

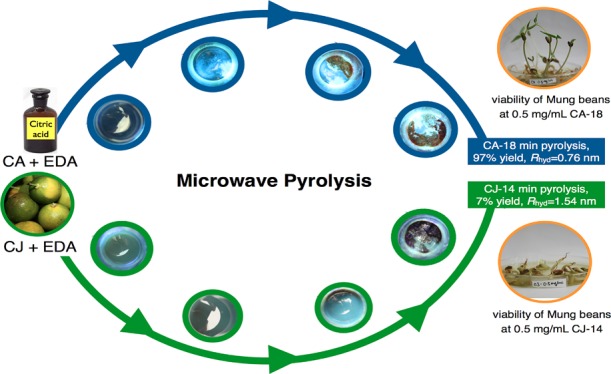
Abstract
Tracking dynamic cellular processes necessitates fluorescent materials that are photostable, biocompatible, water-soluble, nanosized, and nontoxic. In this study, highly fluorescent carbon dots (CDs) were produced from cheap and readily available sources, citric acid (CA) and Philippine citrus (Citrus japonica Thunb.) or calamansi juice (CJ) via a microwave-assisted method. A number of synthetic conditions were investigated systematically to optimize the preparation of CDs from CA and CJ. The formation mechanism, surface chemistry, and photoluminescence of CA-based CDs (CA-CDs) and CJ-based CDs (CJ-CDs) were evaluated after each stage of pyrolysis in detail using different characterization techniques, such as dynamic light scattering, diffusion-ordered spectroscopy, atomic force microscopy, ζ potential, X-ray diffraction, Fourier transform infrared spectroscopy, 1H and 13C nuclear magnetic resonance spectroscopy, and absorption/emission spectroscopy. Gram-scale pyrolysis of CA with ethylenediamine (EDA) and CJ with EDA were carried out to provide CA-CDs (CA-18) within 18 min total pyrolysis time at 97% yield and CJ-CDs (CJ-14) within 14 min total pyrolysis time at 7% yield. Aqueous suspensions of CA-18 and CJ-14 CDs gave comparable bright blue luminescence at 462 nm. CA-CDs were shown to be nontoxic for mung beans up to 2 mg/mL, whereas CJ-CDs with higher surface negative charges inhibited growth above 0.5 mg/mL. This study demonstrates that bright CA- and CJ-CDs can be produced in gram-scale quantities using inexpensive methods. The size, amount, and extent of EDA incorporation are important in contributing to the formation of highly emissive particles.
Introduction
Carbon nanodots, also known as carbon dots (CDs), are a new class of discrete quasispherical amorphous particles richly decorated with surface oxygen/nitrogen groups.1 CDs have become one of the most studied nanomaterials during the past decade due to their promising properties. CDs exhibit unique optical and thermal characteristics, chemical stability, biocompatibility, water solubility, and tunable surface functionalities. These properties enable the application of CDs in the fields of chemical sensing, biological monitoring, drug delivery, catalysis, photovoltaic devices, and optoelectronic devices.1−6 For example, because of the superior optical properties of CDs and their compatibility with different cell lines (e.g., T47D,7 MCF-7, HT-29,8 HeLa,9 and Ehrlich ascites carcinoma10) and sprouts,11 these can potentially replace other photoluminescent materials like fluorescent dye probes and quantum dots (such as, CdSe and PbS) in sensing, cell tracking, and bioimaging.2 Fluorescent dye probes have been traditionally used for imaging but may photobleach after long exposures. Quantum dots became popular choices; however, their high production cost and toxicity inherent in these systems limit their applications in biomedicine.7,8
CDs have been accessed using different methods via the top-down or bottom-up approach.2−5 In the top-down approach, CDs are formed from the breakdown of larger C structures, such as carbon fibers, carbon nanotubes, fullerene, or graphite electrodes, utilizing any of these methods such as arc discharge, laser ablation, electrochemical synthesis, or plasma. Conversely, in the bottom-up approach, CDs are formed from fusion of small molecular precursors or polycyclic aromatics via stepwise organic synthesis, combustion, hydrothermal treatment, acidic oxidation, or microwave or ultrasound methods. Among the different techniques under the bottom-up approach, the microwave method offers a simple, scalable, cost-effective, and facile manner to access CDs.1,3,4,9,12−18
Even with these different techniques, challenge remains in scaleup preparation of photoluminscent CDs in a short period of time.19 Aside from the treatment method and carbon source consideration, scalable highly photoluminescent CDs can be obtained by overcoming the challenges such as carbon aggregation, size, surface chemistry, and exact chemical identity.9,19,20 Size control and uniformity of properties have been previously addressed by post-treatment, such as polyacrylamide gel electrophoresis,21 filtration,8 column chromatography,11,22 or centrifugation10/dialysis.6,9,23 Conversely, surface properties are critical for solubility, as well as altering the electronic density for tuning the optoelectronic properties and for selected applications. Surface properties of CDs can be modified by surface functionalization24−27 with amines9,14,28 or passivation,29,30 doping,31 cross-linking,32 or other post-treatments.
Among the different CDs reported, citric acid (CA) with amines like ethylenediamine (EDA) provided CDs with some of the highest quantum yields (QYs) (i.e., 9%33 using direct pyrolysis; 30.2,14 44,18 and 52%20 using microwave; 8019 and 94%34 using hydrothermal treatment). CDs prepared without the presence of amines show only low emission intensities.14 Because CA has been a well-studied CD precursor, in addition to having high quantum yield in the presence of EDA, we chose CA as our carbon precursor to benchmark the results obtained with another CA-rich carbon source, calamansi juice (CJ, from Citrus japonica Thunb., family of Rutaceae). Calamansi is a citrus fruit found in several regions, such as, Southeast Asia, India, West Indies, and Central and North America. Its juice (CJ) possesses the aroma of orange and the zesty taste of lime and is commonly used as food seasoning or as food additive. CA (50 g/L) is one of the major organic acids found in CJ, whereas the other organic acids, ascorbic (0.36 g/L), malic (2.13 g/L), and succinic (1.48 g/L), are found in trace amounts. CJ of the Philippine variety has a juice titratable acidity (% CA) of 5.66 ± 0.07%.36 Aside from organic acids, CJ also contains sugars (e.g., fructose, glucose, and sucrose) and free and/or bound phenolic acids (e.g., caffeic, p-coumaric, ferulic, and sinapic acids).36 CJ has also been shown, in a preliminary study,37 to possess CDs with higher emission intensities compared to those from other carbon sources, such as orange, dalandan, and suha/pomelo juice (data not shown).
Unique to other studies, we address some of the challenges in CD preparation by a systematic study on how to inexpensively scale up CD production, focusing also on the time required to synthesize these materials via a kitchen microwave. In addition, the formation mechanism of CDs was investigated utilizing two different carbon sources (CA and CJ) as these are pyrolyzed over time. The particle size and surface chemistry of the pyrolysis products were monitored to understand how highly photoluminescent CDs are formed. Hence, the preparation of CDs from CA and CJ was optimized using a microwave-assisted method by varying the microwave power, sample volume, heating time pattern, amines utilized, and the concentration of CA and amines. We also described the approach we utilized to reproducibly scale up the CD synthesis to gram-scale quantities (100 mL of 0.1 g CA/mL deionized (DI) water or CJ) in a short period of time. The mechanism of formation, size, surface chemistry, and photoluminescence of CA-based CDs (CA-CDs) and CJ-based CDs (CJ-CDs) were also investigated via the time study approach using different characterization techniques (i.e., dynamic light scattering (DLS), ζ potential, diffusion-ordered spectroscopy (DOSY), Fourier transform infrared (FT-IR) spectroscopy, 1H and 13C nuclear magnetic resonance (NMR) spectroscopy, ultraviolet–visible (UV–vis) spectroscopy, photoluminescence emission spectroscopy, dispersibility, and stability of the CA- and CJ-CDs at different pHs (acidic, physiologic, and basic)). Finally, we used the viability of mung beans to examine the toxicity of CA and CJ-CDs.
Results and Discussion
Establishing Preliminary CD Synthesis Parameters
CA-CDs were prepared using microwave pyrolysis by varying the microwave power at a fixed concentration of 0.1 g/mL of CA in water. Two settings were selected to determine the microwave power conditions, 50% (equivalent to 430 W) and 70% (equivalent to 630 W). These power settings fall within the range where CDs were obtained in previous studies.9,13,14,16−18 CA solutions of 1, 3, 5, and 7 mL sample volumes were pyrolyzed in a 5 min (min) repetitive heating pattern using a microwave until red-brown solids were obtained. Extreme sample sputtering was observed from the vials with 5 and 7 mL sample volumes, resulting in losses. As a result, 1, 2, and 3 mL initial sample volumes were used and CDs were obtained after four to six times 5 min repetitive heating (Table S1). Using 50 or 70% power at 1, 2, and 3 mL, pale yellow solids were obtained with average product yields between 32 and 87%. Comparable to the microwave power conditions carried out by Zhai14 with CA and EDA at 700 W, Qu17 using CA and urea at 750 W, and Du18 using CA and EDA at 720 W for pyrolysis; the CDs formed using 70% (630 W) power and 3 mL initial sample volume provided the highest photoluminescence intensities with peak maxima at 462 nm. Hence, these conditions were utilized for the succeeding experiments.
Using 70% power and 3 mL of CA stock solution of concentration 0.1 g/mL, we investigated the effect of pyrolysis time pattern on CD synthesis. Since a 5 min repetitive heating used during the initial optimization trapped excessive heat and steam inside the microwave, a 2 min repetitive time pattern was tested. CDs synthesized at 70% power with the 3 mL sample using 0.1 g/mL CA at 2 min repetitive heating provided CDs with highest photoluminescence intensities among the series (Table S1).
Amine-passivated CDs have been shown to provide increased fluorescence emission compared to that from CDs pyrolyzed in the absence of amines.14,33,35 Different amine-passivating agents (primary, secondary, tertiary, and aromatic amines) were tested using the 3 mL sample solution of 1:1 molar ratio (CA/amine) at 70% power and 2 min repetitive heating pattern (Table S1). Diethylamine (CA_DEA; fluorescence emission at 462 nm = 17.1 au) and triethylamine (CA_TEA = 38.6 au) did not lead to significant fluorescence enhancement compared with CDs with no amine (CA_no amine = 12.9 au). However, EDA (CA_EDA = 2130 au) and quinine-passivated samples (CA_quinine = 1338 au) provided enhanced fluorescence emission. From the results, even when CA-quinine provided one of the highest emission intensities, the quinine only pyrolyzed sample (quinine = 14.3 au) inherently possesses low fluorescence emission intensity, this may interfere with the emission results when used as amine passivating agent. Conversely, triethylamine molecules were only N-doped on the CD surface, whereas EDA and DEA have available nitrogen groups that can form amide bonds with the carboxylic acid moieties of CA, in addition to its nitrogen atoms being incorporated into the carbon core during heating.14 Thus, these amines enhanced the photoluminescence of the CDs, with EDA giving the highest photoluminesence enhancement effect on the CD due to its dual role as N-doping precursors and surface passivation agents. The presence of amide bonds is evident in the FT-IR analysis (Figure S1).
EDA, one of the widely used passivating agents, has been shown to produce bright blue fluorescent CDs with CA.14,18,19,33,35 The UV–vis spectrum provides a weak absorption band at 353 nm and a peak at 200 nm with a corresponding shoulder peak at 237 nm, whereas the CA-only sample gives a low absorption peak at 200 nm and no peak at 353 nm (Figure 1A). The CA-EDA sample solution is clear under normal light but produced a bright blue light under UV (365 nm). A photoluminescence emission peak at 462 nm (recorded highest peak) was observed upon excitation of an aqueous solution of CA-EDA at 355 nm, which is in contrast to the absence of peak at 462 nm for CA-only samples (Figure 1B). The coexistence of N-doping and the formation of surface amide groups contributed to the fluorescence enhancement of the CDs. This enhancement was also higher than that provided by DEA due to the presence of more amine groups capable of forming surface amide groups with CDs. It is notable that solutions obtained from pyrolyzed CA-only (12.9 au) or EDA-only (5.4 au) samples provided low fluorescence emission intensities but the combined pyrolysis of the two gave samples with high photoluminescence intensity. In a different study, EDA also significantly enhanced the CD photoluminescence intensity, from an emission intensity of less than 25 au to 3686 au (see CA-1G vs CAEDA-1G, Table S1). This indicated that EDA effectively doped and passivated the CA-CDs, which resulted in high fluorescence emissions. With these findings, only EDA was utilized as the amine additive in the next experiments.
Figure 1.

Representative (A) UV–vis absorption spectrum of 0.1 g CD/mL DI water and (B) photoluminescence emission spectrum of 1.2 mg CD/mL DI water. CA CDs were prepared with EDA (348 μL of EDA and 0.1 g/mL CA in DI water, for 10 mL) and without EDA (0.1 g/mL DI water) using 70% microwave power at 2 min repetitive heating and 3 mL sample volume (see Table S1).
The amount of carbon source (i.e., the concentration of CA stock solution) and the concentration of passivating agent (EDA) were varied. The CDs were synthesized at varying CA concentrations: 0.5, 1, 1.5, 3, and 5 g CA in DI water (10 mL) while keeping the EDA concentration constant, i.e., 348 μL. Pyrolysis was conducted using 70% power, 3 mL sample volume, and 2 min repetitive heating. Photoluminescence intensity slightly increased from sample CAEDA-0.5G (0.5 g CA/10 mL of DI water) (3609 au) to CA-EDA-1G (3686 au) and then decreased significantly as the CA concentration was increased to 1.5 g CA/10 mL of DI water (CAEDA-1.5G, 2496 au). Using CAEDA-3G and CAEDA-5G, low emission intensities of less than 200 au were obtained (Table S1). These results indicate that increasing the CA amount does not provide CDs with enhanced emission. CAEDA-1G provided CDs with the highest photoluminescence intensity and a higher yield of 100% (calculated based on the amount of CA) compared with 60% for CAEDA-0.5G.
The presence of EDA not only significantly enhances the CD emission intensity but also shortens the pyrolysis time from 22 min (CA-0.5G) to 16 min (CAEDA-0.5G), from 32 min (CA-1G) to 22 min (CAEDA-1G), from 26 min (CA-1.5G) to 16 min (CAEDA-1.5G), from 18 min (CA-3G) to 12 min (CAEDA-3G), and from 22 min (CA-5G) to 12 min (CAEDA-5G). Addition of EDA shortens the synthesis time for at least 6–10 min (Table S1).
Likewise, the amount of EDA was varied while keeping the CA concentration constant at 0.1 g/mL in DI water (10 mL) at 1:0.25 (0.1 g/mL CA solution/86.9 μL EDA, CAEDA-0.25), 1:0.5 (0.1 g/mL:173.37 μL, CAEDA-0.50), 1:1.00 (0.1 g/mL:348 μL, CAEDA-1.00), 1:1.50 (0.1 g/mL:521 μL, CAEDA-1.50), and 1:2.00 (0.1 g/mL:695 μL, CAEDA-2.00). CDs synthesized with 1:1 CA/EDA molar ratio (CAEDA-1.00) provided the highest photoluminescence intensity at 462 nm with 3686 au, followed by CAEDA-2.00 (2945 au) and then CAEDA-1.50 (2418 au). CAEDA-0.25 and CAEDA-0.50 gave CDs with low emission intensities (<400 au) (Table S1).
CJ-CDs were also prepared at varying amounts of EDA concentration (Table S1). CDs were pyrolyzed using pure CJ and EDA at 70% power, 3 mL sample volume, and 2 min repetitive heating with the CJ/EDA ratios of 1:0.25 (10 mL CJ/86.9 μL EDA, CJEDA-0.25), 1:0.50 (10 mL:173.37 μL, CJEDA-0.50), 1:1.00 (10 mL:348 μL, CJEDA-1.00), 1:1.50 (10 mL:521 μL, CJEDA-1.50), and 1:2.00 (10 mL:695 μL, CJEDA-2.00). The photoluminescence intensity increased with increasing amine concentration from CJEDA-0.25 (41 au) to CJEDA-1.50 (2285 au) but decreased as the amine concentration was further increased.
Gram-Scale Synthesis of CA- and CJ-CDs
Access to gram-scale quantities of CDs having uniform particle size and surface chemistry with high photoluminescent emission remains a challenge.19 Herein, we varied the sample volume from small scale 3 mL in small vials, to 50 and 100 mL in large beakers to examine the large-scale synthesis. Pyrolysis of the small-scale sample, CAEDA-3 mL, took longer (22 min), followed by CAEDA-50 mL (12 min) and CAEDA-100 mL (18 min) (Table S2). Pyrolysis of a larger amount of sample volume gave higher amounts of CA CDs, starting from CAEDA-3 mL, 0.316 g (100% yield), to CAEDA-50 mL, 4.88 g (98% yield), and CAEDA-100 mL, 9.93 g (99% yield) (Table S2). Both the UV absorption and photoluminescence emission intensities of CA-CDs produced using CAEDA-50 mL (emission at 462 nm = 3461 au) and CAEDA-100 mL (3759 au) were comparable with those of CAEDA-3 mL (3578 au) (Figure 2). The repeatability of the synthesis was evaluated using six trials of CAEDA solutions at 100 mL, with a total heating time of 18 min at 2 min repetitive heating (CA-18). The results showed that the yields for the six trials are almost quantitative (Table S3). All of the CA with EDA-pyrolyzed samples showed characteristic absorbance peaks at 200 with a shoulder peak at around 240 nm and a broad peak at 350 nm and photoluminescence emission between 420 and 600 nm with a maximum emission intensity at 462 nm (Figure 3). The average photoluminescence emission for the six samples was 3724 ± 165 au. Even though the CDs obtained can be easily dispersed in water, the effect of centrifugation on the spectroscopic properties of the samples was also examined because smaller particles were shown to provide higher photoluminescent emission than that from coarser ones.23 Hence, separation of “bigger” particles through centrifugation should be beneficial. The same CA CDs from trials 1–5 (repeatability test) were used for this study, except that the samples were subjected to centrifugation. Results showed no visible particles settled and the emission intensity of the resulting CA CD supernatant (average = 3670 ± 247 au) was comparable to that of samples that were not subjected to centrifugation. In addition, CDs subjected to a dialysis membrane (Spectra/Por 7, 1000 molecular weight cut-off) for 48 h also provided comparable emission intensities for both samples subjected to or without dialysis.37 Thus, these results showed that CA-CDs can be reproduced in gram quantities.
Figure 2.

Comparison of CA-CDs synthesized with EDA at bulk (100 and 50 mL) and small (3 mL) scales using 70% microwave power, 2 min repetitive heating, 0.1 g/mL CA in DI water/3480 μL EDA/100 mL solution. (A) UV absorption spectra of CA-CDs at 1.21 × 10–5 g CD/mL DI water at small (3 mL) and bulk (100 and 50 mL) scales. (B) Photoluminescence emission spectra of 1.2 mg CD/mL DI water small-scale CA-CDs (3 mL) and bulk CA-CDs (100 and 50 mL). Samples were excited using a 355 nm laser (see Table S2).
Figure 3.

Absorption and emission plots of gram-scale synthesis of six samples of CA-18 and CJ-14 CDs. CDs were synthesized using 100 mL starting volume in the presence of EDA. (A) UV absorption spectra of 1.21 × 10–5 g CA-18 CD/mL DI water from different trials. (B) Photoluminescence emission spectra of 1.2 mg CA-18 CD/mL DI water. (C) UV absorption spectra of 1.21 × 10–5 g CJ-14 CD/mL DI water from different trials. (D) Photoluminescence emission spectra of 1.2 mg CJ-14 CD/mL DI water. Samples were excited using a 355 nm laser (see Table S3).
Gram-scale synthesis of CJ-CDs was carried out using 100 mL sample volume with 1:1 molar ratio (100 mL CJ/3480 μL EDA) and pyrolyzed at 70% power (630 W) at 2 min repetitive heating. Results showed that freshly extracted CJ has a higher emission intensity at 462 nm (3743 au) compared to that of the old CJ (3077 au), which was extracted for at least 2 weeks and kept refrigerated before pyrolysis. Repeatability study was carried out using six trials containing fresh CJ obtained from different calamansi fruit sources and EDA. Pyrolysis was carried out for 14 min at 2 min repetitive heating (CJ-14). Dark brown solids weighing ∼7 g and an average yield of 7% were obtained from the pyrolysis of the different trials (Table S3). Similar to CA-CDs, CJ-CDs also provided photoluminescence emission between 420 and 600 nm with the highest emission point at 462 nm and an intensity ranging from 3500 to 3900 au from the different samples (average = 3816 ± 164 au) (Figure 3D). From these results, CJ-CDs were prepared in gram quantities with emission intensities higher than those of CA CDs. This is also supported by the higher quantum yield for CJ-14 (Φ = 2.0%) compared to that for CA-18 (Φ = 1.7%) CDs using quinine sulfate as standard.
Heating Time Study of the Formation of CA- and CJ-CDs
Understanding the stages of transition toward the formation of CDs during microwave pyrolysis may allow facile identification of the parameters required to achieve desired material properties. Herein, we are interested in examining the properties such as the functionality (the chemical identities of the CDs); the size, as the raw materials transform to bright and highly photoluminescent CDs; and the mechanism of formation of CDs. Several studies have reported on the possible mechanisms associated with photoluminescence:30 quantum confinement, excitons of carbon,38 free zigzag sites and/or edge defects,39 emissive traps due to the presence of surface and/or molecular state determined by hybridization of the carbon backbone and functional moieties present upon surface passivation,13,19,29,40 conjugated π domains41 on the surface or at the core of the CDs,5,42 and the crosslink-enhanced emission effect.32 However, the exact mechanism of formation and the photoluminescence behavior of CDs from CA or CJ with EDA are not completely clear.
Hence, a heating time study was performed where solution samples that were not heated and those heated at increments of 2–4 min toward the conversion to CDs were analyzed. For CA, selected samples were pyrolyzed at 2 min increment for a total of 4, 8, 10, 12, 14, 16, 18, and 20 min, whereas for CJ, the samples were pyrolyzed at 2 min increment for a total of 4, 8, 10, 12, 14, and 16 min. Of the microwave power, 70% was used for both CA and CJ. The samples were analyzed using UV–vis absorption and photoluminescence spectroscopy. The dispersibility and pH stability tests for the CDs were performed. We further used FT-IR and NMR spectroscopy to identify the evolution of surface functional groups of CDs in the process of pyrolysis. The size, charge, and state of the CDs were determined using dynamic light scattering (DLS), diffusion-ordered spectroscopy (DOSY), atomic force microscopy (AFM), ζ potential, and X-ray diffraction (XRD).
As the pyrolysis time increased from CA-0 (no heating) to CA-12 (CA sample pyrolyzed for a total of 12 min), the samples showed little to no fluorescence when exposed under UV light (365 nm) (Figure 4A). After subjecting the sample to 14, 16, 18, or 20 min pyrolysis (Figure 4A UV; CA-14, CA-16, CA-18, or CA-20, respectively), the aqueous suspensions of the samples visualized under UV light showed bright blue fluorescent solutions. Water has completely evaporated, and sample CA-14 was already dry after pyrolysis. For CA-16, burnt residues were already evident under normal light and the amount of residue increased over time. All pyrolyzed CA samples gave a broad UV absorption peak at 200 nm with a shoulder peak at 237 nm, which can be ascribed to the π-to-π* transition of C=C bonds or the carbonic core for CA-EDA (Figure S2A). The peak at 237 nm for CA-0 provided an absorption of 0.18. This peak dipped to a minimum with CA-12 (0) and peaked at CA-18 (0.30). However, sample CA-20 provided an absorbance of 0.06, less than that of CA-18. These results indicated the changes occurring on the carbonic core of the particles. However, even when CA-14 provided a bright blue light under UV, only the samples pyrolyzed for more than 18 min (CA-18) provided a weak n-to-π* absorption band at 353 nm, indicative of the surface group (C=O) (Figure S2A). Photoluminescence emission spectra provided the signature peak at 462 nm when the pyrolyzed samples (CA-14, -16, -18, and -20) were excited at 355 nm (Figure S2B). This is similar to the results observed by Du18 and Zhu19 for CDs obtained via microwave and hydrothermal pyrolysis using CA and EDA. Samples CA-18 (3819 au) and CA-20 (3964 au) provided solutions with high photoluminescence intensities among the series (Figure S2B). However, the photoluminescence intensity for CA-20 was observed to be higher and the intensity of the UV absorption band at 350 nm was lower compared to that of CA-18. DOSY results showed that only CA-18 CDs were formed (Figure S7); this may suggest that further heating resulted into a more photoluminescent system, but this does not mean that CDs are produced. Conversely, CJ samples provided bright blue fluorescence under UV light only after subjecting the sample to 10, 12, 14, or 16 min total pyrolysis time (Figure 4B UV; CJ-10, CJ-12, CJ-14, or CJ-16, respectively). From samples CJ-12 to CJ-14, the yellow CJ was converted into a dark brown viscous solid (Figure 4B, normal). The UV absorbance peaks at 237 and 350 nm for CJ-12, CJ-14, and CJ-16 have comparable absorbance intensities. Conversely, this weak band at 350 nm was not evident for the other CJ samples (Figure S2C). Strong photoluminescence emission at 462 nm was apparent only for samples pyrolyzed for a total time of 12, 14, and 16 min, with the highest emission peak obtained with CJ-14 at 3778 au (Figure S2D). Pyrolyzed CA samples were obtained as off white to tan-colored solids, whereas yellow to brown solids were obtained for CJ samples. It is gratifying to note that CA and CJ samples fluoresce under UV light both in aqueous and in dry solid states, similar to that reported by Xu et al.43 using CDs produced with calcium citrate and urea.
Figure 4.

Time study photo of (A) CA and (B) CJ CD solutions after respective heating time viewed under normal light (top) and UV light at 365 nm (bottom). The CA samples (100 mL solution of 0.1 g CA and 3480 μL EDA in DI water) were pyrolyzed at 2 min increment for a total of 4, 8, 10, 12, 14, 16, 18, and 20 min; and CJ samples (100 mL of CJ and 3480 μL EDA) were pyrolyzed at 2 min increment for a total of 4, 8, 10, 12, 14, and 16 min using 70% microwave power (630 W).
The dispersibility of CA-18 and CJ-14 CDs in different solvents was explored using water (H2O), dimethyl sulfoxide (DMSO), dimethylformamide (DMF), acetone, methanol (MeOH), ethanol (EtOH), tetrahydrofuran (THF), hexane, toluene, chloroform (CHCl3), and methylene chloride (CH2Cl2) at room temperature. These solvents were added to the CDs to obtain 1 mg/mL solutions, separately. CA-18 CDs were readily dispersible in water, DMSO, and DMF, providing clear solutions in 1 mL solvent (Figure S3A). However, CJ-14 CDs were dispersible only in water (Figure S3B). The other test solvents provided two separate phases. This suggested that the surface of the CA-18 and CJ-14 CDs are hydrophilic.
The pH stability can provide indirect information regarding the photoluminescence origin of the sample.30 The UV absorbance and photoluminescence emission of CA-18 and CJ-14 CDs were obtained by suspending the CDs at pHs 2, 7.4, and 12 (Figure S4). The CDs were suspended separately, and the absorbance was taken at the noted time (0, 0.5, 2, 4, 8, and 24 h) (Figure S5). The absorbance of CA-18 dispersed in neutral and basic environments for 0.5 h does not vary over time (λmax = 349 nm) (Figure S4A). However, when CA-18 was suspended at pH 2, a slight red shift to λmax = 353 nm compared to the results for pHs 7.4 and 12 was obtained (Figure S4A). CA-18 CDs exhibited high photoluminescence emission under neutral and basic pH environments (highest emission peak at 462 nm for pHs 7.4 and 12 and at 464 nm with shoulder peak for pH 2); however, the photoluminescence intensity was slightly lower at pH 2 (Figure S4B). Even though, there was a decrease in the observed intensity at pH 2, this is not as dramatic as the results reported by Zhou et al.20 and Liu et al.,13 wherein the presence of emissive traps on the surface of the CDs obtained from polyol resulted into low emission intensities at pHs 2 and 12. Conversely, Jin et al.24 reported that the protonation or deprotonation of the functional groups on the CD can lead to shift in photoluminescence using graphene quantum dots. Absorption results of CJ-14 CDs suspended at pHs 7.4 and 12 were shown to be stable over time (λmax = 353 nm for pH 7.4 and 351 for pH 12) and those of CDs suspended at pH 2 showed a slight red shift compared to the results for pHs 7.4 and 12 (λmax = 358 nm) (Figure S4C). The photoluminescence emission of CJ-14 CDs at pH 2 (464 nm) was comparable to that at pHs 7.4 and 12 (462 nm), where a shoulder peak was evident for all of the samples (Figure S4D).
The FT-IR spectra of CA samples confirmed that polar groups such as O–H (3450 cm–1), N–H (3200 cm–1), and carbonyl groups (vide infra) are present on the CD surfaces (Figure S1A), rendering CA-18 CDs dispersible in water, DMSO, and DMF (Figure S3A). For CA-0, C=O stretching at 1720 cm–1, indicative of the presence of acid moieties, disappeared when the sample was pyrolyzed for a longer time. Instead, a peak at 1640 cm–1, characteristic of the amide C=O stretching, appeared starting at CA-14, and this peak became more prominent over time. Small peaks characteristic of N–H bending (1550 cm–1) and C–NH–C stretching (1420 cm–1) were also observed. The disappearance of O–H and N–H stretching may be due to dehydrolysis of COOH moieties from CA, followed by amide bond formation between CA and EDA. For CA-20, the longer pyrolysis time may have led to the carbonization of the material. However, even when UV absorption at 350 nm for CA-20 was less intense than that for CA-18, its emission intensity (3964 au) was higher than that for CA-18 (3819 au) (Figures S2A and S2B).
The FT-IR spectra of CJ samples (CJ-0 to CJ-14) revealed that samples possessed hydrophilic groups, such as O–H (3414 cm–1), N–H (3238 cm–1), carbonyl, and COC groups (1192 cm–1) (Figure S1B). Peaks characteristic of N–H bending (1535 cm–1) and C–NH–C stretching (1406 cm–1) were also observed. Similar to CA-0, CJ-0 showed a C=O stretch at 1734 cm–1, indicative of the presence of acid groups, which disappeared when the sample was pyrolyzed. The peak at 1638 cm–1, characteristic of the amide C=O stretch, appeared starting at CJ-10. This peak became more prominent as the sample was heated for a total time of 12 and 14 min (CJ-12 and CJ-14). Unlike CA, CJ is composed of various sugars, free and bound phenolic acids, and organic acids found in the CJ juice matrix, which provided the complexity in the spectra.
The 1H and 13C NMR shifts of the CDs prepared from the pyrolysis of CA (CA-0 to CA-20) and CJ (CJ-0 to CJ-16) were monitored for the functional groups present as the samples were pyrolyzed. From 0 to 14 min, prominent 1H NMR peaks can be observed at 2.6 and 2.75 ppm and at 3.38 ppm, which corresponded to methylene peaks of CA and EDA, respectively (Figures 5, S6A, and Table S4). This illustrates that the chemical environment surrounding the methylene protons for both CA and EDA did not significantly change. Between 8 and 14 min (CA-8 to CA-14), very small amount of new peaks was observed since very little condensation reaction has proceeded. In addition, water has not completely evaporated from the sample after these were subjected to pyrolysis (Figure 4A). However, these peaks at 2.6, 2.75, and 3.38 ppm disappear as CA dehydrolyzes during CA condensation with other CA or EDA molecules (see CA-16, CA-18, and CA-20 in Figure 5A and Table S4). Several small peaks are observed between 2.5 and 4.2 ppm starting at CA-8. These peaks along 2.5–4.2 ppm become more prominent after the sample has been subjected to at least 14 min pyrolysis (CA-14 to C-20) (Figure 5A).
Figure 5.

NMR spectra of CA-CD time study in D2O. (A) 1H NMR spectra and (B) 13C NMR spectra at the 30–80 ppm and 140–200 ppm regions. Each sample was obtained from microwave pyrolysis of 100 mL of 0.1 g/mL CA in DI water and 3480 μL EDA at 4, 8, 12, 14, 16, 18, and 20 min using 70% microwave power (630 W) at 2 min repetitive heating. Stacked NMR graphs were plotted for these samples: CA-0 (sample with no heating), CA-4, 8, 12, 14, 16, 18, and 20 (sample with the indicated total heating time in min) (see Table S4).
13C NMR spectra also show the disappearance of the peak at 78 ppm, indicative of the absence of methine proton of CA (Figure 5B, CA-18). The small peaks along 40–50 ppm may be due to the formation of a carbon framework attached to electron withdrawing atoms like oxygen or nitrogen (C–H, C–NR2, C–C=O, and −OCH3). However, the peaks along 150–185 ppm may be attributed to the presence of oxygen groups attached to the aromatic rings; conjugated alkenes attached to a carbonyl group, such as amide, ester, and acid moieties (R–CONR2, R–COOR, and R–COOH); or conjugated imine groups. IR also confirmed the disappearance of acid C=O stretching at 1720 cm–1 and the appearance of amide C=O stretching at 1640 cm–1 when the samples were pyrolyzed for at least 14 min (CA-14, Figure S1A). By CA-20, new set of peaks similar to those of CA and EDA (CA-0 sample) appeared, but these are slightly downshifted at 2.6–2.8, 2.8–2.9, and 3.4 ppm. One of the methylene peaks, at 48 ppm, disappeared, indicating that CA may have been converted to a different functional moiety. When EDA was incorporated into the CA carbon network as the solution was heated beyond 14 min total pyrolysis time, the presence of multiple amine groups in EDA also allowed its facile incorporation on the surface of the CDs. Both types of incorporation and the pyrolysis conditions increase the chance of formation of emissive traps, arising from the functional moieties and/or change in the hybridization of the carbon core and conjugated π domains on the surface or at the core of the CDs. Because higher photoluminescence intensities were obtained from these samples (CA-14 to C-20), this may be due to the synergistic effect of both the surface state and the carbogenic core, as reported by Qu et al.34
1H NMR spectrum of CJ with EDA before pyrolysis, CJ-0, also provides peaks at 2.52–2.69 and 3.27 ppm, which correspond to the α-methylene protons of CA in CJ and EDA, respectively (Figure S6). 13C NMR peaks at 182 and 185 ppm correspond to the carbonyl groups, at 79 ppm corresponds to the CA methine carbon, at 49 ppm is for the CA methylene carbon, and at 40 ppm is for the EDA methylene carbon. Both 1H NMR and 13C NMR spectra ascertain the presence of CA in CJ in significant quantities. However, different from CA 1H NMR and 13C NMR spectra, small peaks between 2.5 and 4.5 ppm in CJ-0 may be due to the functional moieties from the sugars (fructose, glucose, and sucrose) present in the CJ matrix. These peaks were also observed along the 38–80 ppm region of 13C NMR spectrum (Figure 6B and Table S4). Because other components in CJ, such as the free and bound phenolic acids and other organic acids, are present in smaller quantities, the peaks corresponding to these compounds are not visible. Between 0 and 10 min heating, only three peaks were predominant in 1H NMR spectrum (2.52–2.69 and 3.27 ppm). Additional small peaks were observed in 1H NMR spectrum of CJ-12, indicative of the presence of the methylene or methine carbon attached to C=O or directly to electron-withdrawing atoms like oxygen or nitrogen in the sample. In addition, the peaks at 2.58–2.73 ppm in CJ-12 became broad, unlike in the CA-18 sample, where these two peaks (corresponding to CA) disappeared as the sample was pyrolyzed for a longer period of time. These peaks remained present in CJ-14 and CJ-16, presumably due to the methylene peaks of unreacted acid groups. However, this can also indicate the presence of methylene protons attached to a carbon adjacent to an amide group arising from condensation between CA and EDA. These new amide carbonyl peaks also appeared upfield around 165–175 ppm, whereas the acid carbonyl peaks were evident along 182 and 185 ppm in the 13C NMR spectra of CJ-12, CJ-14, and CJ-16 (Figure 6B). Furthermore, additional peaks appeared along the 40–50 and 70–80 regions in the 13C NMR spectra of CJ-12, CJ-14, and CJ-16; these peaks can be attributed to the methyl, methylene, or methine groups attached to electron-withdrawing atoms like oxygen or nitrogen and the methylene or methine groups belonging to cyclic rings, which are attached to a carbonyl moiety or an aromatic ring. The 13C NMR spectra of CJ-12, CJ-14, and CJ-16 along the 150–185 ppm region are similar to those of CA-18, wherein the peaks can be attributed to aromatic rings possessing oxygen moieties, alkenes attached to a ring and/or conjugated to a carbonyl carbon, and conjugated imine groups. Some of these groups arise from the concomitant formation of ring-structured carbon from CA in CJ with EDA or due to the reaction of existing functionalities found in the CJ matrix with EDA, and these peaks can be observed along the 5.5 ppm region and above of 1H NMR spectrum and >150 ppm region of 13C NMR spectrum.
Figure 6.

NMR spectra of CJ-CD time study in D2O. (A) 1H NMR spectra and (B) 13C NMR spectra at the 30–80 ppm and 140–200 ppm regions. Each sample was obtained from microwave pyrolysis of 100 mL of CJ and 3480 μL of EDA at 4, 8, 10, 12, 14, and 16 min using 70% microwave power (630 W) at 2 min repetitive heating. Stacked NMR graphs were plotted for these samples: CJ-0 min (sample with no heating), CJ-4, 8, 10, 12, 14, and 16 (sample with the indicated total heating time in minutes) (see Table S4).
The CA (CA-0-CA-20) and CJ (CJ-0-CJ-16) time study samples were subjected to dynamic light scattering (DLS) and diffusion-ordered NMR spectroscopy (DOSY). The CA samples did not provide any DLS signal until CA-18, which has a hydrodynamic radius (Rh) of 0.76 nm. This indicates that the CA CDs formed slowly with time and the CDs were only observed after the sample was pyrolyzed for 18 min. Because most of the CA samples did not give positive signals when subjected to light scattering experiments, the size of the particles was also ascertained using DOSY (Figure S7). The DOSY experiment was performed in the CA-20 sample, which provided particle size ranging from 0.1 to 0.65 nm. This supported the observation that no large CDs were formed from the CA time study samples because the size range is along that of small molecules. However, AFM results revealed that CA-18 is mostly composed of spherical particles with height in the range of 6–8 nm. It is possible that the particles aggregated on the mica sheet when the samples were drop-casted and air-dried, thereby providing bigger particle sizes compared to those in DLS results (Figure 7A). ζ Potential experiments also confirmed that CA-18 exhibited a ζ potential of −5.68 mV. However, other CA samples showed near-zero ζ potential, which confirmed the absence of CDs in the samples. For CJ samples, the hydrodynamic radius increased from 1.07 nm (CJ-0) to 9.25 nm (CJ-4). The size of the particles gradually decreased from 4.20 nm (CJ-8) to 2.84 nm (CJ-10), 1.15 nm (CJ-12), 1.54 nm (CJ-14), and 0.88 nm (CJ-16) (Figure 7C). The observed results may be due to the aggregation of pyrolyzed sugars, free and bound phenolic acids, and organic acids found in the juice matrix, and then these big carbon particles break up into smaller CD particles during further pyrolysis. Because CJ is composed of a mixture of different compounds that can form and aggregate differently compared with CA, the presence of nonuniformly shaped particles is evident in the CJ-14 micrograph (Figure 7B). CJ-14 has big particles with height in the range of 6–9 nm and a number of smaller particles of 1–4 nm. The ζ of the CJ samples decreases from −12.99 mV (CJ-0) to around −8 to 9 mV (−9.02 mV for CJ-14 and −8.32 mV for CJ-16), which indicated that the functional moieties carrying negative charges are disrupted as pyrolysis of the CJ samples progresses. In addition, because the ζ values of the CDs are <10 mV, these particles have a higher chance to form aggregates.
Figure 7.

AFM image of CDs on the mica substrate with the associated height profile: (A) CA-18 and (B) CJ-14 CDs. (C) Size distribution of CJ CDs obtained using DLS with the sample concentration of 2 mg/mL in water. All samples were filtered through membrane filters with a pore size of 450 nm prior to analysis. (D) XRD patterns of CA-4, CA-8, CA-12, CA-16, CA-18, and CJ-14 CDs.
The XRD patterns of CA-4 to CA-12 revealed highly crystalline samples (Figure 7D). The presence of a number of peaks may indicate a complex mixture of crystalline components. When the samples were heated for 16 and 18 min total time (CA-16 and CA-18), the XRD patterns displayed a broad peak along 25°, which can be attributed to the presence of amorphous or highly disordered carbon atoms.44 Conversely, the XRD pattern revealed that CJ-14 is highly amorphous (Figure 7D). However, there are some crystalline peaks present in the sample.
Toxicity Study of CA-18 and CJ-14 CDs
Mung beans were used to assess the toxicity of the CDs.45 Increasing CA-18 and CJ-14 CD concentrations ranging from 0.1 to 0.5, 1, 1.2, 2, 5, and 10 mg CD/mL DI water were used to grow mung beans, with DI water as control. The growth of the beans was monitored at 0, 24, 48, 72, and 96 h. The results showed that the beans tolerated up to 2 mg/mL CA-18 CDs. At 5 and 10 mg/mL concentration, no observable growth took place (Figure S8). This may be due to the toxicity attributed to the higher concentration of CDs in the system. Although the beans sprouted within 24 h, the growth did not occur after that. Mung beans grown using CJ-14 CD solutions were observed to be more dose-dependent compared to CA-CDs. CJ-14 CDs inhibited the growth of the mung beans beyond 0.5 mg/mL (Figure S8). At CJ-14 CD concentrations of 2 mg/mL and above, no observable growth took place, although the beans sprouted within 24 h. Li et al.11 have shown that using 0.1 g/mL is optimum for cultivating bean sprout using different carbon nanomaterials, such as carbon quantum dots, graphene quantum dots, graphene oxide, and single-walled carbon nanotubes. Similar to what was observed in this study, the toxicity increased when the concentrations of these four carbon nanomaterials were elevated and at prolonged cultivation time.
After 96 h, one bean sprout per concentration dish was taken and photographed under normal light and UV light (365 nm). As shown in Figure S9A–D, bean sprouts grown in CA-18 and CJ-14 CDs at concentrations 0.5 mg/mL and above exhibited fluorescence along their stems. This suggests that the CDs (Rh CA-18 = 0.76 nm and Rh CJ-14 = 1.54 nm) were transported inside the cytoderm of the bean sprout. Sprouts grown in pure water and 0.1 mg/mL CDs showed no fluorescence.
The length of the sprouts for each bean was also measured after 144 h. The average length of nine sprouts per test concentration and its corresponding standard deviation are summarized in Table S5. CA-18 CDs were shown to be less toxic than CJ-14 CDs because the growth of the beans was promoted (Figure S9E). The beans grown using CA-18 CDs at 1 mg/mL DI water were longer (average length = 16.4 ± 2.7 cm) compared with those grown using the control (13.4 ± 5.9 cm). However, Qu et al.17 and Xu et al.43 showed that sprouts grown at 1.5 mg/mL CDs prepared using CA-urea and calcium citrate–urea, respectively, were nontoxic and did not hinder plant growth. CA-18 CDs were shown to be nontoxic up to 2 mg/mL, whereas CJ-14 CDs inhibited growth at concentrations 0.5 mg/mL and above. From the results, it is clear that CJ-14 CDs are more toxic than CA-18 CDs. The toxicity of CJ-14 may arise from the material size, nonuniform particle shape, more negative surface charge, and higher crystallinity compared to those of CA-18 and less likely due to the material composition of the particle’s core. Li et al.45 reported that CDs with positive surface charge translocate from the roots to the stems and leaves of the mung beans through the vascular system via the apoplastic pathway. Although the surface charge dictates the differential internalization and subcellular localization of the CDs, cells rarely use different uptake routes for cationic or anionic particles.46,47 Functionalized carbon particles possessing carbonyl (C=O), carboxyl (COOH), and/or hydroxyl (OH) groups on the surface are more toxic to cells (lung tumor) compared with their nonfunctionalized counterparts.48 Charged particles such as anionic particles (carboxyl functionalized) have been shown to cause intracellular damage (induce apoptosis), whereas cationic particles (amine functionalized) induce membrane damage.47
Conclusions
A straightforward and inexpensive gram-scale preparation of CDs from readily available sources was described. CA and Philippine citrus calamansi juice (CJ) CDs were prepared using microwave pyrolysis of 1:1 molar ratio (0.1 g CA/mL DI water (100 mL)/3.48 mL EDA) or CJ 1:1 (100 mL CJ/3.48 mL EDA) using 70% (630 W) at 2 min repetitive heating. The gram-scale pyrolysis for CA and EDA was carried out for a total pyrolysis time of 18 min to provide CA-18 (Rh = 0.76 nm and ζ = −5.68 mV) in 97% yield and for a total pyrolysis time of 14 min for CJ and EDA to provide CJ-14 (Rh = 1.54 nm and ζ = −9.02 mV) in 7% yield. Sample CJ-14 has a nonuniform particle shape and broader size distribution compared to those of spherical CA-18. XRD results also revealed the difference between CA-18 (amorphous) and CJ-14 (amorphous and crystalline). The CDs were shown to be stable at pHs 7.4 and 12, but the emission slightly red-shifted at pH 2 for CA-18. CA-18 CDs promoted mung bean growth and were shown to be nontoxic up to 2 mg/mL. However, CJ-14 CDs inhibited mung bean growth at concentrations 0.5 mg/mL and above. Herein, we demonstrated that although the surface functional groups and the carbogenic cores of CA and CJ systems evolved differently using microwave pyrolysis, the final carbon dots showed very similar optical properties. The surface functionalities of the carbon dots from the IR and NMR results do vary when comparing CA and CJ systems, and this leads to very different toxicity. Taken together, these results indicate that the pyrolysis conditions, the particle size, and the surface groups are important in obtaining highly emissive particles from a pure or mixture sample source.
Methods
Materials
The following reagents were used for this study: citric acid monohydrate (CA; J.T. Baker, RG), calamansi juice (CJ; obtained from various markets within Metro Manila), ethylenediamine (Sigma-Aldrich, RG), triethylamine (Merck, RG), diethylamine (Unilab, RG), quinine (Merck, RG), potassium bromide (HiMedia, RG), D2O (Cambridge isotope laboratories, Inc.), d6-dimethyl sulfoxide oxide (DMSO) (Cambridge Isotope Laboratories, Inc.), sodium hydroxide (RCL Labscan, RG), hydrochloric acid (Macron Chemicals), potassium hydrogen phthalate (Merck, RG), and deionized water (conductivity = 0.1 μS/cm). All chemicals were used without further purification.
Gram-Scale Synthesis of CA-18 and CJ-14 CDs
CA stock solution was prepared with 1:1 molar ratio of CA/EDA (100 mL of 0.1 g/mL CA in DI water/3480 μL EDA). Two solutions (50 and 100 mL) were pyrolyzed using a microwave oven at 70% power (630 W) and at 2 min repetitive heating until red-brown products were obtained. Samples were transferred into vials and subjected to lyophilization (Eyela FDU-2200) at −80 °C and 20 Pa for at least 24 h. Dried samples were subjected to UV–vis (UV-1800 Shimadzu spectrophotometer equipped with a W–I2 lamp using 0.7 mL quartz cuvettes) and photoluminescence emission (Ocean Optics, USB 2000 spectrometer and 355 nm UV Crystal PL 2003 Q Switch Laser equipped with an OOIBase32 software set at 50 ms integration time, five units average, five units boxcar, and 1 ms strobe frequency) experiments using 1 × 10–5 g CD/mL DI water and 1.2 mg CD/mL DI water, respectively. For gram-scale synthesis of aminated CA-CDs, six trials were carried out to ensure repeatability. Subsequently, these samples were subjected to freeze-drying and analyzed using a similar procedure as detailed above. For gram-scale synthesis of CJ-CDs, six trial samples of 100 mL of freshly extracted juice containing EDA from the stock solution (100 mL CJ/3480 μL EDA) were heated in a microwave oven using 70% power (630 W) at 2 min repetitive heating until red-brown products were obtained. Calamansi fruits were obtained from different sources: trial 1, Rosario market, Pasig City; trial 2, Del Monte market, Quezon City; trial 3, Pure Gold (Quezon Avenue branch), Quezon City; trial 4, combined samples from Del Monte and Pure Gold, Quezon City; trial 5, SM Savemore (Santolan branch), Pasig City; trial 6, combination of juices from trials 1 to 5.
Quantum Yield Measurements19
Quinine sulfate in 0.1 M H2SO (quantum yield, QY = 0.54) was chosen as a standard. The QYs of the CDs (CA-18 and CJ-14, in 0.1 M H2SO) were calculated using eq 1, where Φ is the QY, I is the measured integrated emission intensity, and η is the refractive index of the solvent. The subscript “st” refers to the standard with known QY and “s” for the sample. To minimize reabsorption effects, the absorption of the solutions in the fluorescence cuvette was less than 0.10 at the excitation wavelength (355 nm). The integrated photoluminescence intensities of quinine sulfate (st) or CDs (s) were plotted against the corresponding absorbance values using several solutions where the concentration was kept below 0.1 at excitation wavelength. The absolute values were calculated using the slope of the quinine sulfate (I = 25 243), CA-18 (I = 785.55), and CJ-14 (I = 939.86) from the plots and the QY (Φ = 54%) of the quinine sulfate. For these solutions, ηs/ηst = 1.
| 1 |
Heating Time Study of the Formation of CA- and CJ-CDs
The CA solution (100 mL of 0.1 g/mL CA in DI water/3480 μL EDA) or 1:1 molar ratio of CJ with EDA (100 mL CJ/3480 μL EDA) was subjected to gram-scale CD synthesis (see gram-scale synthesis). The samples were heated in a microwave using 70% power (630 W) at increments of 2 min for a total time of 0, 4, 8, 12, 14, 16, 18, and 20 min, whereas CJ samples were pyrolyzed at increments of 2 min for a total of 0, 4, 8, 10, 12, 14, and 16 min. The samples were subjected to lyophilization to remove any traces of water, and the CDs were characterized using UV–vis, photoluminescence emission, FT-IR (Shimadzu IP Prestige/IRAffinity/FT-IR-8000 series using KBr pellet, 2% w/w), and NMR (JEOL-400 MHz for 1H and 100 MHz for 13C NMR using D2O or d6-DMSO, 0.75 mL) spectroscopy. The product yields for the CA or CJ CDs were calculated using the dried mass of CDs over the mass of CA or CJ (juice) used (eq 2).
| 2 |
Dispersibility of CA-18 and CJ-14 CDs
The dispersibilities of CA CDs produced after total heating time of 18 min (CA-18) and of CJ CDs produced after total heating time of 14 min (CJ-14) were tested against different solvents: water (H2O), dimethyl sulfoxide (DMSO), dimethylformamide (DMF), acetone, methanol (MeOH), ethanol (EtOH), tetrahydrofuran (THF), hexane, toluene, chloroform (CHCl3), and methylene chloride (CH2Cl2) at room temperature. The CDs were dispersed in the test solvents to obtain 1 mg/mL solutions.
pH Stability of CA-18 and CJ-14 CDs
The pH stabilities of CA-18 and CJ-14 CDs from the gram-scale study were tested (pH 700; Eutech Instruments). Solutions with 1.2 × 10–5 g CD/mL were prepared by dispersing CDs in DI water at pHs 2, 7.4, and 12 (adjusted using HCl and NaOH). UV absorption of these samples was monitored at 0, 0.5, 2, 4, 8, and 24 h upon dispersion. Two trial samples were prepared for each sample. Solutions of 1.2 mg CD/mL DI water dispersion were also prepared by mixing the CDs in DI water at pHs 2, 7.4, and 12 (adjusted using HCl and NaOH). Photoluminescence emission of the different suspensions was measured after dispersion. Two trials were carried out for each sample.
Size, Surface Charge, and State of CA and CJ Time Study Samples
The size of sample was measured via dynamic light scattering (DLS) and diffusion-ordered spectroscopy (DOSY). DLS analysis was carried out using an ALV/CGS-3 MD goniometer system, consisting of a 22 nW He–Ne laser (emitting vertically polarized light with a wavelength of 632.8 nm) and an avalanche photodiode detector located at an angle of 90°. The samples were prepared by suspending the CD samples in water (2 mg/mL), and the samples were filtered using a filter paper with a pore size of 450 nm to remove the dust prior to analysis. The data was obtained by taking the number-weighted size. DOSY tests were carried out on a Bruker Avance 300 MHz spectrometer. The diffusion coefficient (D) was determined, and this was utilized to obtain the hydrodynamic radius by employing the Stokes–Einstein equation (eq 3).
| 3 |
where D is the diffusion coefficient measured from DOSY; k is the Boltzmann constant; T is the temperature (300 K); η is the viscosity of solvent (D2O); and R is the hydrodynamic radius.
The surface charge of CDs was characterized via ζ potential using ZetaPlus (Brookhaven Instruments) equipped with a SR-516-type electrode. The CD samples were prepared by suspending the CDs in 0.1 mM KCl to provide CD solutions with 2 mg/mL concentration. All of the samples were filtered using filters with a pore size of 450 nm prior to analysis. Three runs were obtained and averaged for each sample, and the ζ potential values were calculated using Smoluchowski fits to the data.
Atomic Force Microscopy (AFM) Imaging
The powdered samples were dispersed in deionized water, drop-casted on freshly cleaved mica surfaces, and air-dried. The samples were imaged via noncontact mode using a Park Systems XE-70 Atomic Force Microscope.
X-ray Powder Diffraction (XRD)
XRD patterns were recorded using an Ultima IV X-ray diffractometer (Rigaku, Japan), with a Cu Kα radiation source. The powdered samples were placed on a silicon low-background sample holder and subjected to a θ–2θ scan.
Toxicity Study of CA-18 and CJ-14 CDs
This study was adopted from the work of Li et al.,11 Qu et al.,17 and Xu et al.43 with the following modifications: CA-18 and CJ-14 CD solutions with concentrations of 0.1, 0.5, 1, 1.2, 2, 5, and 10 mg/mL were prepared. For each sample, Petri dishes with 10 mung bean seeds were soaked in 15 mL of CD solutions. A total of 45 mL of CD solution was added to each corresponding dish (at 5, 10, 15, and 15 mL increments after 24, 48, 72, and 96 h, respectively). The samples were observed and photographed at 0, 24, 48, 72, and 96 h. The length of the stem of the mung bean was measured after 144 h. Sample size (n = 9) of the sprouts was measured with each plotted data as the average. Beans that did not sprout were labeled as 0 cm.
Acknowledgments
The authors acknowledge the Department of Science and Technology, Philippines, for the scholarship to J.E.S. and Srinivas Thanneeru for the DOSY experiment.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b00551.
Establishing CA and CJ CD synthesis parameters; FTIR spectra of CA and CJ samples after subjecting to time study; yield and emission intensity of gram-scale synthesis of CA-18 and CJ-14 CDs; reproducibility data for bulk (100 mL) CA-18 and CJ-14 CD synthesis; photoluminescence emission and UV–vis absorption spectra of CA and CJ samples after subjecting to time study; dispersibility test for CA-18 and CJ-14 CDs; photoluminescence emission and UV–vis absorption spectra of CA-18 and CJ-14 CDs after dispersion in pH 2, 7.4, and 12 solutions; UV–vis absorption spectra of CA-18 and CJ-14 CDs at different pHs when CDs were suspended for 0, 0.5, 2, 4, 8, and 24 h; tabulated NMR shifts of CA and CJ samples after subjecting to time study; DOSY spectrum of CA-20; photo of mung beans cultivated in CA-18 and CJ-14 CDs; photo of mung beans grown using different concentrations of CA-18 and CJ-14 CDs after 96 h; and length of mung bean sprouts grown in CA-18 and CJ-14 CDs after 144 h (PDF)
The authors declare no competing financial interest.
Dedication
This article is dedicated to the memory of Dr. Rosa Taboada.
Supplementary Material
References
- Wang Y.; Hu A. Carbon quantum dots: synthesis, properties and applications. J. Mater. Chem. C 2014, 2, 6921–6939. 10.1039/c4tc00988f. [DOI] [Google Scholar]
- Baker S. N.; Baker G. A. Luminescent Carbon Nanodots: Emergent Nanolights. Angew. Chem., Int. Ed. 2010, 49, 6726–6744. 10.1002/anie.200906623. [DOI] [PubMed] [Google Scholar]
- Zheng X. T.; Ananthanarayanan A.; Luo K. Q.; Chen P. Glowing graphene quantum dots and carbon dots: properties, syntheses, and biological applications. Small 2015, 11, 1620–1636. 10.1002/smll.201402648. [DOI] [PubMed] [Google Scholar]
- da Silva J. C. G. E.; Gonçalves H. M. R. Analytical and bioanalytical applications of carbon dots. TrAC, Trends Anal. Chem. 2011, 30, 1327–1336. 10.1016/j.trac.2011.04.009. [DOI] [Google Scholar]
- Li H.; Kang Z.; Liu Y.; Lee S.-T. Carbon nanodots: synthesis, properties and applications. J. Mater. Chem. 2012, 22, 24230–24253. 10.1039/c2jm34690g. [DOI] [Google Scholar]
- Wei J.; Shen J.; Zhang X.; Guo J.; Pan J.; Hou X.; Zhang H.; Wang L.; Feng B. Simple one-step synthesis of water-soluble fluorescent carbon dots derived from paper ash. RSC Adv. 2013, 3, 13119–13122. 10.1039/c3ra41751d. [DOI] [Google Scholar]
- Peng J.; Gao W.; Gupta B. K.; Liu Z.; Romero-Aburto R.; Ge L.; Song L.; Alemany L. B.; Zhan X.; Gao G.; Vithayathil S. A.; Kaipparettu B. A.; Marti A. A.; Hayashi T.; Zhu J.-J.; Ajayan P. M. Graphene Quantum Dots Derived from Carbon Fibers. Nano Lett. 2012, 12, 844–849. 10.1021/nl2038979. [DOI] [PubMed] [Google Scholar]
- Yang S.-T.; Wang X.; Wang H.; Lu F.; Luo P. G.; Cao L.; Meziani M. J.; Liu J.-H.; Liu Y.; Chen M.; Huang Y.; Sun Y.-P. Carbon Dots as Nontoxic and High-Performance Fluorescence Imaging Agents. J. Phys. Chem. C 2009, 113, 18110–18114. 10.1021/jp9085969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaisare M. L.; Talib A.; Khan M. S.; Pardey S.; Wu H.-F. Synthesis of fluorescent carbon dots via microwave carbonization of citric acid in presence of tetraoctylammonium ion, and their application to cellular bioimaging. Microchim. Acta 2015, 182, 2173–2181. 10.1007/s00604-015-1541-5. [DOI] [Google Scholar]
- Ray S. C.; Saha A.; Jana N. R.; Sarkar R. Fluorescent Carbon Nanoparticles: Synthesis, Characterization, and Bioimaging Application. J. Phys. Chem. C 2009, 113, 18546–18551. 10.1021/jp905912n. [DOI] [Google Scholar]
- Li X.; Zhou Z.; Lu D.; Dong X.; Xu M.; Wei L.; Zhang Y. The effect of pristine carbon-based nanomaterial on the growth of green gram sprouts and pH of water. Nanoscale Res. Lett. 2014, 9, 583. 10.1186/1556-276X-9-583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.; Li Z.; Xu M.; Ma Y.; Zhang J.; Su Y.; Gao F.; Wei H.; Zhang L. Controllable Synthesis of Fluorescent Carbon Dots and Their Detection Application as Nanoprobes. Nano-Micro Lett. 2013, 5, 247–259. 10.1007/BF03353756. [DOI] [Google Scholar]
- Liu Y.; Xiao N.; Gong N.; Wang H.; Shi X.; Gu W.; Ye L. One-step microwave-assisted polyol synthesis of green luminescent carbon dots as optical nanoprobes. Carbon 2014, 68, 258–264. 10.1016/j.carbon.2013.10.086. [DOI] [PubMed] [Google Scholar]
- Zhai X.; Zhang P.; Liu C.; Bai T.; Li W.; Dai L.; Liu W. Highly luminescent carbon nanodots by microwave-assisted pyrolysis. Chem. Commun. 2012, 48, 7955–7957. 10.1039/c2cc33869f. [DOI] [PubMed] [Google Scholar]
- Guan W.; Gu W.; Ye L.; Guo C.; Su S.; Xu P.; Xue M. Microwave assisted polyol synthesis of carbon nitride dots from folic acid for cell imaging. Int. J. Nanomed. 2014, 9, 5071–5078. 10.2147/IJN.S68575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra S.; Chandra S.; Kundu T.; Banerjee R.; Pramanik P.; Goswami A. Rapid microwave synthesis of fluorescent hydrophobic carbon dots. RSC Adv. 2012, 2, 12129–12131. 10.1039/c2ra21048g. [DOI] [Google Scholar]
- Qu S.; Wang X.; Lu Q.; Liu X.; Wang L. A Biocompatible Fluorescent Ink Based on Water-Soluble Luminescent Carbon Nanodots. Angew. Chem., Int. Ed. 2012, 51, 12215–12218. 10.1002/anie.201206791. [DOI] [PubMed] [Google Scholar]
- Du F.; Zeng F.; Ming Y.; Wu S. Carbon dots-based fluorescent probes for sensitive and selective detection of iodide. Microchim. Acta 2013, 180, 453–460. 10.1007/s00604-013-0954-2. [DOI] [Google Scholar]
- Zhu S.; Meng Q.; Wang L.; Zhang J.; Song Y.; Jin H.; Zhang K.; Sun H.; Wang H.; Yang B. Highly photoluminescent carbon dots for multicolor patterning, sensors and bioimaging. Angew. Chem., Int. Ed. 2013, 52, 3953–3957. 10.1002/anie.201300519. [DOI] [PubMed] [Google Scholar]
- Zhou M.; Zhou Z.; Gong A.; Zhang Y.; Li Q. Synthesis of highly photoluminescent carbon dots via citric acid and Tris for iron (III) ions sensors and bioimaging. Talanta 2015, 143, 107–113. 10.1016/j.talanta.2015.04.015. [DOI] [PubMed] [Google Scholar]
- Liu H.; Ye T.; Mao C. Fluorescent Carbon Nanoparticles Derived from Candle Soot. Angew. Chem., Int. Ed. 2007, 46, 6473–6475. 10.1002/anie.200701271. [DOI] [PubMed] [Google Scholar]
- Vinci J. C.; Ferrer I. M.; Seedhouse S. J.; Bourdon A. K.; Reynard J. M.; Foster B. A.; Bright F. V.; Colón L. A. Hidden Properties of Carbon Dots Revealed After HPLC Fractionation. J. Phys. Chem. Lett. 2013, 4, 239–243. 10.1021/jz301911y. [DOI] [PubMed] [Google Scholar]
- Sahu S.; Behera B.; Maiti T. K.; Mohapatra S. Simple one-step synthesis of highly luminescent carbon dots from orange juice: application as excellent bio-imaging agents. Chem. Commun. 2012, 48, 8835–8837. 10.1039/c2cc33796g. [DOI] [PubMed] [Google Scholar]
- Jin S. H.; Kim D. H.; Jun G. H.; Hong S. H.; Jeon S. Tuning the Photoluminescence of Graphene Quantum Dots through the Charge Transfer Effect of Functional Groups. ACS Nano 2013, 7, 1239–1245. 10.1021/nn304675g. [DOI] [PubMed] [Google Scholar]
- Zhu B.; Sun S.; Wang Y.; Deng S.; Qian G.; Wanga M.; Hu A. Preparation of carbon nanodots from single chain polymeric nanoparticles and theoretical investigation of the photoluminescence mechanism. J. Mater. Chem. C 2013, 1, 580–586. 10.1039/C2TC00140C. [DOI] [Google Scholar]
- Peng H.; Travas-Sejdic J. Simple Aqueous Solution Route to Luminescent Carbogenic Dots from Carbohydrates. Chem. Mater. 2009, 21, 5563–5565. 10.1021/cm901593y. [DOI] [Google Scholar]
- Zheng H.; Wang Q.; Long Y.; Zhang H.; Huang X.; Zhu R. Enhancing the luminescence of carbon dots with a reduction pathway. Chem. Commun. 2011, 47, 10650–10652. 10.1039/c1cc14741b. [DOI] [PubMed] [Google Scholar]
- Bourlinos A. B.; Zbořil R.; Petr J.; Bakandritsos A.; Krysmann M.; Giannelis E. P. Luminescent Surface Quaternized Carbon Dots. Chem. Mater. 2012, 24, 6–8. 10.1021/cm2026637. [DOI] [Google Scholar]
- Sun Y.-P.; Zhou B.; Lin Y.; Wang W.; Fernando K. A. S.; Pathak P.; Meziani M. J.; Harruff B. A.; Wang X.; Wang H.; Luo P. G.; Yang H.; Kose M. E.; Chen B.; Veca L. M.; Xie S.-Y. Quantum-Sized Carbon Dots for Bright and Colorful Photoluminescence. J. Am. Chem. Soc. 2006, 128, 7756–7757. 10.1021/ja062677d. [DOI] [PubMed] [Google Scholar]
- Zhu S.; Song Y.; Zhao X.; Shao J.; Zhang J.; Yang B. The photoluminescence mechanism in carbon dots (graphene quantum dots, carbon nanodots and polymer dots): current state and future perspective. Nano Res. 2015, 8, 355–381. 10.1007/s12274-014-0644-3. [DOI] [Google Scholar]
- Sun Y.-P.; Wang X.; Lu F.; Cao L.; Meziani M. J.; Luo P. G.; Gu L.; Veca L. M. Doped Carbon Nanoparticles as a New Platform for Highly Photoluminescent Dots. J. Phys. Chem. C 2008, 112, 18295–18298. 10.1021/jp8076485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S.; Wang L.; Zhou N.; Zhao X.; Song Y.; Maharjan S.; Zhang J.; Lu L.; Wang H.; Yang B. The crosslink enhanced emission (CEE) in non-conjugated polymer dots: from the photoluminescence mechanism to the cellular uptake mechanism and internalization. Chem. Commun. 2014, 50, 13845–13848. 10.1039/C4CC05806B. [DOI] [PubMed] [Google Scholar]
- Dong Y.; Shao J.; Chen C.; Li H.; Wang R.; Chi Y.; Lin X.; Chen G. Blue Luminescent Graphene Quantum Dots and Graphene Oxide Prepared by Tuning the Carbonization Degree of Citric Acid. Carbon 2012, 50, 4738–4743. 10.1016/j.carbon.2012.06.002. [DOI] [Google Scholar]
- Qu D.; Zheng M.; Zhang L.; Zhao H.; Xie Z.; Jing X.; Haddad R. E.; et al. Formation mechanism and optimization of highly luminescent N-doped graphene quantum dots. Sci. Rep. 2014, 4, 5294 10.1038/srep05294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.; Sheng Z.; Han H.; Zou M.; Li C. Facile synthesis of fluorescent carbon dots using watermelon peel as a carbon source. Mater. Lett. 2012, 66, 222–224. 10.1016/j.matlet.2011.08.081. [DOI] [Google Scholar]
- Wai C.Chemical Components and Aromatic Profiles of Citrus and Coffee in Asia. Ph.D. Dissertation, National University of Singapore, Singapore, 2013. [Google Scholar]
- Amit S.Synthesis of Fluorescent Carbon Dots from Calamansi. Bachelor of Science in Chemistry Thesis, Ateneo De Manila University, Philippines, 2015. [Google Scholar]
- Zhou J.; Booker C.; Li R.; Zhou X.; Sham T.-K.; Sun X.; Ding Z. An Electrochemical Avenue to Blue Luminescent Nanocrystals from Multiwalled Carbon Nanotubes (MWCNTs). J. Am. Chem. Soc. 2007, 129, 744–745. 10.1021/ja0669070. [DOI] [PubMed] [Google Scholar]
- Pan D.; Zhang J.; Li Z.; Wu M. Hydrothermal Route for Cutting Graphene Sheets into Blue-Luminescent Graphene Quantum Dots. Adv. Mater. 2010, 22, 734–738. 10.1002/adma.200902825. [DOI] [PubMed] [Google Scholar]
- Mochalin V. N.; Gogotsi Y. Wet Chemistry Route to Hydrophobic Blue Fluorescent Nanodiamond. J. Am. Chem. Soc. 2009, 131, 4594–4595. 10.1021/ja9004514. [DOI] [PubMed] [Google Scholar]
- Zhu S.; Zhang J.; Qiao C.; Tang S.; Li Y.; Yuan W.; Li B.; Tian L.; Liu F.; Hu R.; Gao H.; Wei H.; Zhang H.; Sun H.; Yang B. Strongly green-photoluminescent graphene quantum dots for bioimaging applications. Chem. Commun. 2011, 47, 6858–6860. 10.1039/c1cc11122a. [DOI] [PubMed] [Google Scholar]
- Tang L.; Ji R.; Cao X.; Lin J.; Jiang H.; Li X.; Teng K. S.; Luk C. M.; Zeng S.; Hao J.; Lau S. P. Deep Ultraviolet Photoluminescence of Water-Soluble Self-Passivated Graphene Quantum Dots. ACS Nano 2012, 6, 5102–5110. 10.1021/nn300760g. [DOI] [PubMed] [Google Scholar]
- Xu M.; He G.; Li Z.; He F.; Gao F.; Su Y.; Zhang L.; Yang Z.; Zhang Y. A green heterogeneous synthesis of N-doped carbon dots and their photoluminescence applications in solid and aqueous states. Nanoscale 2014, 6, 10307–10315. 10.1039/C4NR02792B. [DOI] [PubMed] [Google Scholar]
- Mitra S.; Chandra S.; Patra P.; Pramanik P.; Goswami A. Novel fluorescent matrix embedded carbon quantum dots for the production of stable gold and silver hydrosols. J. Mater. Chem. 2011, 21, 17638–17641. 10.1039/c1jm13858h. [DOI] [Google Scholar]
- Li W.; Zheng Y.; Zhang H.; Liu Z.; Su W.; Chen S.; Liu Y.; Zhuang J.; Lei B. Phytotoxicity, Uptake, and Translocation of Fluorescent Carbon Dots in Mung Bean Plants. ACS Appl. Mater. Interfaces 2016, 8, 19939–19945. 10.1021/acsami.6b07268. [DOI] [PubMed] [Google Scholar]
- Asati A.; Santra S.; Kaittanis C.; Perez J. M. Surface-Charge-Dependent Cell Localization and Cytotoxicity of Cerium Oxide Nanoparticles. ACS Nano 2010, 4, 5321–5331. 10.1021/nn100816s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fröhlich E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int. J. Nanomed. 2012, 7, 5577–5591. 10.2147/IJN.S36111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrez A.; Kasas S.; Salicio V.; Pasquier N.; Seo J. W.; Celio M.; Catsicas S.; Schwaller B.; Forró L. Cellular Toxicity of Carbon-Based Nanomaterials. Nano Lett. 2006, 6, 1121–1125. 10.1021/nl060162e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.