

Abstract
Early detection of cancer plays a crucial role in disease prognosis. It requires the recognition and quantification of low amounts of specific molecular biomarkers, either free or transported inside nanovesicles, through the development of novel sensitive diagnostic technologies. In this context, we have developed a nanoarray platform for the noninvasive quantification of cancer biomarkers circulating in the bloodstream. The assay is based on molecular manipulation to create functional spots of surface-immobilized binders and differential topography measurements. It is label-free and requires just a single binder per antigen, and when it is implemented with fluorescence labeling/readout, it can be used for epitope mapping. As a benchmark, we focused on the plasma release of Her2 extracellular domain (ECD), a proposed biomarker for the progression of Her2-positive tumors and response to anticancer therapies. By employing robust, easily engineered camelid nanobodies as binders, we measured ECD-Her2 concentrations in the range of the actual clinical cutoff value for Her2-positive breast cancer. The specificity for Her2 detection was preserved when it was measured in parallel with other potential biomarkers, demonstrating a forthcoming implementation of this approach for multiplexing analysis. Prospectively, this nanorarray platform may be customized to allow for the detection of promising new classes of circulating biomarkers, such as exosomes and microvesicles.
Introduction
The past decade has witnessed the development of diagnostic devices based on diverse physical principles for the highly sensitive, noninvasive, and fast detection of disease-related biomolecules.1−3 In this assorted framework, nanotechnological breakthroughs in terms of integration and miniaturization of the various bioassays2 have pushed beyond the limitations of conventional approaches. Notable examples are miniaturized electrochemical and surface plasmon resonance (SPR)-based sensors, functional nanoparticles, and protein/DNA nanoarrays that can offer enhanced precision and significantly faster measurements than those achieved with current technologies.4−7 Miniaturized, integrated, and multiplexed strategies would be of particular utility when moving in the direction of noninvasive “liquid biopsy”, i.e., the detection of reliable biomarkers (proteins, nucleic acids, nano- and microvesicles, and circulating cells) directly in the bloodstream of patients to monitor early stage tumor occurrence as well as its evolution during therapy.
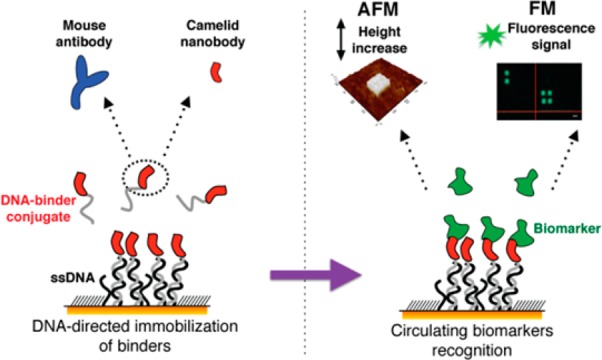
We propose here an atomic force microscopy (AFM)-based miniaturized bioaffinity test for the quantification of blood-circulating protein biomarkers. This approach capitalizes on other studies performed by our group1,8,9 and is based on the precise confinement of high-affinity antigen-binding molecules on a surface by means of AFM nanografting of single-stranded DNA (ssDNA) and DNA-directed immobilization (DDI) of conjugated binders, following the scheme in Figure 1.
Figure 1.

Schematic representation of the DDI strategy to immobilize antibodies on a microsized DNA-based biosensor.
In particular, we focused our attention on the detection of the blood-released portion of human epidermal growth factor receptor 2 (Her2). Her2 is a member of the epithelial growth factor receptor (EGFR/ErbB) family, which is involved in many biological processes, such as signal transduction, regulation of cell adhesion, regulation of transcription, positive regulation of cell growth/proliferation, and negative regulation of apoptosis.10 It is normally expressed at low levels in the epithelial cells of many tissues, whereas gene amplification and overexpression of Her2 are observed in many types of cancer, such as breast, lung, ovarian, and gastric.11 In breast cancer, this amplification defines a subtype of cancer (Her2-positive cancers) with a unique gene signature that is maintained during progression.12,13 The ability to reveal this “signature” at extremely low concentrations may be used for early stage cancer detection and/or to monitor and follow cancer progression to tune personalized drug treatments.
The increase in membrane concentration (crowding) of Her2 induced by receptor overexpression might be the leading cause of aberrant, ligand-independent homodimerization or heterodimerization with other ErbB receptors, which in turn causes proteolytic cleavage of the Her2 extracellular domain (ECD), known as shedding,14 and downstream signaling activation.15,16 Anti-Her2-positive cancer therapies based on humanized antibodies, such as trastuzumab and pertuzumab, are focused on blocking Her2 dimerization and/or shedding and/or Her2-dependent signals.17 Although it is strongly discussed in the clinic, the lack of suitable technologies to detect circulating ECD-Her2 prevents this molecule from being considered a proper biomarker at present. A precise quantification of the level of ECD-Her2 released and circulating freely in the bloodstream or as nanovesicle (exosome) cargo might boost the ability to detect Her2-positive tumors early in their development after diagnosis and allow tumor response to Her2-targeted therapy to be monitored.
So far, enzyme-linked immunosorbent assays (ELISAs) constitute the only FDA-approved clinical method to evaluate the amount of circulating ECD-Her2 in the serum of patients with both primary breast cancer and metastatic disease16 and to monitor their response to therapeutic treatments.18,19 With this method, ECD-Her2 has been found in 30% of primary breast tumors and 50% of metastatic ones.20 However, ELISA bears some limitations, such as the use of expensive labels and the need for multiple antibodies for the same analyte, in addition to the difficulty of implementing it in multiplexing analysis. This last point is crucial, given that other soluble forms of ECD-Her2 could derive from alternative splicing (e.g., herstatin and p100). The ability to detect multiple biomarkers is strongly required to unequivocally correlate these quantitative measurements with tumor status and progression.
In this specific context, we propose to exploit the full potential of our innovative nanoarray platforms. To move toward clinical applications, we pushed their sensitivity for antigen detection, exploiting different binders with different affinity/recognition epitopes. With respect to ELISA, AFM nanoarrays enable multiplexing, require no labeling, and entail a single type of binder for analysis. In particular, we used two different mouse IgG monoclonal antibodies (Abs) selected for their high binding affinity to regions outside the epitope recognized by trastuzumab, which makes them suitable for monitoring the disease during therapeutic drug treatment. Then, we explored alternative binders such as high-affinity single-domain nanobodies (VHH) (12–15 kDa) selected to bind epitopes in the Her2 hinge region.21,22 With respect to antibodies, nanobodies are more stable and can be easily engineered with specific tags and fusion partners to meet diagnostics needs, contributing to an overall reduction in the cost of the device.22,23 Moreover, nanobodies can be selected to avoid cross-reaction with the human anti-mouse antibodies (HAMA) that are present in human serum and could limit the efficacy of antigen quantification.
Nanoarray detection performance was tested for a broad range of Her2 concentrations in buffer and then in standardized human serum. To demonstrate the multiplexing capabilities of this approach, we evaluated the simultaneous recognition of Her2 and urokinase plasminogen activator (uPA), which participate in cell migration, angiogenesis, embryogenesis, tumor cell dissemination, and metastasis in many tumors. Increased levels of uPA have been reported in primary breast cancer patients and correlate with tumor aggressiveness.24 To prove the reliability to our method, we validated the AFM nanoarray with SPR and fluorescence microscopy (FM) measurements. We also used different combinations of available Her2 binders to build a sandwich assay to identify, via combined AFM and FM measurements, the epitope involved in the specific binding interaction.
Results
Her2 Detection with MGR2 and MGR3 Antibodies
Initially, we used two mouse monoclonal antibodies, MGR2 and MGR3, for to set up and validate the nanoarray. Both antibodies recognize ECD-Her2 with high specificity and have been thoroughly characterized in immunoprecipitation experiments and by immunohistochemistry using Her2-overexpressing cell lines.25,26 A competitive immunoassay as an epitope binning experiment demonstrated that MGR2 and MGR3 bind to independent epitopes on the ECD-Her2 protein surface, which in turn differ from the epitope bound by trastuzumab.25
Toward the realization of the nanoassay, we first conjugated the two antibodies with ssDNA by means of a commercial click chemistry kit that exploits amine groups available on the surface of an antibody to covalently anchor the DNA. Such a bioconjugation method is a highly efficient (>80% of the protein is conjugated), three-step procedure in which both protein and DNA are first modified with specific chemical groups that “click” together. The produced conjugate is extremely stable, and the degree of DNA–protein functionalization is then quantifiable by UV spectrophotometry. To avoid multiple strands attached to a single Ab, sterically limiting the binding affinity to the target, we optimized the original conjugation protocol, obtaining two–three strands per protein, on average, and never more than four.
For nanoscale functionalization, we combined DNA nanografting, fine-tuning the process to optimize the density of patterned biomolecules,8 and the immobilization of semisynthetic Abs–DNA conjugates through DDI with high efficiency, site-selectivity, and reversibility.27,28 We then capitalized on AFM topographic imaging to visualize any change in the surface topography of the nanopatches occurring upon interaction with the antigen, with subnanometric resolution, and to quantify the number of biorecognition events, as successfully demonstrated in previous work published by our group.1,8,27,29,30 The height of the patch is measured at each step of the fabrication (i.e., ssDNA nanografting, DDI of Abs–DNA conjugates, antigen binding) with respect to the surrounding self-assembled monolayer (SAM) of biorepellent alkanethiols through topographic AFM images. The unchanged roughness over the SAM throughout the entire process is an indication of SAM antifouling action.31−33
The binding of 10 nM ECD-Her2 over MGR2 and MGR3 antibody nanostructures is reported in Figure 2. Images of the nanopatches (Figure 2A,D) show that the height increases at each step of the assay until antigen binding (the brighter the color, the higher the nanoassembly height versus the reference SAM). Relative height variations are visualized that show the topographic line profiles (Figure 2B,E) and are reported as absolute values (Figure 2C,F).
Figure 2.

ECD-Her2 nanoarray detection by MGR2 (A–C) and MGR3 (D–F) Abs. (A, D) AFM images across the nanopatches after DNA immobilization, antibody loading via DDI, and binding of the antigen. (B, E) Relative topographic line profiles from the AFM images in panels A and D (light blue, DNA nanografting; red, Ab-conjugate immobilization via DDI; green, 10 nM ECD-Her2 binding). (C, F) Mean and SD values of the patch heights measured from the line profiles across the nanopatches, which correspond to the colored areas in images A and D at each step of the experiment.
In both cases, the average height of the ssDNA nanopatches (Figure 2B,E, blue lines) is close to 3.0 nm. This experimental value is in agreement with the expected output due to the “low-density” nanografting conditions (S/A = 0.3–0.6), in which the vertically standing thiolated DNA molecules are below the highest packing limit (around 5 nm34) and are therefore not fully vertically stretched by repulsive electrostatic forces.35 This DNA density has been previously identified as the optimal one to accommodate the steric hindrance of the subsequently immobilized antibodies and to make their binding site easily accessible to the analyte in solution (Figure S4).
Immobilization of the DNA–antibody conjugate provides a height increase of roughly 4–5 nm (Figure 2B,E, red lines). The further increase in the height after incubation with ECD-Her2 (Figure 2B,E, green lines) clearly indicates the efficient antibody-dependent capture of the antigen.
Next, the nanodevice was assessed for its ability to quantitatively measure the target. We calculated the affinity calibration curve for both MGR2 and MGR3, with the assumption that the variation in height across the nanopatches is proportional to the amount of analyte bound to the ligand. Variations in height were plotted versus ECD-Her2 concentration (ranging from 200 pM to 100 nM in the case of MGR2 and from 1 to 300 nM in the case of MGR3; Figure 3).
Figure 3.

ECD-Her2 binding curves for MGR2–DNA (left) and MGR3–DNA (right) conjugates immobilized on the nanoarray. Average values of the variation in height (Δh) detected on the nanopatches (n = 4) are plotted versus ECD-Her2 concentration. Data were analyzed using the Hill equation with sigmoidal fitting, and KD values were determined.
The sigmoidal distribution of data is in agreement with the saturable binding isotherm for a receptor–ligand binding equilibrium and was fitted with the Hill equation,36 obtaining a dissociation constant KD in the low nanomolar range for both MGR2 (KD = 3.3 ± 0.9 nM) and MGR3 (KD = 17.0 ± 10.7 nM). From Figure 3, we determined a limit of blank (LoB) value of about 1 nM for the higher affinity MGR2 and 3 nM for MGR3.
To validate the nanoarray results, we set up a standard ELISA assay in which different concentrations of Abs were added to ECD-Her2-coated wells (Figure S6). The dissociation constant values found with this assay (MGR2: KD = 2.3 ± 1.3 nM; MGR3: KD = 23.0 ± 6.2 nM) were in very good agreement with those found with the nanoarray (confidence level (CL) = 0.74 and 0.69, respectively). SPR experiments were also performed to evaluate whether functionalization affects an antibody’s affinity for its antigen. The immobilization conditions used on the nanoarray were reproduced by attaching biotinylated ssDNAs to a streptavidin-modified dextran matrix. The Ab conjugated with the complementary DNA was then linked by DDI to the matrix, and then the ECD-Her2 analyte was fluxed. We tested the conjugate with the higher affinity Ab, MGR2, and found that the KD was in the low nanomolar range (17.1 ± 1.1). The different experimental setup explains the much higher KD value obtained with SPR with respect to the nanoarray and ELISA; nonetheless, the low nanomolar KD value confirms that efficient antigen recognition is enabled in a nanoarray-like configuration.
Her2 Detection with EM1 Nanobody
As an alternative to conventional antibodies, we exploited camelid nanobodies, which are recombinant molecules that can be easily engineered at precise and unique residues to avoid multiple and heterogeneous labeling and loss of activity. Moreover, due to their reduced dimensions, nanobodies can be used to prepare functional surfaces with higher ligand densities compared to that with conventional antibodies, which simultaneously increases the active detection surface and avoids steric hindrance. In particular, we recently showed the advantage of producing nanobodies specialized for ECD-Her2 biorecognition with a free C-terminal cysteine available for single-point maleimide functionalization.37 This approach enables functionalization using residues not involved in the Ab paratope, which consequently prevents modifications of the Ab–antigen binding features. The llama nanobody EM1 was selected in vitro from a naïve library38 using the same strategy reported in Djender et al.39 It binds to an ECD-Her2 epitope close to the one recognized by trastuzumab. EM1 was expressed with a free C-terminal cysteine and covalently linked with a maleimide–ssDNA construct. We first measured the binding properties of 1:1 EM1–ssDNA conjugates by SPR, finding a KD of 3.4 ± 0.3 nM. We then proceeded with nanobody–DNA conjugate nanoscale immobilization on a gold surface with the same DDI approach used for the MGR2 and MGR3 antibodies.
The first two preliminary steps (grafting ssDNA and hybridizing the DNA–nanobody conjugate) were optimized according to the small dimensions (2 × 2 × 3 nm3; ∼15 kDa) of VHHs. Notably, the density of grafted ssDNA sequences has a great impact on the hybridization of the conjugate and consequently on the detection of Her2. Whereas low DNA densities are needed to avoid steric hindrance effects in the case of full antibodies, voids should not be left in the nanografted DNA SAM for nanobodies; the nanobody itself could hide in such voids, becoming unavailable to interact with the analyte. On the contrary, at high DNA density, the nanostructures are well packed and the binder stands up over the surface DNA layer in the right configuration. This explains why, as shown in Figure S5, the conditions for high-performance target detection were reached only at high density. Tuning the DNA density and DDI conditions, depending on the dimensions and the specific characteristics of the binder, to optimize detection performance in the immunoassay is a unique feature of AFM nanografting.
Having optimized the immobilization conditions, we performed ECD-Her2 detection with EM1 conjugate nanostructures, following the same experimental procedure as that used for the Abs (ssDNA grafting, DDI-driven conjugate immobilization, and target binding). As a representative case, we report the results related to the detection of 1 nM ECD-Her2 (Figure 4).
Figure 4.

Detection of ECD-Her2 on the nanoarray. Images of nanopatches on the gold surface (A) and their topographical profiles (B). (C) Mean and SD values of the height measured across the nanopatches at each step of the experiment (light blue, grafting of ssDNA; red, hybridization of VHH–DNA conjugate; and green, 1 nM ECD-Her2 incubation).
The measured height of the “high-density” grafted ssDNA is 4.5 ± 0.2 nm, a value that describes the highest packing nanopattern in which the thiolated DNA molecules stand vertically.34
As provided for the MGR2 and MGR3 Abs, we determined the EM1–ECD-Her2 binding affinity curve with different analyte concentrations ranging from 200 pM to 10 nM (Figure 5).
Figure 5.
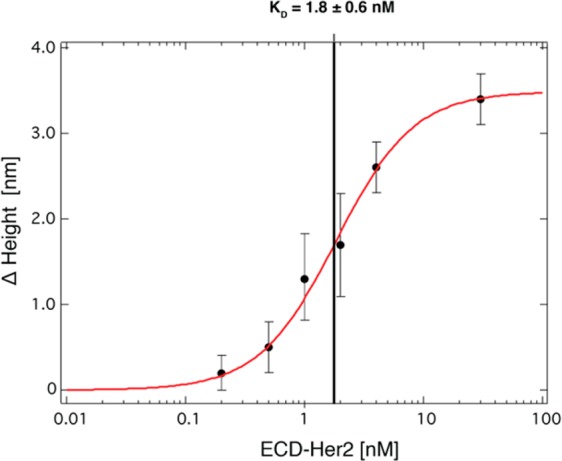
Binding curve of ECD-Her2 and EM1 nanobody conjugate immobilized on the nanoarray surface. Average changes in height (Δ height) values on nanopatches (n = 8) are plotted versus ECD-Her2 concentration; the data were analyzed with the Hill using equation sigmoidal fitting, and KD values were determined.
We fitted data with the Hill equation in this case as well, determining a dissociation constant of KD = 1.8 ± 0.6 nM, which is lower than that for MGR2. The S/N ratio improves substantially upon moving from antibody to nanobody binders, probably because the higher density DNA layer underneath increases the mechanical robustness of the device; the read-out, in fact, is based on a mechanical probe, which still reflects the mechanical resistance of the surface even though it is operated at minimum force. Overall, the limit of sensitivity (LoB) decreases to about 200 pM, pushing the nanoarray’s sensitivity to a level comparable to the cutoff value of 15 ng/mL that is commonly used in the clinic for Her2-positive breast cancer assessment.40,41
Her2 Detection in Standardized Human Serum
As an intermediate step toward using this approach for detection in blood serum samples, we tested the capability of the nanoarray to recognize the target of interest in human serum. We chose to work with standardized human serum first, which mimics the serum of human patients in terms of protein content (IgG antibody and albumin). First, we assessed nonspecific protein binding to the functionalized patches and/or to the surrounding thiol carpet. As shown in Figure 6A, we observed a negligible increase in the patch height (about 0.2 ± 0.4 nm) upon moving from PBS to standardized human serum. Also, the roughness of the biorepellent SAM carpet did not change significantly (rms = 0.23 and 0.25 nm before and after serum incubation, respectively). Instead, upon addition of 1 nM ECD-Her2 to the serum, we observed a relevant patch height increase of 1.3 nm (Figure 6A). This value is the same as the differential height increase (Δh) extracted for an ECD-Her2 concentration of 1 nM from the calibration curve obtained with the recombinant protein in buffer solution. This proves that our nanoarray is capable of filtering the background noise of a biological sample and recognizing a specific biomarker of interest.
Figure 6.

ECD-Her2 detection in standardized human serum. (A) Nanopatch height variation (n = 8) on the nanobody-immobilized nanoarray incubated with standardized human serum with or without 1 nM ECD-Her2. (B) Extrapolation of the height increase expected from the nanobody-based calibration curve.
Multiplexed Detection
To further challenge the capability of the nanoarray and to prove its validity for use in clinical practice, we performed a preliminary multiplex analysis, testing the simultaneous detection of ECD-Her2 and uPA, another relevant breast cancer biomarker. As shown in Figure 7, we grafted ssDNA SAM nanopatches of two different DNA sequences (SH-cF9 and SH-cF5) in serial order, enabling the immobilization of two binders (VHH EM1 and αuPA Ab)—each of them conjugated by means of its complementary ssDNA (F9 and F5)—specific for independent antigens. We then performed a selective detection of the different targets, adding each of them in sequence to the solution.
Figure 7.

Multiplex detection of two different biomarkers (ECD-Her2 and uPA) on the same surface. (A) Schematic of DDI immobilization of binders on the DNA-based biosensor and subsequent detection of the antigens. Height variations of the two separated sets of DNA nanopatch SAMs (cF5 and cF9) after each biomolecule incubation step (in the following sequential order: VHH EM1-F9 conjugate, αuPA Ab-F5; ECD-Her2 antigen, and uPA antigen) are visualized by topographic profiles (B) and histograms (C); mean and SD height values (n = 4) are reported in the tables. Columns where no binding was expected (and therefore no height increase) are gray.
The profile across the grafted SH-cF5 and SH-cF9 ssDNA patches was acquired first (black profiles in Figure 7B, left and right, respectively). Then, we inserted the VHH-F9 conjugate (red curves in Figure 7B), observing that only the patches on the right show a height increase, whereas across the patches on the left, the red profile almost overlaps with the previously acquired black curve. This demonstrated the specificity of the DDI process. Similarly, after adding the uPA Ab-F5 (blue curves in Figure 7B), we see a height increase corresponding to the cF5 patches (panel B, left) but not corresponding to the cF9 patches (panel B, right). Finally, the loading of uPA was sensitively measurable only across the uPA Ab-F5 patches (green profiles in Figure 7B), and the last incubation with ECD-Her2 was detectable only across the VHH-F9 patches (yellow profiles in Figure 7B), completing the demonstration of the specificity of our nanoarray.
The unchanged roughness of the thiol SAM carpet at each step further indicated that nonspecific binding over the surface did not occur, proving again the validity of the TOEG6 SAM carpet as a reference for height measurements. Two different antigens were incubated over the surface at the same concentration (20 nM); the different Δh values (uPA = 2.7 nm; ECD-Her2 = 4 nm) observed can be explained by the different dimensions of the two proteins (53 and 72 kDa, respectively).
The high selectivity of the binding of the two Ab–Ag pairs working in parallel is a promising result that indicates the possibility of implementing the multiplexing nanoarray under real conditions.
Fluorescence Experiments
So far, we have demonstrated that our nanoarrays work in a sensitive and antigen-specific manner, with clear advantages over ELISA platforms because the nanoarrays use small volumes and, more importantly, require only one binder per antigen. To perform an additional validation of our nanoarray, we compared the single protein binder topographic measurement approach and the conventional nanoarray-based sandwich approach with a fluorescent readout. The goal is to label the nanostructures with a fluorescent molecule as unequivocal proof of the presence of the target bound on the surface.
Moreover, we also aim to demonstrate that a fluorescence-integrated sandwich nanoarray allows epitope binning of the binders to be performed by analyzing the pattern of simultaneous or competitive action of different pairs of binders.
As a first approach, we used an “indirect” sandwich configuration (Figure 8A), more similar to classical ELISA setups. We first immobilized the Abs on the surface, and after ECD-Her2 capture, we added another Ab specific for another independent epitope. The fluorescent signal was obtained using a fluorescently labeled secondary α-mouse Ab. From Figure 8, it can be seen that the match between topographic and fluorescence data in this indirect nanosandwich approach is very good.
Figure 8.

Fluorescent visualization of ECD-Her2 captured by the nanoarray. (A) Schematic of the molecular nanoassembly on a gold surface together with AFM images of the patches at each step of the incubation (sequentially: ssDNA, trastuzumab–DNA conjugate, 200 pM ECD-Her2, and MGR2) at different densities of grafted ssDNA (indicated with different colors, see the text). Scale bar 2 μm. (B) Fluorescent image of the patches described in (A) after incubation with anti-mouse Ab labeled with Alexa488 fluorophore. Scale bar 2 μm. (C) Height increase values recorded with AFM topographic analysis after each incubation step on the patches and fluorescence intensity values measured as pixel counts. The plot shows fluorescence–AFM topography correlation (number of independent patches: four for each grafted ssDNA density).
The successive incubation steps were as follows: (i) ssDNA, (ii) trastuzumab–DNA conjugates, (iii) ECD-Her2, (iv) MGR2, and (v) secondary α-mouse fluorescent Ab. Notably, we confirmed, by means of fluorescence measurements, that even a low topographic height increase (0.2–0.5 nm) corresponds to a measurable fluorescent signal, validating the occurrence of binding of target at very low concentrations (200 pM). Moreover, as demonstrated by the values reported in the tables and in the AFM–fluorescence correlation graph in Figure 8c, the fluorescence signal scales linearly with the topographic signal and with the density of ssDNA grafted on the surface, which in turn correlates with the binder density and therefore the total amount of captured target molecules. This not only validates the quantitative analysis performed through topographic measurements across the nanopatches of the nanoarray but also confirms that our single-binder assay retains all the benefits of ELISA with the advantages of simplicity, lower cost, and greater efficiency.
In addition, the fluorescence-integrated sandwich-nanoarray setup might be useful to obtain important information about protein epitopes recognized by different binders. For this, we used the same indirect approach described above as well as a simplified “direct” sandwich approach by exploiting the easy functionalization of the VHH EM1. As the first binder, we used a mouse/humanized Ab, whereas the second one was GFP-conjugated VHH. The two configurations are reported in Figure 9 together with fluorescence images relative to different combinations of binders in the sandwich.
Figure 9.

Fluorescence images obtained with different combinations in the Ab/VHH “sandwich” assay. For images related to the indirect configuration (A–C), the first Ab of the sandwich is indicated in purple and the second is in blue; for the direct configuration (D–F), the first Ab of the sandwich is indicated in purple. Next to each fluorescence image is reported the schematic representation of the position of the patches (violet squares). Scale bar 2 μm.
We noticed that the fluorescence signal coming from the secondary antibody is generally sharper compared to the GFP fluorescent signal on the VHH. This could be related to the different fluorescence quantum yield, to the amplification effect due to the secondary Ab, or to the different dimensions of the two systems, with VHH being 5 times shorter than the Abs and therefore closer to the gold surface, which could partially quench the fluorescence signal.
With this in mind, we used the VHH-related direct configuration (Figure 9D–F) to test its relative epitope for ECD-Her2 recognition. While a fluorescence signal was visible when it was coupled to MGR2 (Figure 9D) and pertuzumab (Figure 9F), no fluorescence was observable when it was coupled to trastuzumab (Figure 9E). This fact indicated that VHH EM1 and trastuzumab bind on close/overlapping epitopes and that these are totally independent from the one recognized by pertuzumab, as reported in the literature.42 Concerning MGR2, whose epitope has not been mapped yet, our preliminary experiments indicate simultaneous binding with VHH; therefore, MGR2 does not exhibit cross-reactivity with VHH. Translating the results of Figure 9D,E, we conclude that MGR2 and trastuzumab recognize different epitopes. This was confirmed by results obtained with the indirect configuration (Figure 9C), where we observed the presence of the fluorescence signal corresponding to the concomitant binding of MGR2 and trastuzumab, proving that the MGR2-functionalized nanoarray is suitable for monitoring a response to therapeutic treatment with this humanized Ab.
With the indirect assay, we also confirmed the simultaneous binding of MGR2 and MGR3 (Figure 9A). The data in Figure 9B relative to the sandwich binding of MGR2 and VHH are apparently in contradiction with the data in Figure 9D, in which the same binders are used but in reverse order. This could be explained by the fact that, due to the small dimensions of the nanobody, the accessibility of the Ab to the analyte would be limited as a consequence of its proximity to the surface. Combining the evidence from both the direct and indirect configurations with these two binders, we can assume that the binding sites of VHH EM1 and MGR2 on ECD-Her2 are most likely very close. These experiments demonstrate that nanografting-based nanoarrays, beyond their potential as a detection tool, can serve as an alternative approach, complementary to more conventional methods, for identifying the molecular determinants of antibody–antigen recognition.
Discussion
The use of miniaturized devices in liquid biopsy for cancer biomarker detection and therapy monitoring will ultimately improve the outcome of early diagnosis and tumor treatment. Here, we employed a nanoarray based on AFM nanolithography and a topographic readout for the detection of cancer-relevant biomarkers circulating in the bloodstream. We focused on the detection of ECD-Her2, optimizing the choice of binders to increase the detection sensitivity and even using it in multiplexing with uPA, to move toward biomolecular footprinting of the disease.
In particular, we demonstrated that nanoarrays employing panning-selected camelid nanobodies can detect ECD-Her2 at less than 200 pM, which corresponds to the cutoff value of 15 ng/mL currently used in the clinic to discriminate between a healthy and pathological status. We showed that the high affinity of nanobodies can be fully exploited by tuning the surface density of the binders: At variance with classical antibodies, for which steric hindrance can promote anticooperativity of adjacent binders, higher probe densities can be used for smaller binders and indeed help to improve the overall sensitivity of target identification.
We expect that the introduction and optimization of suitable linkers and in silico engineering platforms exploiting novel mixed computational–experimental protocols for boosting the affinity for a cognate antigen43 will substantially improve the use of nanobodies as capture molecules and further decrease the detection limit to the hundreds of femtomolar/low picomolar range. In this direction, we plan to identify in silico customized VHHs for specific (nonimmunogenic) epitopes of Her2 fragments by means of computational design42 and/or high-affinity peptides for chosen epitopes of target proteins by stochastically exploring their sequence and structure space.
In conclusion, we demonstrated that this simple assay has the required sensitivity to be operated in the clinic, allowing detection in small volumes of complex matrices and enabling continuous noninvasive monitoring of therapy from single blood droplets. Moreover, at variance with ELISA, our assay requires a single binder for each biomarker and is label-free, avoiding false results connected to fluorescence interference susceptibility and tremendously reducing diagnostic costs while maintaining high biorecognition specificity and fast readout. On the basis of the achieved results, we put forward the idea of implementing a simplified, automated, cantilever-based readout machine in the clinic for antigen quantification purposes.1
When coupled with frequent, noninvasive monitoring in single blood droplets, our method might then have a crucial impact on therapeutic drug monitoring, simultaneously profiling multiple circulating biomarkers and accounting for the time evolution of the cancer genome and consequent resistance to drug treatment. Similar nanoscale platforms can be generated to anchor selected populations of exosomes, via the binding of a specific transmembrane protein, and to characterize them in terms of other membrane protein components. All of these features heighten the significance of our nanoarray as an attractive platform for personalized cancer diagnostic applications.
Finally, the same nanoarray can serve as a platform for epitope binning. This, combined with fluorescent assays in living cell membranes for colocalization of different receptors, will help to understand the specific functions enabled by selective binding, paving the way to the modular design of synthetic receptors.
Methods
DNA
ssDNA sequences (SH-cF9: SH-(CH2)6-5′-CTTCACGATTGCCACTTTCCAC-3′, F9: NH2-(CH2)6-5′-GTGGAAAGTGGCAATCGTGAAG-3′, SH-cF5: SH-(CH2)6-5′-CTTATCGCTTTATGACCGGACC-3′, F5: NH2-(CH2)6-5′-GGTCCGGTCATAAAGCGATAA-3′) were from Biomers GmbH Ulm, Germany.
Antibodies and Nanobodies
Monoclonal mouse antibodies MGR2 and MGR325,26 were produced at Fondazione IRCCS Istituto Nazionale dei Tumori, Milan. These Abs have been tested in IHC, immuno precipitation (CoIP), and immunoblot assays.25,26
Commercial monoclonal human antibodies trastuzumab and pertuzumab were from Genentech.
Commercial monoclonal αuPA antibody was from MyBioSource.
Single-domain antibodies (VHH) were isolated and produced at the University of Nova Gorica as described previously using a naïve nanobody library,38 a differential panning approach on whole cells, and the cytoplasmic expression of the nanobodies in the presence of sulfhydryl oxidase.39
Proteins and Standardized Serum
Recombinant human ECD-Her2 and uPA were from ACRO Biosystems.
Standardized human serum was from Biseko (Biotest).
Conjugates Production
Antibodies
Conjugation of the MGR2, MGR3, trastuzumab, and pertuzumab antibodies with ssDNA sequence F9 and the αuPA antibody with F5 was performed using a click chemistry reaction using the commercial three-step SoluLink protein–oligo conjugation kit (Figure S1).
Protein Modification
Antibodies at a concentration of 2.5–4.0 mg/mL were buffer exchanged into modification buffer (100 mM phosphate, 150 mM NaCl, pH 8.0) usingZeba desalt spin columns (Pierce Chemical) before adding 10–20 mol equiv of HyNic/mol antibody. The reaction was carried out at room temperature for 1.5 h, and the HyNic-modified antibody was desalted into conjugation buffer (100 mM phosphate, 150 mM NaCl, pH 6.0).
Oligonucleotide Modification
The oligonucleotide was desalted into nuclease-free water using a 5K MWCO VivaSpin diafiltration apparatus, and the OD/μL concentration at 260 nm was adjusted to 0.2–0.5 OD/μL. A volume containing 20 equiv S-4FB was added to the oligonucleotide solution and incubated at room temperature for 2 h. The 4FB-modified oligonucleotide was equilibrated into conjugation buffer (100 mM phosphate, 150 mM NaCl, pH 6.0).
Protein–Oligo Conjugation
Volumes of the antibody and oligonucleotide were mixed. One-tenth of the volume of 10× TurboLink catalyst buffer was added to the conjugation solution, and the reaction was carried out at room temperature for 2 h; the amount of the two components used in this step is strictly related to the concentration and the molar substitution ratio (MSR) of both HyNic-modified protein and 4FB-modified oligonucleotide obtained in the previous steps.
The conjugation reaction was visualized spectrophotometrically by determining the absorbance at A354 due to the formation of the chromophoric conjugate bond. The reaction solution was exchanged with PBS using Zeba columns.
Nanobody
The conjugation of nanobody EM1 with ssDNA sequence F9 was performed by a maleimide reaction (Figure S2): the nanobody, containing a free cysteine at its C-terminus, was diluted to a concentration of 100 μM in Hepes 10 mM pH 7.4 buffer and kept reduced by the addition of TCEP in a 10-fold molar excess for 20 min at room temperature. F9-maleimide was dissolved in TE buffer (Tris 10 mM, EDTA 1 mM) pH 8.0 and then added to the nanobody with a ssDNA:protein molar ratio of 10:1 (250:25 μM:μM). After 2 h at room temperature, the modified nanobody was separated from the reactants and reaction byproducts using a G-25 Illustra microspin column (GE Healthcare Life Science).
Surface Plasmon Resonance
Biacore T100 and Biacore 2000 SPR instruments were used at a constant temperature of 25 °C.
As shown in Figure S3, a biotinylated cF9 sequence (cF9-biotin, 2 μM in PBS buffer) was immobilized over the Biacore SA gold chip surface. A continuous flow (5 μL/min) of PBS buffer (running buffer) was maintained during all experiments. The immobilization through streptavidin–biotin binding was stopped after reaching a binding level of ∼1200 RU, corresponding to an amount of ssDNA on the surface that ensured an efficient attachment of the molecules in the following steps of the experiment. The surface was rinsed twice with a 1 min pulse of 50 mM NaOH solution to remove unbound cF9-biotin.44 The hybridization was carried out by incubation with the conjugates at 100 μM in TE buffer with 1 M NaCl until reaching a binding level of ∼1200 RU. The binding affinity of ECD-Her2 was evaluated by injecting two different concentrations of the protein in running buffer at a flow rate of 30 μL/min for 3 min (association phase) and afterward flushing with running buffer for 10 min (dissociation phase). Since the dissociation phase allows complete detachment of the protein, no regeneration procedure was required.
Binding affinity parameters were determined using the 1:1 Langmuir model in the BIAevaluation 3.1 software.
AFM Measurements
The tip-assisted AFM-based nanolithography used to fabricate DNA nanoarrays and all high-resolution topographic measurements were performed using an XE-100 (Park-Systems) with a custom liquid cell at room temperature. First, a biorepellent SAM of top oligo(ethylene glycol)-terminated alkylthiols (TOEG6: HS-(CH2)11-(OCH2CH2)6-OH, Sigma-Aldrich) was prepared on ultraflat stripped gold surfaces following a modified version of the Ulman procedure.45 A gold sample was soaked in a 300 μM solution of TOEG6 in ethanol for about 24 h. Then, it was rinsed with distilled water and ethanol, dried with a gentle stream of nitrogen, and finally glued inside the AFM liquid cell.
Multiple nanografting assembled monolayers of thiol-modified ssDNA SH-cF9 were prepared by serial AFM-based nanografting inside the TOEG6 SAM.1,8,27,29,46 An AFM tip with sufficient rigidity (MikroMasch NSC 19/no Al, spring constant 0.6 nN nm–1) was operated at high load (set point/force ≈ 100 nN) on areas of 1 μm × 1 μm in order to locally displace the TOEG6 SAMs and to facilitate the exchange with the thiolated ssDNA biomolecules (5 μM in TE buffer 1 M NaCl) present in the liquid cell.
Antibody/nanobody conjugate immobilization was performed via DDI,27,28 incubating the ssDNA SAM with Abs/VHH-F9 at 100 nM in TE buffer with 1 M NaCl. ECD-Her2 binding was promoted through the incubation of the antibody/nanobody nanopatches with a solution containing recombinant protein at different concentrations in PBS buffer (binding curve determination and multiplex measurement) or in Biseko (detection in standardized human serum).
Topographic height variations of the nanopatches at each step of the experiment were measured with AFM in gentle contact mode in liquid using a softer AFM tip (MikroMasch CSC 38/no Al, spring constant 0.03 nN nm–1), at a cantilever speed of 1 Hz, in buffer solution (TE buffer, Tris 10 mM, EDTA 1 mM). To minimize perturbations, the applied force during measurement was the minimum stable value (>0.1 nN). We already proved that under these conditions nanostructures can be imaged and their height with respect to the SAM carpet (Δh) can be measured.29 The AFM topography analysis results were expressed as mean ± SD obtained from measurements performed on at least four independent patches (n ≥ 4).
The fabrication parameters of the nanografting process were systematically tuned, consequently modifying the density of ssDNA molecules adsorbed in the nanopatch.1,8,27 In particular, it was possible to control the nanoscale DNA surface by modifying the number of scanning lines during nanografting over the area, a parameter described by the S/A ratio (where S is the scanned area and A is the area of the final patch).35
Fluorescence Imaging
Fluorescence measurements were performed both on direct and indirect sandwich configurations. Monoclonal IgG antibodies specific for ECD-Her2 (MGR2, transtuzumab, and pertuzumab) were directly immobilized via DDI on the surface, incubating the ssDNA nanopatches with the immunoconjugate at 100 nM in TE buffer 1 M NaCl. ECD-Her2 binding to immobilized antibodies was obtained by incubating nanopatches with a solution containing ECD-Her2 at 100 pM (nanoarray validation) or 10 nM (epitope mapping studies).
With respect to direct measurements, the EM1–GFP fusion construct (1 μM) was incubated for 1 h over the surface; indirect measurements were performed using a second Ab specific for ECD-Her2 at the saturating concentration of 500 nM and then the secondary fluorescent α-mouse_Alexa488 Ab (Thermo Fisher Scientific) diluted 1:500 in PBS buffer, which was incubated over the surface for 30 min. After repeated washing steps with PBS buffer, the gold sample was mounted on a glass coverslip with Vectashield H-1400 mounting media (Vector).
Fluorescent images were acquired on an inverted epifluorescence microscope (Nikon Eclipse TiU) using 20× (NA = 0.45) and 40× (NA = 0.70) air objectives and a dichroic filter for FITC (Nikon; excitation: 465–495 nm; DM: 505 nm; emission: 515–555 nm). 1600 × 1200 pixel images were collected using a color digital camera and controller (Nikon DS-Fi2 and Digital Sight DS-L2) at acquisition times ranging from 1 to 10 s.
ELISA assay
The indirect ELISA assay was performed first by coating the ECD-Her2 antigen to a PVC microtiter plate. The protein was diluted to a final concentration of 100 μg/mL in carbonate buffer (100 mM, pH 9.6), and 50 μL was used to coat the wells of the microplate overnight at 4 °C. Then, the coating solution was removed, and the plate washed three times with PBS. The coated wells were blocked with 1% BSA in PBS (blocking buffer) for 2 h at room temperature. The plate was washed twice in PBS, and serial dilutions (from 100 pM to 1 μM) of primary antibody were added. The plate was washed three times with PBS after a 4 h incubation at 4 °C, and horse radish peroxidase (HRP)-conjugated secondary antibody (Sigma) diluted 1:2000 in blocking buffer was added. After the incubation (1 h at 4 °C), binding was assessed by adding the substrate 3,3′,5,5′-tetramethylbenzidine (TMB, Pierce). The reaction was stopped with a solution of 1 M HCl after 15 min of incubation at room temperature, and the optical density was read at 450 nm.
Statistical Analysis
The LoB is the highest measured test result likely to be observed (typically at 95% certainty) for a sample containing no analyte. Values above the LoB are not consistent with the absence of analyte; hence, LoB frequently replaces the analytical sensitivity of a method. It is calculated as follows:
Acknowledgments
This work was supported by a project financed by the Cross-Border Cooperation Programme Italy–Slovenia 2007–2013 (Project PROTEO, Code N. CB166) (to E.A., A.d.M. and L.C.), by a grant from the Associazione Italiana per la Ricerca sul Cancro (AIRC) (AIRC 5 per mille 2011, No. 12214) (to L.C.), and by a FIRB 2011 grant “Nanotechnological Approaches toward Tumor Theragnostic” (to E.A., Pi.P., and L.C.).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b00284.
Schematics of the antibody–DNA and nanobody–DNA conjugation reactions and SPR experimental approach; histograms; and indirect ELISA data (PDF)
Author Present Address
○ (E.A.) Department of Medical Biochemistry and Biophysics, Karolinska Institutet, Scheeles väg, 17177 Stockholm, Sweden.
Author Contributions
E.A. designed the experiments and performed the AFM, SPR, immunofluorescence measurements, and conjugation experiments; Pa.P. helped with AFM measurements; A.B. and Pi.P. helped with AFM measurements, experimental design, and data analysis; D.S. performed fluorescence measurements; E.T. produced and provided the specific antibodies and helped with the experimental design; A.d.M. produced and provided the nanobody and helped with the experimental design; L.C. designed the experiments and coordinated the work; E.A. and L.C. wrote the manuscript, and all authors contributed to its revision and gave approval to the final version.
The authors declare no competing financial interest.
Supplementary Material
References
- Ganau M.; et al. A DNA-based nano-immunoassay for the label-free detection of glial fibrillary acidic protein in multicell lysates. Nanomedicine 2015, 11, 293–300. 10.1016/j.nano.2014.04.006. [DOI] [PubMed] [Google Scholar]
- Powers A. D.; Palecek S. P. Protein analytical assays for diagnosing, monitoring, and choosing treatment for cancer patients. J. Healthc. Eng. 2012, 3, 503–534. 10.1260/2040-2295.3.4.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhandare N.; Narayana A. Applications of Nanotechnology in Cancer: A Literature Review of Imaging and Treatment. J. Nucl. Med. Radiat. Ther. 2014, 5, 195. 10.4172/2155-9619.1000195. [DOI] [Google Scholar]
- Heath J. R.; Davis M. E.; Hood L. Nanomedicine Targets CANCER: Viewing each human body as a system of interacting molecular networks and targeting disruptions in the system with nanoscale technologies can transform how disease is understood, attacked and possibly prevented. Sci. Am. 2009, 300, 44–51. 10.1038/scientificamerican0209-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahfoud O. K.; Rakovich T. Y.; Prina-Mello A.; Movia D.; Alves F.; Volkov Y. Detection of ErbB2: nanotechnological solutions for clinical diagnostics. RSC Adv. 2014, 4, 3422–3442. 10.1039/C3RA45401K. [DOI] [Google Scholar]
- Bohunicky B.; Mousa S. A. Biosensors: the new wave in cancer diagnosis. Nanotechnol., Sci. Appl. 2010, 4, 1–10. 10.2147/NSA.S13465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y. E.; Kwak J. W.; Park J. W. Nanotechnology for early cancer detection. Sensors 2010, 10, 428–455. 10.3390/s100100428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco A.; Bano F.; Parisse P.; Casalis L.; DeSimone A.; Micheletti C. Hybridization in nanostructured DNA monolayers probed by AFM: theory versus experiment. Nanoscale 2012, 4, 1734. 10.1039/c2nr11662f. [DOI] [PubMed] [Google Scholar]
- Nkoua Ngavouka M. D.; Capaldo P.; Ambrosetti E.; Scoles G.; Casalis L.; Parisse P. Mismatch detection in DNA monolayers by Atomic Force Microscopy and Electrochemical Impedance Spectroscopy. Beilstein J. Nanotechnol. 2016, 7, 220–227. 10.3762/bjnano.7.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez C.; Shiff R. HER 2: Biology, Detection, and Clinical Implications. Arch. Pathol. Lab. Med. 2011, 135, 55–62. 10.1043/2010-0454-RAR.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slamon D. J.; Clark G. M.; Wong S. G.; Levin W. J.; Ullrich A.; McGuire W. L. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- Kallioniemi O. P.; et al. ERBB2 amplification in breast cancer analyzed by fluorescence in situ hybridization. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 5321–5325. 10.1073/pnas.89.12.5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson J.; et al. HER2 expression in breast cancer primary tumours and corresponding metastases. Original data and literature review. Br. J. Cancer 2004, 90, 2344–2348. 10.1038/sj.bjc.6601881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan P. J.; Kumogai T.; Berezov A.; Murali R.; Greene M. HER2/Neu: mechanisms of dimerization/oligomerization. Oncogene 2000, 19, 6093–6101. 10.1038/sj.onc.1203967. [DOI] [PubMed] [Google Scholar]
- Moasser M. M. The oncogene HER2; Its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene 2007, 26, 6469–6487. 10.1038/sj.onc.1210477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsé C.; Gauchez A. S.; Jacot W.; Lamy P. J. HER2 shedding and serum HER2 extracellular domain: Biology and clinical utility in breast cancer. Cancer Treat. Rev. 2012, 38, 133–142. 10.1016/j.ctrv.2011.03.008. [DOI] [PubMed] [Google Scholar]
- Swain S. M.; et al. CLEOPATRA Study Group. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N. Engl. J. Med. 2015, 372, 724–34. 10.1056/NEJMoa1413513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajria G.; Chandarlapaty S. HER2-amplified breast cancer: mechanisms of trastuzumab resistance and novel targeted therapies. Expert Rev. Anticancer Ther. 2011, 11, 263–275. 10.1586/era.10.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali S. M.; et al. Serum HER-2/neu and Relative Resistance to Trastuzumab-based Therapy in Patients With Metastatic Breast Cancer. Cancer 2008, 113, 1294–1301. 10.1002/cncr.23689. [DOI] [PubMed] [Google Scholar]
- Aurilio G.; et al. Serum HER2 extracellular domain levels and HER2 circulating tumor cell status in patients with metastatic breast cancer. Future Oncol. 2016, 12, 2001–2008. 10.2217/fon-2016-0081. [DOI] [PubMed] [Google Scholar]
- Desmyter A.; Spinelli S.; Roussel A.; Cambillau C. Camelid nanobodies: killing two birds with one stone. Curr. Opin. Struct. Biol. 2015, 32, 1–8. 10.1016/j.sbi.2015.01.001. [DOI] [PubMed] [Google Scholar]
- De Marco A. Biotechnological applications of recombinant single-domain antibody fragments. Microb. Cell Fact. 2011, 10, 44. 10.1186/1475-2859-10-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goode J.; Dillon G.; Millner P. A. The Development and Optimisation of Nanobody Based Electrochemical Immunosensors for IgG. Sens. Actuators, B 2016, 234, 478–484. 10.1016/j.snb.2016.04.132. [DOI] [Google Scholar]
- Duffy M. J.; McGowan P. M.; Harbeck N.; Thomssen C.; Schmitt M. uPA and PAI-1 as biomarkers in breast cancer: validated for clinical use in level-of-evidence-1 studies. Breast Cancer Res. 2014, 16, 428. 10.1186/s13058-014-0428-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centis F.; et al. p185 HER2/neu epitope mapping with murine monoclonal antibodies. Hybridoma 1992, 11, 267–276. 10.1089/hyb.1992.11.267. [DOI] [PubMed] [Google Scholar]
- Tagliabue E.; et al. Selection of monoclonal antibodies which induce internalization and phosphorylation of p185HER2 and growth inhibition of cells with Her2/neu gene amplification. Int. J. Cancer 1991, 47, 933–937. 10.1002/ijc.2910470625. [DOI] [PubMed] [Google Scholar]
- Bano F.; et al. Toward multiprotein Nanoarrays using nanografting and DNA directed immobilization of proteins. Nano Lett. 2009, 9, 2614–2618. 10.1021/nl9008869. [DOI] [PubMed] [Google Scholar]
- Carter P.; et al. Humanization of an anti-p185her2 antibody for human cancer-therapy. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 4285–4289. 10.1073/pnas.89.10.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M.; Amro N. A.; Chow C. S.; Liu G. Y. Production of Nanostructures of DNA on Surfaces. Nano Lett. 2002, 2, 863–867. 10.1021/nl025626x. [DOI] [Google Scholar]
- Niemeyer C. M.; Boldt L.; Ceyhan B.; Blohm D. DNA-Directed immobilization: efficient, reversible, and site-selective surface binding of proteins by means of covalent DNA-streptavidin conjugates. Anal. Biochem. 1999, 268, 54–63. 10.1006/abio.1998.3017. [DOI] [PubMed] [Google Scholar]
- Prime K.; Whitesides G. Self-Assembled Organic Monolayers: Model Systems for Studying Adsorption of Proteins at Surfaces. Science 1991, 252, 1164–1167. 10.1126/science.252.5009.1164. [DOI] [PubMed] [Google Scholar]
- Pale-Grosdemange C.; Simon E. S.; Prime K. L.; Whitesides G. M. Formation of self assembled monolayer by chemisorption of derivatives of oligo(ethylene glycol) of structure HS(CH2)11(OCH2CH2)mOH on gold. J. Am. Chem. Soc. 1991, 113, 12–20. 10.1021/ja00001a002. [DOI] [Google Scholar]
- Solano I.; et al. Spectroscopic Ellipsometry meets AFM nanolithography: about hydration of bio-inert oligo(ethylene glycol)-terminated Self Assembled Monolayers on gold. Phys. Chem. Chem. Phys. 2015, 17, 28774. 10.1039/C5CP04028K. [DOI] [PubMed] [Google Scholar]
- Nkoua Ngavouka M. D.; Bosco A.; Casalis L.; Parisse P. Determination of Average Internucleotide Distance in Variable Density ssDNA Nanobrushes in the Presence of Different Cations Species. Macromolecules 2014, 47, 8748. 10.1021/ma501712a. [DOI] [Google Scholar]
- Mirmomtaz E.; et al. Quantitative study of the effect of coverage on the hybridization efficiency of surface-bound DNA nanostructures. Nano Lett. 2008, 8, 4134–4139. 10.1021/nl802722k. [DOI] [PubMed] [Google Scholar]
- Copeland R. A.Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis, 2nd ed.; Wiley-VCH, 2000; pp 76–108. [Google Scholar]
- Zou T.; et al. Nanobody-functionalized PEG-b-PCL polymersomes and their targeting study. J. Biotechnol. 2015, 214, 147–155. 10.1016/j.jbiotec.2015.09.034. [DOI] [PubMed] [Google Scholar]
- Monegal A.; et al. Immunological applications of single- domain llama recombinant antibodies isolated from a naive library. Protein Eng., Des. Sel. 2009, 22, 273–280. 10.1093/protein/gzp002. [DOI] [PubMed] [Google Scholar]
- Djender S.; et al. Bacterial cytoplasm as an effective cell compartment for producing functional VHH-based affinity reagents and Camelidae IgG-like recombinant antibodies. Microb. Cell Fact. 2014, 13, 140. 10.1186/s12934-014-0140-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo L. N.; et al. Highly sensitive detection of HER2 extracellular domain (ECD) in the serum of breast cancer patients by piezoelectric microcantilevers (PEMS). Anal. Chem. 2011, 83, 3392–3397. 10.1021/ac103301r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam L.; McAndrew N.; Yee M.; Fu T.; Tchou G. J.; Zhang H. Challenges in the clinical utility of the serum test for HER2 ECD. Biochim. Biophys. Acta, Rev. Cancer 2012, 1826, 199–208. 10.1016/j.bbcan.2012.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin M. C.; et al. Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell 2004, 5, 317–328. 10.1016/S1535-6108(04)00083-2. [DOI] [PubMed] [Google Scholar]
- Soler M. A.; de Marco A.; Fortuna S. Predicting the yields of engineered nanobodies by means of molecular dynamics simulations and docking. Sci. Rep. 2016, 6, 34869. 10.1038/srep34869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D.; et al. Label-Free and high-sensitive detection of Salmonella using a Surface Plasmon Resonance DNA-based biosensor. J. Biotechnol. 2012, 160, 123–128. 10.1016/j.jbiotec.2012.03.024. [DOI] [PubMed] [Google Scholar]
- Gupta O.; Loos K.; Korniakov A.; Spagnoli C.; Cowman M.; Ulman A. Facile route to ultraflat SAM-protected gold surfaces by “amphiphile splitting. Angew. Chem., Int. Ed. 2004, 43, 520–523. 10.1002/anie.200352249. [DOI] [PubMed] [Google Scholar]
- Liu M.; Amro N. A.; Liu G. Nanografting for surface Physical Chemistry. Annu. Rev. Phys. Chem. 2008, 59, 367–386. 10.1146/annurev.physchem.58.032806.104542. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.