scholar]
- Antibiotic resistance, fact sheet, October 2015. http://www.who.int/mediacentre/factsheets/antibiotic-resistance/en/.
- Centers for Disease Control and Prevention (CDC). Antibiotic/Antimicrobial Resistance. https://www.cdc.gov/drugresistance/Antibiotic/AntimicrobialResistance.
- Worldwide Country Situation Analysis: Response to Antimicrobial Resistance; World Health Organization, 2015. [Google Scholar]
- Shanson D. C. Septicaemia in patients with AIDS. Trans. R. Soc. Trop. Med. Hyg. 1990, 84, 14–16. 10.1016/0035-9203(90)90449-o. [DOI] [PubMed] [Google Scholar]
- Elkin S.; Geddes D. Pseudomonal infection in cystic fibrosis: the battle continues. Expert Rev. Anticancer Ther. 2003, 1, 609–618. 10.1586/14787210.1.4.609. [DOI] [PubMed] [Google Scholar]
- Stover C. K.; Pham X. Q.; Erwin A. L.; Mizoguchi S. D.; Warrener P.; Hickey M. J.; Brinkman F. S.; Hufnagle W. O.; Kowalik D. J.; Lagrou M.; Garber R. L.; Goltry L.; Tolentino E.; Westbrock-Wadman S.; Yuan Y.; Brody L. L.; Coulter S. N.; Folger K. R.; Kas A.; Larbig K.; Lim R.; Smith K.; Spencer D.; Wong G. K.; Wu Z.; Paulsen I. T.; Reizer J.; Saier M. H.; Hancock R. E.; Lory S.; Olson M. V. Complete genome sequence of Pseudomonas aeruginosa PA01, an opportunistic pathogen. Nature 2000, 406, 959–964. 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- Tokajian S.; Timani R.; Issa N.; Araj G. Molecular characterization, multiple drug resistance, and virulence determinants of Pseudomonas aeruginosa isolated from Lebanon. Br. Microbiol. Res. J. 2012, 2, 243–250. 10.9734/BMRJ/2012/2217. [DOI] [Google Scholar]
- Gellatly S. L.; Hancock R. E. Pseudomonas aeruginosa: new insights into pathogenesis and host defenses. Pathog. Dis. 2013, 67, 159–173. 10.1111/2049-632X.12033. [DOI] [PubMed] [Google Scholar]
- Lambert P. A. Mechanisms of antibiotic resistance in Pseudomonas aeruginosa. J. R. Soc. Med. 2002, 95, 22–26. [PMC free article] [PubMed] [Google Scholar]
- Iglewski B. H.; Sadoff J. C. Toxin inhibitors of protein synthesis: production, purification, and assay of Pseudomonas aeruginosa toxin A. Methods Enzymol. 1979, 60, 780–793. 10.1016/S0076-6879(79)60071-X. [DOI] [PubMed] [Google Scholar]
- Wick M. J.; Frank D. W.; Storey D. G; Iglewski B. H. Structure, function, and regulation of Pseudomonas aeruginosa exotoxin A. Annu. Rev. Microbiol. 1990, 44, 335–363. 10.1146/annurev.mi.44.100190.002003. [DOI] [PubMed] [Google Scholar]
- Lyczak J. B.; Cannon C. L.; Pier G. B. Establishment of Pseudomonas aeruginosa infection: lessons from a versatile opportunist. Microbes Infect. 2000, 2, 1051–1060. 10.1016/S1286-4579(00)01259-4. [DOI] [PubMed] [Google Scholar]
- Liu P. V. The roles of various fractions of Pseudomonas aeruginosa in its pathogenesis - 3: identity of the lethal toxins produced in vitro and in vivo. J. Infect. Dis. 1966, 116, 481–489. 10.1093/infdis/116.4.481. [DOI] [PubMed] [Google Scholar]
- Collier R. J. Understanding the mode of action of diphtheria toxin: a perspective on progress during the 20th century. Toxicon 2001, 39, 1793–1803. 10.1016/S0041-0101(01)00165-9. [DOI] [PubMed] [Google Scholar]
- Fitzgerald D.; Morris R. E.; Saelinger C. B. Receptor-mediated internalization of Pseudomonas toxin by mouse fibroblasts. Cell 1980, 21, 867–873. 10.1016/0092-8674(80)90450-X. [DOI] [PubMed] [Google Scholar]
- Andersen G. R.; Nissen P.; Nyborg J. Elongation factors in protein biosynthesis. Trends Biochem. Sci. 2003, 28, 434–441. 10.1016/S0968-0004(03)00162-2. [DOI] [PubMed] [Google Scholar]
- Jørgensen R.; Yates S. P.; Teal D. J.; Nilsson J.; Prentice G. A.; Rod Merrill A.; Andersen G. R. Crystal structure of ADP-ribosylated ribosomal translocase from Saccharomyces cerevisiae. J. Biol. Chem. 2004, 279, 45919–45925. 10.1074/jbc.M406218200. [DOI] [PubMed] [Google Scholar]
- Moehring J. M.; Moehring T. J. The post-translational trimethylation of diphthamide studied in vitro. J. Biol. Chem. 1988, 263, 3840–3844. [PubMed] [Google Scholar]
- Liu S.; Bachran C.; Gupta P.; Miller-Randolph S.; Wang H.; Crown D.; Zhang Y.; Wein A. N.; Singh R.; Fattah R.; Leppla S. H. Diphthamide modification on eukaryotic elongation factor 2 is needed to assure fidelity of mRNA translation and mouse development. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 13817–13822. 10.1073/pnas.1206933109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz P. A.; Ulloque R.; Kihara G. K.; Zheng H.; Kinzy T. G. Translation elongation factor 2 anticodon mimicry domain mutants affect fidelity and diphtheria toxin resistance. J. Biol. Chem. 2006, 281, 32639–32648. 10.1074/jbc.M607076200. [DOI] [PubMed] [Google Scholar]
- Schaffrath R.; Stark M. J. R. Decoding the biosynthesis and function of diphthamide, an enigmatic modification of translation elongation factor 2 (EF2). Microb. Cell Fact. 2014, 1, 203–205. 10.15698/mic2014.06.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon N. C.; Aktories K.; Barbieri J. T. Novel bacterial ADP-ribosylating toxins: structure and function. Nat. Rev. Microbiol. 2014, 12, 599–611. 10.1038/nrmicro3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautman P.; Gupta P.; Sharma P.; Dangwal P. Virtuous aspects of vicious bacterial toxins. J. Chem. Pharm. Res. 2015, 8, 279–289. [Google Scholar]
- Oppenheimer N. J.; Bodley J. W. Diphtheria Toxin. Site and configuration of ADP-ribosylation of diphthamide in elongation factor 2. J. Biol. Chem. 1981, 256, 8579–8581. [PubMed] [Google Scholar]
- Yates S. P.; Jørgensen R.; Andersen G. R.; Merrill A. R. Stealth and mimicry by deadly bacterial toxins. Trends Biochem. Sci. 2006, 31, 123–133. 10.1016/j.tibs.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Iglewski B. H.; Liu P. V.; Kabat B. Mechanism of action of Pseudomonas aeruginosa exotoxin A: adenosine diphosphate-ribosylation of mammalian elongation factor 2 in vitro and in vivo. Infect. Immun. 1977, 15, 138–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong S.; Merrill A. R. Toward the elucidation of the catalytic mechanism of the mono-ADP-ribosyltransferase activity of Pseudomonas aeruginosa exotoxin A. Biochemistry 2004, 43, 183–194. 10.1021/bi034772u. [DOI] [PubMed] [Google Scholar]
- Armstrong S.; Yates S. P.; Merrill A. R. Insight into the catalytic mechanism of Pseudomonas aeruginosa exotoxin A. Studies of toxin interaction with eukaryotic elongation factor-2. J. Biol. Chem. 2002, 277, 46669–46675. 10.1074/jbc.M206916200. [DOI] [PubMed] [Google Scholar]
- Jørgensen R.; Merrill A. R.; Yates S. P.; Marquez V. E.; Schwan A. L.; Boesen T.; Andersen G. R. Exotoxin A–eEF2 complex structure indicates ADP ribosylation by ribosome mimicry. Nature 2005, 436, 979–984. 10.1038/nature03871. [DOI] [PubMed] [Google Scholar]
- Parik S. L.; Schramm V. L. Transition state structure for ADP-ribosylation of eukaryotic elongation factor 2 catalyzed by diphtheria toxin. Biochemistry 2004, 43, 1204–1212. 10.1021/bi035907z. [DOI] [PubMed] [Google Scholar]
- Van Ness B. G.; Howard J. B.; Bodley J. W. ADP-Ribosylation of Elongation Factor 2 by Diphtheria Toxin. NMR spectra and proposed structures of ribosyl-diphthamide and its hydrolysis products. J. Biol. Chem. 1980, 255, 10710–10716. [PubMed] [Google Scholar]
- Van Ness B. G.; Howard J. B.; Bodley J. W. ADP-ribosylation of elongation factor 2 by Diphtheria toxin. Isolation and properties of the novel ribosyl-amino acid and its hydrolysis products. J. Biol. Chem. 1980, 255, 10717–10720. [PubMed] [Google Scholar]
- Sitikov A. S.; Davydova E. K.; Bezlepkina T. A.; Ovchinnikov L. P.; Spirin A. S. Eukaryotic elongation factor 2 loses its non-specific affinity for RNA and leaves polyribosomes as a result of ADP-ribosylation. FEBS Lett. 1984, 176, 406–410. 10.1016/0014-5793(84)81207-7. [DOI] [PubMed] [Google Scholar]
- Choe S.; Bennet M. J.; Fujii G.; Curmi P. M. G.; Kantardjieff K. A.; Collier R. J.; Eisenberg D. The crystal structure of diphtheria toxin. Nature 1992, 357, 216–222. 10.1038/357216a0. [DOI] [PubMed] [Google Scholar]
- Holmes R. K. Biology and molecular epidemiology of diphtheria toxin and the tox gene. J. Infect. Dis. 2000, 181, S156–S167. 10.1086/315554. [DOI] [PubMed] [Google Scholar]
- Murphy J. R. Mechanism of diphtheria toxin catalytic domain delivery to the eukaryotic cell cytosol and the cellular factors that directly participate in the process. Toxins 2011, 3, 294–308. 10.3390/toxins3030294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jørgensen R.; Purdy A. E.; Fieldhouse R. J.; Kimber M. S.; Bartlett D. H.; Merrill A. R. Cholix toxin, a novel ADP-ribosylating factor from Vibrio cholerae. J. Biol. Chem. 2008, 283, 10671–10678. 10.1074/jbc.M710008200. [DOI] [PubMed] [Google Scholar]
- Carroll S. F.; Collier R. J. NAD binding site of diphtheria toxin: identification of a residue within the nicotinamide subsite by photochemical modification with NAD. Proc. Natl. Acad. Sci. U.S.A. 1984, 81, 3307–3311. 10.1073/pnas.81.11.3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll S. F.; Collier R. J. Active site of Pseudomonas aeruginosa exotoxin A. Glutamic acid 553 is photolabeled by NAD and shows functional homology with glutamic acid 148 of diphtheria toxin. J. Biol. Chem. 1987, 262, 8707–8711. [PubMed] [Google Scholar]
- Altaee M. F.; Nafee S. K.; Hamza S. J. Evaluation for the cytotoxic Effect of exotoxin a produced by Pseudomonas aeruginosa on mice by using cytogenetic parameters. Curr. Microbiol. 2013, 1, 257–261. [Google Scholar]
- Allured V. S.; Collier R. J.; Carroll S. F.; McKay D. B. Structure of exotoxin A of Pseudomonas aeruginosa at 3.0-Angstrom resolution. Proc. Natl. Acad. Sci. U.S.A. 1986, 83, 1320–1324. 10.1073/pnas.83.5.1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M.; Dyda F.; Benhar I.; Pastan I.; Davies D. R. The crystal structure of Pseudomonas aeruginosa exotoxin domain III with nicotinamide and AMP: Conformational differences with the intact toxin. Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 9308–9312. 10.1073/pnas.92.20.9308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fieldhouse R. J.; Jørgensen R.; Lugo M. R.; Merrill A. R. The 1.8 A Cholix Toxin crystal structure in complex with NAD+ and evidence for a new kinetic model. J. Biol. Chem. 2012, 287, 21176–21188. 10.1074/jbc.M111.337311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugo M. R.; Merrill A. R. The father, son and cholix toxin: the third member of the DT group mono-ADP-ribosyltransferase toxin family. Toxins 2015, 7, 2757–2772. 10.3390/toxins7082757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M.; Dyda F.; Benhar I.; Pastan I.; Davies D. R. Crystal structure of the catalytic domain of Pseudomonas exotoxin A complexed with a nicotinamide adenine dinucleotide analog: Implications for the activation process and for ADP ribosylation. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 6902–6906. 10.1073/pnas.93.14.6902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell C. E.; Eisenberg D. Crystal structure of Diphtheria toxin bound to nicotinamide adenine dinucleotide. Biochemistry 1996, 35, 1137–1149. 10.1021/bi9520848. [DOI] [PubMed] [Google Scholar]
- Blanke S. R.; Huang K.; Wilson B. A.; Papini E.; Covacci A.; Collier R. J. Active-site mutations of the diphtheria toxin catalytic domain: role of histidine-21 in nicotinamide adenine dinucleotide binding and ADP-Ribosylation of Elongation Factor 2. Biochemistry 1994, 33, 5155–5161. 10.1021/bi00183a019. [DOI] [PubMed] [Google Scholar]
- Lukac M.; Collier R. J. Pseudomonas aeruginosa exotoxin A: effects of mutating tyrosine-470 and tyrosine-481 to phenylalanine. Biochemistry 1988, 27, 7629–7632. 10.1021/bi00420a009. [DOI] [PubMed] [Google Scholar]
- Ehrlich P.Beiträge zur theorie und praxis der histologischen färbung; Leipzig University, 1878. [Google Scholar]
- Ehrlich P. Die wertbemessung des diphterieheilserums und deren theoretische grundlagen. Klin. Jahrb. 1897, 6, 299–326. [Google Scholar]
- Strebhardt K.; Ullrich A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat. Rev. Cancer 2008, 8, 473–480. 10.1038/nrc2394. [DOI] [PubMed] [Google Scholar]
- Medina-Franco J. L.; Giulianotti M. A.; Welmaker G. S.; Houghten R. A. Shifting from the single to the multitarget paradigm in drug discovery. Drug Discovery Today 2013, 18, 495–501. 10.1016/j.drudis.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth B. L.; Sheffler D. J.; Kroeze W. K. Magic shotguns versus magic bullets: selectively non-selective drugs for mood disorders and schizophrenia. Nat. Rev. Drug Discovery 2004, 3, 353–359. 10.1038/nrd1346. [DOI] [PubMed] [Google Scholar]
- Zimmermann G. R.; Lehar J.; Keith C. T. Multi-target therapeutics: when the whole is greater than the sum of the parts. Drug Discovery Today 2007, 12, 34–42. 10.1016/j.drudis.2006.11.008. [DOI] [PubMed] [Google Scholar]
- Chen Y. Z.; Zhi D. G. Ligand–protein inverse docking and Its potential use in the computer search of protein targets of a small molecule. Proteins 2001, 43, 217–226. . [DOI] [PubMed] [Google Scholar]
- Grinter S. Z.; Liang Y.; Huang S. Y.; Hyder S. M.; Zou X. An inverse docking approach for identifying new potential anti-cancer targets. J. Mol. Graphics Modell. 2011, 29, 795–799. 10.1016/j.jmgm.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahler S.; Tietze S.; Totzke F.; Kubbutat M.; Meijer L.; Vollmar A. M.; Apostolakis J. Inverse in silico screening for identification of kinase inhibitor targets. Chem. Biol. 2007, 14, 1207–1214. 10.1016/j.chembiol.2007.10.010. [DOI] [PubMed] [Google Scholar]
- Wang W.; Zhou X.; He W.; Fan Y.; Chen Y.; Chen X. The interprotein scoring noises in glide docking scores. Proteins 2012, 80, 169–183. 10.1002/prot.23173. [DOI] [PubMed] [Google Scholar]
- Xie L.; Xie L.; Bourne P. E. Structure-based systems biology for analyzing off-target binding. Curr. Opin. Struct. Biol. 2011, 21, 189–199. 10.1016/j.sbi.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erić S.; Ke S.; Barata T.; Solmajer T.; Antic Stankovic J.; Juranic Z.; Savic V.; Zloh M. Target fishing and docking studies of the novel derivatives of aryl-aminopyridines with potential anticancer activity. Bioorg. Med. Chem. 2012, 20, 5220–5228. 10.1016/j.bmc.2012.06.051. [DOI] [PubMed] [Google Scholar]
- Molecular Operating Environment (MOE); Chemical Computing Group: Montreal, Canada, 2015. [Google Scholar]
- Goodford P. J. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J. Med. Chem. 1985, 28, 849–857. 10.1021/jm00145a002. [DOI] [PubMed] [Google Scholar]
- Boobbyer D. N. A.; Goodford P. J.; McWhinnie P. M.; Wade R. C. New hydrogen-bond potentials for use in determining energetically favorable binding sites on molecules of known structure. J. Med. Chem. 1989, 32, 1083–1094. 10.1021/jm00125a025. [DOI] [PubMed] [Google Scholar]
- Feinstein W. P.; Brylinski M. Calculating an optimal box size for ligand docking and virtual screening against experimental and predicted binding pockets. J. Cheminf. 2015, 7, 18. 10.1186/s13321-015-0067-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards F. M. Areas, volumes, packing, and protein structure. Annu. Rev. Biophys. Bioeng. 1977, 6, 151–176. 10.1146/annurev.bb.06.060177.001055. [DOI] [PubMed] [Google Scholar]
- Kuntz I. D.; Blaney J. M.; Oatley S. J.; Langridge R.; Ferrin T. E. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982, 161, 269–288. 10.1016/0022-2836(82)90153-X. [DOI] [PubMed] [Google Scholar]
- Maestro, version 10.3; Schrödinger, LLC: New York, 2016.
- Casey F. P.; Pihan E.; Shields D. C. Discovery of small molecule inhibitors of protein-protein interactions using combined ligand and target score normalization. J. Chem. Inf. Model. 2009, 49, 2708–2717. 10.1021/ci900294x. [DOI] [PubMed] [Google Scholar]
- Lauro G.; Romano A.; Riccio R.; Bifulco G. Inverse virtual screening of antitumor targets: pilot study on a small database of natural bioactive compounds. J. Nat. Prod. 2011, 74, 1401–1407. 10.1021/np100935s. [DOI] [PubMed] [Google Scholar]
- Lauro G.; Masullo M.; Piacente S.; Riccio R.; Bifulco G. Inverse Virtual Screening allows the discovery of the biological activity of natural compounds. Bioorg. Med. Chem. 2012, 20, 3596–3602. 10.1016/j.bmc.2012.03.072. [DOI] [PubMed] [Google Scholar]
- Yates S. P.; Merrill A. R. A catalytic loop within Pseudomonas aeruginosa exotoxin A modulates its transferase activity. J. Biol. Chem. 2001, 276, 35029–35036. 10.1074/jbc.M105002200. [DOI] [PubMed] [Google Scholar]
- Vigers G. P. A.; Rizzi J. P. Multiple active site corrections for docking and virtual screening. J. Med. Chem. 2004, 47, 80–89. 10.1021/jm030161o. [DOI] [PubMed] [Google Scholar]
- Weiss M. S.; Blanke S. R.; Collier R. J.; Eisenberg D. Structure of the isolated catalytic domain of Diphtheria toxin. Biochemistry 1995, 34, 773–781. 10.1021/bi00003a010. [DOI] [PubMed] [Google Scholar]
- Protein Data Bank. http://www.rcsb.org/pdb/home/home.do.
- DOCK 6.7; University of California: San Francisco, 2015. [Google Scholar]
- Lang P. T.; Brozell S. R.; Mukherjee S.; Pettersen E. F.; Meng E. C.; Thomas V.; Rizzo R. C.; Case D. A.; James T. L.; Kuntz I. D. DOCK 6: combining techniques to model RNA-small molecule complexes. RNA 2009, 15, 1219–1230. 10.1261/rna.1563609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Maier J. A.; Martinez C.; Kasavajhala K.; Wickstrom L.; Hauser K. E.; Simmerling C. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates S. P.; Taylor P. J.; Jøergensen R.; Ferraris D.; Zhang J.; Andersen G. R.; Merrill A. R. Structure-function analysis of water-soluble inhibitors of the catalytic domain of exotoxin A from Pseudomonas aeruginosa. Biochem. J. 2005, 385, 667–675. 10.1042/BJ20041480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turgeon Z.; White D.; Jørgensen R.; Visschedyk D.; Fieldhouse R. J.; Mangroo D.; Merrill A. R. Yeast as a tool for characterizing mono-ADP-ribosyltransferase toxins. FEMS Microbiol. Lett. 2009, 300, 97–106. 10.1111/j.1574-6968.2009.01777.x. [DOI] [PubMed] [Google Scholar]
- Turgeon Z.; Jørgensen R.; Visschedyk D.; Edwards P. R.; Legree S.; McGregor C.; Fieldhouse R. J.; Mangroo D.; Schapira M.; Merrill A. R. Newly discovered and characterized antivirulence compounds inhibit bacterial mono-ADP-ribosyltransferase toxins. Antimicrob. Agents Chemother. 2011, 55, 983–991. 10.1128/AAC.01164-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.; Cui T.; Hu L.; Wang Z.; Li Z.; He Z. G. Cyclic diguanylate monophosphate directly binds to human siderocalin and inhibits its antibacterial activity. Nat. Commun. 2015, 6, 8330 10.1038/ncomms9330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S.; Balius T. E.; Rizzo R. C. Docking validation resources: protein family and ligand flexibility experiments. J. Chem. Inf. Model. 2010, 50, 1986–2000. 10.1021/ci1001982. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.