

Abstract
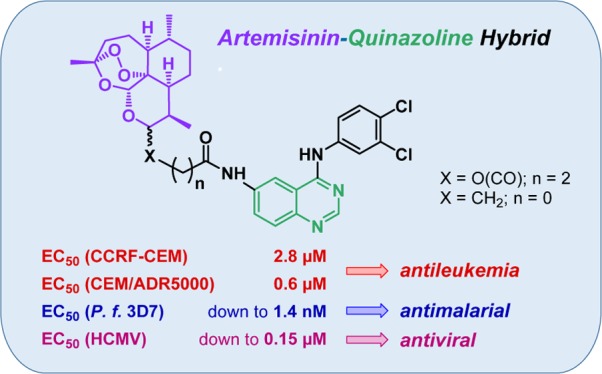
Many quinazoline derivatives have been synthesized over the last few decades with great pharmacological potential, such as antimalarial, anti-inflammatory, antimicrobial, anticancer, and antiviral. But so far, no quinazoline–artemisinin hybrids have been reported in the literature. In the present study, five novel quinazoline–artemisinin hybrids were synthesized and evaluated for their in vitro biological activity against malarial parasites (Plasmodium falciparum 3D7), leukemia cells (CCRF-CEM and CEM/ADR5000), and human cytomegalovirus. Remarkably, hybrid 9 (EC50 = 1.4 nM), the most active antimalarial compound of this study, was not only more potent than artesunic acid (EC50 = 9.7 nM) but at the same time more active than the clinically used drugs dihydroartemisinin (EC50 = 2.4 nM) and chloroquine (EC50 = 9.8 nM). Furthermore, hybrids 9 and 10 were the most potent compounds with regard to anticytomegaloviral activity (EC50 = 0.15–0.21 μM). They were able to outperform ganciclovir (EC50 = 2.6 μM), which is the relevant standard drug of antiviral therapy, by a factor of 12–17. Moreover, we identified a new highly active quinazoline derivative, compound 14, that is most effective in suppressing cytomegalovirus replication with an EC50 value in the nanomolar range (EC50 = 50 nM). In addition, hybrid 9 exhibited an antileukemia effect similar to that of artesunic acid, with EC50 values in the low micromolar range, and was 45 times more active toward the multidrug-resistant CEM/ADR5000 cells (EC50 = 0.5 μM) than the standard drug doxorubicin.
1. Introduction
The quinazoline scaffold, which consists of two fused six-membered aromatic rings benzene and pyrimidine, plays a fundamental role in today’s discovery and design of new drugs due to its wide-ranging pharmacological properties, such as antimicrobial,1−3 antimalarial,4,5 anti-inflammatory,6−8 anticancer,9−11 and antiviral.12,13 One of the first quinazoline derivatives, which was found to have very promising biological efficacy, was the natural occurring alkaloid febrifugine (1) (Figure 1). The compound was isolated 60 years ago from the Chinese plant aseru (Dichroa febrifuga Lour), which has been applied as an antimalarial agent in traditional Chinese medicine for over 2000 years.14−16 Since the discovery of febrifugine (1), the quinazoline moiety became one of the most studied motifs in medicinal chemistry, and as a consequence, a great variety of quinazoline derivatives exhibiting diverse biological activities have been synthesized in the past few decades.17−21 Selected examples are displayed in Figure 1: gefitinib (2a), an epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI), which is already used clinically for the treatment of non-small-cell lung carcinoma;22,23 lapatinib (2b), an orally active drug against breast cancer;24 and two other synthetic 4-anilinoquinazoline derivatives: compound 3, which possesses anticytomegaloviral activity,13 and compound 4, which is effective even in vivo against malarial parasites.5
Figure 1.

Biologically active quinazoline derivatives: febrifugine (1) (antimalarial), gefitinib (2a), lapatinib (2b) (anticancer), and two other synthetic 4-anilinoquinazoline derivatives 3 (anti-human cytomegalovirus (anti-HCMV)) and 4 (antimalarial), as well as the structure of artemisinin (5).
To enhance the pharmacological properties of the available quinazoline drugs and to counter the spread of resistance, especially toward malaria and cancer, we intended to apply the so-called molecular hybridization, which is a relatively new concept in drug design and development, wherein at least two different pharmacophoric moieties originating from different bioactive substances are combined into one single structure.25−28 The resulting hybrids have, in many cases, improved pharmacological properties compared to those of their parent drugs, such as increased biological efficacy, reduced undesired side effects, a modified selectivity profile (lower toxicity), better bioavailability, and sometimes even completely new biological features that were absent in the parent compounds.28−32 The second pharmacologically active substance we chose besides quinazoline for the hybridization was artemisinin (5), which is an enantiomerically pure sesquiterpene with a 1,2,4-trioxane heterocycle and was first isolated in 1972 from the Chinese medicinal plant Artemisia annua L. by Youyou Tu (Nobel Prize 2015).33 Artemisinin was selected by us not only because of its known antimalarial34−37 and anticancer potential38,39 but also due to our promising experiences in the past with artemisinin-based hybrids.40−44
In this study, we report the synthesis of five novel quinazoline–artemisinin hybrids 6–10 (Figure 2), which were investigated for their inhibitory potency against the malarial parasite Plasmodium falciparum 3D7 strain, the leukemia cell lines CCRF-CEM and CEM/ADR5000, and HCMV.
Figure 2.

Hybrids 6–10 applied in this work for biological tests against P. falciparum 3D7 parasites, HCMV, and leukemia cell lines CCRF-CEM and CEM/ADR5000.
2. Results and Discussion
2.1. Chemistry
Quinazoline derivatives 11, 12, and 14 (Figure 3, Scheme 1), which were necessary for the synthesis of hybrids 6−10 (Scheme 2), were prepared in analogy to a literature-known procedure developed by Wang et al. in 2014.45 Applying the Steglich esterification reaction, it was possible to couple artesunic acid (16), a semisynthetic derivative of artemisinin (5), directly with quinazoline derivative 11 containing a phenol moiety, yielding the desired hybrid 6 in 60% (Scheme 2). The reaction was performed using N,N′-dicyclohexylcarbodiimide (DCC) and 4-(dimethylamino)-pyridine (DMAP) as coupling agents and a mixture of CH3CN and ethyl acetate (EtOAc) as the solvent.
Figure 3.

Precursors for hybrids presented in this work.
Scheme 1. Synthesis of Quinazoline-Derived Precursors 13 and 15.

Scheme 2. Synthesis Route for Hybrids 6–10.

To be able to synthesize the other quinazoline–artemisinin hybrids, 7–10 (Scheme 2), further modifications of quinazoline derivatives 12 and 14 were required (Scheme 1). Quinazoline 12 was converted into an alcohol, 13, by N-alkylation of the secondary amine using 3-bromopropanol as the reagent, K2CO3 as the base, TBAI as the additive, and CH3CN as the solvent. After refluxing the reaction mixture overnight, the product could be isolated in 22% yield. Conversion of the aromatic nitro group of new quinazoline 14 to the corresponding primary amine was realized by hydrazine monohydrate-mediated reduction catalyzed by Raney-Ni.
This procedure, which is already reported in the literature in context of the synthesis of other simple aromatic amino compounds,46 afforded quinazoline amine 15 in 87% yield. These two quinazoline precursors, 13 and 15, were then coupled with either artesunic acid (16) or artemisinin-derived carboxylic acid 17 to furnish hybrids 7–10: Steglich conditions (DCC, DMAP) afforded ester hybrids 7/8 in 35/49% yield, and standard amide coupling (EDCI/1-hydroxybenzotriazole (HOBt), DIPEA) gave amide hybrids 9/10 in 64/68% yield. Artemisinin-derived acid 17 was synthesized from dihydroartemisinin (DHA) according to the literature (Scheme 2)47 and was chosen as the starting material for the hybrid synthesis, as it belongs to the group of C-10 nonacetals, which are known to be more hydrolytically stable than common artemisinin derivatives, such as artesunic acid (16).48
The stability of all our new target compounds 6–10 was tested by heating them for 20 h at 60 °C. After utilizing these conditions, less than 5% decomposition was observed via 1H NMR spectroscopy, indicating that all synthesized hybrids are sufficiently stable.
2.2. Biological Evaluation and Discussion
2.2.1. Cytotoxicity toward P. falciparum 3D7
The antimalarial activity of all synthesized hybrids 6–10, as well as their parent compounds artesunic acid (16) and quinazoline derivatives 12 and 14, was evaluated in vitro against P. falciparum 3D7 (Table 1).
Table 1. EC50 Values for Chloroquine, Dihydrartemisinin, Artesunic Acid (16), Quinazoline Derivatives 12 and 14, and Hybrids 6–10 Tested against P. falciparum 3D7 Parasites.
| compound | molecular weight (g/mol) | 3D7 EC50 (nM) |
|---|---|---|
| chloroquinea | 319.87 | 9.8 ± 2.8 |
| DHAa | 284.25 | 2.4 ± 0.4 |
| artesunic acid (16) | 384.42 | 9.7 |
| 12 | 290.15 | 3177 ± 443 |
| 14 | 335.14 | 148 ± 8.2 |
| 6 | 603.67 | 3.8 ± 1 |
| 7 | 714.64 | 14.4 ± 0.3 |
| 8 | 656.60 | 15.3 ± 1.9 |
| 9 | 671.57 | 1.4 ± 0.4 |
| 10 | 613.54 | 39.9 ± 0.8 |
EC50 values have been previously reported.44
In general, it can be said that all hybrids exhibit excellent antimalarial efficacy with EC50 values within the nanomolar range (1.4–39.9 nM). This is quite remarkable because their corresponding quinazoline precursors 12 and 14 only exhibit antimalarial activities in the low micromolar and moderate nanomolar ranges (EC50 = 148–3177 nM). All hybrids except for hybrid 10 (EC50 = 39.9 nM) had a similar or even higher activity than that of the tested reference compound, artesunic acid (16) (EC50 = 9.7 nM), and clinically used chloroquine (EC50 = 9.8 nM). The most active compounds in this study concerning antimalarial efficacy were hybrids 6 (EC50 = 3.8 nM) and 9 (EC50 = 1.4 nM), which were comparable to or even more active than the clinically used drug DHA (EC50 = 2.4 nM). On analyzing the structure–activity relationship of the hybrids, interesting observations can be made. C-10 acetal hybrid 9 is 30 times more active than hybrid 10, which contains a C-10 nonacetal-linked artemisinin subunit. In consequence, a C-10 acetal linkage seems to be beneficial for antimalarial activity in the case of quinazoline–artemisinin hybrids.
A contrary effect was observed in connection with artemisin-derived dimers,49 which supports the assumption that the underlying mechanism of action might be different. The free secondary amine group of 4-anilinoquinazoline-derived hybrids seems to play an important role with regard to antimalarial efficacy, as hybrids 7 and 8, in which the artemisinin moiety is attached to the N-atom of the aniline subunit, are approximately up to 10 times less active than hybrids 6 and 9. Therefore, the functionalized aromatic subunits in 4-anilinoquinazoline derivatives should be used for hybridization, and the secondary amine should be left untouched.
2.2.2. Inhibitory Activity against HCMV in Primary Human Fibroblasts
The antiviral activity of compounds was investigated for HCMV, strain AD169-GFP. This recombinant virus expresses the green fluorescent protein (GFP) that can be reliably quantitated as a reporter of viral replication (Table 2). Infection experiments were carried out with cultures of primary human foreskin fibroblasts (HFFs), and measurements of antiviral activity were performed according to an already published protocol.50−52 Used as two reference compounds, artesunic acid (16) and ganciclovir both exerted strong anti-HCMV activity at EC50 levels of 3.80 and 2.60 μM, respectively, and were compared to that of the novel quinazoline–artemisinin hybrids. Four of five hybrids analyzed showed anti-HCMV activity in the submicromolar range with EC50 values of 0.86 μM (hybrid 7), 0.65 μM (hybrid 8), 0.21 μM (hybrid 9), and 0.15 μM (hybrid 10; the fifth compound, hybrid 6, was less active but still showed anti-HCMV activity in the low micromolar range, with EC50 = 5.89 μM). Thus, these hybrids were up to 17-fold more effective than the reference drug ganciclovir.
Table 2. EC50 Values of Anti-HCMV Activity (AD169-GFP) Displayed in Virus-Infected HFFs: Artesunic Acid (16) Compared with Five Chemically Different Quinazoline–Artemisinin Hybrids 6–10 and Their Precursors 12–14.
| compound | molecular weight (g/mol) | HCMV EC50 (μM) |
|---|---|---|
| ganciclovira | 255.23 | 2.60 ± 0.50 |
| artemisinin (5)b | 282.42 | >10 |
| DHAb | 284.35 | >10 |
| artesunic acid (16)a | 384.42 | 3.80 ± 0.40 |
| 12 | 290.15 | 2.01 ± 0.10 |
| 13 | 348.23 | >10 |
| 14 | 335.14 | 0.05 ± 0.02 |
| 6 | 603.67 | 5.89 ± 1.07 |
| 7 | 714.64 | 0.86 ±0.20 |
| 8 | 656.60 | 0.65 ± 0.03 |
| 9 | 671.57 | 0.21 ± 0.12 |
| 10 | 613.54 | 0.15 ± 0.05 |
We concluded from these data that a similar structure–activity trend refers to the anti-HCMV activity as already previously stated for antimalarial efficacy. Hybrids 7 and 8, in which the secondary amine group of the 4-anilinoquinazoline derivatives were used for hybridization, were less active than hybrids 9 and 10 with the free secondary amine group. This effect was even more pronounced for quinazoline precursors 12 and 13: quinazoline derivative 12 possessed an efficacy comparable to that of ganciclovir (EC50 = 2.01 μM), whereas compound 13 was completely inactive (EC50 > 10 μM). However, unlike antimalarial efficacy, C-10 nonacetal-linked artemisinin-derived hybrids 8 and 10 were more potent against HCMV than their corresponding C-10 acetal-linked counterparts (hybrids 7 and 9).
The highest antiviral efficacy in vitro among the tested hybrids was noted for hybrid 10; therefore, it is tempting to speculate whether the specific chemical nature of this hybrid compound may combine in an optimized manner the antiviral mechanisms displayed by artesunic acid and quinazolines. Previous reports demonstrated that the antiviral mode of action of artesunic acid is based on its signaling-inhibitory potential, particularly its interference with the canonical NF-κB (RelA/p65) pathway,55−57 whereas quinazolines typically act on the basis of a direct catalytic inhibition of viral and cellular protein kinases.57−60 For these compounds, no or only modest signs of cytotoxicity were detectable on HFFs up to a concentration of 10 μM, as routinely monitored by light and fluorescence microscopy, thus strongly arguing for a specific antiviral effect. Combined, these results indicate a strong anti-HCMV potential of quinazoline–artemisinin hybrids.
Surprisingly, the most active compound tested in this study against HCMV was new quinazoline 14 with an EC50 value in the nanomolar range (50 nM). The compound exerted a 50-fold higher activity than the standard drug ganciclovir and also 40-fold higher activity than other tested quinazoline derivatives 12 and 13.
2.2.3. Cytotoxicity toward Sensitive CCRF-CEM and Multidrug-Resistant CEM/ADR5000 Leukemia Cells
The most promising hybrids in this study, 9 and 10, and its precursor, 14, were tested for a potential in vitro antileukemia effect in sensitive wild-type CCRF-CEM and multidrug-resistant P-glycoprotein-overexpressing CEM/ADR5000 cells (Table 3). Hybrid 10 and precursor 14 showed no promising antileukemia behavior. In contarst, hybrid 9 with EC50 values of 2.8 μM (CCRF-CEM) and 0.5 μM (CEM/ADR5000) exhibited an antileukemia effect similar to that of artesunic acid. Especially, the activity against the multidrug-resistant CEM/ADR5000 cells is quite promising, as it is 45 times higher than that of doxorubicin (EC50 = 23.27 μM). This is important as drug resistance is one of the biggest challenges to be overcome in current cancer treatment.
Table 3. EC50 Values for Quinazoline–Artemisinin Hybrids 9/10 and Its Precursor 14 in Sensitive Wild-Type CCRF-CEM and Multidrug-Resistant P-Glycoprotein-Overexpressing CEM/ADR5000 Cells.
3. Conclusions
In conclusion, five quinazoline–artemisinin-derived hybrids, 6–10, were successfully synthesized and investigated for the first time for their antimalarial, antiviral, and antileukemia activity. All novel quinazoline–artemisinin hybrids showed high antimalarial efficacy against the P. falciparum 3D7 strain (EC50 = 1.4–39.9 nM), which was better or comparable to that of artesunic acid (EC50 = 9.7 nM). Remarkably, hybrid 9 (EC50 = 1.4 nM) was even more active than the clinically used drugs DHA (EC50 = 2.4 nM) and chloroquine (EC50 = 9.8 nM). With EC50 values of 0.15–0.21 μM, compounds 9 and 10 were most potent in the inhibition of HCMV replication in primary cell cultures, surpassing the antiviral activity of ganciclovir by factors 12–17. In the course of this study, we also characterized a novel, highly active new quinazoline 14 that was most active with an EC50 in the nanomolar range (HCMV EC50 = 50 nM). Moreover, hybrid 9 showed an antileukemia effect similar to that of artesunic acid with EC50 values in the low micromolar range and was 45 times more active toward the multidrug-resistant CEM/ADR5000 cells (EC50 = 0.6 μM) than the standard drug doxorubicin. These promising results further underline the high potential of the hybridization concept for further investigations and hybrid-based drug design.
4. Experimental Section
4.1. Chemistry
4.1.1. Synthesis of Hybrid Molecules: General
All reactions were performed in flame-dried glassware under a nitrogen atmosphere. If necessary, the synthesized hybrids were further purified after column chromatography via reprecipitation from CH2Cl2 in n-hexane to yield a pure compound for elemental analysis and biological tests. EtOAc was purchased as an anhydrous solvent. CH2Cl2 was dried initially over CaCl2 and then distilled from P2O5. All other solvents were purified by distillation using rotary evaporation or were purchased in high-performance liquid chromatography-quality. Reagents obtained from commercial sources were used without further purification. Thin-layer chromatography was performed on precoated aluminum silica gel SIL G/UV254 plates (Macherey-Nagel & Co.). The detection occurred via fluorescence quenching or development in a phosphomolybdic acid solution (10% in EtOH). All products were dried under high vacuum (10–3 mbar). 1H NMR and 13C NMR spectra were recorded at room temperature (rt) either on a Bruker Avance spectrometer operating at 300 and 400 MHz, respectively, or on a JEOL JNM GX 400 spectrometer operating at 400 MHz. ESI mass spectra were recorded on a Bruker micrOTOF II focus time-of-flight mass spectrometer (TOF MS) or a Shimadzu Axima Confidence MALDI-TOF MS without a matrix. IR spectra were recorded on a Varian IR-660 apparatus. The absorption is indicated in wave numbers [cm–1]. Elemental analysis (C, H, N), carried out with a Euro EA 3000 (Euro Vector) machine and an EA 1119 CHNS, CE machine, is within ±0.50% of the calculated values, confirming a purity of >95%. Artesunic acid (16) and DHA were obtained from ABCR (Karlsruhe, Germany).
4.1.2. General Procedure for Hybrids 6–8
To the corresponding quinazoline alcohol, 11 or 13, in CH3CN (and anhydrous EtOAc) with 4 Å molecular sieves, DMAP and artesunic acid (16) or artemisinin-derived acid 17 (prepared according to the literature procedures,47 see Scheme 2) were added consecutively, and the reaction mixture was cooled to 0 °C. Subsequently, DCC was added, and the reaction mixture was warmed to rt and stirred for 24, 48, or 53 h. Subsequently, DCU was removed by filtration, and the solvent was removed under reduced pressure. The crude product was purified by column chromatography, and hybrids 6 and 7 were obtained as pale yellow solids and hybrid 8 as a pale yellow oil.
4.1.3. Quinazoline–Artesunic Acid Hybrid 6
Quinazoline alcohol 11 (177 mg, 0.74 mmol, 1.0 equiv), CH3CN (13.0 mL), EtOAc (2.5 mL), DMAP (45.4 mg, 0.37 mmol, 50 mol %), artesunic acid (16) (373 mg, 0.97 mmol, 1.3 equiv), DCC (200 mg, 0.97 mmol, 1.3 equiv). Column conditions: CH2Cl2/MeOH 80:1 → CH2Cl2/MeOH 40:1. Yield: 269 mg, 0.45 mmol, 60%, off-white solid. Rf = 0.49 (CH2Cl2/MeOH 20:1, UV and phosphomolybdic acid). 1H NMR (400 MHz, CDCl3): δ = 0.82 (d, J = 7.1 Hz, 3H), 0.93 (d, J = 5.8 Hz, 3H), 0.96–1.05 (m, 1H), 1.19–1.51 (m, 7H), 1.56–1.63 (m, 1H), 1.63–1.77 (m, 2H), 1.82–1.91 (m, 1H), 1.96–2.04 (m, 1H), 2.28–2.40 (m, 1H), 2.51–2.61 (m, 1H), 2.72–2.97 (m, 4H), 5.41 (s, 1H), 5.80 (d, J = 9.9 Hz, 1H), 7.05–7.11 (m, 2H), 7.52–7.59 (m, 1H), 7.68–7.74 (m, 2H), 7.75–7.81 (m, 1H), 7.87–7.97 (m, 2H), 7.98–8.04 (m, 1H), 8.70 (s, 1H) ppm. 13C NMR (100 MHz, CDCl3): δ = 12.0, 20.2, 21.9, 24.5, 25.9, 29.0, 29.2, 31.8, 34.0, 36.2, 37.2, 45.2, 51.5, 80.1, 91.5, 92.4, 104.5, 115.0, 120.9, 122.0, 123.1, 126.8, 128.3, 133.1, 135.7, 147.1, 149.2, 154.4, 157.7, 170.9, 171.0 ppm. MS (ESI): m/z = 604 ([M + H]+); HRMS (ESI): calcd for [C33H38N3O8]+ 604.2653, found 604.2656. FT-IR (ATR): ν̃ = 3386 (w), 2924 (w), 2878 (w), 2358 (w), 2350 (w), 2020 (w), 1747 (s), 1620 (w), 1572 (s), 1528 (s), 1507 (s), 1494 (s), 1416 (m), 1405 (m), 1358 (m), 1308 (w), 1249 (w), 1195 (s), 1165 (m), 1135 (s), 1099 (m), 1033 (s), 1008 (s), 920 (w), 875 (m), 842 (w), 766 (m), 732 (w), 679 (w), 595 (w), 515 (w) cm–1. Anal. Calcd for C33H37N3O8·0.3CH2Cl2: C, 63.57; H, 6.02; N, 6.68; found: C, 63.32; H, 5.98; N, 6.75.
4.1.4. Quinazoline–Artesunic Acid Hybrid 7
Quinazoline alcohol 13 (56.0 mg, 0.16 mmol, 1.0 equiv), CH3CN (5.0 mL), DMAP (9.50 mg, 0.08 mmol, 48 mol %), artesunic acid (16) (80.5 mg, 0.21 mmol, 1.3 equiv), DCC (43.1 mg, 0.21 mmol, 1.3 equiv). Column conditions: hexanes/EtOAc 3:1 → hexanes/EtOAc 1:2. Yield: 40.0 mg, 0.06 mmol, 35%, yellow solid. Rf = 0.20 (hexanes/EtOAc 1:4, UV and phosphomolybdic acid). 1H NMR (400 MHz, CDCl3): δ = 0.82 (d, J = 7.1 Hz, 3H), 0.92 (d, J = 5.5 Hz, 3H), 0.94–1.08 (m, 1H), 1.17–1.44 (m, 7H), 1.53–1.77 (m, 5H), 1.78–2.00 (m, 2H), 2.07–2.14 (m, 1H), 2.31 (td, J = 13.8, 3.8 Hz, 1H), 2.46–2.59 (m, 1H), 2.61–2.83 (m, 4H), 3.94–4.4.08 (m, 2H), 4.25–4.36 (m, 1H), 5.29 (s, 1H), 5.73 (d, J = 9.9 Hz, 1H), 6.94 (dd, J = 8.4, 2.2 Hz, 1H), 7.14–7.21 (m, 1H), 7.23 (d, J = 2.2 Hz, 1H), 7.31–7.43 (m, 2H), 7.55–7.64 (m, 1H), 7.75 (s, 1H), 8.39 (d, J = 8.1 Hz, 1H) ppm. 13C NMR (100 MHz, CDCl3): δ = 12.0, 20.2, 21.9, 24.5, 25.8, 27.7, 29.0, 29.2, 29.7, 31.7, 34.0, 36.1, 37.2, 45.1, 46.4, 51.5, 60.9, 80.1, 91.4, 92.3, 104.4, 113.8, 120.8, 122.3, 124.3, 125.5, 126.2, 127.6, 130.1, 132.0, 132.3, 137.4, 150.2, 153.2, 171.4, 171.7 ppm. MS (ESI): m/z = 714 ([M + H]+); HRMS (ESI): calcd for [C36H42Cl2N3O8]+ 714.2344, found 714.2362. FT-IR (ATR): ν̃ = 2953 (w), 2924 (m), 2872 (w), 2358 (w), 2249 (w), 2013 (w), 1736 (s), 1614 (s), 1578 (m), 1546 (s), 1462 (s), 1401 (m), 1376 (m), 1312 (w), 1243 (m), 1155 (m), 1129 (m), 1098 (m), 1011 (s), 908 (w), 875 (m), 843 (w), 804 (m), 755 (s), 729 (s), 667 (m) 545 (w) cm–1. Anal. Calcd for C36H41Cl2N3O8·1.1H2O: C, 58.87; H, 5.93; N, 5.72; found: C, 58.96; H, 5.57; N, 5.67.
4.1.5. Quinazoline–Artemisinin Hybrid 8
Quinazoline alcohol 13 (50.0 mg, 0.14 mmol, 1.0 equiv), CH3CN (4.5 mL), DMAP (8.77 mg, 0.07 mmol, 50 mol %), artemisinin-derived acid 17 (61.0 mg, 0.19 mmol, 1.3 equiv), DCC (38.6 mg, 0.19 mmol, 1.3 equiv). Column conditions: hexanes/EtOAc 1:19. Yield: 46.0 mg, 0.07 mmol, 49%, pale yellow oil. 1H NMR (400 MHz, CDCl3): δ = 0.85 (d, J = 7.5 Hz, 3H), 0.93 (d, J = 5.9 Hz, 3H), 1.20–1.29 (m, 6H), 1.58–1.68 (m, 4H), 1.76–1.91 (m, 3H), 1.94–2.02 (m, 3H), 2.24–2.34 (m, 1H), 2.38–2.45 (m, 1H), 2.55–2.77 (m, 2H), 4.00–4.37 (m, 4H), 4.70–4.77 (m, 1H), 5.23 (s, 1H), 6.96 (d, J = 6.7 Hz, 1H), 7.19 (d, J = 8.3 Hz, 1H), 7.25 (s, 1H), 7.33 (d, J = 8.5 Hz, 1H), 7.39 (t, J = 7.6 Hz, 1H), 7.59 (t, J = 7.2 Hz, 1H), 7.81 (s, 1H), 8.41 (bs, 1H) ppm. 13C NMR (100 MHz, CDCl3): δ = 13.1, 14.2, 20.1, 21.2, 24.6, 24.7, 25.9, 27.7, 29.7, 34.3, 36.1, 36.4, 37.4, 44.1, 46.2, 52.2, 60.4, 60.7, 72.3, 80.8, 89.0, 92.2, 103.4, 113.9, 122.5, 124.4, 126.3, 127.7, 130.1, 131.9, 132.5, 137.3, 150.3, 171.7 ppm. MS (ESI): m/z = 656 ([M + H]+); HRMS (ESI): calcd for [C34H40Cl2N3O6]+ 656.2289, found: 656.2304. FT-IR (ATR): ν̃ = 2925 (w), 2873 (w), 2333 (w), 2201 (w), 1969 (w), 1733 (s), 1613 (s), 1577 (m), 1545 (s), 1462 (s), 1401 (m), 1377 (m), 1311 (w), 1271 (w), 1243 (w), 1219 (w), 1169 (m), 1124 (m), 1092 (m), 1047 (s), 1011 (s), 940 (w), 874 (m), 821 (w), 756 (s), 689 (w), 669 (w), 617 (w), 528 (w), 479 (w), 442 (w), 421 (w) cm–1. Anal. Calcd for C34H39Cl2N3O6·2.5H2O: C, 58.20, H, 6.32, N, 5.99; found: C, 57.78, H, 5.87, N, 5.72.
4.1.6. General Procedure for Hybrids 9 and 10
A solution of artesunic acid (16) or artemisinin-derived acid 17 and HOBt in dry DMF (2.0 mL) was cooled to 0 °C. EDCI·HCl was added to the reaction mixture at 0 °C under N2. After stirring the reaction mixture for 10 min, a solution of quinazoline amine 15 in dry DMF (5.0 mL) and DIPEA were added subsequently at 0 °C under N2. The resulting reaction mixture was slowly warmed to rt and stirred overnight for 22 h or 3 days. After this time period, EtOAc (30 mL) and H2O (30 mL) were added. The two phases were separated, and the water phase was extracted with EtOAc (3 × 25 mL). The combined organic layers were washed with H2O (3 × 15 mL) and brine (20 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give a yellow oil. The crude product was purified by gradient column chromatography to obtain hybrids 9 and 10 as off-white solids.
4.1.7. Quinazoline–Artesunic Acid Hybrid 9
Artesunic acid (16) (94.5 mg, 0.25 mmol, 1.0 equiv) HOBt (33.2 mg, 0.25 mmol, 1.0 equiv), EDCI·HCl (47.2 mg, 0.25 mmol, 1.0 equiv), quinazoline amine 15 (75.0 mg, 0.25 mmol, 1.0 equiv), DIPEA (40.2 μL, 31.7 mg, 0.25 mmol, 1.0 equiv). Column conditions: CH2Cl2/MeOH 9.8:0.2 → CH2Cl2/MeOH 9.5:0.5. Yield: 112 mg, 0.17 mmol, 68%, off-white solid. Rf = 0.24 (CH2Cl2/MeOH 9.5:0.5, UV and phosphomolybdic acid). 1H NMR (MeOD + drops of CDCl3, 300 MHz): δ = 0.83 (d, 3H, J = 6.9 Hz), 0.93 (d, 3H, J = 6.0 Hz), 0.99–1.09 (m, 1H), 1.18–1.52 (m, 8H), 1.53–1.64 (m, 1H), 1.65–1.81 (m, 2H), 1.82–1.95 (m, 1H), 1.96–2.10 (m, 1H), 2.32 (td, 1H, J = 13.5 Hz, 2.4 Hz), 2.45–2.58 (m, 1H), 2.73–2.90 (m, 4H), 5.44 (s, 1H), 5.75 (d, 1H, J = 9.9 Hz), 7.42 (d, 1H, J = 8.7 Hz), 7.62–7.78 (m, 3H), 8.06 (d, 1H, J = 2.4 Hz), 8.54 (s, 1H), 8.61 (d, 1H, J = 1.8 Hz) ppm. 13C NMR (MeOD + drops of CDCl3, 75.5 MHz): δ = 11.2, 19.3, 21.3, 24.0, 24.7, 28.7, 30.4, 31.2, 33.4, 35.6, 36.7, 44.7, 60.0, 79.7, 91.1, 91.9, 104.1, 110.0, 115.1, 121.1, 123.3, 126.2, 127.4, 129.7, 131.7, 136.4, 138.1, 145.5, 152.6, 157.4, 170.8, 171.4 ppm. MS (ESI): m/z = 671 ([M + H]+), 693 ([M + Na]+); HRMS (ESI): calcd for [C33H36Cl2N4O7Na]+ 693.1853, found 693.1862. FT-IR (ATR): ν̃ = 3337 (w), 2929 (w), 2873 (w), 2479 (w), 2196 (w), 2033 (w), 1745 (m), 1692 (m), 1567 (m), 1533 (s), 1517 (s), 1475 (s), 1450 (m), 1415 (s), 1377 (s), 1318 (m), 1284 (m), 1230 (w), 1153 (s), 1132 (s), 1096 (w), 1053 (m), 1007 (s), 926 (m), 900 (w), 875 (m), 844 (m), 800 (m), 680 (m), 636 (w), 574 (w), 532 (m), 502 (m), 434 (m) cm–1. Anal. Calcd for C33H36Cl2N4O7: C, 59.02; H, 5.40; N, 8.34; found: C, 58.48; H, 5.58; N, 8.10.
4.1.8. Quinazoline–Artemisinin Hybrid 10
Artemisinin-derived acid 17 (104 mg, 0.32 mmol, 1.2 equiv) HOBt (43.1 mg, 0.32 mmol, 1.2 equiv), EDCI·HCl (61.2 mg, 0.32 mmol, 1.2 equiv), quinazoline amine 15 (81.0 mg, 0.27 mmol, 1.0 equiv), DIPEA (90.0 μL, 68.4 mg, 0.53 mmol, 2.0 equiv). Column conditions: CH2Cl2/MeOH 9.9:0.1 → CH2Cl2/MeOH 9.7:0.3. Yield: 104 mg, 0.17 mmol, 64%, off-white solid. Rf = 0.40 (CH2Cl2/MeOH 9.5:0.5, UV and phosphomolybdic acid). 1H NMR (MeOD + drops of CDCl3, 300 MHz): δ = 0.91–0.98 (m, 7H), 1.20–1.49 (m, 8H), 1.62–1.76 (m, 2H), 1.77–1.88 (m, 1H), 1.89–2.08 (m, 2H), 2.29 (td, 1H, J = 14.0 Hz, 3.0 Hz), 2.50 (dd, 1H, J = 15.2 Hz, 2.1 Hz), 2.70–2.84 (m, 2H), 4.82–4.92 (m, 1H), 5.53 (s, 1H), 7.39 (d, 1H, J = 8.7 Hz), 7.48–7.53 (m, 2H), 7.62 (d, 1H, J = 9.0 Hz), 7.69 (dd, 1H, J = 8.9 Hz, 2.7 Hz), 8.09 (d, 1H, J = 2.4 Hz), 8.50 (s, 1H), 8.61 (d, 1H, J = 2.1 Hz) ppm. 13C NMR (MeOD + drops of CDCl3, 75.5 MHz): δ = 13.1, 20.4, 25.2, 25.3, 26.1, 30.7, 34.9, 36.9, 38.0, 38.6, 44.5, 52.7, 72.4, 81.6, 90.3, 104.1, 111.1, 116.1, 122.0, 124.2, 127.1, 127.5, 128.4, 130.7, 132.7, 137.0, 139.2, 146.6, 153.8, 158.3, 171.8 ppm. MS (ESI): m/z = 635 ([M + Na]+); HRMS (ESI): calcd for [C31H34Cl2N4O5Na]+ 635.1799, found 635.1800. FT-IR (ATR): ν̃ = 3321 (w), 2928 (w), 2874 (w), 2175 (w), 2004 (w), 1673 (m), 1597 (m), 1572 (m), 1527 (s), 1474 (s), 1416 (s), 1380 (s), 1318 (m), 1287 (m), 1264 (m), 1224 (m), 1192 (m), 1126 (m), 1091 (m), 1041 (m), 1010 (m), 938 (m), 918 (m), 873 (m), 838 (m), 681 (m), 645 (w), 614 (w), 575 (w), 531 (m), 503 (m), 438 (m) cm–1. Anal. Calcd for C33H36Cl2N4O7: C, 60.69; H, 5.59; N, 9.13; found: C, 60.19; H, 5.55; N, 9.03.
4.2. Cytotoxicity Studies against P. falciparum 3D7 Strains
4.2.1. P. falciparum culture
P. falciparum 3D7 parasites were cultured in type A-positive human erythrocytes at a hematocrit of 5% in RPMI 1640 supplemented with 25 mM HEPES, 0.1 mM hypoxanthine, 50 μg/mL gentamycin, and 0.5% albumax I. Cultures were incubated at 37 °C under controlled atmospheric conditions of 5% O2, 3% CO2, and 92% N2 at 95% relative humidity.
4.2.2. In Vitro Antimalarial Activity Assay
Cultures used in cell proliferation assays were synchronized by sorbitol treatment.62 Concentrations to inhibit parasite growth by 50% (EC50) were determined using the SYBR Green I malaria drug sensitivity assay.63 Aliquots of 100 μL of a cell suspension containing ring stages at a parasitemia of 0.2% and a hematocrit of 2% were added to the wells of 96-well microtiter plates. Plates were incubated for 72 h in the presence of different drug concentrations. Subsequently, cells of each well were lysed with 100 μL of lysis buffer (40 mM Tris, pH 7.5, 10 mM ethylenediaminetetraacetate, 0.02% saponin, 0.08% Triton X-100) containing 8.3 μM SYBR Green. Plates were incubated for 1 h in the dark at rt under constant mixing before fluorescence (excitation wavelength 485 nm; emission wavelength >520 nm) was determined using a microtiter plate fluorescence reader (Victor X4; Perkin Elmer). Drugs were serially diluted to 1:3 with the initial drug concentrations being 243 nM for chloroquine, artesunic acid (16), and its derivates and 81 nM for DHA and its derivates. Each drug concentration was examined in triplicate and repeated at least three times. Uninfected erythrocytes (hematocrit 2%) and infected erythrocytes without the drug served as controls and were investigated in parallel. Percent growth was calculated as described by Beez.64 Data were analyzed using the SigmaPlot (version 12.0; Hill function, three parameters) and Sigma Stat programs.
4.3. HCMV GFP-Based Replication Assay
The HCMV GFP-based replication assay was carried out over a duration of 7 days (multiround HCMV infection) with primary HFFs infected with a GFP-expressing recombinant HCMV (AD169-GFP), as described before.50,52,54,57 All data represent mean values of determinations in quadruplicate (HCMV infections performed in duplicate, GFP measurements of total cell lysates performed in duplicate). Processing and evaluation of data were performed by the use of Excel (means and standard deviations).
4.4. Cytotoxicity Studies against Leukemia Cells
4.4.1. Cell Culture
Human leukemic CCRF-CEM and the P-glycoprotein expressing CEM/ADR5000 cells were obtained from the University of Jena (Department for Pediatrics, University of Jena, Germany) and were cultivated in the RPMI 1640 medium supplemented with 10% (v/v) inactivated fetal calf serum and 1% penicillin/streptomycin at 37 °C with 5% CO2 in a humidified atmosphere (95% relative humidity). CEM/ADR5000 cells were treated with 5000 ng/mL doxorubicin once per week to keep them resistant.65 The multidrug resistance profile of CEM/ADR5000 has been reported.66,67 Cells were passaged twice a week and used for experiments in the logarithmic phase.
4.4.2. Cell Viability Assay
CCRF-CEM or CEM/ADR5000 cells were seeded at an appropriate density (10 000 cells/well) in a 96-well plate with a total volume of 200 μL. Compounds were added in varying concentrations [0.001, 0.003, 0.01, 0.03, 0.1, 0.3, 1, 3, 10, and 100 μM]. Each concentration was tested six times within each experiment, and each experiment was repeated three times. In addition, CEM/ADR5000 cells were tested with doxorubicin alone and in combination with the three derivatives (10 μM) and verapamil [0.1, 0.3, 1, 3, 10, and 100 μM]. After 72 h at 37 °C and 5% CO2, 20 μL of resazurin 0.01% w/v in ddH2O was added to each well, and there was further incubation of the plates for 4 h. The plates were measured using an excitation wavelength of 544 nm and an emission wavelength of 590 nm. The test compound concentrations required to inhibit 50% of cell proliferation were represented by EC50 values, calculated from the dose–response curves.
Acknowledgments
We gratefully acknowledge the financial support from Deutsche Forschungsgemeinschaft (DFG) by grants TS 87/16-3 and MM 1289/7-1/7-3. We also thank the Interdisciplinary Center for Molecular Materials (ICMM), the Graduate School Molecular Science (GSMS) for research support, as well as the Wilhelm Sander-Stiftung (Grant Nos. 2014.019.1 and 2011.085.2), Bayerische Forschungsstiftung (Forschungsverbund ForBIMed—Biomarker in der Infektionsmedizin, I1/M.M.-C.H.), and Emerging Fields Initiative (EFI) “Chemistry in Live Cells” supported by Friedrich-Alexander-Universität Erlangen-Nürnberg for funding. Financial support by the German Academic Exchange Service DAAD (research fellowship for Dr. Mohammad M. Ibrahim) is also gratefully acknowledged.
Glossary
Abbreviations
- DCC
N,N′-dicyclohexylcarbodiimide
- DCE
1,2-dichloroethane
- DCU
N,N′-dicyclohexylurea
- DHA
dihydroartemisinin
- DIEA
N,N-diisopropylethylamine
- DMAP
4-(dimethylamino)-pyridine
- equiv
equivalent
- EtOAc
ethyl acetate
- GFP
green fluorescent protein
- HCMV
human cytomegalovirus
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HFF
human foreskin fibroblasts
- HOBt
1-hydroxybenzotriazole
- TBAI
tetrabutylammonium iodide
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b00310.
Experimental conditions and procedures for quinazoline precursors 11–15 and artemisinin derivatives 17–19; spectral data of quinazoline precursors 11–15 and artemisinin derivatives 17–19; recorded spectra of target compounds 6–10, quinazoline precursors 11–15, and artemisinin derivatives 17–19 (PDF)
Author Contributions
# T.F. and C.R. contributed equally to this work.
Author Contributions
The manuscript was written through contribution of all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Maggio B.; Daidone G.; Raffa D.; Plescia S.; Mantione L.; Cutuli V. M. C.; Mangano N. G.; Caruso A. Synthesis and pharmacological study of ethyl 1-methyl-5-(substituted 3,4-dihydro-4-oxoquinazolin-3-yl)-1H-pyrazole-4-acetates. Eur. J. Med. Chem. 2001, 36, 737–742. 10.1016/S0223-5234(01)01259-4. [DOI] [PubMed] [Google Scholar]
- Grover G.; Kini S. G. Synthesis and evaluation of new quinazolone derivatives of nalidixic acid as potential antibacterial and antifungal agents. Eur. J. Med. Chem. 2006, 41, 256–262. 10.1016/j.ejmech.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Rohini R.; Shanker K.; Reddy P. M.; Ho Y. P.; Ravinder V. Mono and bis-6-arylbenzimidazo[1,2-c]quinazolines: a new class of antimicrobial agents. Eur. J. Med. Chem. 2009, 44, 3330–3339. 10.1016/j.ejmech.2009.03.022. [DOI] [PubMed] [Google Scholar]
- Verhaeghe P.; Azas N.; Gasquet M.; Hutter S.; Ducros C.; Laget M.; Rault S.; Rathelot P.; Vanelle P. Synthesis and antiplasmodial activity of new 4-aryl-2-trichloromethylquinazolines. Bioorg. Med. Chem. Lett. 2008, 18, 396–401. 10.1016/j.bmcl.2007.10.027. [DOI] [PubMed] [Google Scholar]
- Madapa S.; Tusi Z.; Mishra A.; Srivastava K.; Pandey S. K.; Tripathi R.; Puri S. K.; Batra S. Search for new pharmacophores for antimalarial activity. Part II: Synthesis and antimalarial activity of new 6-ureido-4-anilinoquinazolines. Bioorg. Med. Chem. 2009, 17, 222–234. 10.1016/j.bmc.2008.11.005. [DOI] [PubMed] [Google Scholar]
- Alagarsamy V.; Murugesan S.; Sheorey R. V. Synthesis and pharmacological investigation of novel 3-(benzyl)-2-substituted amino-3H-quinazolin-4-ones as analgesic and anti-inflammatory agents. Med. Chem. Res. 2008, 17, 564–577. 10.1007/s00044-008-9098-z. [DOI] [Google Scholar]
- Alagarsamy V.; Solomon V. R.; Murugan M.; Sankaranarayanan R.; Periyasamy P.; Deepa R.; Anandkumar T. D. Synthesis of 3-(2-pyridyl)-2-substituted-quinazolin-4(3H)-ones as new analgesic and anti-inflammatory agents. Biomed. Pharmacother. 2008, 62, 454–461. 10.1016/j.biopha.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Smits R. A.; Adami M.; Istyastono E. P.; Zuiderveld O. P.; van Dam C. M. E.; de Kanter F. J. J.; Jongejan A.; Coruzzi G.; Leurs R.; de Esch I. J. P. Synthesis and QSAR of Quinazoline Sulfonamides As Highly Potent Human Histamine H4 Receptor Inverse Agonists. J. Med. Chem. 2010, 53, 2390–2400. 10.1021/jm901379s. [DOI] [PubMed] [Google Scholar]
- Chandrika P. M.; Yakaiah T.; Rao A. R. R.; Narsaiah B.; Reddy N. C.; Sridhar V.; Rao J. V. Synthesis of novel 4,6-disubstituted quinazoline derivatives, their anti-inflammatory and anti-cancer activity (cytotoxic) against U937 leukemia cell lines. Eur. J. Med. Chem. 2008, 43, 846–852. 10.1016/j.ejmech.2007.06.010. [DOI] [PubMed] [Google Scholar]
- Marvania B.; Lee P.-C.; Chaniyara R.; Dong H.; Suman S.; Kakadiya R.; Chou T.-C.; Lee T.-C.; Shah A.; Su T.-L. Design, synthesis and antitumor evaluation of phenyl N-mustard-quinazoline conjugates. Bioorg. Med. Chem. 2011, 19, 1987–1998. 10.1016/j.bmc.2011.01.055. [DOI] [PubMed] [Google Scholar]
- Wang H. Lapatinib for the treatment of breast cancer in the People’s Republic of China. OncoTargets Ther. 2014, 7, 1367–1373. 10.2147/OTT.S60586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herget T.; Freitag M.; Morbitzer M.; Kupfer R.; Stamminger T.; Marschall M. Novel chemical class of pUL97 protein kinase-specific inhibitors with strong anticytomegaloviral activity. Antimicrob. Agents Chemother. 2004, 48, 4154–4162. 10.1128/AAC.48.11.4154-4162.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleiss M.; Eickhoff J.; Auerochs S.; Leis M.; Abele S.; Rechter S.; Choi Y.; Anderson J.; Scott G.; Rawlinson W.; Michel D.; Ensminger S.; Klebl B.; Stamminger T.; Marschall M. Protein kinase inhibitors of the quinazoline class exert anti-cytomegaloviral activity in vitro and in vivo. Antiviral Res. 2008, 79, 49–61. 10.1016/j.antiviral.2008.01.154. [DOI] [PubMed] [Google Scholar]
- Coatney G. R.; Cooper W. C.; Culwell W. B.; White W. C.; Imboden C. A. Jr. <tep-common:author-query>AQ2: Please provide a DOI number for ref 14 or indicate if one doesn&#x2019;t exist.</tep-common:author-query>Studies in Human Malaria. XXV. Trial of Febrifugine, an Alkaloid obtained from <genus-species>Dichroa febrífuga</genus-species> Lour., against the Chesson Strain of <genus-species>Plasmodium vivax</genus-species>. J. Natl. Malar. Soc. 1950, 9, 183–186. [PubMed] [Google Scholar]
- Fishman M.; Cruickshank P. A. Febrifugine antimalarial agents. I. Pyridine analogs of febrifugine. J. Med. Chem. 1970, 13, 155–156. 10.1021/jm00295a050. [DOI] [PubMed] [Google Scholar]
- Jiang S.; Zeng Q.; Gettayacamin M.; Tungtaeng A.; Wannaying S.; Lim A.; Hansukjariya P.; Okunji C. O.; Zhu S.; Fang D. Antimalarial Activities and Therapeutic Properties of Febrifugine Analogs. Antimicrob. Agents Chemother. 2005, 49, 1169–1176. 10.1128/AAC.49.3.1169-1176.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvam T. P.; Kumar P. V. Quinazoline Marketed drugs - A Review. Res. Pharm. 2011, 1, 1–21. [Google Scholar]
- Wang D.; Gao F. Quinazoline derivatives: synthesis and bioactivities. Chem. Cent. J. 2013, 7, 95. 10.1186/1752-153X-7-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asif M. Chemical Characteristics, Synthetic Methods, and Biological Potential of Quinazoline and Quinazolinone Derivatives. Int. J. Med. Chem. 2014, 2014, 395637 10.1155/2014/395637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravez S.; Castillo-Aguilera O.; Depreux P.; Goossens L. Quinazoline derivatives as anticancer drugs: a patent review (2011 - present). Expert Opin. Ther. Pat. 2015, 25, 789–804. 10.1517/13543776.2015.1039512. [DOI] [PubMed] [Google Scholar]
- Jafari E.; Khajouei M. R.; Hassanzadeh F.; Hakimelahi G. H.; Khodarahmi G. A. Quinazolinone and quinazoline derivatives: recent structures with potent antimicrobial and cytotoxic activities. Res. Pharm. Sci. 2016, 11, 1–14. [PMC free article] [PubMed] [Google Scholar]
- Pao W.; Miller V.; Zakowski M.; Doherty J.; Politi K.; Sarkaria I.; Singh B.; Heelan R.; Rusch V.; Fulton L.; Mardis E.; Kupfer D.; Wilson R.; Kris M.; Varmus H. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 13306–13311. 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chonan M.; Narita N.; Tominaga T. Total regression of brain metastases in non-small cell lung cancer patients harboring EGFR mutations treated with gefitinib without radiotherapy: two case reports. BMC Res. Notes 2016, 9, 2. 10.1186/s13104-015-1834-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackey K. E. Lessons from the drug discovery of lapatinib, a dual ErbB1/2 tyrosine kinase inhibitor. Curr. Top. Med. Chem. 2006, 6, 435–460. 10.2174/156802606776743156. [DOI] [PubMed] [Google Scholar]
- Mehta G.; Singh V. Hybrid systems through natural product leads: An approach towards new molecular entities. Chem. Soc. Rev. 2002, 31, 324–334. 10.1039/b204748a. [DOI] [PubMed] [Google Scholar]
- Tietze L. F.; Bell H. P.; Chandasekhar S. Natural Product Hybrids as New Leads for Drug Discovery. Angew. Chem., Int. Ed. 2003, 42, 3996–4028. 10.1002/anie.200200553. [DOI] [PubMed] [Google Scholar]
- Gademann K. Natural Product Hybrids. Chimia 2006, 60, 841–845. 10.2533/chimia.2006.841. [DOI] [Google Scholar]
- Viegas-Junior C.; Danuello A.; da Silva Bolzani V.; Barreiro E. J.; Fraga C. A. Molecular hybridization: a useful tool in the design of new drug prototypes. Curr. Med. Chem. 2007, 14, 1829–1852. 10.2174/092986707781058805. [DOI] [PubMed] [Google Scholar]
- Walsh J. J.; Coughlan D.; Heneghan N.; Gaynor C.; Bell A. A novel artemisinin–quinine hybrid with potent antimalarial activity. Bioorg. Med. Chem. Lett. 2007, 17, 3599–3602. 10.1016/j.bmcl.2007.04.054. [DOI] [PubMed] [Google Scholar]
- Meunier B. Hybrid molecules with a dual mode of action: dream or reality?. Acc. Chem. Res. 2008, 41, 69–77. 10.1021/ar7000843. [DOI] [PubMed] [Google Scholar]
- Tsogoeva S. B. Recent Progress in the Development of Synthetic Hybrids of Natural or Unnatural Bioactive Compounds for Medicinal Chemistry. Mini-Rev. Med. Chem. 2010, 10, 773–793. 10.2174/138955710791608280. [DOI] [PubMed] [Google Scholar]
- Miranda D.; Capela R.; Albuquerque I. S.; Meireles P.; Paiva I.; Nogueira F.; Amewu R.; Gut J.; Rosenthal P. J.; Oliveira R.; Mota M. M.; Moreira R.; Marti F.; Prudêncio M.; O’Neill P. M.; Lopes F. Novel Endoperoxide-Based Transmission-Blocking Antimalarials with Liver- and Blood-Schizontocidal Activities. ACS Med. Chem. Lett. 2014, 5, 108–112. 10.1021/ml4002985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Y. The discovery of artemisinin (qinghaosu) and gifts from Chinese medicine. Nat. Med. 2011, 17, 1217–1220. 10.1038/nm.2471. [DOI] [PubMed] [Google Scholar]
- Antimalaria studies on Qinghaosu. Chin. Med. J. 1979, 12, 811–816. [PubMed] [Google Scholar]
- Klayman D. L. Qinghaosu (artemisinin): an antimalarial drug from China. Science 1985, 228, 1049–1055. 10.1126/science.3887571. [DOI] [PubMed] [Google Scholar]
- Li Y.; Wu Y. L. An over four millennium story behind Qinghaosu (artemisinin): a fantastic antimalarial drug from a traditional Chinese herb. Curr. Med. Chem. 2003, 10, 2197–2230. 10.2174/0929867033456710. [DOI] [PubMed] [Google Scholar]
- Miller L. H.; Su X. Artemisinin: Discovery from the Chinese Herbal Garden. Cell 2011, 146, 855–858. 10.1016/j.cell.2011.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efferth T. Molecular pharmacology and pharmacogenomics of artemisinin and its derivatives in cancer cells. Curr. Drug Targets 2006, 7, 407–421. 10.2174/138945006776359412. [DOI] [PubMed] [Google Scholar]
- Singh N. P.; Lai H. C. Artemisinin induces apoptosis in human cancer cells. Anticancer Res. 2004, 24, 2277–2280. [PubMed] [Google Scholar]
- Horwedel C.; Tsogoeva S. B.; Wei S. W.; Efferth T. Cytotoxicity of Artesunic Acid Homo- and Heterodimer Molecules toward Sensitive and Multidrug-Resistant CCRF-CEM Leukemia Cells. J. Med. Chem. 2010, 53, 4842–4848. 10.1021/jm100404t. [DOI] [PubMed] [Google Scholar]
- Reiter C.; Hermann A.; Çapcı A.; Efferth T.; Tsogoeva S. B. New artesunic acid homodimers: Potent reversal agents of multidrug resistance in leukemia cells. Bioorg. Med. Chem. 2012, 20, 5637–5641. 10.1016/j.bmc.2012.07.015. [DOI] [PubMed] [Google Scholar]
- Reiter C.; Çapcı Karagöz A.; Fröhlich T.; Klein V.; Zeino M.; Viertel K.; Held J.; Mordmüller B.; Emirdağ Öztürk S.; Anıl H.; Efferth T.; Tsogoeva S. B. Synthesis and study of cytotoxic activity of 1,2,4-trioxane- and egonol-derived hybrid molecules against Plasmodium falciparum and multidrug-resistant human leukemia cells. Eur. J. Med. Chem. 2014, 75, 403–412. 10.1016/j.ejmech.2014.01.043. [DOI] [PubMed] [Google Scholar]
- Reiter C.; Fröhlich T.; Gruber L.; Hutterer C.; Marschall M.; Voigtländer C.; Friedrich O.; Kappes B.; Efferth T.; Tsogoeva S. B. Highly potent artemisinin-derived dimers and trimers: Synthesis and evaluation of their antimalarial, antileukemia and antiviral activities. Bioorg. Med. Chem. 2015, 23, 5452–5458. 10.1016/j.bmc.2015.07.048. [DOI] [PubMed] [Google Scholar]
- Reiter C.; Fröhlich T.; Zeino M.; Marschall M.; Bahsi H.; Leidenberger M.; Friedrich O.; Kappes B.; Hampel F.; Efferth T.; Tsogoeva S. B. New efficient artemisinin derived agents against human leukemia cells, human cytomegalovirus and Plasmodium falciparum: 2nd generation 1,2,4-trioxane-ferrocene hybrids. Eur. J. Med. Chem. 2015, 97, 164–172. 10.1016/j.ejmech.2015.04.053. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Wang C.; Sun Y.; Zhang N.; Liu Z.; Liu J. A novel strategy to the synthesis of 4-anilinoquinazoline derivatives. Tetrahedron 2014, 70, 906–913. 10.1016/j.tet.2013.12.028. [DOI] [Google Scholar]
- Yuste F.; Saldaña M.; Walls F. Selective reduction of aromatic nitro compounds containing o- and n-benzyl groups with hydrazine and raney nickel. Tetrahedron Lett. 1982, 23, 147–148. 10.1016/S0040-4039(00)86770-2. [DOI] [Google Scholar]
- Stocks P. A.; Bray P. G.; Barton V. E.; Al-Helal M.; Jones M.; Araujo N. C.; Gibbons P.; Ward S. A.; Hughes R. H.; Biagini G. A.; Davies J.; Amewu R.; Mercer A. E.; Ellis G.; O’Neill P. M. Evidence for a Common Non-Heme Chelatable-Iron-Dependent Activation Mechanism for Semisynthetic and Synthetic Endoperoxide Antimalarial Drugs. Angew. Chem. Int. Ed. 2007, 46, 6278–6283. 10.1002/anie.200604697. [DOI] [PubMed] [Google Scholar]
- Posner G. H.; Ploypradith P.; Parker M. H.; O’Dowd H.; Wou S.-H.; Northrop J.; Krasavin M.; Dolan P.; Kensler T. W.; Xie S.; Shapiro T. A. Antimalarial, antiproliferative, and antitumor activities of artemisinin-derived, chemically robust, trioxane dimers. J. Med. Chem. 1999, 42, 4275–4280. 10.1021/jm990363d. [DOI] [PubMed] [Google Scholar]
- Fröhlich T.; Çapcı Karagöz A.; Reiter C.; Tsogoeva S. B. Artemisinin-Derived Dimers: Potent Antimalarial and Anticancer Agents. J. Med. Chem. 2016, 59, 7360–7388. 10.1021/acs.jmedchem.5b01380. [DOI] [PubMed] [Google Scholar]
- Marschall M.; Freitag M.; Weiler S.; Sorg G.; Stamminger T. Recombinant Green Fluorescent Protein-Expressing Human Cytomegalovirus as a Tool for Screening Antiviral Agents. Antimicrob. Agents Chemother. 2000, 44, 1588–1597. 10.1128/AAC.44.6.1588-1597.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marschall M.; Niemann I.; Kosulin K.; Bootz A.; Wagner S.; Dobner T.; Herz T.; Kramer B.; Leban J.; Vitt D.; Stamminger T.; Hutterer C.; Strobl S. Assessment of drug candidates for broad-spectrum antiviral therapy targeting cellular pyrimidine biosynthesis. Antiviral Res. 2013, 100, 640–648. 10.1016/j.antiviral.2013.10.003. [DOI] [PubMed] [Google Scholar]
- Hutterer C.; Eickhoff J.; Milbradt J.; Korn K.; Zeitträger I.; Bahsi H.; Wagner S.; Zischinsky G.; Wolf A.; Degenhart C.; Unger A.; Baumann M.; Klebl B.; Marschall M. A novel CDK7 inhibitor of the Pyrazolotriazine class exerts broad-spectrum antiviral activity at nanomolar concentrations. Antimicrob. Agents Chemother. 2015, 59, 2062–2071. 10.1128/AAC.04534-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaptein S. J.; Efferth T.; Leis M.; Rechter S.; Auerochs S.; Kalmer M.; Bruggeman C. A.; Vink C.; Stamminger T.; Marschall M. The anti-malaria drug artesunate inhibits replication of cytomegalovirus in vitro and in vivo. Antiviral Res. 2006, 69, 60–69. 10.1016/j.antiviral.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Chou S.; Marousek G.; Auerochs S.; Stamminger T.; Milbradt J.; Marschall M. The unique antiviral activity of artesunate is broadly effective against human cytomegaloviruses including therapy-resistant mutants. Antiviral Res. 2011, 92, 364–368. 10.1016/j.antiviral.2011.07.018. [DOI] [PubMed] [Google Scholar]
- Efferth T.; Romero M. R.; Wolf D. G.; Stamminger T.; Marin J. J.; Marschall M. The antiviral activities of artemisinin and artesunate. Clin. Infect. Dis. 2008, 47, 804–811. 10.1086/591195. [DOI] [PubMed] [Google Scholar]
- Ho W. E.; Peh H. Y.; Chan T. K.; Wong W. S. Artemisinins: pharmacological actions beyond anti-malarial. Pharmacol. Ther. 2014, 142, 126–139. 10.1016/j.pharmthera.2013.12.001. [DOI] [PubMed] [Google Scholar]
- Hutterer C.; Niemann I.; Milbradt J.; Fröhlich T.; Reiter C.; Kadioglu O.; Bahsi H.; Zeitträger I.; Wagner S.; Einsiedel J.; Gmeiner P.; Vogel N.; Wandinger S.; Godl K.; Stamminger T.; Efferth T.; Tsogoeva S. B.; Marschall M. The broad-spectrum antiinfective drug artesunate interferes with the canonical nuclear factor kappa B (NF-kappaB) pathway by targeting RelA/p65. Antiviral Res. 2015, 124, 101–109. 10.1016/j.antiviral.2015.10.003. [DOI] [PubMed] [Google Scholar]
- Herget T.; Marschall M.. Recent Developments in Anti-Herpesviral Therapy Based on Protein Kinase Inhibitors. In New Concepts of Antiviral Therapy; Holzenburg A., Bogner E., Eds.; Springer US: Boston, MA, 2006; pp 351–371. [Google Scholar]
- Marschall M.; Stamminger T. Molecular targets for antiviral therapy of cytomegalovirus infections. Future Microbiol. 2009, 4, 731–742. 10.2217/fmb.09.40. [DOI] [PubMed] [Google Scholar]
- Hutterer C.; Hamilton S.; Steingruber M.; Zeitträger I.; Bahsi H.; Thuma N.; Naing Z.; Orfi Z.; Orfi L.; Socher E.; Sticht H.; Rawlinson W.; Chou S.; Haupt V. J.; Marschall M. The chemical class of quinazoline compounds provides a core structure for the design of anticytomegaloviral kinase inhibitors. Antiviral Res. 2016, 134, 130–143. 10.1016/j.antiviral.2016.08.005. [DOI] [PubMed] [Google Scholar]
- Saab A. M.; Guerrini A.; Sacchetti G.; Maietti S.; Zeino M.; Arend J.; Gambari R.; Bernadi F.; Efferth T. Phytochemical analysis and cytotoxicity towards multidrug-resistant leukemia cells of essential oils derived from Lebanese medicinal plants. Planta Med. 2012, 78, 1927–1931. 10.1055/s-0032-1327896. [DOI] [PubMed] [Google Scholar]
- Lambros C.; Vanderberg J. P. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J. Parasitol. 1979, 65, 418–420. 10.2307/3280287. [DOI] [PubMed] [Google Scholar]
- Smilkstein M.; Sriwilaijaroen N.; Kelly J. X.; Wilairat P.; Riscoe M. Simple and inexpensive fluorescence-based technique for highthroughput antimalarial drug screening. Antimicrob. Agents Chemother. 2004, 48, 1803–1806. 10.1128/AAC.48.5.1803-1806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beez D.; Sanchez C. P.; Stein W. D.; Lanzer M. Genetic predisposition favors the acquisition of stable artemisinin resistance in malaria parasites. Antimicrob. Agents Chemother. 2011, 55, 50–55. 10.1128/AAC.00916-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmig A.; Gekeler V.; Neumann M.; Frese G.; Handgretinger R.; Kardos G.; Diddens H.; Niethammer D. Susceptibility of Multidrug-resistant Human Leukemia Cell Lines to Human Interleukin 2-activated Killer Cells. Cancer Res. 1990, 50, 6793–6799. [PubMed] [Google Scholar]
- Gillet J.-P.; Efferth T.; Steinbach D.; Hamels J.; de Longueville F.; Bertholet V.; Remacle J. Microarray-based detection of multidrug resistance in human tumor cells by expression profiling of ATP-binding cassette transporter genes. Cancer Res. 2004, 64, 8987–8993. 10.1158/0008-5472.CAN-04-1978. [DOI] [PubMed] [Google Scholar]
- Efferth T.; Konkimalla V. B.; Wang Y.-F.; Sauerbrey A.; Meinhardt S.; Zintl F.; Mattern J.; Volm M. Prediction of Broad Spectrum Resistance of Tumors towards Anticancer Drugs. Clin. Cancer Res. 2008, 14, 2405–2412. 10.1158/1078-0432.CCR-07-4525. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.