

Abstract
We have recently identified a new class of filamenting temperature-sensitive mutant Z (FtsZ)-interacting compounds that possess a 2,4,6-trisubstituted pyrimidine–quinuclidine scaffold with moderate antibacterial activity. Employing this scaffold as a molecular template, a compound library of amine-linked 2,4,6-trisubstituted pyrimidines with 99 candidates was successfully established by employing an efficient convergent synthesis designed to explore their structure–activity relationship. The results of minimum inhibitory concentration (MIC) assay against Staphylococcus aureus strains and cytotoxicity assay against the mouse L929 cell line identified those compounds with potent antistaphylococcal properties (MIC ranges from 3 to 8 μg/mL) and some extent of cytotoxicity against normal cells (IC50 ranges from 6 to 27 μM). Importantly, three compounds also exhibited potent antibacterial activities against nine clinically isolated methicillin-resistant S. aureus (MRSA) strains. One of the compounds, 14av_amine16, exhibited low spontaneous frequency of resistance, low toxicity against Galleria mellonella larvae, and the ability to rescue G. mellonella larvae (20% survival rate at a dosage of 100 mg/kg) infected with a lethal dose of MRSA ATCC 43300 strain. Biological characterization of compound 14av_amine16 by saturation transfer difference NMR, light scattering assay, and guanosine triphosphatase hydrolysis assay with purified S. aureus FtsZ protein verified that it interacted with the FtsZ protein. Such a property of FtsZ inhibitors was further confirmed by observing iconic filamentous cell phenotype and mislocalization of the Z-ring formation of Bacillus subtilis. Taken together, these 2,4,6-trisubstituted pyrimidine derivatives represent a novel scaffold of S. aureus FtsZ inhibitors.
1. Introduction
The inexorable rise in the incidence of serious bacterial infections caused by multiple antibiotic-resistant bacteria in health care- and community-associated settings has become a pressing threat of public health worldwide.1 Of particular concern is the rise in the incidence of methicillin-resistant Staphylococcus aureus (MRSA) infections. MRSA can cause a wide range of illnesses, from mild skin and wound infections to pneumonia and bloodstream infections that cause sepsis and death. The Centers for Disease Control and Prevention of the United States estimates that over 80 000 severe MRSA infections occur annually, resulting in 11 000 deaths.2 This scenario has driven the search for novel classes of antistaphylococcal agents which act on novel bacterial drug targets.
The bacterial cell division machinery has been considered as an important field for exploring potential novel drug targets of antibacterial agents.3 The filamenting temperature-sensitive mutant Z (FtsZ) protein undoubtedly represents one of the well-characterized and exploitable antibacterial drug targets.4−10 FtsZ is a cytoplasmic protein and highly conserved tubulin-like guanosine triphosphatase (GTPase), playing an important role in bacterial cell division. In order for bacteria to carry out cell division, FtsZ monomers are required to localize mid cells through the precise positioning of cell division site positioning proteins and self-polymerize into single stranded straight protofilaments by means of head-to-tail association that curve upon the hydrolysis of guanosine triphosphate (GTP) molecules.11 Consecutive lateral contacts between FtsZ protofilaments produce FtsZ bundles, which eventually lead to the formation of a contractile ring called Z-ring at the mid cell. Following the subsequent involvement of other downstream cell division proteins, Z-ring contraction and depolymerization complete the cell division process to furnish identical daughter cells. Small molecules interfering the initial stage of the FtsZ polymerization are capable of blocking bacterial cell division, causing the abrogation of bacterial cell viability eventually. These types of compounds have great potential to be developed as efficacious antimicrobial agents with a novel mode of action for clinical applications. Several high-resolution X-ray crystal structures of FtsZ homologues have been reported.12 These results contributed to the knowledge regarding the general organization of the FtsZ protein structure, which is known to comprise two independent folding domains (Figure 1). The N-terminal domain forms a nucleotide-binding site (GTP-binding site, the upper red circle of Figure 1), whereas the C-terminal domain contains a flexible loop (T7 loop). Both the domains were interconnected via a long central helix 7 (H7) of high rigidity.
Figure 1.

Crystal structure of S. aureus FtsZ (PDB ID: 4DXD) with labeled GTP-binding site (red circle), T7 loop, central H7, and amino acids at positions 193, 196, and 263. The black oval indicates the binding site of PC190723. The chemical structures of PC190723, zantrin Z3, and FtsZ inhibitors 1–7 are shown.
Many FtsZ-interacting compounds have been discovered and reported to bind either the GTP-binding site or a cleft formed by the H7, T7 loop, and C-terminal β sheet (Figure 1, black oval). Some exhibit potent antibacterial activity with minimum inhibitory concentration (MIC) at the micromolar range. PC190723 (Figure 1, top left)13−15 and its prodrugs 1a,161b,17−19 and benzamide 2(20) have been demonstrated to exhibit in vitro and in vivo efficacy in a murine infection model. Moreover, X-ray crystallographic analysis revealed that PC190723 binds to a narrow cleft formed by the H7, T7 loop, and C-terminal β sheet (Figure 1, black oval).21 However, analysis of PC190723 drug-resistant mutants across various MRSA strains revealed that all PC190723 drug-resistant isolates had multiple mutations, resulting in amino acid substitutions that mapped to the FtsZ protein.22 These mutations mainly occurred at amino acid positions 193, 196, and 263 (Figure 1), which accounted for over 90% of PC190723 drug-resistant mutants. These results suggested that amino acid substitutions can alter slightly the overall shape of the binding pocket without interfering the normal function of FtsZ. Nevertheless, this change resulted in PC190723 no longer binding to this pocket, therefore causing drug resistance. Such findings may hinder the potential of PC190723 and other related compounds23 from being developed into agents that exhibit the potential to target the same binding pocket for further clinical development. On the other hand, several compounds targeting the GTP-binding site of FtsZ have also been demonstrated to exhibit potent antibacterial activity, including zantrin Z3,24,25 natural product chrysophaentin A 3,26 C8-substituted GTP analogues 4,27,28 berberine analogues 5,29,30 and naphthol derivatives 6(31,32) (Figure 1, right). Surprisingly, among these inhibitors, no drug-resistant mutants have been reported in the literature so far, presumably because of the fact that the GTP-binding site is very important for recognizing the GTP molecules. Amino acid substitutions at this binding pocket may cause improper recognition of GTP molecules and thus hinder normal GTP hydrolysis process, therefore losing the energy source to drive the polymerization of FtsZ monomers. We reasoned that specially designed small molecules, which can mimic and compete with GTP molecules to bind the GTP-binding site of FtsZ, may have greater potential to be developed as antimicrobial agents without acquiring drug resistance.
To take advantage of this idea, we have employed a combined approach of high-throughput virtual screening and biological evaluation of natural product libraries to discover compounds with new chemical scaffolds that target the GTP-binding site of the FtsZ protein. Our previous study identified a new class of FtsZ inhibitor 7 bearing a 2,4,6-trisubstituted pyrimidine and a chiral aminoquinuclidine moiety (Figure 1, bottom right).33 This class of compounds has been demonstrated to inhibit the GTPase hydrolysis activity of S. aureus FtsZ at a low micromolar IC50 value with moderate antimicrobial activity against S. aureus and Escherichia coli. Compound 7a also exhibited a strong synergistic effect against various MRSA and vancomycin-resistant Enterococcus faecium strains when combined with clinically used β-lactam antibiotics.34 However, the structural complexity of the chiral quinuclidine scaffold and limited compound availability from commercial sources have prevented further development. In the present study, we report a design and efficient synthesis of a novel amine-linked 2,4,6-trisubstituted pyrimidine compound library with the aims of simplifying the complex quinuclidine scaffold with simple amines and improving their antimicrobial activity. A promising compound exhibiting potent antimicrobial activity has been selected for further investigation regarding its interaction with the S. aureus FtsZ protein. Because of its structural novelty, potent antimicrobial activity, and high selectivity against S. aureus, a new series of such compounds may represent a promising starting point for further investigation.
2. Results and Discussion
2.1. Compound Library Design and Synthesis
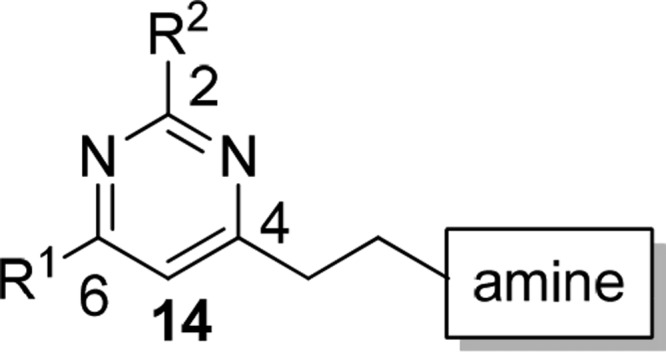
Molecular docking study of compound 7 and GTP molecules using S. aureus FtsZ revealed that the 2,4,6-trisubstituted pyrimidine moiety of 7 occupied exactly the same binding pocket as the guanosine moiety of GTP molecules through an extensive network of hydrogen bonds with the FtsZ protein, suggesting that the pyrimidine moiety is crucial for binding.33 Therefore, our molecular design strategy is to retain the pyrimidine moiety and replace the chiral quinuclidine moiety at position 4 with various substituted amine moieties, which offer desirable compound flexibility and physicochemical property.35 At the same time, various substituents will be installed at positions 2 and 6. As there was no structural information to guide the compound design, compound 7 was therefore divided into two parts (pyrimidine head and amine tail), as outlined in Figure 1. A compound library would then be designed and synthesized to sequentially interrogate the pyrimidine head and the amine tail using convergent synthesis. With the preliminary structure–activity relationship (SAR) in hand, compounds combining the promising structural motifs would then be synthesized by dedicated synthesis. On the basis of this approach, eight pyrimidine heads (Scheme 1) and 39 secondary amine tails (Chart 1) were designed and synthesized. Cyclic amine tails (for a seven-member homopiperazine ring, amine01–23, or for a six-member piperazine ring, amine24–34) and linear amine tails (amine35–39) were included in this study for comparison purpose. A general synthesis route of target compounds 14 is outlined in Scheme 1. The pyrimidine heads were easily obtained by the coupling of various acid chlorides 8a–d with silyl group-protected terminal alkyne 9 in the presence of triethylamine under Sonogashira conditions, followed by the subsequent addition of excess amidinium hydrochloride salts 10v–z in one pot, allowing a straightforward access to 2,4,6-trisubstituted pyrimidines 11 under mild conditions and in good yields.36 It is worthy to note that the use of unprotected terminal alkyne 9 should be avoided because of the relatively low yield of product. Deprotection of the silyl group of 11 by acidic treatment of concentrated hydrochloric acid in methanol afforded alcohols 12 in quantitative yield, which were further treated with triphenylphosphine and carbon tetrabromide to furnish the bromides 13 in high yield (two steps). It is also worthy to note that bromides 13 were found to be unstable at high temperatures (>50 °C) or under prolonged exposure of weakly basic medium at room temperature, affording the elimination product of 4-vinyl pyrimidine. It is recommended that bromides 13 should be prepared freshly and used immediately for the next step. The compound library of amine-linked 2,4,6-trisubstituted pyrimidines 14 was then constructed by mixing the freshly prepared bromides 13 with various amine derivatives amine01–39 (Chart 1) in acetonitrile (ACN) at room temperature. By employing this approach, a small library of amine-linked 2,4,6-trisubstituted pyrimidines with 99 candidates was successfully constructed for SAR study. Similarly, pyrimidines 14 with a sulfonamide group were also synthesized for SAR study, and their synthesis route is depicted in Scheme 2. Treatment of selected bromides 13ay and 13az with excess 1-Boc-homopiperazine afforded Boc-protected pyrimidines 14 (14ay_amine40 and 14az_amine40), respectively, in high yield, which were further acidified with trifluoroacetic acid (TFA) in dichloromethane (DCM) to furnish pyrimidines 14 (14ay_amine41 and 14az_amine41) with a secondary amino group. Pyrimidines 14 (14az_amine42–47) with a sulfonamide group were successfully synthesized in good yield by treating various commercially available sulfonyl chlorides with amine 14az_amine41.
Scheme 1.

(a) (i) 2 mol % Pd(PPh3)2Cl2, 4 mol % CuI, NEt3, tetrahydrofuran (THF), rt 1–2 h and (ii) Na2CO3, reflux 14 h; (b) concn HCl, MeOH, rt 2 h; (c) PPh3, CBr4, THF, rt 3–4 h; and (d) amine01–39 (refer to Chart 1 for chemical structures), ACN, rt 24 h.
Chart 1. Chemical Structures of Amines Used in This Study.

Scheme 2.

(a) 1-Boc-homopiperazine, ACN, rt, 14 h; (b) TFA, DCM, 0 °C, 2 h; and (c) various sulfonyl chlorides, NEt3, DCM, 0 °C, 3 h.
2.2. Evaluation of Antimicrobial Activities, SAR, and Cytotoxicity against Normal Cells
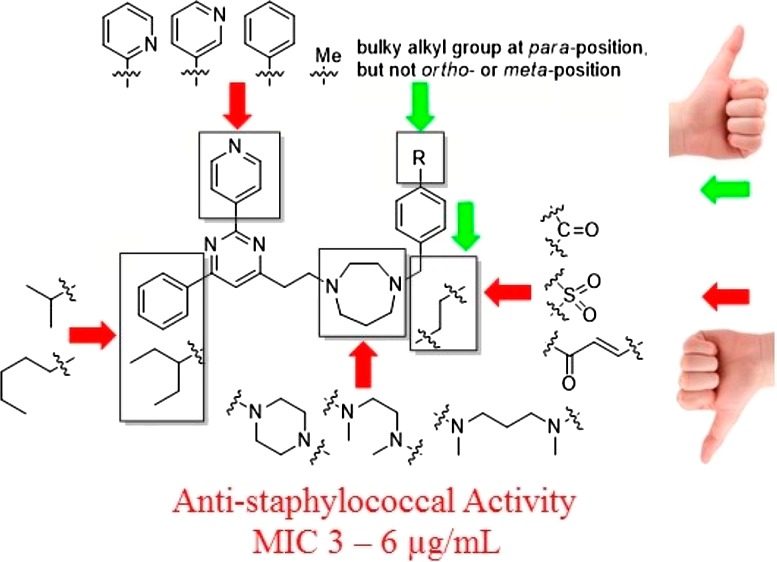
The compound library of 2,4,6-trisubstituted pyrimidines 14 was then evaluated simultaneously for (1) their antimicrobial activities against the Gram-positive S. aureus strain ATCC 29213 and the Gram-negative E. coli strain ATCC 25922 by measuring their MIC, which is the lowest concentration of a compound that prevents the visible growth of bacteria in a broth dilution susceptibility test according to the Clinical and Laboratory Standards Institute (CLSI) guidelines,37 and (2) their cytotoxicity against normal mouse fibroblasts L929 by measuring their IC50, which is the concentration of a compound that is required for 50% inhibition. Parent compounds 7a and 7b (Figure 1, bottom right) were selected as a positive control. Their antimicrobial activities against both the bacterial strains ranged from 24 to 64 μg/mL, demonstrating only moderate antibacterial activity and poor selectivity of killing between the two bacterial strains (Table 1, entries 1 and 2). By employing progressive MIC screening together with cytotoxicity assay, we have successfully identified several compounds that displayed superior antimicrobial activity and selectivity against S. aureus. The results of MIC screening and cytotoxicity assay are summarized in Table 1, in which only compounds with MIC values against S. aureus 29213 less than 8 μg/mL are shown. Compounds with MIC values higher than 8 μg/mL are shown in the Supporting Information (Table S1). In general, among all newly synthesized derivatives, most of them displayed potent inhibitory activities against the growth of S. aureus along with some extent of cytotoxicity against normal cells L929. The low IC50 values of L929 indicate that the compounds are very toxic and may have unselective interaction with protein targets other than FtsZ, causing high cytotoxicity against normal cells. However, all of them exhibited very weak inhibitory activities against the growth of E. coli even at a concentration of 64 μg/mL. Such a weak activity against E. coli may be attributed to their poor penetration ability into the cytoplasm of Gram-negative bacteria, which have an outer membrane of low permeability. From the compounds listed in Table 1, seven compounds displaying superior potency with MIC values at 3–4 μg/mL were identified, namely, 14dv_amine16, 14dv_amine06, 14dv_amine20, 14dv_amine07, 14dv_amine08, 14av_amine16, and 14av_amine20 (Table 1, entries 3–9). Compared with compound 7b, their potencies were dramatically improved by 16–21-fold. Interestingly, these compounds possess the common structural features of a 4-pyridyl (4-Py) group at position 2 and a cyclic seven-member homopiperazine ring substituted with a benzyl group at position 4. Detailed SAR analysis of 2,4,6-trisubstituted pyrimidine 14 derivatives is depicted in Figure 2. For the pyrimidine head, the 4-Py group of R2 of 14av_amine16 was very important for potent antibacterial activity. Replacing this functional group with others such as 3-pyridyl of 14ay_amine16, 2-pyridyl of 14az_amine16, phenyl of 14ax_amine16, and methyl groups of 14aw_amine16 resulted in a weak antibacterial activity, implying that the nitrogen atom of the 4-Py group is crucial for maintaining potent antibacterial activity. For R1 group of the pyrimidine head, both the phenyl of 14dv_amine16 and the 3-pentyl group of 14av_amine16 were favorable substituents. The less bulky isopropyl group of 14bv_amine16 and the linear n-pentyl group of 14cv_amine16 both caused poor antibacterial activity. In general, pyrimidine head bearing 4-Py group of R2 and phenyl or 3-pentyl groups of R1 exhibited the most potent antibacterial activity. For SAR analysis of the amine tail (14av_amine01–14av_amine39), there were two important structural features that gave rise to potent antibacterial activity: (1) the cyclic seven-member homopiperazine ring with a substituted benzyl group and (2) the benzyl group substituted with a bulky group (tert-butyl of 14av_amine07, trifluoromethyl of 14av_amine16, isopropyl of 14av_amine06, and bromo of 14av_amine08) at the para position, but not at the meta or ortho position, of 14av_amine17–18. Replacing the amine tail with a cyclic six-member piperazine ring of 14av_amine24, linear 1,2-diamine of 14av_amine35–36, or 1,3-diamine of 14av_amine37–38 resulted in a poor antibacterial activity. It is also worthy to note that replacing the benzylic carbon with a rigid functional group, such as carbonyl group of 14av_amine19, sulfonyl group of 14az_amine43–44, or α,β-unsaturated ketone group of 14av_amine21–23, caused a poor antibacterial activity. These results suggested that a more flexible and freely rotatable functional group should be installed at this position. This observation was further supported by the low MIC value of compound 14av_amine20, in which the benzylic carbon was replaced with a freely rotatable CH2CH2 group.
Table 1. MIC and IC50 Values of 2,4,6-Trisubstituted Pyrimidines against S. aureus 29213, E. coli ATCC 25922, and L929 Murine Fibroblast Cell Lines as Well as Calculated Selectivity Index (SI)a.
| MIC# |
||||||||
|---|---|---|---|---|---|---|---|---|
| entry | compound no. | R1 | R2 | amine no. | S. aureus | E. coli | L929 IC50* | SI^ |
| 1 | 7a | N.A. | N.A. | N.A. | 24 (51) | 32 | 36 ± 2 | 0.7 |
| 2 | 7b | N.A. | N.A. | N.A. | 64 (125) | 64 | N.D. | N.D. |
| 3 | 14dv_amine16 | phenyl | 4-Py | 16 | 3 (5) | >64 | 7 ± 1 | 1.4 |
| 4 | 14dv_amine06 | phenyl | 4-Py | 06 | 3 (5) | >64 | 6 ± 1 | 1.2 |
| 5 | 14dv_amine20 | phenyl | 4-Py | 20 | 3 (5) | >64 | 6 ± 1 | 1.2 |
| 6 | 14dv_amine07 | phenyl | 4-Py | 07 | 3 (5) | >64 | 6 ± 1 | 1.2 |
| 7 | 14dv_amine08 | phenyl | 4-Py | 08 | 3 (5) | >64 | 7 ± 1 | 1.4 |
| 8 | 14av_amine16 | 3-pentyl | 4-Py | 16 | 4 (8) | >64 | 8 ± 1 | 1.0 |
| 9 | 14av_amine20 | 3-pentyl | 4-Py | 20 | 4 (8) | >64 | 7 ± 1 | 0.9 |
| 10 | 14cv_amine06 | n-pentyl | 4-Py | 06 | 6 (10) | >64 | 7 ± 2 | 0.7 |
| 11 | 14cv_amine16 | n-pentyl | 4-Py | 16 | 6 (10) | >64 | 9 ± 1 | 0.9 |
| 12 | 14dv_amine12 | phenyl | 4-Py | 12 | 6 (10) | >64 | 9 ± 1 | 0.9 |
| 13 | 14dv_amine10 | phenyl | 4-Py | 10 | 6 (10) | >64 | 8 ± 1 | 0.8 |
| 14 | 14av_amine28 | 3-pentyl | 4-Py | 28 | 6 (10) | >64 | 27 ± 1 | 2.7 |
| 15 | 14av_amine38 | 3-pentyl | 4-Py | 38 | 6 (10) | >64 | 14 ± 1 | 1.4 |
| 16 | 14av_amine39 | 3-pentyl | 4-Py | 39 | 6 (10) | >64 | 9 ± 1 | 0.9 |
| 17 | 14av_amine24 | 3-pentyl | 4-Py | 24 | 6 (12) | >64 | 15 ± 2 | 1.3 |
| 18 | 14av_amine35 | 3-pentyl | 4-Py | 35 | 6 (12) | >64 | 10 ± 1 | 0.8 |
| 19 | 14av_amine13 | 3-pentyl | 4-Py | 13 | 6 (13) | >64 | 10 ± 1 | 0.8 |
| 20 | 14av_amine06 | 3-pentyl | 4-Py | 06 | 8 (16) | >64 | 8 ± 2 | 0.5 |
| 21 | 14av_amine07 | 3-pentyl | 4-Py | 07 | 8 (16) | >64 | 9 ± 2 | 0.6 |
| 22 | 14av_amine08 | 3-pentyl | 4-Py | 08 | 8 (16) | >64 | 8 ± 2 | 0.5 |
| 23 | 14av_amine17 | 3-pentyl | 4-Py | 17 | 8 (16) | >64 | 15 ± 1 | 0.9 |
| 24 | 14av_amine18 | 3-pentyl | 4-Py | 18 | 8 (16) | >64 | 18 ± 2 | 1.1 |
N.A.: not applicable; N.D.: not determined; # MIC, μg/mL; for S. aureus, the number in the parentheses is the MIC value calculated in the unit of μM; * concentration of a compound that is required for 50% inhibition (IC50), μM, N = 1–3 independent experiments, and the values are presented as mean ± standard error of mean; and ^ SI, it was calculated using the formula L929 IC50 (μM)/MIC value of S. aureus (μM).
Figure 2.
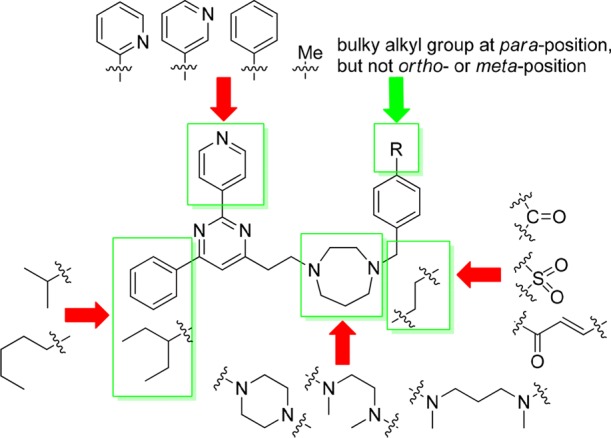
Summary of the SAR study. The red arrows indicate unfavorable substituents. The green arrow and the green boxes indicate favorable substituents.
Encouraged by these promising results, three compounds, namely, 14av_amine16, 14dv_amine16, and 14dv_amine06, were further selected to test against nine clinically isolated bacterial strains including S. aureus ATCC 29247, which is an ampicillin-resistant strain, S. aureus ATCC BAA-41, ATCC BAA-1717, ATCC BAA-1720, and ATCC 43300 which are methicillin-resistant strains, and four USA300 strains (#417, #757, #1799, and #2690), which are the predominant strain type of community-associated MRSA in the United States. PC19072338 and compound 5(29) were also synthesized according the literature for a positive control. The MIC screening results are summarized in Table 2. All compounds were found to retain potent antibacterial activities against these antibiotic-resistant strains with MIC values ranging from 1 to 6 μg/mL. Compounds 14av_amine16, 14dv_amine16, and 14dv_amine06 displayed a potent antibacterial activity against MRSA, with MIC values of 3–6 μg/mL, which are very close to those of reported FtsZ inhibitors (5 and PC190723). Compared with methicillin, which is the clinically used antibiotic with MIC values higher than 64 μg/mL against most of the MRSA, these pyrimidine derivatives exhibited significantly lower MIC values and thus exhibited the potential to be developed into new antistaphylococcal agents in the future. It is worthy to mention that compound 7 did not exhibit any antibacterial activity against these clinically isolated S. aureus strains (MIC > 48 μg/mL).
Table 2. MIC Values of Selected Pyrimidines against Nine Clinically Isolated S. aureus Strainsa.
| MIC (μg/mL) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| compound no. | strain 1 | strain 2 | strain 3 | strain 4 | strain 5 | strain 6 | strain 7 | strain 8 | strain 9 |
| PC190723 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| 5 | 2 | 2 | 2 | 2 | 4 | 2 | 2 | 2 | 2 |
| 14av_amine16 | 4 | 4 | 6 | 6 | 6 | 6 | 6 | 6 | 6 |
| 14dv_amine16 | 3 | 3 | 6 | 6 | 6 | 6 | 6 | 6 | 6 |
| 14dv_amine06 | 3 | 3 | 6 | 6 | 6 | 6 | 6 | 6 | 6 |
| methicillin | N.D. | >64 | >64 | >64 | >64 | >64 | 64 | 64 | 32 |
N.D.: not determined; strain 1: ATCC 29247; strain 2: ATCC BAA-1717; strain 3: ATCC BAA-1720; strain 4: ATCC BAA-41; strain 5: ATCC 43300; strain 6: USA300 #417; strain 7: USA300 #757; strain 8: USA300 #1799; and strain 9: USA300 #2690.
2.3. Investigation of the Potential for the Development of Resistance and in Vivo Biological Evaluation
A crucial attribute of potential antimicrobial agents is that the spontaneous development of drug resistance is not easily attained. To evaluate the spontaneous resistance rate of these compounds in MRSA, compound 14av_amine16 was selected for the study against the MRSA strain ATCC 43300. We plated 1 × 109 cells on agar plates containing 1% dimethyl sulfoxide (DMSO) or 8× the MIC of compound 14av_amine16 and observed the degree of bacterial growth. As shown in Figure 3A (left), almost confluent bacterial growth was observed for the DMSO-treated bacterial cells, yet no colony was observed for the plates treated with compound 14av_amine16 (Figure 3A, right) after 48 h incubation, implying that the rate of emergence of spontaneous resistant mutants was as low as <1 × 109. This low frequency of spontaneous resistance implies a reduced probability of rapid development of resistance against compound 14av_amine16 in clinical practice.
Figure 3.
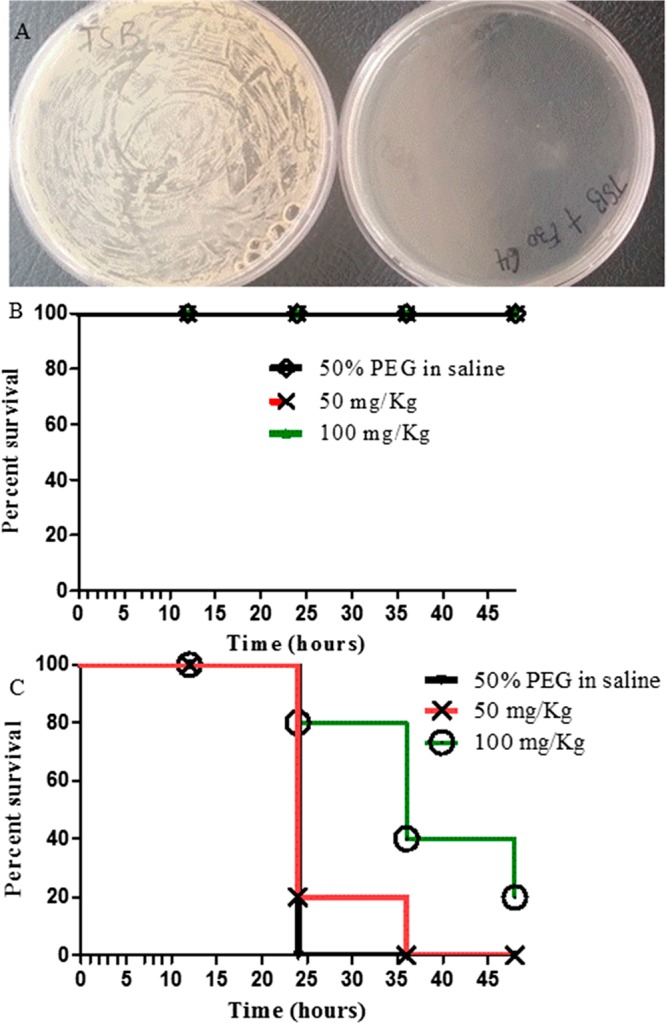
Frequency of resistance (FOR) study (A) and Kaplan–Meier survival analysis of G. mellonella larvae following the injection of various concentrations of compound 14av_amine16 without (B) and with (C) lethal dose of MRSA ATCC 43300 inoculation. Larvae were considered dead if they did not respond to physical stimuli. Data presented are the mean of three independent experiments.
As a preliminary measure of in vivo toxicity and efficacy, we next tested our lead compound 14av_amine16 in a Galleria mellonella model, which is an easy and inexpensive in vivo model with no ethical constraints for investigating the antibacterial activity of a compound.39 To examine the feasibility of using G. mellonella for compound 14av_amine16 toxicity study, we examined the ability of compound 14av_amine16 to kill G. mellonella larvae. Various concentrations of compound 14av_amine16 dissolved in 50% poly(ethylene glycol) (PEG) in saline (0, 50, and 100 mg/kg) were injected into the hemocoel of last-instar G. mellonella larvae, and survival was scored over time. All concentrations of compound 14av_amine16 tested did not kill the G. mellonella larvae during a 48 h period (Figure 3B). These data indicate that the formulation of 50% PEG in saline and compound 14av_amine16 are nontoxic and safe against G. mellonella larvae. Next, we investigated the efficacy of compound 14av_amine16 against G. mellonella larvae infected with MRSA ATCC 43300 (Figure 3C). Inoculation of lethal dose of 2.5 × 106 CFU/larva of MRSA ATCC 43300 led to a significant death rate. All larvae injected with 50% PEG in saline (0 mg/kg) died within a 24 h infection period. Injection of compound 14av_amine16 at a dose of 50 mg/kg to the infected larvae was found to prolong the survival time of the infected larvae to 36 h. Encouragingly, we observed 20% survival rate of the infected larvae over the infection period with increased dosage with this agent at 100 mg/kg. Compared to the vehicle group, it was found to be highly significant (p < 0.05). Further increased dosage did not lead to improved efficacy (data not shown). These data indicated that compound 14av_amine16 is capable of preventing or delaying the lethal effect of MRSA ATCC 43300 in G. mellonella larvae. This suggests that compound 14av_amine16 has strong potential for animal studies in future.
2.4. Saturation Transfer Difference NMR Study
Saturation transfer difference (STD) NMR spectroscopy is a powerful and unique tool that can detect the magnetization that was transferred from a protein to a bound ligand proton. It is commonly used to detect the interaction between low-molecular-weight compounds and large biomolecules.40 To get further insights into the interaction between compound 14av_amine16 and S. aureus FtsZ protein, STD NMR spectroscopy was employed to characterize the binding properties and identify the epitopes of small molecules that interact with a protein receptor. S. aureus FtsZ protein was expressed and purified as described in a previous report.29 STD NMR spectroscopy was performed, and the relative degrees of saturation for individual protons of compound 14av_amine16 are displayed in Figure 4, with the integral value of the largest signal set to 100%. The line boarding observed was caused by compound 14av_amine16 being in close contact with the FtsZ protein, resulting in slow tumbling rate of the protein–ligand complex, which also confirmed that compound 14av_amine16 indeed binds to FtsZ protein. All of the protons of compound 14av_amine16 showed some degree of enhancement, demonstrating that the molecule, except the homopiperazine moiety, is bound to the FtsZ protein. The largest amount of saturation transfer was observed for H1, indicating that the pyrimidine ring of compound 14av_amine16 is making more intimate contacts with the FtsZ protein.
Figure 4.

STD effect measured for compound 14av_amine16 binding to S. aureus FtsZ protein. Chemical structure and proton assignments of compound 14av_amine16 (upper panel); 1D (STD-off) spectrum displayed in blue with signal assignment (middle panel); and STD spectrum displayed in red (lower panel), and STD effects (ISTD/I0) for each proton are reported.
2.5. S. aureus FtsZ Polymerization and GTPase Hydrolysis Assay
Next, we sought to determine whether the binding of compound 14av_amine16 to S. aureus FtsZ protein led to a change in the polymerization activity and GTPase activity of the protein itself. To assay the polymerization activity, an in vitro light scattering assay, in which FtsZ polymerization was detected in solution by a time-dependent increase in light scattering as reflected by an increase in solution absorbance at 600 nm, was employed. Figure 5 shows the relative time-dependent absorbance at 600 nm of S. aureus FtsZ in the presence of compound 14av_amine16 at concentrations ranging from 0 to 30 μM. It was clearly demonstrated that compound 14av_amine16 potently suppressed the self-polymerization of S. aureus FtsZ protein, with the magnitude of these suppressing effects increasing with increasing compound concentration. Compared with the vehicle (1% DMSO), complete inhibition of FtsZ polymerization was observed at 30 μM of compound 14av_amine16. In addition, we also investigated the inhibitory effect of compound 14av_amine16 on GTPase hydrolysis activity according to the protocol described previously.33 Compounds 7a and 7b inhibited the GTPase hydrolysis activity with IC50 values at 73 and 189 μM, respectively. However, compound 14av_amine16 at 50 and 75 μM displayed only moderate inhibition of the GTPase hydrolysis activity at about 20 ± 3% and 25 ± 4%, respectively. GTPase hydrolysis activity at higher compound concentrations (>80 μM) cannot be measured because of the poor compound solubility, which causes precipitation in aqueous medium. Nonetheless, it seems very likely that compound 14av_amine16 suppresses the self-polymerization of the FtsZ protein, probably via disrupting the GTPase hydrolysis activity of the FtsZ protein.
Figure 5.
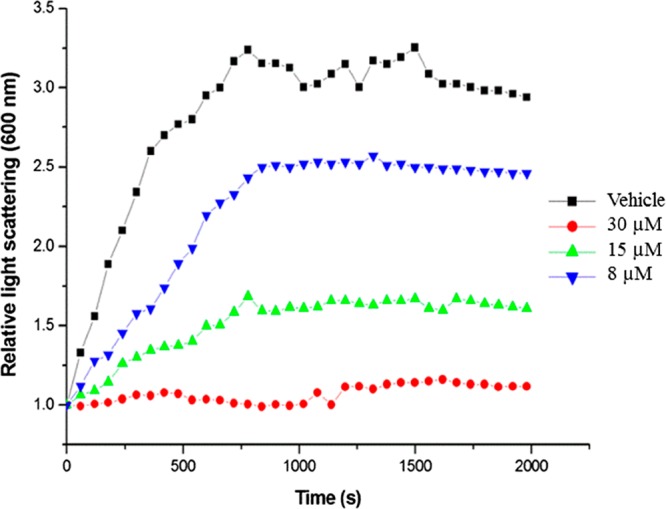
Effect of compound 14av_amine16 at various concentrations on the kinetics of S. aureus FtsZ polymerization. The experiments were performed in triplicate, with the symbols indicating the mean value (N = 3).
2.6. Effects on the Bacillus subtilis 168 Cell Morphology and Localization of the Z-Ring
The underlying mode of action of the antibacterial activity of compound 14av_amine16 was further investigated by the microscopic observation of a rod-shaped B. subtilis 168 cell morphology. Compound 14av_amine16 exhibited antibacterial activity against B. subtilis 168 with an MIC value at 12 μM. Treatment of B. subtilis cells at a sublethal concentration of compound 14av_amine16 significantly increased the cell length with average cell length > 20 μm (Figure 6B) as compared with the DMSO-treated cells (cell length < 5 μm, Figure 6A). Interestingly, such a phenomenon of cell elongation was also observed for other FtsZ inhibitors reported, strongly suggesting that 14av_amine16 interacts with the FtsZ protein in vivo. The iconic filamentous cell phenotype of FtsZ inhibitors was believed to be caused by the disruption and mislocalization of the Z-ring. To further confirm that, a fluorescence microscopic analysis of dynamic Z-ring formation in B. subtilis 168 was carried out by using a functional green fluorescent protein-tagged FtsZ in B. subtilis 168. In the absence of compound (DMSO-treated), fluorescent foci corresponding to the Z-ring formation were observed at the mid cell. Each cell possesses only one fluorescent focus, indicating the proper Z-ring formation and localization (Figure 6C, red arrow). By contrast, upon exposure to compound 14av_amine16, the bacteria cell lacked mid-cell foci. Instead, the FtsZ protein was randomly distributed as multiple discrete foci (Figure 6E, red arrows) throughout the whole elongated cell, demonstrating that compound 14av_amine16 caused obvious mislocalization of the Z-ring.
Figure 6.

Effects of DMSO or compound 14av_amine16 treated on the cell morphology of B. subtilis 168 (A,B), B. subtilis 168 with a functional green fluorescent protein-tagged FtsZ (C,E), and their corresponding phase-contrast microscopic pictures (D,F).
3. Conclusions
In this study, a total of 99 amine-linked 2,4,6-trisubstituted pyrimidines, many displaying potent antistaphylococcal activity and some extent of cytotoxicity against normal cells, has been synthesized systematically by varying the substitutions in 2, 4, and 6 positions. These compounds also exhibited potent antibacterial activities against clinically isolated MRSA strains. These promising results led to the efficacy testing of the lead compound 14av_amine16, which revealed a very low spontaneous FOR and limited toxicity against G. mellonella larvae. Investigation of the mode of action suggests that compound 14av_amine16 exerted its antibacterial activity by interacting with S. aureus FtsZ protein and suppressing its polymerization, resulting in obvious mislocalization of the Z-ring formation. In summary, these newly synthesized 2,4,6-trisubstituted pyrimidine derivatives represent a novel scaffold of FtsZ inhibitors.
4. Experimental Section
4.1. Chemical Synthesis
All NMR and mass spectra were recorded on a Bruker Avance-III spectrometer and a Micromass Q-TOF-2 spectrometer by the electrospray ionization (ESI) mode, respectively. STD NMR experiments were performed on a Bruker Avance III 600 NMR spectrometer equipped with a QCI cryoprobe at 298 K. All reagents and solvents were of reagent grade and were used without further purification unless otherwise stated. The thin-layer chromatography (TLC) plates (silica gel 60F254, 0.25 mm thickness) were purchased from E. Merck. Flash column chromatographic purifications were performed on MN silica gel 60 (230–400 mesh). Compound purity was determined by an Agilent 1100 series high-performance liquid chromatography (HPLC) system installed with a Prep-Sil scalar column (4.6 mm × 250 mm, 5 μm) at an UV detection of 254 nm (reference at 450 nm). All tested compounds were shown to have >95% purity according to HPLC. Amine20 was prepared from homopiperazine and 1-(2-bromoethyl)-4-trifluoromethylbenzene. Amine38 was prepared from N,N-diethyl-1,3-diaminopropane and 4-(trifluoromethyl)benzyl bromide. Other amines used in this study are commercially available.
4.2. General Procedure I for the Synthesis of Pyrimidines 11
To a well-stirred solution of Pd(PPh3)2Cl2 (0.4 mmol) and CuI (0.8 mmol) in degassed THF (150 mL) under a nitrogen atmosphere, were added NEt3 (13 mmol), acid chloride 8 (10 mmol), and alkyne 9 (10 mmol) successively. The reaction mixture was stirred for 3 h until complete conversion as monitored by TLC. After that, Na2CO3 (34 mmol) and amidinium hydrochloride salt 10 (12 mmol) were added to the reaction mixture. The resulting suspension was heated to reflux for 14 h. After cooling to room temperature, the reaction mixture was filtered through a short pad of silica gel to remove excess Na2CO3. The obtained pale brown filtrate was evaporated under reduced pressure to crude oil which was further subjected to flash column chromatography on silica gel with gradient elution (5% ethyl acetate (EA) in Hex to 30% EA in Hex) to furnish the desired product 11.
4.2.1. 4-(2-((tert-Butyldimethylsilyl)oxy)ethyl)-6-(pentan-3-yl)-2-(pyridin-4-yl)pyrimidine (11av)
The titled compound 11av was obtained as a colorless oil (0.32 g, 83% yield) according to the general procedure I described above: 1H NMR (400 MHz, CDCl3): δ 8.76 (d, J = 4.40 Hz, 2H), 8.35 (d, J = 5.87 Hz, 2H), 7.03 (s, 1H), 4.10 (t, J = 6.11 Hz, 2H), 3.01 (t, J = 6.11 Hz, 2H), 2.50–2.64 (m, 1H), 1.69–1.87 (m, 4H), 0.77–0.88 (m, 15H), −0.02 (s, 6H); 13C NMR (101 MHz, chloroform-d): δ 173.8, 168.2, 161.9, 150.3, 145.7, 122.2, 119.9, 61.9, 51.0, 41.2, 27.5, 25.8, 18.2, 12.0, −5.5; LRMS (ESI) m/z: 386 (M+ + H, 100); HRMS (ESI) calcd for C22H36N3OSi (M+ + H), 386.2628; found, 386.2621.
4.3. General Procedure II for the Synthesis of Alcohols 12
To a well-stirred solution of pyrimidine 11 (5 mmol) in methanol (20 mL) at room temperature, was added excess concn hydrochloric acid (5 mL) dropwise. The reaction mixture was stirred for 2 h until complete conversion as monitored by TLC. After that, 2 M NaOH solution was added to neutralize the reaction mixture (pH 7), followed by extraction with DCM. The combined organic layers were dried over MgSO4, filtered, and evaporated to afford the desired product 12, which is pure enough for the next step.
4.3.1. 2-(6-(Pentan-3-yl)-2-(pyridin-4-yl)pyrimidin-4-yl)ethanol (12av)
The titled compound 12av was obtained as a colorless oil (0.27 g, quantitative yield) according to the general procedure II described above: 1H NMR (400 MHz, CDCl3): δ 8.46 (d, J = 4.40 Hz, 2H), 8.15 (d, J = 5.87 Hz, 2H), 6.96 (s, 1H), 5.00 (br, OH, 1H), 4.00 (t, J = 6.11 Hz, 2H), 3.36 (t, J = 6.11 Hz, 2H), 2.56–2.63 (m, 1H), 1.71–1.88 (m, 4H), 0.83 (t, J = 6.11 Hz, 6H); 13C NMR (101 MHz, CDCl3): δ 175.3, 168.5, 161.3, 149.8, 145.8, 122.1, 118.0, 60.1, 40.8, 30.5, 27.9, 12.0; LRMS (ESI) m/z: 272 (M+ + H, 100); HRMS (ESI) calcd for C16H22N3O (M+ + H), 272.1763; found, 272.1750.
4.4. General Procedure III for the Synthesis of Bromides 13
To a well-stirred solution of alcohol 12 (3 mmol) and PPh3 (3.6 mmol) in THF (50 mL) at room temperature, was added carbon tetrabromide (3.6 mmol) once. The reaction mixture was stirred for 3 h until complete conversion as monitored by TLC. The reaction mixture was filtered through a short pad of silica gel. The obtained pale brown filtrate was evaporated under reduced pressure to crude oil which was further subjected to flash column chromatography on silica gel with gradient elution (5% EA in Hex to 20% EA in Hex) to furnish the desired product 13.
4.4.1. 4-(2-Bromoethyl)-6-(pentan-3-yl)-2-(pyridin-4-yl)pyrimidine (13av)
The titled compound 13av was obtained as a colorless oil (0.27 g, 80% yield) according to the general procedure III described above: 1H NMR (400 MHz, CDCl3): δ 8.77 (dd, J = 1.47, 4.40 Hz, 2H), 8.26–8.42 (m, 2H), 6.99 (s, 1H), 3.88 (t, J = 6.60 Hz, 2H), 3.37 (t, J = 6.85 Hz, 2H), 2.51–2.67 (m, 1H), 1.69–1.89 (m, 4H), 0.84 (t, J = 7.34 Hz, 6H); 13C NMR (101 MHz, CDCl3): δ 174.5, 166.5, 162.2, 150.3, 145.4, 122.2, 119.4, 51.0, 40.4, 30.1, 27.5, 12.0; LRMS (ESI) m/z: 334 (M+ + H, 100); HRMS (ESI) calcd for C16H21N3Br (M+ + H), 334.0919; found, 334.0914.
4.5. General Procedure IV for the Synthesis of Pyrimidines 14
To a well-stirred solution of slightly excess amine (1.1 mmol) in ACN (10 mL) at room temperature, was added a freshly prepared bromide 13 (1 mmol) solution in ACN (10 mL) once. The reaction mixture was stirred for 14 h until complete conversion as monitored by TLC. The reaction mixture was evaporated under reduced pressure to crude oil which was further subjected to flash column chromatography on silica gel with gradient elution (1% MeOH in DCM to 5% MeOH in DCM) to furnish the desired product 14.
4.5.1. 1-(2-(6-(Pentan-3-yl)-2-(pyridin-4-yl)pyrimidin-4-yl)ethyl)-4-(4-(trifluoromethyl)benzyl)-1,4-diazepane (14av_amine16)
The titled compound 14av_amine16 was obtained as a colorless oil (0.27 g, 53% yield) according to the general procedure IV described above: 1H NMR (400 MHz, CDCl3): δ 8.72–8.80 (m, 2H), 8.31–8.38 (m, 2H), 7.52–7.60 (m, J = 8.31 Hz, 2H), 7.40–7.49 (m, J = 8.31 Hz, 2H), 6.99 (s, 1H), 3.67 (s, 2H), 2.96–3.08 (m, 4H), 2.83–2.90 (m, 2H), 2.77–2.83 (m, 2H), 2.66–2.72 (m, 4H), 2.57 (tt, J = 5.81, 8.38 Hz, 1H), 1.73–1.87 (m, 6H), 0.83 (t, J = 7.58 Hz, 6H); 13C NMR (101 MHz, CDCl3): δ 173.9, 169.0, 161.9, 150.3, 145.7, 143.9, 128.8, 125.6, 125.2, 125.1, 125.1, 125.1, 122.9, 122.2, 119.1, 62.2, 57.2, 55.3, 55.2, 54.3, 53.9, 51.0, 35.9, 27.8, 27.5, 12.0; LRMS (ESI) m/z: 512 (M+ + H, 100), 534 (M+ + Na, 13); HRMS (ESI) calcd for C29H37N5F3 (M+ + H), 512.3001; found, 512.2986.
4.6. Antimicrobial Susceptibility Tests
The MIC values of all compounds were determined using the broth microdilution procedure in accordance with the CLSI guidelines37 and our previous reports.29,44
4.7. Cytotoxicity (IC50) Assay toward the L929 Cell Line
Standard MTS assay was performed to determine the cytotoxicity of all compounds toward the L929 cells. Briefly, 10 000 cells were mixed with compounds at different concentrations in a final volume of 100 μL in each well of a 96-well plate, followed by 3 days incubation at 37 °C. DMSO at 1% was used as a solvent control. The half-maximal inhibition of the compounds was determined using a CellTiter 96 AQueous assay (Promega). An aliquot of the freshly prepared MTS/phenazine methosulfate mixture at a ratio of 20:1 was added into each well, followed by 2 h incubation at 37 °C. Optical absorbance at 490 nm was measured with a microplate reader. The IC50 values were determined from the dose–response curves of the MTS assay (Prism 4.0). All experiments were performed in triplicates, and the results will be presented as the average of the three independent measurements.
4.8. Expression and Purification of S. aureus FtsZ Protein
Expression and purification of S. aureus FtsZ protein were performed as previously reported, which was stored at −20 °C as a lyophilized powder.29,44 A stock solution of the FtsZ protein was then prepared from the lyophilized powder for subsequent STD NMR study, light scattering assay, and GTPase activity assay.
4.9. STD NMR Study
The STD NMR study of compound 14av_amine16 was performed according to our previous report.42 Group epitope mapping was performed by integrating the STD signal of the individual protons with respect to the strongest STD signal, which was assigned to a value of 100%.
4.10. Light Scattering Assays and GTPase Activity Assays
The light scattering assays and GTPase activity assays were performed as previously described.29,34,44
4.11. FOR Determination
MRSA ATCC 43300 cells were grown to the late-exponential phase (1 × 109 CFU/mL) and spread on agar plates containing 1% DMSO or compound 14av_amine16 at 8 folds of MIC level. The plates were incubated for 48 h to allow resistant mutants to grow. The spontaneous FOR was calculated as the number of resistance colonies divided by the number of CFUs originally plated. The assay was performed in triplicates.
4.12. Evaluation of Toxicity and Antimicrobial Efficacy Using a G. mellonella Model of Infection
A G. mellonella model of S. aureus infection was adopted to test the in vivo toxicity and efficacy of compound 14av_amine16 according to previous reports.41,43 Groups of G. mellonella larvae (N = 10) weighing 200–300 mg were used in these assays. All larvae were incubated at 37 °C, and the mortality rates were assessed at 12 h interval for 48 h after different treatments. All larvae were scored as dead if they showed no signs of movement to physical stimuli. All injections were carried out into the last left proleg of the larvae using a 10 μL Hamilton syringe. For toxicity evaluation, the larvae were injected with 0 (vehicle), 50, and 100 mg/kg compound 14av_amine16 only. For antimicrobial efficacy, the larvae were inoculated with a lethal dose of 10 μL of S. aureus 43300 (2.5 × 106 CFU). All larvae received compound 14av_amine16 of different dosages (0, 50, and 100 mg/kg) 1 h before bacterial inoculation. All data were analyzed for statistical significance using a log rank and χ squared test with 1° of freedom.
4.13. Bacterial Morphology of B. subtilis 168
The bacterial morphology study of B. subtilis 168 was performed under a phase-contrast optical microscope as previously described.29,44
4.14. Z-Ring Visualization of B. subtilis 168
The study of Z-ring visualization of B. subtilis 168 was performed under a fluorescence and phase-contrast microscope using the Olympus FSX100 bio imaging navigator software as previously described.29,44
Acknowledgments
We acknowledge the support by the Research Grants Council of Hong Kong (grant no. 15100115), the Innovation and Technology Commission Hong Kong SAR, and The Hong Kong Polytechnic University.
Glossary
Abbreviations
- MRSA
methicillin-resistant Staphylococcus aureus
- FtsZ
filamenting temperature-sensitive mutant Z
- GTPase
guanosine triphosphatase
- GTP
guanosine triphosphate
- MIC
minimum inhibitory concentration
- VREF
vancomycin-resistant Enterococcus faecium
- SAR
structure–activity relationship
- ACN
acetonitrile
- TFA
trifluoroacetic acid
- DCM
dichloromethane
- STD
saturation transfer difference
- CLSI
Clinical and Laboratory Standards Institute
- CA-MHB
cation-adjusted Mueller Hinton broth
- DMSO
dimethyl sulfoxide
- TLC
thin-layer chromatography
- 4-Py
4-pyridyl
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b00701.
1H NMR, 13C NMR, and mass spectrometry data of all compounds 11–14 and 1H and 13C NMR spectra and MIC values of compounds 14 (PDF)
Author Contributions
K.-F.C. and N.S. contributed equally. The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Klevens R. M.; Morrison M. A.; Nadle J.; et al. Invasive methicillin-resistant staphylococcus aureus infections in the united states. JAMA, J. Am. Med. Assoc. 2007, 298, 1763–1771. 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- http://www.cdc.gov/drugresistance/pdf/ar-threats-2013-508.pdf (accessed July 15, 2017).
- Lock R. L.; Harry E. J. Cell-division inhibitors: new insights for future antibiotics. Nat. Rev. Drug Discovery 2008, 7, 324–338. 10.1038/nrd2510. [DOI] [PubMed] [Google Scholar]
- Sass P.; Brötz-Oesterhelt H. Bacterial cell division as a target for new antibiotics. Curr. Opin. Microbiol. 2013, 16, 522–530. 10.1016/j.mib.2013.07.006. [DOI] [PubMed] [Google Scholar]
- Foss M. H.; Eun Y.-J.; Weibel D. B. Chemical–Biological Studies of Subcellular Organization in Bacteria. Biochemistry 2011, 50, 7719–7734. 10.1021/bi200940d. [DOI] [PubMed] [Google Scholar]
- Schaffner-Barbero C.; Martín-Fontecha M.; Chacón P.; Andreu J. M. Targeting the Assembly of Bacterial Cell Division Protein FtsZ with Small Molecules. ACS Chem. Biol. 2012, 7, 269–277. 10.1021/cb2003626. [DOI] [PubMed] [Google Scholar]
- Ma S.; Ma S. The Development of FtsZ Inhibitors as Potential Antibacterial Agents. ChemMedChem 2012, 7, 1161–1172. 10.1002/cmdc.201200156. [DOI] [PubMed] [Google Scholar]
- Ojima I.; Kumar K.; Awasthi D.; Vineberg J. G. Drug discovery targeting cell division proteins, microtubules and FtsZ. Bioorg. Med. Chem. 2014, 22, 5060–5077. 10.1016/j.bmc.2014.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley K. A.; Santos T. M. A.; Nepomuceno G. M.; Huynh V.; Shaw J. T.; Weibel D. B. Targeting the bacterial division protein FtsZ. J. Med. Chem. 2016, 59, 6975–6998. 10.1021/acs.jmedchem.5b01098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson D. E.; Kim M. B.; Moore J. T.; O’Brien T. E.; Sorto N. A.; Grove C. I.; Lackner L. L.; Ames J. B.; Shaw J. T. Comparison of Small Molecule Inhibitors of the Bacterial Cell Division Protein FtsZ and Identification of a Reliable Cross-Species Inhibitor. ACS Chem. Biol. 2012, 7, 1918–1928. 10.1021/cb300340j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Hsin J.; Zhao L.; Cheng Y.; Shang W.; Huang K. C.; Wang H.-W.; Ye S. FtsZ Protofilaments Use a Hinge-Opening Mechanism for Constrictive Force Generation. Science 2013, 341, 392–395. 10.1126/science.1239248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T.; Han X.; Yu J.; Yao M.; Tanaka I. Structural Change in FtsZ Induced by Intermolecular Interactions between Bound GTP and the T7 Loop. J. Biol. Chem. 2014, 289, 3501–3509. 10.1074/jbc.m113.514901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydon D. J.; Stokes N. R.; Ure R.; Galbraith G.; Bennett J. M.; Brown D. R.; Baker P. J.; Barynin V. V.; Rice D. W.; Sedelnikova S. E.; Heal J. R.; Sheridan J. M.; Aiwale S. T.; Chauhan P. K.; Srivastava A.; Taneja A.; Collins I.; Errington J.; Czaplewski L. G. An inhibitor of FtsZ with potent and selective anti-staphylococcal activity. Science 2008, 321, 1673–1675. 10.1126/science.1159961. [DOI] [PubMed] [Google Scholar]
- Haydon D. J.; Bennett J. M.; Brown D.; Collins I.; Galbraith G.; Lancett P.; Macdonald R.; Stokes N. R.; Chauhan P. K.; Sutariya J. K.; Nayal N.; Srivastava A.; Beanland J.; Hall R.; Henstock V.; Noula C.; Rockley C.; Czaplewski L. Creating an Antibacterial with in Vivo Efficacy: Synthesis and Characterization of Potent Inhibitors of the Bacterial Cell Division Protein FtsZ with Improved Pharmaceutical Properties. J. Med. Chem. 2010, 53, 3927–3936. 10.1021/jm9016366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang S.; Wang C.; Venter H.; Li X.; Wang Y.; Guo L.; Ma R.; Ma S. Synthesis and Biological Evaluation of Novel FtsZ-targeted 3-arylalkoxy-2,6-difluorobenzamides as Potential Antimicrobial Agents. Chem. Biol. Drug Des. 2016, 87, 257–264. 10.1111/cbdd.12658. [DOI] [PubMed] [Google Scholar]
- Kaul M.; Mark L.; Zhang Y.; Parhi A. K.; LaVoie E. J.; Pilch D. S. An FtsZ-Targeting Prodrug with Oral Antistaphylococcal Efficacy In Vivo. Antimicrob. Agents Chemother. 2013, 57, 5860–5869. 10.1128/aac.01016-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul M.; Mark L.; Zhang Y.; Parhi A. K.; LaVoie E. J.; Pilch D. S. Pharmacokinetics and in vivo antistaphylococcal efficacy of TXY541, a 1-methylpiperidine-4-carboxamide prodrug of PC190723. Biochem. Pharmacol. 2013, 86, 1699–1707. 10.1016/j.bcp.2013.10.010. [DOI] [PubMed] [Google Scholar]
- Kaul M.; Mark L.; Zhang Y.; Parhi A. K.; Lyu Y. L.; Pawlak J.; Saravolatz S.; Saravolatz L. D.; Weinstein M. P.; LaVoie E. J.; Pilch D. S. TXA709, an FtsZ-Targeting Benzamide Prodrug with Improved Pharmacokinetics and Enhanced In Vivo Efficacy against Methicillin-Resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2015, 59, 4845–4855. 10.1128/aac.00708-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepak A. J.; Parhi A.; Madison M.; Marchillo K.; VanHecker J.; Andes D. R. In Vivo Pharmacodynamic Evaluation of an FtsZ Inhibitor, TXA-709, and Its Active Metabolite, TXA-707, in a Murine Neutropenic Thigh Infection Model. Antimicrob. Agents Chemother. 2015, 59, 6568–6574. 10.1128/aac.01464-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes N. R.; Baker N.; Bennett J. M.; Berry J.; Collins I.; Czaplewski L. G.; Logan A.; Macdonald R.; MacLeod L.; Peasley H.; Mitchell J. P.; Nayal N.; Yadav A.; Srivastava A.; Haydon D. J. An Improved Small-Molecule Inhibitor of FtsZ with Superior In Vitro Potency, Drug-Like Properties, and In Vivo Efficacy. Antimicrob. Agents Chemother. 2013, 57, 317–325. 10.1128/aac.01580-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T.; Yamane J.; Mogi N.; Yamaguchi H.; Takemoto H.; Yao M.; Tanaka I. Structural reorganization of the bacterial cell-division protein FtsZ from Staphylococcus aureus. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2012, 68, 1175–1188. 10.1107/s0907444912022640. [DOI] [PubMed] [Google Scholar]
- Tan C. M.; Therien A. G.; Lu J.; Lee S. H.; Caron A.; Gill C. J.; Lebeau-Jacob C.; Benton-Perdomo L.; Monteiro J. M.; Pereira P. M.; Elsen N. L.; Wu J.; Deschamps K.; Petcu M.; Wong S.; Daigneault E.; Kramer S.; Liang L.; Maxwell E.; Claveau D.; Vaillancourt J.; Skorey K.; Tam J.; Wang H.; Meredith T. C.; Sillaots S.; Wang-Jarantow L.; Ramtohul Y.; Langlois E.; Landry F.; Reid J. C.; Parthasarathy G.; Sharma S.; Baryshnikova A.; Lumb K. J.; Pinho M. G.; Soisson S. M.; Roemer T. Restoring Methicillin-Resistant Staphylococcus aureus Susceptibility to β-Lactam Antibiotics. Sci. Transl. Med. 2012, 4, 126ra35. 10.1126/scitranslmed.3003592. [DOI] [PubMed] [Google Scholar]
- Kaul M.; Parhi A. K.; Zhang Y.; LaVoie E. J.; Tuske S.; Arnold E.; Kerrigan J. E.; Pilch D. S. A Bactericidal Guanidinomethyl Biaryl That Alters the Dynamics of Bacterial FtsZ Polymerization. J. Med. Chem. 2012, 55, 10160–10176. 10.1021/jm3012728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margalit D. N.; Romberg L.; Mets R. B.; Hebert A. M.; Mitchison T. J.; Kirschner M. W.; RayChaudhuri D. Targeting cell division: Small-molecule inhibitors of FtsZ GTPase perturb cytokinetic ring assembly and induce bacterial lethality. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 11821–11826. 10.1073/pnas.0404439101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nepomuceno G. M.; Chan K. M.; Huynh V.; Martin K. S.; Moore J. T.; O’Brien T. E.; Pollo L. A. E.; Sarabia F. J.; Tadeus C.; Yao Z.; Anderson D. E.; Ames J. B.; Shaw J. T. Synthesis and Evaluation of Quinazolines as Inhibitors of the Bacterial Cell Division Protein FtsZ. ACS Med. Chem. Lett. 2015, 6, 308–312. 10.1021/ml500497s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaza A.; Keffer J. L.; Bifulco G.; Lloyd J. R.; Bewley C. A. Chrysophaentins A–H, Antibacterial Bisdiarylbutene Macrocycles That Inhibit the Bacterial Cell Division Protein FtsZ. J. Am. Chem. Soc. 2010, 132, 9069–9077. 10.1021/ja102100h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcelo F.; Huecas S.; Ruiz-Ávila L. B.; Cañada F. J.; Perona A.; Poveda A.; Martín-Santamaría S.; Morreale A.; Jiménez-Barbero J.; Andreu J. M. Interactions of Bacterial Cell Division Protein FtsZ with C8-Substituted Guanine Nucleotide Inhibitors. A Combined NMR, Biochemical and Molecular Modeling Perspective. J. Am. Chem. Soc. 2013, 135, 16418–16428. 10.1021/ja405515r. [DOI] [PubMed] [Google Scholar]
- Huecas S.; Marcelo F.; Perona A.; Ruiz-Ávila L. B.; Morreale A.; Cañada F. J.; Jiménez-Barbero J.; Andreu J. M. Beyond a Fluorescent Probe: Inhibition of Cell Division Protein FtsZ by mant-GTP Elucidated by NMR and Biochemical Approaches. ACS Chem. Biol. 2015, 10, 2382–2392. 10.1021/acschembio.5b00444. [DOI] [PubMed] [Google Scholar]
- Sun N.; Chan F.-Y.; Lu Y.-J.; Neves M. A. C.; Lui H.-K.; Wang Y.; Chow K.-Y.; Chan K.-F.; Yan S.-C.; Leung Y.-C.; Abagyan R.; Chan T.-H.; Wong K.-Y. Rational Design of Berberine-Based FtsZ Inhibitors with Broad-Spectrum Antibacterial Activity. PLoS One 2014, 9, e97514 10.1371/journal.pone.0097514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domadia P. N.; Bhunia A.; Sivaraman J.; Swarup S.; Dasgupta D. Berberine targets assembly of Escherichia coli cell division protein FtsZ. Biochemistry 2008, 47, 3225–3234. 10.1021/bi7018546. [DOI] [PubMed] [Google Scholar]
- Artola M.; Ruiz-Avila L. B.; Vergoñós A.; Huecas S.; Araujo-Bazán L.; Martín-Fontecha M.; Vázquez-Villa H.; Turrado C.; Ramírez-Aportela E.; Hoegl A.; Nodwell M.; Barasoain I.; Chacón P.; Sieber S. A.; Andreu J. M.; López-Rodríguez M. L. Effective GTP-Replacing FtsZ Inhibitors and Antibacterial Mechanism of Action. ACS Chem. Biol. 2015, 10, 834–843. 10.1021/cb500974d. [DOI] [PubMed] [Google Scholar]
- Ruiz-Avila L. B.; Huecas S.; Artola M.; Vergoñós A.; Ramírez-Aportela E.; Cercenado E.; Barasoain I.; Vázquez-Villa H.; Martín-Fontecha M.; Chacón P.; López-Rodríguez M. L.; Andreu J. M. Synthetic Inhibitors of Bacterial Cell Division Targeting the GTP-Binding Site of FtsZ. ACS Chem. Biol. 2013, 8, 2072–2083. 10.1021/cb400208z. [DOI] [PubMed] [Google Scholar]
- Chan F.-Y.; Sun N.; Neves M. A. C.; Lam P. C.-H.; Chung W.-H.; Wong L.-K.; Chow H.-Y.; Ma D.-L.; Chan P.-H.; Leung Y.-C.; Chan T.-H.; Abagyan R.; Wong K.-Y. Identification of a New Class of FtsZ Inhibitors by Structure-Based Design and in Vitro Screening. J. Chem. Inf. Model. 2013, 53, 2131–2140. 10.1021/ci400203f. [DOI] [PubMed] [Google Scholar]
- Chan F.-Y.; Sun N.; Leung Y.-C.; Wong K.-Y. Antimicrobial activity of a quinuclidine-based FtsZ inhibitor and its synergistic potential with β-lactam antibiotics. J. Antibiot. 2015, 68, 253–258. 10.1038/ja.2014.140. [DOI] [PubMed] [Google Scholar]
- O’Shea R.; Moser H. E. Physicochemical Properties of Antibacterial Compounds: Implications for Drug Discovery. J. Med. Chem. 2008, 51, 2871–2878. 10.1021/jm700967e. [DOI] [PubMed] [Google Scholar]
- Karpov A. S.; Müller T. J. J. Straightforward novel one-pot enaminone and pyrimidine syntheses by coupling-addition-cyclocondensation sequences. Synthesis 2003, 2815–2826. 10.1055/s-2003-42480. [DOI] [Google Scholar]
- Clinical and Laboratory Standards Institute . Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard, 7th ed.; CLSI document M07-A7; Clinical and Laboratory Standards Institute: Wayne, PA, 2006. [Google Scholar]
- Sorto N. A.; Olmstead M. M.; Shaw J. T. Practical Synthesis of PC190723, an Inhibitor of the Bacterial Cell Division Protein FtsZ. J. Org. Chem. 2010, 75, 7946–7949. 10.1021/jo101720y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai C. J.-Y.; Loh J. M. S.; Proft T. Galleria mellonella infection models for the study of bacterial diseases and for antimicrobial drug testing. Virulence 2016, 7, 214–229. 10.1080/21505594.2015.1135289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer M.; Meyer B. Characterization of Ligand Binding by Saturation Transfer Difference NMR Spectroscopy. Angew. Chem., Int. Ed. 1999, 38, 1784–1788. 10.1002/(sici)1521-3773(19990614)38:12<1784::aid-anie1784>3.3.co;2-h. [DOI] [PubMed] [Google Scholar]
- Lam T.; Hilgers M. T.; Cunningham M. L.; Kwan B. P.; Nelson K. J.; Brown-Driver V.; Ong V.; Trzoss M.; Hough G.; Shaw K. J.; Finn J. Structure-Based Design of New Dihydrofolate Reductase Antibacterial Agents: 7-(Benzimidazol-1-yl)-2,4-diaminoquinazolines. J. Med. Chem. 2014, 57, 651–668. 10.1021/jm401204g. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Chan F.-Y.; Sun N.; Lui H.-K.; So P.-K.; Yan S.-C.; Chan K.-F.; Chiou J.; Chen S.; Abagyan R.; Leung Y.-C.; Wong K.-Y. Structure-based design, synthesis, and biological evaluation of isatin derivatives as potential glycosyltransferase inhibitors. Chem. Biol. Drug Des. 2014, 84, 685–696. 10.1111/cbdd.12361. [DOI] [PubMed] [Google Scholar]
- Horn K. S. V.; Burda W. N.; Fleeman R.; Shaw L. N.; Manetsch R. Antibacterial Activity of a Series of N2,N4-Disubstituted Quinazoline-2,4-diamines. J. Med. Chem. 2014, 57, 3075–3093. 10.1021/jm500039e. [DOI] [PubMed] [Google Scholar]
- Lui H. K.Alkoxy- and amino-benzamides as inhibitors of the bacterial cell division protein FtsZ and antibacterial agents. M. Phil. Thesis, The Hong Kong Polytechnic University, Hong Kong, 2014. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.